

Abstract
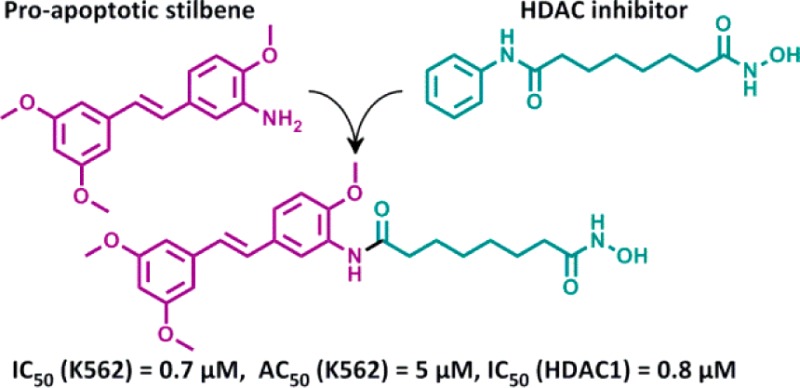
Given our interest in finding potential antitumor agents and in view of the multifactorial mechanistic nature of cancer, in the present work, taking advantage of the multifunctional ligands approach, new chimeric molecules were designed and synthesized by combining in single chemical entities structural features of SAHA, targeting histone deacetylases (HDACs), with substituted stilbene or terphenyl derivatives previously obtained by us and endowed with antiproliferative and pro-apoptotic activity. The new chimeric derivatives were characterized with respect to their cytotoxic activity and their effects on cell cycle progression on different tumor cell lines, as well as their HDACs inhibition. Among the other, trans-6 showed the most interesting biological profile, as it exhibited a strong pro-apoptotic activity in tumor cell lines in comparison with both of its parent compounds and a marked HDAC inhibition.
Keywords: Multifunctional ligands, chimeric compound, stilbene, HDAC inhibition, antiproliferative activity
To address the biological complexity of cancer1 and to develop adequate therapeutic tools, an interesting and fruitful strategy is provided by the development of “multiple ligands”, rationally designed single chemical entities able to modulate multiple altered pathways, overcoming problems like different pharmacokinetics and poor compliance.2−4 In the present report, we applied this paradigm to develop novel multifunctional ligands able to interfere with different molecular pathways involved in neoplastic diseases, specifically targeting some control mechanisms of epigenetics5,6 and cell cycle progression.
Histone deacetylase inhibitors (HDIs)7,8 have emerged as a new class of promising cancer therapeutic agents and their synergistic effects with many other drugs are well documented.9,10 Several HDIs belonging to different chemical classes11 are currently in clinical trials as monotherapy or in combination with chemo- or radiation therapy.12 According to the well-known pharmacophore model for HDIs,13,14 a deacetylase inhibitor should bear a CAP group to interact with the rim of the catalytic tunnel of the enzyme, a connection unit, linking the CAP to a hydrophobic spacer and lying into the tunnel, and an enzyme inhibiting group, able to complex the zinc ion, crucial for the catalysis at the bottom of the tunnel. SAHA (suberoylanilide hydroxamic acid, Vorinostat, Zolinza) (Figure 1A) was the first HDI approved by FDA (in 2006) for the treatment of the rare cancer cutaneous T-cell lymphoma (CTCL).8
Figure 1.

(A) SAHA. (B) Representative terphenyl and stilbene derivatives endowed with pro-apoptotic or differentiating activity.
On the other side, because of our ongoing interest in finding new anticancer agents, we recently synthesized small libraries of stilbenes and terphenyls (privileged structures)15−17 (Figure 1B) able to induce apoptosis and differentiation in leukemia cells, after arresting the cell cycle in G0–G1 phase.18−20 Agents that are able to block and/or kill cancer cells in G0–G1 phase are currently considered of interest because most chemotherapeutic drugs available for the treatment of malignancies act in the S or G2–M phase of the cell cycle, but not in G0–G1, thus allowing a variable percentage of cells in G0–G1 phase to escape from the cytotoxic effects of the therapy.
On the basis of these premises, following the multifunctional ligands approach, we designed and synthesized novel chimeric compounds as potential antitumor agents, by incorporating the linker-Zn2+-binding motif fragment of SAHA into the selected most biologically promising terphenyl 1(19) or stilbenes cis-2 and trans-218 (Figure 1B). A new generation of more powerful HDAC inhibitors could represent an opportunity for a clinical use as single agent, unlike what happens now where the HDAC inhibitors are mainly used in combination with other cytotoxic agents.21
According to Morphy and Rankovic,2,3 the new chimeric compounds 3, 4, cis-6, and trans-6 (Table 1) were “designed in” by linking together the appropriate fragments via a suitable connection unit, while a “merged” approach was applied for chimeric compound 5 in which the selected frameworks were integrated and overlapped in a common structure (Figure S1, Supporting Information). The new derivatives were then characterized with respect to their cytotoxic activity and their effects on cell cycle progression and epigenetics on leukemia Bcr-Abl-expressing K562, monoblastic U937, and breast cancer MCF-7 cell line.
Table 1. IC50 (μM ± SE) and AC50 (μM± SE) of 3, 4, 5, cis-6, and trans-6 and Their Parent Compounds SAHA, 1, cis-2, and trans-2 Evaluated in K562 Cells after 48 h of Treatment.
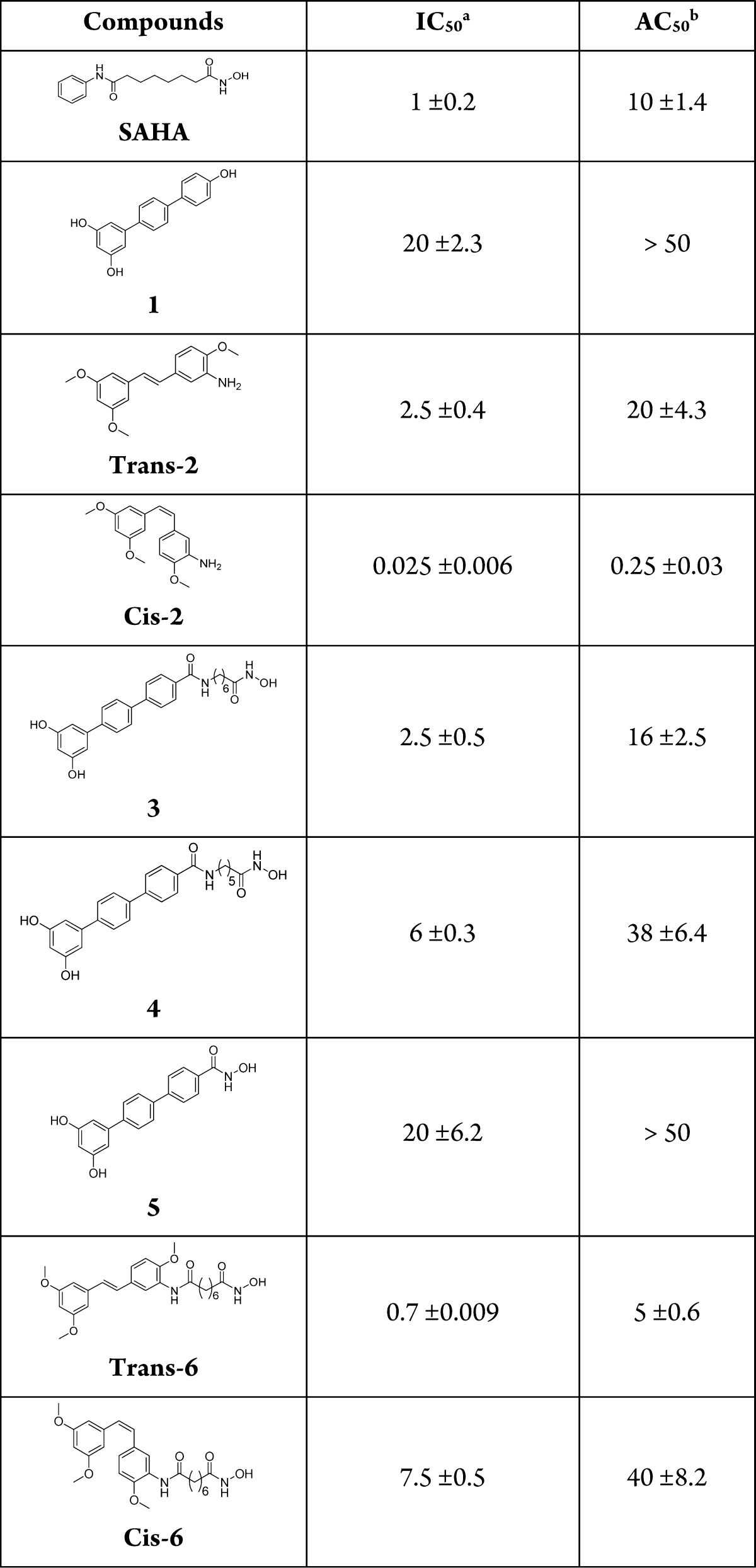
Concentration (μM) able to inhibit 50% cell growth.
Concentration able to induce apoptosis in 50% of cells. All points were tested in triplicate with error bars indicating the standard deviation.
The desired chimeric compounds were synthesized as described in the following. The synthetic strategy for compounds 3 and 4 bearing a terphenyl fragment first consisted of a PyBOP coupling between protected terphenyl acid 7 and the appropriate amino methyl esters 8 and 9. The resulting amides 10 and 11 were deprotected by tetrabutylammonium fluoride (TBAF) giving 12 and 13, which reacted with hydroxylamine hydrochloride in the presence of sodium methoxide as a base to obtain the hydroxamic acid functions, affording the final compounds 3 and 4, respectively (Scheme 1). Similarly, deprotection with TBAF of the terphenyl methyl ester 14 gave the dihydroxy derivative 15, which was treated with hydroxylamine hydrochloride in the same conditions described above to give the desired compound 5 (Scheme 2). Chimeric compounds cis-6 and trans-6 possessing the stilbene fragment were prepared through amidation by use of DCC as coupling reagent between suberic acid monomethyl ester (commercially available) 16 and the aminostilbenes cis-2 and trans-2(18) to obtain amides cis-17 and trans-17, respectively. Conversion of the methyl ester to the hydroxamic acid moiety afforded hybrid compounds cis-6 and trans-6 (Scheme 3). Synthetic strategies for intermediates 7 and 14 (Scheme S1) and 8 and 9 (Scheme S2) are given in the Supporting Information.
Scheme 1.

Scheme 2.

Scheme 3.

Initially, we investigated the antiproliferative and the proapoptotic activities of chimeric derivatives 3, 4, 5, cis-6 and trans-6 in comparison with their parent compounds SAHA, 1, cis-2, and trans-2 on K562 cells (Table 1).
The antiproliferative activity of each compound was evaluated by counting cells with an automatic cell counter; apoptosis was evaluated by annexin V test. The effects of chimeric compounds on K562 cell cycle progression were evaluated by flow cytometry after staining cells with propidium iodide, and they were compared to the effects of the parent compounds alone or in association (Figure S2, Supporting Information). Among the chimeric hydroxamates 3–5 bearing the terphenyl fragment, compound 3 containing the six methylenes motif of SAHA exhibited an increased cytotoxic and proapototic activity with respect to the parent compound 1 (IC50, 2.5 μM vs 20 μM; AC50, 16 μM vs >50 μM, respectively, Table 1), while it was less potent than SAHA. Reducing the alkyl chain to five methylenes as in 4 resulted in a decrease of activity even though still higher if compared to 1, while compound 5 lacking the methylene chain displayed the same activity as parent terphenyl 1. These results suggested that the alkyl hydroxamic chain conferred a better cytotoxic and pro-apototic activity to the terphenyl scaffold.
Unfortunately the cell cycle analysis (Figure S2, Supporting Information) revealed that none of the chimeric compounds 3–5 was able to selectively block cells in G1 phase as 1 or the 1 and SAHA combination as we expected. Interesting results were obtained with trans-6 and cis-6, designed by combining the structural features of SAHA and two stilbenes, trans-2 and cis-2, respectively, previously designed by us and endowed with potent pro-apoptotic activity (Table 1).18 The cytotoxic activity of hydroxamate cis-6 was markedly lower than that of parents cis-2 and SAHA (Table 1). Noteworthy, trans-6 (IC50, 0.7 μM; AC50, 5.0 μM) was more potent as cytotoxic agent than trans-2 (IC50, 2.5 μM; AC50, 20 μM) and SAHA (IC50, 1 μM; AC50, 10 μM). In particular, the ability to induce apoptosis in K562 cells was two times higher than that observed with SAHA (Table 1). The pattern of cell cycle distribution of K562 cells after exposure to trans-6 and cis-6 showed that both compounds behaved similarly to SAHA (block in G1 and G2–M phases) and differently from the parent compounds alone (block in G2–M) or in combination (prevalent block in G1), thus suggesting they could share a mechanism of action similar to that of SAHA (Figure S2, Supporting Information).
Because of its interesting cytotoxic activity on K562 cells, we considered trans-6 the best candidate for further investigations. First, we studied this compound in two different cell lines: the monoblastic U937 and the breast cancer MCF-7 cells. Once again, trans-6 resulted to act similarly to SAHA as it was able to induce a G1- and G2-block in U937 and MCF-7 cells, respectively, with progressive cell death (Figure S3A,B, Supporting Information). To better understand the progressive cell death induction, a cytofluorimetric analysis was performed to discriminate necrosis from apoptosis. As shown in Figure 2, trans-6, similarly to SAHA, triggered an apoptotic death, but with a 2-fold increase. These data corroborated and extended the value of the AC50 shown in Table 1.
Figure 2.

Left: apoptosis/necrosis evaluation, by Annexin V/PI doubling staining by flow cytometric assays on U937 cells treated with 5 μM trans-6 or SAHA (taken as positive control) for 48 h. All points were tested in triplicate with error bars indicating the standard deviation. Right: Western blot for caspase 8 after a stimulation of 24 h for U937 cells and 48 h for MCF-7 cells; compounds were used at concentration of 5 μM. ImageJ was used to quantify protein expression levels. The ERKs signal accounts for equal loading.
To test HDAC inhibitory potential of all new chimeric compounds, enzymatic assays were performed against human recombinant (hr) enzymes (Table 2 and Figure S4A, Supporting Information). Among the terphenyl derivatives, the compound 4, containing the five methylene chain, although exhibited an interesting inhibitory activity against hrHDAC1 (IC50, 0.9 μM), was completely inactive on hrHDAC4 as 3 and 5, demonstrating that the terphenyl scaffold is not a suitable moiety for the anti-HDAC activity. At the same time the stilbene hydroxamate trans-6 and its cis isoform, cis-6, resulted in being strong HDAC1 inhibitors (IC50, 0.8 and 2.1 μM, respectively), and their effect was therefore tested on HDAC4 and 6 (Table 2 and Figure S4B, Supporting Information). Both compounds at 5 μM were also able to inhibit these two enzymes better than SAHA showing good IC50 values (IC50, 9.8 and 7.6 μM for HDAC4 and 1.3 and 1.6 μM for HDAC6, respectively). In particular, trans-6 privileged HDAC1 inhibition. However, cis-6 inhibited all tested enzymes at comparable levels (Figure S4, Supporting Information).
Table 2. Human Recombinant HDAC1 Inhibitory Activity of Compounds 3–5, Compared with SAHA and Human Recombinant HDAC4 and HDAC6 Inhibitory Activity of trans-6, cis-6, and SAHA.
| inhibition at 5 μM (% ± SE)a |
IC50 (μM)
(95% confidence interval) |
|||||
|---|---|---|---|---|---|---|
| compounds | hrHDAC1 | hrHDAC4 | hrHDAC6 | hrHDAC1 | hrHDAC4 | hrHDAC6 |
| SAHA | 68.9 ± 1.73 | 21.5 ± 0.25 | 40.7 ± 0.84 | 0.6 (0.313–3,224) | 7.8 (5.88–12.06) | 5.6 (5.31–7.45) |
| 3 | 20.4 ± 2.12 | 0 | n.d.b | 15.7 (7.675–32.13) | n.d.b | n.d.b |
| 4 | 62.1 ± 1.87 | 0 | n.d.b | 0.9 (0.6941–1.088) | n.d.b | n.d.b |
| 5 | 33.1 ± 2.86 | 0 | n.d.b | 8.9 (6.496–12.34) | n.d.b | n.d.b |
| trans-6 | 91.7 ± 1.19 | 39.1 ± 0.78 | 50.2 ± 1.40 | 0.8 (0.493–1.163) | 9.8 (6.24–15.54) | 1.3 (0.437–3.851) |
| cis-6 | 88.3 ± 2.0 | 68.1 ± 1.3 | 74.7 ± 2.0 | 2.1 (1.207- 3.614) | 7.6 (4.37–13.34) | 1.6 (0.512–5.049) |
All points were tested in triplicate with error bars indicating the standard deviation.
Not determined.
Given the above results, again we considered trans-6 the best candidate for further investigations. According to its HDAC inhibitory action, trans-6 showed the ability to induce a high histone hyper-acetylation in both U937 and MCF-7 cell lines (Figure 3). Western blotting analyses showed a hyper-acetylation of histone H3 in lysines 9 and 14 (H3K9 and 14ac) and of histone H4 in lysine 16 (H4K16ac). Remarkably, the quantification of the signals showed a 6-fold against a 4-fold increase of acetylation of histone H3 for trans-6 compared to its parent compound SAHA, respectively. Taken together, these results demonstrated that trans-6 was able to increase acetylation of lysine residues on the N-terminal histones tails of H3 and H4. Furthermore, trans-6 was also able to trigger a repair pathway, causing the acetylation in lysine 382 of p53 (p53K382ac) and up-regulating the cell-cycle inhibitor p21waf1/cip1 (Figure 3). As HDAC6 assay showed, trans-6, as a HDAC6 inhibitor, induced a clear increment of the acetylation level of the α-tubulin (Figure 3).
Figure 3.

Western blot analyses carried out for the indicated targets in U937 and MCF-7 cells after 24 h of treatment. ImageJ was used to quantify protein expression. Histone H1, H4, and ERKs indicate equal loading. SAHA and trans-6 were used at concentration of 5 μM.
To better address the anticancer activity of trans-6, proliferation and migration analysis were also performed in MCF-7 cells in real-time mode. The inhibitory effects of trans-6 on the proliferation were showed within the 36 h of treatment and were already evident in the early phases after 10 h (Figure 4). This trend was highlighted by the doubling time analysis, which showed a strong decrease of this parameter both for trans-6 and SAHA within the interval 10–18 h (Figure 3A). Migration analyses showed a clear ability of trans-6 to inhibit the tumor cell migration. The effects were more evident if represented in slope values displaying a decrease of 25% within 24 h (Figure 4B).
Figure 4.
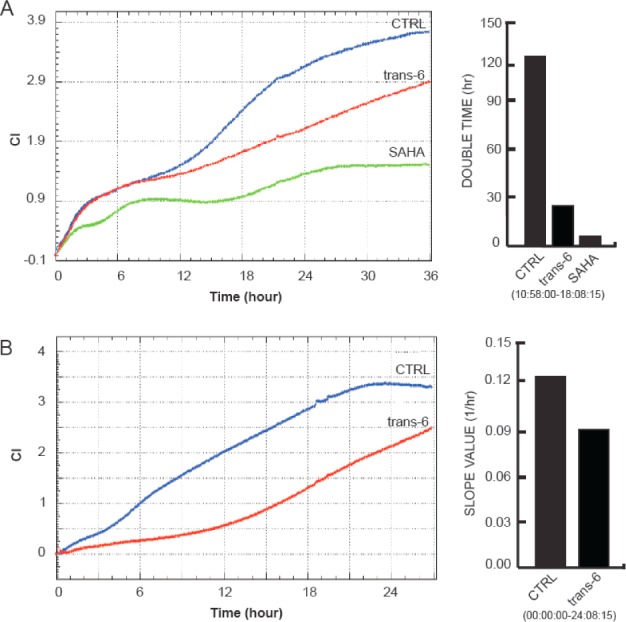
(A) Proliferation analysis in real time of MCF-7 cells treated with 5 μM SAHA or trans-6 as CI (cell index) vs time (left) within 36 h and as doubling time (right) within the interval 10–18 h. (B) Migration analysis in real-time of MCF-7 cells treated with 5 μM trans-6 as CI vs time (left) within 27 h and as slope within 24 h.
On the basis of these results, it appears that the chimeric hydroxamates bearing the terphenyl motif did not enhance the biological activity of the parent compounds, losing the peculiar block in G1 phase and showing a not significant HDAC inhibitory profile. However, the stilbene structure proved to be a valid scaffold for the design of strong HDAC inhibitors. Of interest, the trans configuration of the stilbene architecture seems to be an important feature in conferring a slight selectivity toward the different isoforms of HDACs.
In continuation of our research for innovative antitumor lead candidates, we designed and synthesized some chimeric compounds following the multifunctional ligands approach. Even though the new molecules did not show a pharmacologically chimeric behavior, trans-6, obtained combining the structural features of SAHA and a stilbene derivative previously designed by us, was able to induce a stronger apoptosis in K562, U937 and MCF-7 cells than both the parent compounds. Moreover, it showed a marked HDAC inhibitory action and a clear ability to inhibit the tumor cell migration. Our strategy contributed to explore the chemical space around SAHA, proving the stilbene structure as a valuable CAP group in HDAC pharmacophore. Therefore, trans-6 could be considered a suitable lead structure to develop new agents endowed with a promising anticancer potential. Noteworthy, from a medicinal chemistry point of view, the stilbene scaffold is a privileged structure in which the biological relevance meets the synthetic accessibility, allowing to rapidly obtain variously substituted analogues, making the follow-up studies of the identified hits more efficient.
Experimental Methods
Chemistry
General Chemical Methods
Reaction progress was monitored by TLC on precoated silica gel plates (Kieselgel 60 F254, Merck) and visualized by UV254 light; hydroxamates were viewed by staining with FeCl3 5% aqueous solution. Flash column chromatography was performed on silica gel (particle size 40–63 μM, Merck). When needed, silica was demetalled by suspending and standing overnight in concentrated HCl, filtered and washed several times with Et2O until free of chloride ions, and dried for 48 h at 120 °C. All solvents were distilled prior to use. All reagents were obtained from commercial sources and used without further purification. Unless otherwise stated, all reactions were carried out under an inert atmosphere. Compounds were named relying on the naming algorithm developed by CambridgeSoft Corporation and used in Chem-BioDraw Ultra 11.0. 1H NMR and 13C NMR spectra were recorded on Varian Gemini at 400 and 100 MHz, respectively. Chemical shifts (δH) are reported relative to TMS as internal standard. Mass spectrum was recorded on a V.G. 7070E spectrometer or on a Waters ZQ 4000 apparatus operating in electrospray (ES) mode. Purity of compounds was determined by elemental analyses; purity for all the tested compounds was ≥95% (see Supporting Information).
General Procedure for Chimeric Compounds 3–5, cis-6, and trans-6
To cooled solutions of the appropriate methyl esters 12, 13, 19, cis-20, and trans-20 (1 equiv) in MeOH/THF 2/1 (5 mL), hydroxylamine hydrochloride NH2OH·HCl (10 equiv) and sodium methylate solution 30% in MeOH (12.4 equiv) were added. The reaction mixture was stirred for 3 to 24 h at room temperature then cooled in an ice bath and acidified with 6 N HCl to pH 4. Water was added to dissolve the salt, and the mixture was concentrated in vacuo to remove MeOH/THF. The aqueous phase was extracted with EtOAc (3 × 10 mL) and purified by flash chromatography on demetalled silica gel.
(trans)-N1-(5-(3,5-Dimethoxystyryl)-2-methoxyphenyl)-N8-hydroxyoctanediamide (trans-6)
Derivative trans-17 (0.17 g, 0.37 mmol) was allowed to react according to the general procedure. The reaction mixture was stirred for 3 h at room temperature. After the workup, the precipitated product was filtered and washed with Et2O to yield trans-6 (0.11 g, yield 64%) as white powder. 1H NMR (400 MHz, CD3OD) δ 1.38–1.39 (m, 4H), 1.60–1.64 (m, 2H), 1.70 (t, J = 7.2, 2H), 2.06–2.10 (m, 2H), 2.41–2.44 (m, 2H), 3.78 (s, 6H), 3.87 (s, 3H), 6.34–6.35 (m, 1H), 6.65 (d, J = 2.4 Hz, 2H), 6.94–6.98 (m, 2H), 7.02–7.06 (m, 1H), 7.24–7.25 (m, 1H), 8.18 (d, J =1.6 Hz, 1H) ppm; 13C NMR (100 MHz, CD3OD) δ 26.6, 26.7, 29.8, 33.7, 37.7, 55.7, 56.4, 100.6, 105.3, 111.9, 121.1, 124.9, 128.2, 128.4, 129.7, 131.3, 140.9, 141.0, 151.3, 162.5, 174.7 ppm; MS (ES) m/z 479 [M + Na]+.
Acknowledgments
This work was supported by a PRIN2009 and PRIN2012 Grant from MiUR, Italy; EU: the Blueprint (contract no. 282510), Epigenomics Flagship Project EPIGEN (MIUR-CNR); and the Italian Association for Cancer Research (AIRC no. 11812).
Supporting Information Available
Spectroscopic data for final compounds 3–5 and cis-6, and synthesis and characterization of compounds 7–15 and cis- and trans-17, elemental analyses, detailed biological protocols; Schemes S1 and S2, Table S1, and Figures S1–S4. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Supplementary Material
References
- Hanahan D.; Weinberg R. A. Hallmarks of cancer: the next generation. Cell 2011, 144, 646–674. [DOI] [PubMed] [Google Scholar]
- Morphy R.; Kay C.; Rankovic Z. From magic bullets to designed multiple ligands. Drug Discovery Today 2004, 9, 641–651. [DOI] [PubMed] [Google Scholar]
- Morphy R.; Rankovic Z. Designing multiple ligands: medicinal chemistry strategies and challenges. Curr. Pharm. Des. 2009, 15, 587–600. [DOI] [PubMed] [Google Scholar]
- Peters J. U. Polypharmacology: foe or friend?. J. Med. Chem. 2013, 56, 8955–8971. [DOI] [PubMed] [Google Scholar]
- Nebbioso A.; Pereira R.; Khanwalkar H.; Matarese F.; Garcìa-Rodrìguez J.; Miceli M.; Logie C.; Kedinger V.; Ferrara F.; Stunnenberg H. G.; De Lera A. R.; Gronemeyer H.; Altucci L. Death receptor pathway activation and increase of ROS production by the triple epigenetic inhibitor UVI5008. Mol. Cancer Ther. 2011, 10, 2394–2404. [DOI] [PubMed] [Google Scholar]
- Conte M.; Altucci L. Molecular pathways: the complexity of the epigenome in cancer and recent clinical advances. Clin. Cancer Res. 2012, 18, 5526–5534. [DOI] [PubMed] [Google Scholar]
- Barneda-Zahonero B.; Parra M. Histone deacetylases and cancer. Mol. Oncol. 2012, 6, 579–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ververis K.; Hiong A.; Karagiannis T. C.; Licciardi P. V. Histone deacetylase inhibitors (HDACIs): multitargeted anticancer agents. Biol.: Targets Ther. 2013, 7, 47–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carew J. S.; Giles F. J.; Nawrocki S. T. Histone deacetylase inhibitors: mechanisms of cell death and promise in combination cancer therapy. Cancer Lett. 2008, 269, 7–17. [DOI] [PubMed] [Google Scholar]
- Ai T.; Cui H.; Chen L. Multi-targeted histone deacetylase inhibitors in cancer therapy. Curr. Med. Chem. 2012, 19, 475–487. [DOI] [PubMed] [Google Scholar]
- Carafa V.; Miceli M.; Altucci L.; Nebbioso A. Histone deacetylase inhibitors: a patent review (2009–2011). Expert Opin. Ther. Pat. 2013, 23, 1–17. [DOI] [PubMed] [Google Scholar]
- Benedetti R.; Conte M.; Altucci L. Targeting HDACs in diseases: where are we?. Antioxid. Redox Signaling 2014, 10.1089/ars.2013.5776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mai A.; Massa S.; Rotili D.; Cerbara I.; Valente S.; Pezzi R.; Simeoni S.; Ragno R. Histone deacetylation in epigenetics: an attractive target for anticancer therapy. Med. Res. Rev. 2005, 25, 261–309. [DOI] [PubMed] [Google Scholar]
- Miller T. A.; Witter D. J.; Belvedere S. Histone deacetylase inhibitors. J. Med. Chem. 2003, 46, 5097–5116. [DOI] [PubMed] [Google Scholar]
- Liu J. K. Natural terphenyls: developments since 1877. Chem. Rev. 2006, 106, 2209–2223. [DOI] [PubMed] [Google Scholar]
- Yin H.; Hamilton A. D. Strategies for targeting protein–protein interactions with synthetic agents. Angew. Chem., Int. Ed. Engl. 2005, 44, 4130–4163. [DOI] [PubMed] [Google Scholar]
- Davis J. M.; Tsou L. K.; Hamilton A. D. Synthetic non-peptide mimetics of alpha-helices. Chem. Soc. Rev. 2007, 36, 326–334. [DOI] [PubMed] [Google Scholar]
- Roberti M.; Pizzirani D.; Simoni D.; Rondanin R.; Baruchello R.; Bonora C.; Buscemi F.; Grimaudo S.; Tolomeo M. Synthesis and biological evaluation of resveratrol and analogues as apoptosis-inducing agents. J. Med. Chem. 2003, 46, 3546–3554. [DOI] [PubMed] [Google Scholar]
- Roberti M.; Pizzirani D.; Recanatini M.; Simoni D.; Grimaudo S.; Di Cristina A.; Abbadessa V.; Gebbia N.; Tolomeo M. Identification of a terphenyl derivative that blocks the cell cycle in the G0-G1 phase and induces differentiation in leukemia cells. J. Med. Chem. 2006, 49, 3012–3018. [DOI] [PubMed] [Google Scholar]
- Pizzirani D.; Roberti M.; Cavalli A.; Grimaudo S.; Di Cristina A.; Pipitone R. M.; Gebbia N.; Tolomeo M.; Recanatini M. Antiproliferative agents that interfere with the cell cycle at the G1 → S transition: further development and characterization of a small library of stilbene-derived compounds. ChemMedChem 2008, 3, 345–355. [DOI] [PubMed] [Google Scholar]
- Taddei M.; Cini E.; Giannotti L.; Giannini G.; Battistuzzi G.; Vignola D.; Vesci L.; Cabri W. Lactam based 7-amino suberoylamide hydroxamic acids as potent HDAC inhibitors. Bioorg. Med. Chem. Lett. 2014, 24, 61–64. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.