

Abstract
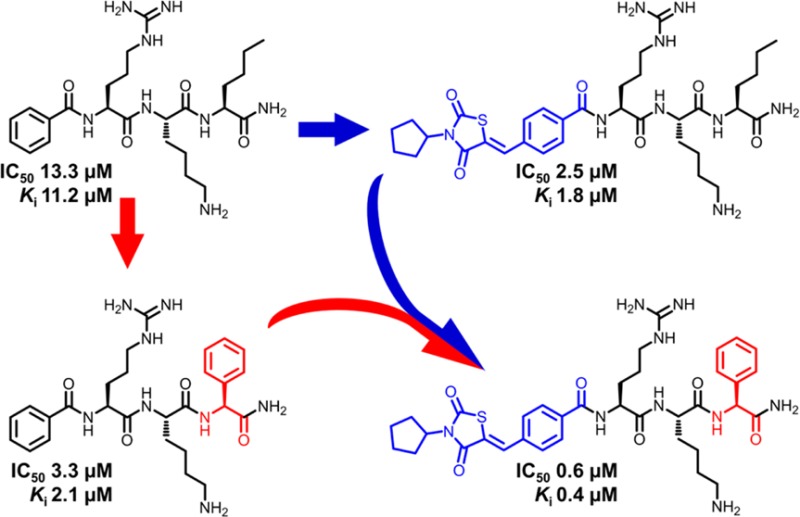
Dengue virus protease is a promising target for the development of antiviral drugs. We describe here a two-step rational optimization that led to the discovery of the potent inhibitor 35 with nanomolar binding affinity at dengue protease serotype 2 (IC50 = 0.6 μM, Ki = 0.4 μM). First, a large number of natural and non-natural amino acids were screened at the C-terminal position of the previously reported, canonical peptide sequence (Cap-Arg-Lys-Nle-NH2). Compared to the reference compound 1 (Bz-Arg-Lys-Nle-NH2, IC50 = 13.3 μM), a 4-fold higher inhibitory potential was observed with the incorporation of a C-terminal phenylglycine (compound 9, IC50 = 3.3 μM). Second, we applied fragment merging of 9 with the previously reported thiazolidinedione peptide hybrid 33 (IC50 = 2.5 μM). This approach led to the fusion of two inhibitor-fragments with micromolar affinity into a 20-fold more potent, competitive inhibitor of dengue protease.
Keywords: Dengue virus, protease inhibitor, peptide, fragment merging
Dengue virus (DENV), with its four common serotypes (DENV 1–4), is considered the most important disease-causing arbovirus in tropical and subtropical regions and a major public health concern. A recent study estimates a global infection burden of 390 million cases in 2010, a more than 3-fold higher number than reported previously by the World Health Organization.1,2
Increasing prevalence of dengue infections worldwide has been associated with the geographical spread of Aedes mosquitoes, the primary transmitting vector, probably as a result of global warming and climate changes.3 Currently, vaccines or antiviral agents against DENV remain unavailable.
Viral proteases are extremely relevant for the development of antiviral drugs.4 Highly successful examples are the hepatitis C virus (HCV) NS3 protease inhibitors, such as boceprevir and telaprevir,5 and the HIV protease inhibitors.6 DENV protease is regarded as a similarly promising target for drug discovery efforts against dengue virus infections.7 The enzyme is critical for the viral replication cycle. It posttranslationally cleaves the viral polyprotein, encoded by a single stranded RNA, into three structural and seven nonstructural (NS) proteins. The proteolytically competent NS3-NS2B complex consists of the NS3 serine protease domain and the hydrophilic core sequence of NS2B as cofactor.8,9 Published inhibitors against DENV protease range from small molecule nonpeptidic inhibitors,10−13 so far not capable of achieving sufficient target inhibition, to substrate-mimicking peptidic inhibitors.14−18 The latter class benefits from improved molecular recognition because of its similarity to the natural substrate, which offers higher selectivity and activity, reaching the low micromolar range and even the nanomolar range when combined with an electrophilic warhead.18 However, the promising in vitro binding affinities come at the expense of pharmacokinetic properties, such as membrane permeability and metabolic stability, which is challenged by the increased polarity through the incorporation of highly basic residues and the peptidic character of the compounds.19 The first reported small peptidic inhibitors against DENV protease were N-benzoyl-capped di-, tri-, and tetrapeptides linked to C-terminal electrophilic warheads that bind covalently to the catalytic serine. The most active derivatives incorporated trifluoromethyl ketone and boronic acid as electrophiles, and reached nanomolar binding affinities of 850 and 43 nM, respectively.17,18 The substrate-based peptide sequence used in these inhibitors relied on the amino acid preference for the subsites S1–S4 in the protease binding cavity to achieve high affinity. These were two basic residues, particularly two arginines, as P1 and P2,20 lysine as P3, and norleucine as P4.21 Against this background, we investigated the activity of different retro, retro-inverse, semiretro-inverse, and nonretro dipeptides and tripeptides.22 The retro tetrapeptide Arg-Arg-Lys-Nle-NH2 was found to be less active than the tripeptide Arg-Lys-Nle-NH2 with only one arginine residue. Considering the added value on the pharmacokinetic profile of having one basic residue less in the sequence, we proceeded with the Arg-Lys-Nle sequence as the basis for our following work. Alternative arrangements or modifications of the basic amino acids resulted in lower activity. Our group reported two generations of peptide-hybrid dengue protease inhibitors. In these molecules, an N-terminal capping group is combined to a short peptide sequence with the aim to extend the interactions toward additional binding subsites. This cap was altered from an arylcyanoacrylamide in the first generation22 to N-substituted 5-arylidenethiazolidinone (thiazolidinediones and rhodanines) in the second generation.23 The modification was associated with increased membrane permeability, in vitro binding affinity, and in cellulo antiviral activity.
Docking studies of the peptide-hybrid inhibitors conducted on the crystal structural of DENV 3 serotype suggested that the lysine and arginine residues bind to the S1 and S2 pockets, respectively, the C-terminal norleucine to S1′ and the N-terminal cap to S3.23 The suggested positioning of the C-terminal norleucine in S1′ stands in contrast to the previously reported substrate preferences of S1′. We therefore decided to perform an extensive investigation of this position, and to explore a large variety of alternative C-terminal residues. A fragment merging strategy was subsequently applied, where the most active and two moderately active sequences were merged with two peptide-hybrid inhibitors with optimized N-capping moiety.
To explore the C-terminal position, a total of 30 N-benzoyl-capped tripeptides with the sequence Bz-Arg-Lys-X-NH2, where X is the variable C-terminal residue, were synthesized. Benzoate was initially chosen as a small, commercially available N-terminal cap to allow straightforward synthesis. All target sequences were evaluated by homogeneous fluorimetric assays for their inhibitory potential against DENV and WNV proteases and thrombin. Additionally, a HPLC-based DENV protease assay was performed for all sequences to confirm the results and exclude false-positive values.24 The reference inhibitor with a C-terminal norleucine 1 was included for comparison.
The selection of alternative C-terminal residues focused mostly on hydrophobic amino acids, in addition to the substrate-mimicking serine and the structurally related threonine. One reason is that we expected hydrophobic groups to balance the otherwise relatively pronounced hydrophilicity of the molecule. The second reason is related to the previously reported docking structure, which suggested that norleucine can be positioned in the S1′ binding pocket.23 Prime side substrate specificity has been reported to favor small polar residues, particularly serine at P1′,21,25 with hydrophobic amino acids lacking flexibility being not advantageous for proteolytic activity.
All tripeptides in Table 1 show lower percentage inhibition to WNV protease in comparison to DENV protease. Furthermore, all sequences have only negligible activity against thrombin. The selectivity of the compounds between the two closely related viral targets, and particularly between the dengue protease and thrombin, indicates a highly specific target–ligand recognition and low promiscuity.
Table 1. Inhibitory Activity of N-Benzoyl-Capped Tripeptides with Various C-Terminal Amino Acids, Possessing the General Structure (Bz-Arg-Lys-X-NH2) against DENV and WNV Proteases and Thrombina.
| DENV |
WNV | THR | |||
|---|---|---|---|---|---|
| X | %b | IC50 (μM)c | %d | %e | |
| 1 | Nle | 68.122 | 13.3(23) | 7.722 | 5.522 |
| 2 | Ser | 57.9 | 30.1 | 21.9 | 6.2 |
| 3 | Phe | 80.7 | 11.9 | 18.2 | 7.2 |
| 4 | Ala | 46.6 | 35.6 | 26.0 | 7.2 |
| 5 | Gly | 70.4 | 21.0 | 44.6 | 3.5 |
| 6 | Met | 45.0 | 47.5 | 11.8 | 6.2 |
| 7 | Nrv | 62.0 | 25.2 | 8.5 | n.i. |
| 8 | Tle | 18.8 | 137.2 | 11.7 | n.i. |
| 9 | Phg | 95.0 | 3.3 | 39.3 | n.i. |
| 10 | Tic | 41.6 | 65.8 | 17.8 | n.i. |
| 11 | d-Nle | 72.7 | 14.1 | 16.1 | 4.9 |
| 12 | 2-Abz | 62.0 | 15.8 | 8.7 | 6.4 |
| 13 | Trp | 74.1 | 17.6 | 32.8 | n.i. |
| 14 | Tyr | 82.3 | 7.4 | 12.9 | 7.9 |
| 15 | β-Ala | 32.9 | 109.3 | 19.8 | n.i. |
| 16 | Aib | 55.0 | 46.7 | 13.7 | n.i. |
| 17 | Pro | 20.0 | 191.9 | 3.6 | n.i. |
| 18 | 3-Abz | 27.0 | 96.6 | 23.0 | 5.6 |
| 19 | 4-Abz | 27.5 | 92.2 | 17.9 | 9.2 |
| 20 | Pra | 72.5 | 17.5 | 14.3 | 5.2 |
| 21 | Pip | 13.7 | 159.2 | 12.1 | n.i. |
| 22 | 3-Pal | 61.3 | 30.1 | 12.5 | n.i. |
| 23 | 2-Pal | 53.0 | 44.3 | 9.8 | n.i. |
| 24 | (4-NO2)Phe | 66.6 | 17.5 | 21.3 | n.i. |
| 25 | (4-CN)Phe | 71.7 | 14.4 | 15.9 | n.i. |
| 26 | Cha | 80.0 | 8.2 | 19.1 | n.i. |
| 27 | Chg | 71.7 | 17.4 | 12.1 | n.i. |
| 28 | Leu | 63.2 | 22.7 | 18.4 | 5.9 |
| 29 | Ile | 56.6 | 28.2 | 14.9 | 4.6 |
| 30 | Thr | 32.1 | 98.6 | 16.5 | n.i. |
| 31 | Val | 60.0 | 28.8 | 18.3 | 4.9 |
n.i. = no inhibition. All measurements were carried out in triplicate. Standard deviations were below 10%.
Percent inhibition of DENV NS2B-NS3 protease serotype 2 (enzyme 100 nM, inhibitor 50 μM, substrate 50 μM, Km = 105 μM).
IC50 values against DENV NS2B-NS3 protease serotype 2 were determined for a substrate concentration of 50 μM.
Percent inhibition of WNV NS2B-NS3 protease (enzyme 150 nM, inhibitor 50 μM, substrate 50 μM, Km = 212 μM).
Percent inhibition of thrombin (enzyme 10 nM, inhibitor 25 μM, substrate 50 μM, Km = 16 μM).
Both d- and l-norleucine sequences showed similar activity, indicating that amino acid configuration plays no role in activity at this position. The phenylglycine-containing sequence 9 was identified as the most active, with 4-fold higher inhibitory potential (IC50 = 3.3 μM, Ki = 2.1 μM) than 1 (IC50 = 13.3 μM, Ki = 11.2 μM). Extending the side chain by one carbon atom in phenylalanine 3 and its substituted derivatives 14, 24, and 25 caused a drop in the inhibitory activity, which was most noticeable in the case of the bioisosteric pyridine analogues of phenylalanine 22 and 23. A moderate decrease in activity was seen for the cyclohexylglycine (27) and 2-amino benzoic acid derivatives (12). The loss of activity was more pronounced in the cases of 3- and 4-amino benzoic acid, 18 and 19, respectively. Branched leucine 28 and isoleucine 29 showed lower inhibition in comparison to linear norleucine, with tert-leucine (8) being least active. Nearly all affinity is lost with cyclic amino acids such as proline 17 (IC50 = 191.9 μM) and pipecolic acid 21 (IC50 = 159.2 μM). Combining the cyclic amino acid to an aromatic ring (10) restored a part of the activity. The peptide sequence incorporating serine 2 had an IC50 of 30.1 μM, and the one with branched threonine 30 was approximately three times less active.
In the context of norleucine modifications as P4 residue, other researchers17 reported equipotent inhibitors in the case of substitutions at P4 with either alanine or phenylalanine, which implied minimal contribution of P4 in the enzyme binding. Inversion of configuration into d-norleucine resulted in a nearly 2-fold lower binding affinity.17 The contradiction of our data with these findings lends further support to the notion that the C-terminal residue does not bind to the S4 pocket.
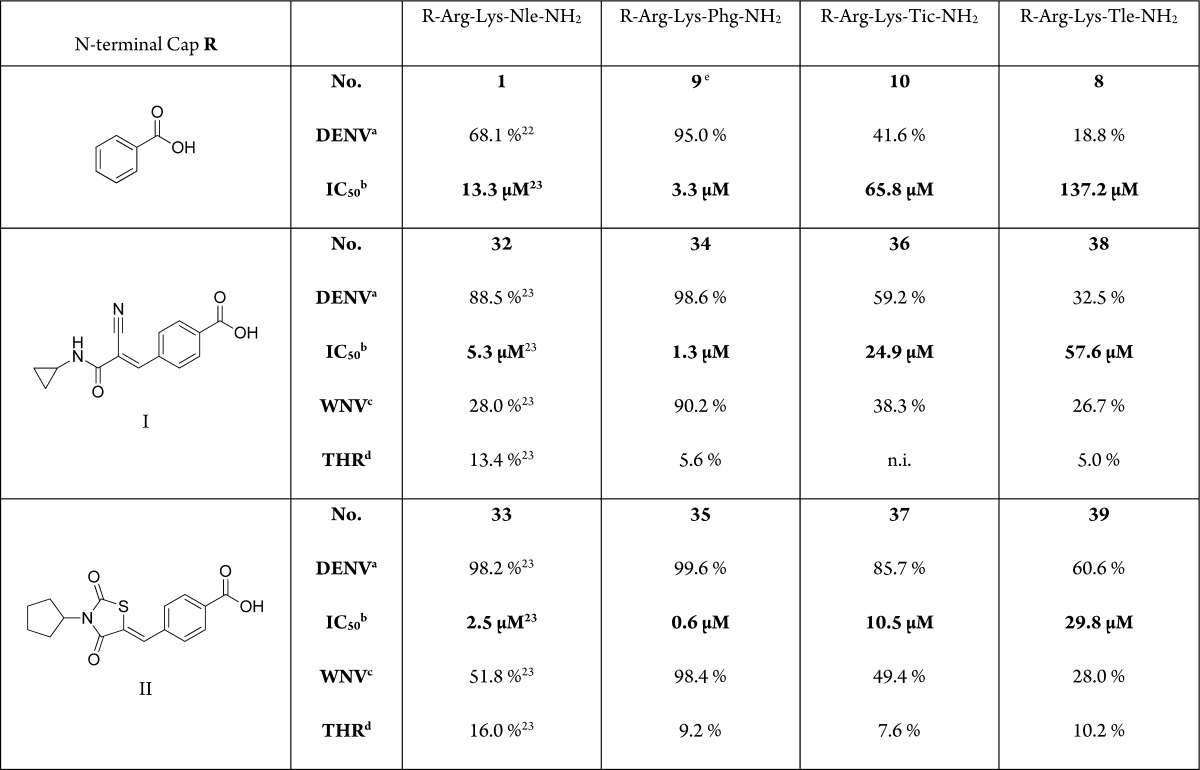
Fragment-based drug design is a tool for drug discovery that witnessed considerable advances in recent years.26 The basic concept is to optimize interactions of smaller building blocks, or fragments, with the target protein and to subsequently integrate these fragments into a single molecular entity that is expected to have a higher binding affinity, which corresponds to the sum of the individual interactions.27 The technique encompasses different strategies: fragment evolution, merging, or linking. Proteases usually possess extended binding sites spanning multiple amino acid-specific subsites and are hence ideally suited for fragment-based drug discovery.28 To apply this approach, we chose three sequences displaying different levels of activity (9, 8, and 10) and merged the C-terminal residues with the N-terminal caps of the optimized peptide hybrids 32 and 33 (Table 2). The overlapping basic residues were not modified.
Table 2. Inhibitory Activity of Peptide Hybrids against DENV and WNV Proteases and Thrombinf.
Percent inhibition of DENV NS2B-NS3 protease serotype 2 (enzyme 100 nM, inhibitor 50 μM, substrate 50 μM, Km = 105 μM).
IC50 values against DENV NS2B-NS3 protease serotype 2 were determined for a substrate concentration of 50 μM.
Percent inhibition of WNV NS2B-NS3 protease (enzyme 150 nM, inhibitor 50 μM, substrate 50 μM, Km = 212 μM).
Percent inhibition of thrombin (enzyme 10 nM, inhibitor 25 μM, substrate 50 μM, Km = 16 μM).
Compound 9Ki at WNV NS2B-NS3 protease: 49 μM, IC50 58 μM. No inhibition of thrombin.
n.i. = no inhibition. All measurements were carried out in triplicate. Standard deviations were below 10%.
In comparison to 1, C-terminal residue optimization from norleucine to phenylglycine provided a 4-fold improvement in the inhibitory potential, while the N-terminal-cap optimization resulted in more than 2-fold improvement with the N-cyclopropyl cyanoacrylamide cap I and 5-fold improvement with the N-cyclopentyl thiazolidinedione cap II. These groups were chosen as the most promising caps characterized in our previous work in terms of potency and selectivity.22,23,29 The thiazolidinedione cap was preferred over the rhodanine cap because the latter is often considered as a fragment with promiscuous binding properties,30 although the generality of this view has been challenged.31
Merging the subpocket-specific fragments, associated with the enhancement in inhibitory potency for our most active peptide hybrids 9 (optimized C-terminus) and 33 (optimized N-terminus), resulted in the 20-fold more potent inhibitor 35 (IC50 = 0.6 μM, Ki = 0.4 μM). This indicates that the generated molecule inherited the most relevant target interactions of the parent sequences. The other merged peptide hybrids displayed a similar tendency with enzymatic inhibitory potential (IC50) being proportional to that of the corresponding N-benzoyl-capped tripeptide and the similarly capped norleucine-containing sequence. The increased affinity is in accordance with the concept of fragment-merging, in that the activity of composite compounds depends on that of the parent molecules or fragments. Using the aprotinin competition assay,32 specific binding of 35 to the DENV protease active site was demonstrated through a concentration-dependent nonlinear decrease of the fluorescence, which was significantly restored following addition of aprotinin (data not shown). The same was shown for our previously reported peptide hybrids.23 Altogether, 35 is the first reported peptidic DENV protease inhibitor without incorporation of highly electrophilic target binding moiety that reaches a binding affinity in the nanomolar range.
Using a homology model of the DENV 2 protease based on the published X-ray structure of the DENV 3 protease in complex with a substrate-based inhibitor (3U1I),33 we performed docking studies to predict the binding mode of our most active compound 35 using AD Vina34 (cf. Supporting Information for details). The proposed binding mode is presented in Figure 1 with the electrostatic potential mapped on the solvent-accessible surface of the protein. The most important interactions between protein and ligand are highlighted. There was a total number of 18 protein residues involved in interactions with the ligand within 3.9 Å, including the catalytic triad (see Table S1 in the Supporting Information). Contrary to our expectations, the phenylglycine of the ligand is placed in the S1 pocket, arginine is in S2, lysine is in the region between S1, S2, and S4, and the N-terminal cap is in the S3 and S4 pockets. Phenylglycine is located in a region where it is able to make a parallel π–π stacking interaction with Tyr215 and a T-shape π–π stacking interaction with Tyr204 of the NS3 domain. Replacing phenylglycine by tert-leucine has a very detrimental effect on the activity. The docking study for 39 showed that tert-leucine occupied the same binding pocket as phenylglycine. Since tert-leucine is not able to participate in π–π stacking interactions and is more bulky, this could explain the significant decrease in the observed activity. Moreover, phenylglycine can form two intermolecular hydrogen bonding interactions with the side-chain of Ser189 and with the backbone of Gly205 as an acceptor and as a donor, respectively. As expected, the arginine residue of the ligand interacts strongly with Asp129 via charge–charge interaction. The side-chain of Lys is close enough to the backbone of the ligand to form an intramolecular hydrogen bond with the oxygen of the benzoyl group in cap II. In addition, the oxygen of the backbone of lysine participates in a hydrogen bond as an acceptor with the Gly207 backbone (S2). The N-terminal cap II interacts with several residues from the cofactor and also with a hydrophobic region formed by Val208 and Val209 from the protease domain.
Figure 1.
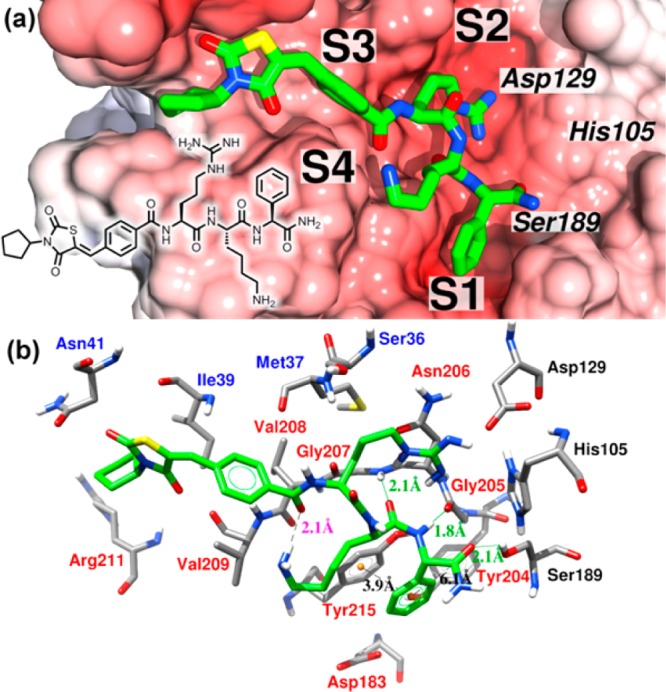
Close-up view of the docked complex. The binding mode of the ligand 35 obtained using AD Vina is shown in green.34 (a) Electrostatic potential calculated using the program APBS with the linear Poisson–Boltzmann equation, contoured at ±11.0 kT/e and mapped on the solvent-accessible surface.35 Negatively charged surface areas are shown in red. The positions of the binding pockets S1, S2, S3, and S4 and the catalytic triad are indicated. The protein is presented in the serine protease standard orientation, i.e., looking into the active site cleft. (b) Detailed view of the main interactions presented in the proposed binding mode. Labels of the NS2B and NS3 residues are shown in blue and red, respectively, labels for the catalytic triad are black, distances of intermolecular hydrogen bonds are labeled in green, ligand intramolecular hydrogen bonds are in magenta, and the centroids of the aromatic rings, involved in π–π stacking interactions, are colored in orange with respective distances labeled in black. The graphic was generated using Chimera.36
Docking experiments on the cyanoacrylamide derivatives presented here indicated that a pose can be adopted in which the nitrile group could react with the catalytic serine to form reactive intermediates and thus inhibit the dengue protease irreversibly (see Figure S2 in the Supporting Information). However, as yet, we did not find experimental evidence for such a binding mode.
In the present work, we extensively investigated and optimized the C-terminal residue of peptide-hybrid inhibitors of dengue protease. Using a fragment merging approach, we combined the activity-enhancing fragments from two peptide-hybrid inhibitors with inhibitory potential in the micromolar range and achieved a significant improvement of activity into the upper nanomolar range. This molecule is the first reported nanomolar active peptide-based DENV protease inhibitor that does not incorporate a highly electrophilic warhead. A competitive and specific binding mode of 35 with the DENV protease binding site was confirmed. Docking studies suggest that π–π stacking and hydrogen bonding interactions with phenylglycine can be responsible for the high affinity of the ligand. The N-terminal cap moiety appears to be anchored at a hydrophobic region formed by cofactor residues and two valines from the protease domain. The applied strategy opens broad perspectives toward the development of highly affine DENV protease inhibitors through the merging of structural fragments that target different protease subsites.
Acknowledgments
We thank Heiko Rudy for measuring ESI high resolution spectra, Lena Weigel for performing LC-MS analytics, and Yasmin El Sherif for technical support. M.A.M.B. appreciates financial support from the German Academic Exchange Service. S.M.V. appreciates financial support by a research fellowship from the National Council for Scientific and Technological Development (CNPq), Brazil. The project was sponsored by the Deutsche Forschungsgemeinschaft, KL-1356/3-1.
Glossary
ABBREVIATIONS
- Abz
aminobenzoic acid
- Aib
2-aminoisobutyric acid
- Cha
cyclohexylalanine
- Chg
cyclohexylglycine
- DENV
dengue virus
- FRET
Förster (fluorescence) resonance energy transfer
- Nle
norleucine
- Nrv
norvaline
- 2-Pal
3-(2-pyridyl)alanine
- 3-Pal
3-(3-pyridyl)alanine
- Pip
pipecolic acid
- Phg
phenylglycine
- Pra
propargylglycine
- Tic
1,2,3,4-tetrahydroisoquinoline-3-carboxylic acid
- Tle
tert-leucine
- THR
thrombin
- WNV
West Nile virus
Supporting Information Available
Chemical reagents and synthesis of peptide hybrids, HRMS, NMR, GSH reactivity and HPLC data for all synthesized compounds, sequence alignment and homology modeling, quality of the homology model and docking procedures. This material is available free of charge via the Internet at http://pubs.acs.org. For details regarding expression and purification of viral proteases, fluorimetric protease assays, IC50, and Ki determination, please refer to our previous publications.
The authors declare no competing financial interest.
Supplementary Material
References
- Bhatt S.; Gething P. W.; Brady O. J.; Messina J. P.; Farlow A. W.; Moyes C. L.; Drake J. M.; Brownstein J. S.; Hoen A. G.; Sankoh O.; Myers M. F.; George D. B.; Jaenisch T.; Wint G. R. W.; Simmons C. P.; Scott T. W.; Farrar J. J.; Hay S. I. The global distribution and burden of dengue. Nature 2013, 4967446504–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitka M. Dengue more prevalent than previously thought. JAMA 2013, 309181882–1882. [DOI] [PubMed] [Google Scholar]
- Bonizzoni M.; Gasperi G.; Chen X.; James A. A. The invasive mosquito species Aedes albopictus: current knowledge and future perspectives. Trends Parasitol. 2013, 299460–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skorenski M.; Sienczyk M. Viral proteases as targets for drug design. Curr. Pharm. Des. 2013, 1961126–53. [DOI] [PubMed] [Google Scholar]
- Au J. S.; Pockros P. J. Novel Therapeutic Approaches for Hepatitis C. Clin. Pharmacol. Ther. 2014, 95178–88. [DOI] [PubMed] [Google Scholar]
- De Clercq E. Anti-HIV drugs: 25 compounds approved within 25 years after the discovery of HIV. Int. J. Antimicrob. Agents 2009, 334307–320. [DOI] [PubMed] [Google Scholar]
- Lescar J.; Luo D.; Xu T.; Sampath A.; Lim S. P.; Canard B.; Vasudevan S. G. Towards the design of antiviral inhibitors against flaviviruses: The case for the multifunctional NS3 protein from Dengue virus as a target. Antiviral Res. 2008, 80294–101. [DOI] [PubMed] [Google Scholar]
- Valle R. P. C.; Falgout B. Mutagenesis of the NS3 protease of Dengue virus type 2. J. Virol. 1998, 721624–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falgout B.; Pethel M.; Zhang Y. M.; Lai C. J. Both nonstructural proteins NS2B and NS3 are required for the proteolytic processing of dengue virus nonstructural proteins. J. Virol. 1991, 6552467–2475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aravapalli S.; Lai H.; Teramoto T.; Alliston K. R.; Lushington G. H.; Ferguson E. L.; Padmanabhan R.; Groutas W. C. Inhibitors of Dengue virus and West Nile virus proteases based on the aminobenzamide scaffold. Bioorg. Med. Chem. 2012, 20134140–4148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai H.; Sridhar Prasad G.; Padmanabhan R. Characterization of 8-hydroxyquinoline derivatives containing aminobenzothiazole as inhibitors of dengue virus type 2 protease in vitro. Antiviral Res. 2013, 97174–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiew K. C.; Dou D.; Teramoto T.; Lai H.; Alliston K. R.; Lushington G. H.; Padmanabhan R.; Groutas W. C. Inhibition of Dengue virus and West Nile virus proteases by click chemistry-derived benz[d]isothiazol-3(2H)-one derivatives. Bioorg. Med. Chem. 2012, 2031213–1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomlinson S. M.; Watowich S. J. Anthracene-based inhibitors of dengue virus NS2B-NS3 protease. Antiviral Res. 2011, 892127–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung D.; Schroder K.; White H.; Fang N. X.; Stoermer M. J.; Abbenante G.; Martin J. L.; Young P. R.; Fairlie D. P. Activity of recombinant Dengue 2 virus NS3 protease in the presence of a truncated NS2B co-factor, small peptide substrates, and inhibitors. J. Biol. Chem. 2001, 2764945762–45771. [DOI] [PubMed] [Google Scholar]
- Prusis P.; Junaid M.; Petrovska R.; Yahorava S.; Yahorau A.; Katzenmeier G.; Lapins M.; Wikberg J. E. S. Design and evaluation of substrate-based octapeptide and non substrate-based tetrapeptide inhibitors of dengue virus NS2B–NS3 proteases. Biochem. Biophys. Res. Commun. 2013, 4344767–772. [DOI] [PubMed] [Google Scholar]
- Schüller A.; Yin Z.; Brian Chia C. S.; Doan D. N. P.; Kim H. K.; Shang L.; Loh T. P.; Hill J.; Vasudevan S. G. Tripeptide inhibitors of dengue and West Nile virus NS2B-NS3 protease. Antiviral Res. 2011, 92196–101. [DOI] [PubMed] [Google Scholar]
- Yin Z.; Patel S. J.; Wang W. L.; Chan W. L.; Ranga Rao K. R.; Wang G.; Ngew X.; Patel V.; Beer D.; Knox J. E.; Ma N. L.; Ehrhardt C.; Lim S. P.; Vasudevan S. G.; Keller T. H. Peptide inhibitors of dengue virus NS3 protease. Part 2: SAR study of tetrapeptide aldehyde inhibitors. Bioorg. Med. Chem. Lett. 2006, 16140–43. [DOI] [PubMed] [Google Scholar]
- Yin Z.; Patel S. J.; Wang W. L.; Wang G.; Chan W. L.; Rao K. R. R.; Alam J.; Jeyaraj D. A.; Ngew X.; Patel V.; Beer D.; Lim S. P.; Vasudevan S. G.; Keller T. H. Peptide inhibitors of dengue virus NS3 protease. Part 1: Warhead. Bioorg. Med. Chem. Lett. 2006, 16136–39. [DOI] [PubMed] [Google Scholar]
- Lim S. P.; Wang Q. Y.; Noble C. G.; Chen Y. L.; Dong H.; Zou B.; Yokokawa F.; Nilar S.; Smith P.; Beer D.; Lescar J.; Shi P. Y. Ten years of Dengue drug discovery: Progress and prospects. Antiviral Res. 2013, 1002500–519. [DOI] [PubMed] [Google Scholar]
- Yusof R.; Clum S.; Wetzel M.; Murthy H. M. K.; Padmanabhan R. Purified NS2B/NS3 serine protease of Dengue virus type 2 exhibits cofactor NS2B dependence for cleavage of substrates with dibasic amino acids in vitro. J. Biol. Chem. 2000, 275149963–9969. [DOI] [PubMed] [Google Scholar]
- Li J.; Lim S. P.; Beer D.; Patel V.; Wen D.; Tumanut C.; Tully D. C.; Williams J. A.; Jiricek J.; Priestle J. P.; Harris J. L.; Vasudevan S. G. Functional profiling of recombinant NS3 proteases from all four serotypes of Dengue virus using tetrapeptide and octapeptide substrate libraries. J. Biol. Chem. 2005, 2803128766–28774. [DOI] [PubMed] [Google Scholar]
- Nitsche C.; Behnam M. A. M.; Steuer C.; Klein C. D. Retro peptide-hybrids as selective inhibitors of the Dengue virus NS2B-NS3 protease. Antiviral Res. 2012, 94172–79. [DOI] [PubMed] [Google Scholar]
- Nitsche C.; Schreier V. N.; Behnam M. A. M.; Kumar A.; Bartenschlager R.; Klein C. D. Thiazolidinone–peptide hybrids as Dengue virus protease inhibitors with antiviral activity in cell culture. J. Med. Chem. 2013, 56218389–8403. [DOI] [PubMed] [Google Scholar]
- Nitsche C.; Klein C.. Fluorimetric and HPLC-Based Dengue Virus Protease Assays Using a FRET Substrate. In Antiviral Methods and Protocols; Gong E. Y., Ed.; Humana Press: New York, 2013; Vol. 1030, pp 221–236. [DOI] [PubMed] [Google Scholar]
- Chambers T. J.; Hahn C. S.; Galler R.; Rice C. M. Flavivirus genome organization, expression, and replication. Annu. Rev. Microbiol. 1990, 441649–688. [DOI] [PubMed] [Google Scholar]
- Joseph-McCarthy D.; Campbell A. J.; Kern G.; Moustakas D. Fragment-based lead discovery and design. J. Chem. Inf. Model. 2014, 543693–704. [DOI] [PubMed] [Google Scholar]
- Hajduk P. J.; Greer J. A decade of fragment-based drug design: strategic advances and lessons learned. Nat. Rev. Drug Discovery 2007, 63211–219. [DOI] [PubMed] [Google Scholar]
- Erlanson D. A. Fragment-based lead discovery: A chemical update. Curr. Opin. Biotechnol. 2006, 176643–652. [DOI] [PubMed] [Google Scholar]
- Nitsche C.; Steuer C.; Klein C. D. Arylcyanoacrylamides as inhibitors of the Dengue and West Nile virus proteases. Bioorg. Med. Chem. 2011, 19247318–7337. [DOI] [PubMed] [Google Scholar]
- Baell J. B.; Holloway G. A. New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J. Med. Chem. 2010, 5372719–2740. [DOI] [PubMed] [Google Scholar]
- Mendgen T.; Steuer C.; Klein C. D. Privileged scaffolds or promiscuous binders: A comparative study on rhodanines and related heterocycles in medicinal chemistry. J. Med. Chem. 2011, 552743–753. [DOI] [PubMed] [Google Scholar]
- Bodenreider C.; Beer D.; Keller T. H.; Sonntag S.; Wen D.; Yap L.; Yau Y. H.; Shochat S. G.; Huang D.; Zhou T.; Caflisch A.; Su X.-C.; Ozawa K.; Otting G.; Vasudevan S. G.; Lescar J.; Lim S. P. A fluorescence quenching assay to discriminate between specific and nonspecific inhibitors of dengue virus protease. Anal. Biochem. 2009, 3952195–204. [DOI] [PubMed] [Google Scholar]
- Noble C. G.; Seh C. C.; Chao A. T.; Shi P. Y. Ligand-bound structures of the Dengue virus protease reveal the active conformation. J. Virol. 2012, 861438–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trott O.; Olson A. J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 312455–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker N. A.; Sept D.; Joseph S.; Holst M. J.; McCammon J. A. Electrostatics of nanosystems: Application to microtubules and the ribosome. Proc. Natl. Acad. Sci. U.S.A. 2001, 981810037–10041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen E. F.; Goddard T. D.; Huang C. C.; Couch G. S.; Greenblatt D. M.; Meng E. C.; Ferrin T. E. UCSF Chimera: A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25131605–1612. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.