

Abstract

Novel sources of antibiotics are needed to address the serious threat of bacterial resistance. Accordingly, we have launched a structure-based drug design program featuring a desmethylation strategy wherein methyl groups have been replaced with hydrogens. Herein we report the total synthesis, molecular modeling, and biological evaluation of 4-desmethyl telithromycin (6), a novel desmethyl analogue of the third-generation ketolide antibiotic telithromycin (2) and our final analogue in this series. While 4-desmethyl telithromycin (6) was found to be equipotent with telithromycin (2) against wild-type bacteria, it was 4-fold less potent against the A2058G mutant. These findings reveal that strategically replacing the C4-methyl group with hydrogen (i.e., desmethylation) did not address this mechanism of resistance. Throughout the desmethyl series, the sequential addition of methyls to the 14-membered macrolactone resulted in improved bioactivity. Molecular modeling methods indicate that changes in conformational flexibility dominate the increased biological activity; moreover, they reveal 6 adopts a different conformation once bound to the A2058G ribosome, thus impacting noncovalent interactions reflected in a lower MIC value. Finally, fluorescence polarization experiments of 6 with E. coli ribosomes confirmed 6 is indeed binding the ribosome.
Keywords: Total synthesis, ketolide antibiotics, antibiotic resistance, telithromycin, molecular modeling, desmethyl analogues
Antibiotic resistance represents a formidable 21st century public health threat.1 The onset of bacterial resistance to established drugs has outpaced the introduction of new FDA-approved antibacterial agents such that a postantibiotic era is imminent. To further aggravate the situation, economic pressures have forced many pharmaceutical companies to terminate their antimicrobial research programs; therefore, new antibiotics are desperately needed.2 To tackle this problem, we devised a strategy wherein desmethyl (i.e., CH3 → H) analogues of telithromycin (TEL, 2),3 an FDA-approved third generation ketolide antibiotic, were accessed via chemical synthesis (Figure 1). Currently, we have accomplished the synthesis, molecular modeling, and biological evaluation of 4,8,10-tridesmethyl TEL (3),4,5 4,10-didesmethyl TEL (4),6 and 4,8-didesmethyl congener (5)7 against wild-type and macrolide-resistant bacteria, which were found to be active (vide infra). Herein, we report the results of 4-desmethyl TEL (6), our final desmethyl analogue in the series.
Figure 1.

Structures of erythromycin (1), telithromycin (TEL, 2), and de novo analogues 4,8,10-tridesmethyl TEL (3), 4,10-didesmethyl TEL (4), 4,8-didesmethyl TEL (5), and 4-desmethyl TEL (6).
Macrolide antibiotics target the bacterial ribosome by reversibly binding the 50S subunit in the peptidyl transferase center, which ultimately blocks protein synthesis.8,9 Telithromycin (2) is a third generation analogue of flagship macrolide antibiotic erythromycin (1) and has been used clinically since 2004.3
The three mechanisms of antibacterial resistance include (1) modification of the drug, (2) efflux of the drug, and (3) modification of the ribosome by N-methylation at the 6-amino position of A2058 by erm enzymes or by point-mutation (i.e., A2058G).
Our desmethylation strategy was inspired by Steitz’s structural studies of macrolides 1 and 2 cocrystallized with 50S ribosomal subunits of the archaeon Haloarcula marismortui (Hm).10 While eubacteria possess an adenine at position 2058, all archea possess a guanine (Escherichia coli numbering) and thus do not efficiently bind the macrolides. Alternatively, the mutation of 2058 from guanine to adenine at position 2058 (i.e., G2058A) in the 23S rRNA enables binding to 2, which allowed the structure of telithromycin bound to the HmA2058 mutant at 2.6 Å resolution to be obtained (Figure 2A). Thus, Steitz revealed that bacterial A2058G mutations effect their resistance by (1) abrogating hydrogen bonding to desosamine’s C-2′ hydroxyl (Figure 2A) and (2) causing a steric clash between the C-4 methyl group on the macrolide framework and the C-2 amine of guanine (Figure 2B). We therefore hypothesized that replacing the C-4 methyl group with hydrogen (i.e., desmethylation) would avoid an attendant steric clash. Binding of 4-desmethyl TEL would be maintained since other residues within the ribosome (e.g., 2059) can also form hydrogen bonds with desosamine’s 2′-OH group, coupled with a salt bridge with the phosphate backbone of G2505.9,11
Figure 2.
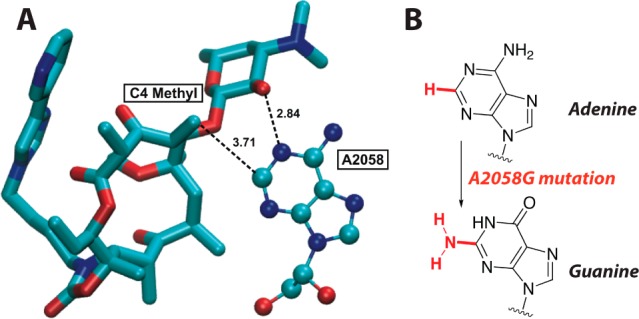
(A) Telithromycin and A2058 interactions in H. marismortui with select distances in Angstroms (Steitz et al. PDB 1YIJ). (B) Steric consequences of A2058G mutation. Image produced with VMD.12
To test our desmethylation hypothesis, we embarked on a total synthesis of 6 wherein evaluation against a panel of wild-type and resistant bacterial strains would follow (i.e., minimum inhibitory concentrations, or MICs).13 In addition, molecular modeling tools would also be employed to assist in the interpretation of the results.
Lessons learned during the syntheses of desmethyl congeners 3–54−7,14 greatly facilitated the successful procurement of 6, which is the only analogue that directly tests our hypotheses that desmethylation at C4 will address the A2058G mechanism of resistance. The total synthesis of 6 began by preparing C10–C13 fragment 8 (Scheme 1A) and C1–C9 fragment 14 (Scheme 1B). The former was easily prepared by the chemoselective acetylation of diol 7, which was employed in the synthesis of 4,8-didesmethyl TEL (5)7 via an intramolecular Nozaki–Hiyama–Kishi (NHK) reaction strategy.15 Tactics employed in the synthesis of the 4,10-didesmethyl TEL (4)6 were modified such that the p-methoxybenzyl (PMB) ether was recruited as the C5-hydroxy protecting group as opposed to the original triethylsilyl (TES) ether. This modification was made to add orthogonality into our protecting group scheme (vide infra).
Scheme 1. (A) Synthesis of C10–C13 Fragment 8 and (B) C1–C9 Fragment 14.

Reagents and conditions: (a) Ac2O, Et3N, DMAP, CH2Cl2, 0 °C to rt, 12 h, 87%; (b) NaH, PMBCl, DMF, 0 °C to rt, 65%; (c) LDA, MeI, THF, −78 °C; (d) LDA, (C6H5)3CCO2H, THF, −78 °C, 45% over two steps; (e) LiAlH4, THF, −45 °C to rt, 3 h, 92%; (f) TBSCl, imidazole, CH2Cl2, 0 °C to rt, 2 h; (g) 2,6-DTBMP, MeOTf, CH2Cl2, 48 h, 72% over two steps; (h) Raney-Ni, EtOH, H2, 6 h, 75%; (i) DMSO, (COCl)2, Et3N, CH2Cl2, −78 °C; (j) (R)-4-benzyl-3-propionyl-2-oxazolidinone, n-Bu2BOTf, Et3N, CH2Cl2, 2 h, 74% over two steps (dr > 20:1); (k) TBSOTf, 2,6-lutidine, CH2Cl2, 0 °C, 30 min, 95%; (l) CSA, MeOH, 0 °C, 2 h, 88%.
The synthesis of fragment 14 began with known lactone 9.6 Protection of the C5-hydroxyl group as its PMB ether was accomplished with NaH and PMBCl to afford 10 in 65% yield. Stereoselective installation of the C8-methyl group followed a two-step sequence. Treatment of 10 with LDA generated an enolate that was quenched with MeI. Subsequent regeneration of the lithium enolate and quenching with bulky proton source triphenylacetic acid, which was superior to previously used pivalic acid, afforded 11 in 45% yield over two steps.6 Lactone reduction with LiAlH4, protection of the primary alcohol as its TBS ether, and methylation of the tertiary C5 carbinol delivered 12 in 66% yield over three steps. Chemoselective reduction of the C3-O-benzyl ether over the C5-O-PMB ether was accomplished with Raney Ni in 75% yield.16 Swern oxidation17 of the primary alcohol to the aldehyde was followed by the Evans aldol reaction,18 which furnished 13a in 74% yield (dr > 20:1) over two steps. Protection of the C3-hydroxyl group as its TBS ether proceeded smoothly in 95% yield. Chemoselective removal of the primary C9-O-TBS ether with CSA in methanol afforded fragment 14 in 88% yield.19
With fragment 14 in hand, we set about coupling it with fragment 8 via an intermolecular NHK reaction to access the entire C1–C14 framework of 4-desmethyl TEL (6), as shown in Scheme 2. To this end, we oxidized the primary C9 carbinol with the Dess–Martin periodinane (DMP).20 The addition of CrCl2 and NiCl2 (100:1) to a solution of the purified aldehyde and vinyl iodide 8 in degassed DMSO at rt followed by DMP-mediated oxidation of the diastereomeric mixture of C9 alcohols delivered enone 15 in 45% yield over three steps. It is important to note that the acetate moiety in 8 was critical in order to execute the NHK coupling reaction.
Scheme 2. Intermolecular Nozaki–Hiyama–Kishi Reaction to Join Fragments 8 and 14 and Yamaguchi Macrolactonization.

Reagents and conditions: (a) DMP, pyridine, CH2Cl2, 2 h; (b) CrCl2, NiCl2, 8, DMSO, 48 h; (c) DMP, pyridine, CH2Cl2, 3 h, 45% over three steps; (d) H2O2, LiOH, THF/H2O (4:1), 0 °C to rt, 48 h, 87%; (e) C6H2Cl3COCl, i-Pr2NEt, DMAP, PhH, 12 h, 65%.
The next goal in the synthesis of 6 was preparing the requisite macroketolactone 17 (Scheme 2). To accomplish this, we employed the venerable Yamaguchi macrolactonization reaction.21,22 Accordingly, removal of the Evans auxiliary in 15 with LiOOH afforded 87% yield of the requisite acid 16. Subjection of 16 to Yamaguchi’s protocol delivered a satisfactory 65% yield of macroketolactone 17.
At this stage, the site- and stereoselective glycosylation of desosamine with donor 19(23) at the C5-hydroxyl position of 17 was pursued (Scheme 3). Again, experience garnered from the synthetic campaigns of desmethyl congeners 3–5 proved critical in the deployment of both strategy and tactics.4−7 Specifically, we found that deprotection of the C5-hydroxyl protecting group was accompanied by ketalization of the C5-OH onto the C9-ketone; moreover, if the tertiary carbinol at C12 was not protected, glycosylation occurred at this position over the desired C5 site. To remedy these issues, we reduced the C9-ketone under Luche conditions in 96% yield (dr = 4.6:1) and protected both the C9- and C12-hydroxyls as TMS ethers with TMSOTf and 2,6-lutidine.24 While we had originally employed a TES ether at the C9-hydroxyl in the 4,10-didesmethyl synthesis, the additional methyl at C10 made both protection and removal of the TES group more recalcitrant.6 Thus, we made recourse to the smaller TMS ether that ultimately suited our purposes. Removal of the C5-PMB ether was accomplished with 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) in 76% yield over two steps to set the stage for the stereoselective glycosylation with donor 19. The addition of AgOTf and 2,6-di-tert-butyl-4-methylpyridine (DTBMP) to a solution of 19 and acceptor 18 furnished glycosylated product 20 in 70% yield as a single stereoisomer by virtue of the C2′-O-methoxycarbonyl (Mc) protecting group.
Scheme 3. Synthesis of 4-Desmethyl Telithromycin (6).

Reagents and conditions: (a) CeCl3·7H2O, NaBH4, MeOH, −15 °C to rt, 45 min, 96% (dr = 4.6:1); (b) 2,6-lutidine, TMSOTf, CH2Cl2, −78 °C, 30 min; (c) DDQ, H2O, CH2Cl2, 0 °C, 30 min, 76% over 2 steps; (d) 19, AgOTf, 2,6-DTBMP, 4 Å MS, PhMe/CH2Cl2, 12 h, 70%; (e) HF·Pyr, THF/Pyr, 0 to 15 °C, 3 h; (f) DMP, CH2Cl2, 3 h, 67% over two steps; (g) NaH, CDI, DMF/THF, −20 to 0 °C, 45 min; (h) 22, MeCN/H2O, 72 h, 61% over two steps; (i) TAS-F, DMF/H2O, 14 h; (j) Me2S, NCS, Et3N, CH2Cl2, −20 °C, 53% over 2 steps; (k) MeOH, rt, 14 h, 67%.
With the glycosylated macrolactone framework fully assembled, we set about installing the C11–C12 oxazolidinone moiety bearing the pyridyl-imidazole side-chain employing the Baker protocol.25 To this end, we first adjusted the oxidation state at C9 by removing the trimethylsilyl (TMS) ethers at C9 and C12 with HF·pyridine and treating the resulting diol with DMP to afford enone 21 in 67% over two steps. Treatment of 21 with NaH and 1,1′-carbonyldiimidazole (CDI) furnished an activated carbamate that when treated with known butylamine 22(26) effected a domino carbamoylation/intramolecular aza-Michael sequence to form oxazolidinone 23 in 61% over two steps.
Endgame for 4-desmethyl TEL (6) began with an adjustment of the C-3 oxidation state. Removal of the TBS protecting group was realized with TAS-F.27 Oxidation of the intermediary carbinol using the Corey–Kim method furnished the C3-ketone in 53% yield over two steps.28 Removal of the Mc protecting group on the 2′-hydroxyl position of desosamine was accomplished by methanolysis, which delivered target compound 6 in 67% yield.
With 4-desmethyl TEL (6) in hand, we were uniquely positioned to directly test our desmethylation hypothesis that replacing the 4-methyl group with hydrogen would avoid the attendant steric clash with ribosomes bearing the A2058G mutation. To this end, we screened the biological activity of 6 against Escherichia coli and Staphylococcus aureus in MIC assays using TEL (2) as comparator and compared it to the other desmethyl analogues (Table 1).13 While both compounds were inactive against A2058G, A2058T, and ermA mutant strains (i.e., entries 1, 4, and 5, respectively), they were active against and E. coli WT and A2058G mutants. In fact, 4-desmethyl analogue 6 was equipotent with TEL (2) against E. coli WT (entry 2). However, it was also 4-fold less potent than 2 against the A2058G mutant, which altogether does not support our hypothesis. Curiously, the tridesmethyl analogue 3 was weakly active against an S. aureus A2058T mutant (entry 4). A notable trend among the demsethyl analogues is that as methyls are added (i.e., 3 → 6), the bioactivity improves. This is likely due to the biasing of the macrolactone conformation (i.e., avoiding syn-pentane interactions) and an increase in van der Waals contacts with the macrolide binding site.
Table 1. Minimum Inhibitory Concentration (MIC) Values in μg/mL for 4,8,10-Tridesmethyl TEL (3); 4,10-Didesmethyl TEL (4); 4,8-Didesmethyl TEL (5); 4-Desmethyl TEL (6); and Telithromycin (2)a.
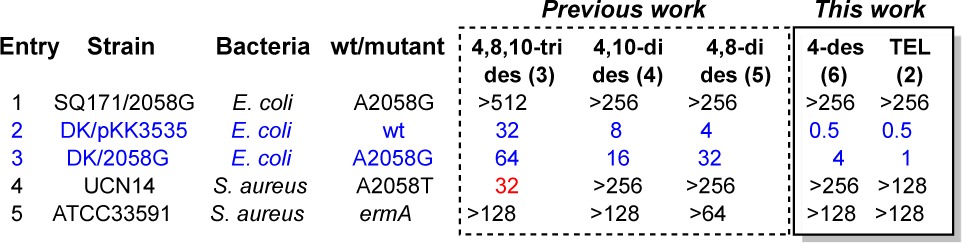
SQ171/2058G contain a pure population of mutant ribosomes; DK/pKK3535 refers to a tolC(−) strain with WT ribosomes; DK/2058 refers to a tolC(−) strain with a mixed population of WT and mutant ribosomes (ca. 1:1); tolC is an outer-membrane efflux pump that recognizes antibacterial drugs.
To help rationalize these results, we employed molecular modeling of telithromycin (2) and 4-desmethyl telithromycin (6) both in solution and bound to the E. coli ribosome (PDB 3OAT).11 Using Hamiltonian replica exchange molecular dynamics (HREX MD) simulations29−31 of 6 and 2 in solution, probability distributions of select distances within the compounds were measured to compare their conformational flexibilities using the conformationally sampled pharmacophore (CSP) approach (Figure 3). These data were compared to distributions of distances for desmethyl analogues from our previous works.4,6,7 Notably, as methyl groups are removed from the macrolactone ring, the distributions become more broad indicating an increase in the conformational flexibility of the desmethyl analogues compared to telithromycin (2). Consistent with this, 6 had a CSP probability distribution the most similar to 2. For the distances in panel B, 6 samples a slightly more extended conformation than 2, while the other distances show a high degree of overlap. In the context of MIC values, the increase in conformational flexibility observed for the di- and tridesmethyl analogues appears to be inversely related to bioactivity, which is supported by the tridesmethyl conformer having the highest MIC and highest conformational flexibility followed by the didesmethyl analogues. The MIC value for 2 and 6 are the same, which is supported by the high degree of overlap in the ensemble of conformations sampled by both conformers. However, their activity against the A2058G mutant (entry 4) is less straightforward to interpret. As per our hypothesis, we expected 6 to be more active against the A2058G mutant, yet its MIC value was 4-fold greater. As mentioned, the removal of methyl groups may decrease the van der Waals (VDW) interactions between the macrolide and the binding pocket. Though the similarity in the bioactivity between 2 and 6 in the WT suggests that this may not be a predominant factor for the 4-desmethyl conformer, it may impact the VDW interactions in the A2058G mutant, in which the conformation of the RNA interacting with the macrolide is slightly different as shown in a previous molecular dynamics (MD) study.32 Therefore, the interaction energy, which approximates the VDW and electrostatic enthalpic contributions, between the compounds and the ribosome were calculated using MD simulations of the telithromycin-bound E. coli ribosome (Table 2). Interestingly, the interaction energy between G2058 and both 2 and 6 is 2.5 to 3 kcal/mol less favorable than the WT A2058 interaction, which does not support our original hypothesis that removing the methyl group would enhance the interaction with 2058 due to a loss of a steric clash. Compared to 2, the interaction energy of 6 with the ribosome is similar (within standard deviation) indicating that the removal of the C4-methyl group does not greatly impact the VDW interactions with the ribosome. In addition, the interactions of 2 and 6 with the erm-related N6-methylations [e.g., monomethyladenine with the methyl directed toward desosamine (MAD1), monomethyladenine with the methyl directed away from desosamine (MAD2), and dimethyladenine (DMAD)] are similar, consistent with their similar MIC values (Table 1).
Figure 3.
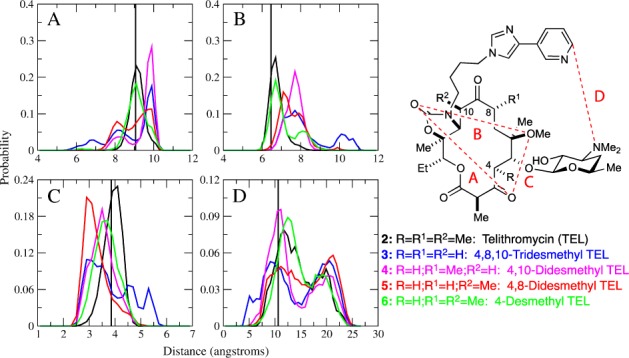
CSP probability distributions for TEL (2, black); 4,8,10-tridesmethyl TEL (3, blue); 4,10-didesmethyl TEL (4, purple); 4,8-didesmethyl TEL (5, red); and 4-desmethyl TEL (6, green). The vertical line corresponds to the crystallographic distances from PDB 1YIJ. Atom pairs represented in A through D are shown in the inset figure.
Table 2. Interaction Energies (kcal/mol) between Telithromycin (2) and the E. coli Crystal Structure (PDB 3OAT)a.
| telithromycin |
C4-desmethyl |
|||
|---|---|---|---|---|
| ribosome | all | base 2058 | all | base 2058 |
| WT | –9.2 (1.0) | –1.8 (0.4) | –7.7(1.0) | –1.4 (0.6) |
| A2058G | –8.5 (1.1) | 1.2 (0.5) | –8.6 (0.9) | 1.1 (0.3) |
| MAD1 | –9.7 (1.1) | –2.1 (0.5) | –7.3 (0.8) | –2.2 (0.3) |
| MAD2 | –11.7 (1.5) | –3.1 (0.5) | –7.5(1.2) | –2.1 (0.6) |
| DMAD | –6.9(1.4) | –0.9 (0.4) | –7.5 (0.9) | –0.9 (0.3) |
Calculations were performed for 2 and 6 with the WT, A2058G, MAD1, MAD2, and DMAD ribosomes. Interaction energies were calculated for selected ligand atoms with all the bases/residues within the truncated ribosome (see Supporting Information) as well as with base 2058 alone. Selected ligand atoms include the neutral group surrounding the C4-methyl [C3(=O)–C4(H2)–C5]. Units in kcal/mol. Standard deviation shown in parentheses. C4-des refers to 4-desmethyl telithromycin (6).
The CSP approach was also applied to the ligand-bound simulations (Figure 4). For 6 in the A2058G mutant, the probability distribution in panels A and B are shifted to 1 to 2 Å larger, suggesting that the macrolactone ring of 6 is in a more extended conformation in the mutant as compared to WT, while the conformations of 2 in the WT and A2058G are similar. This suggests that 6 adopts a slightly different conformation once is it bound within the A2058G ribosome, which may impact interactions with the ribosome and hence its MIC value. Of note are the distances in panel D showing them to differ by a few Angstroms from each other. This is consistent with our previous work showing the imidazole–pyridine moiety to be dynamic within the binding pocket.32
Figure 4.
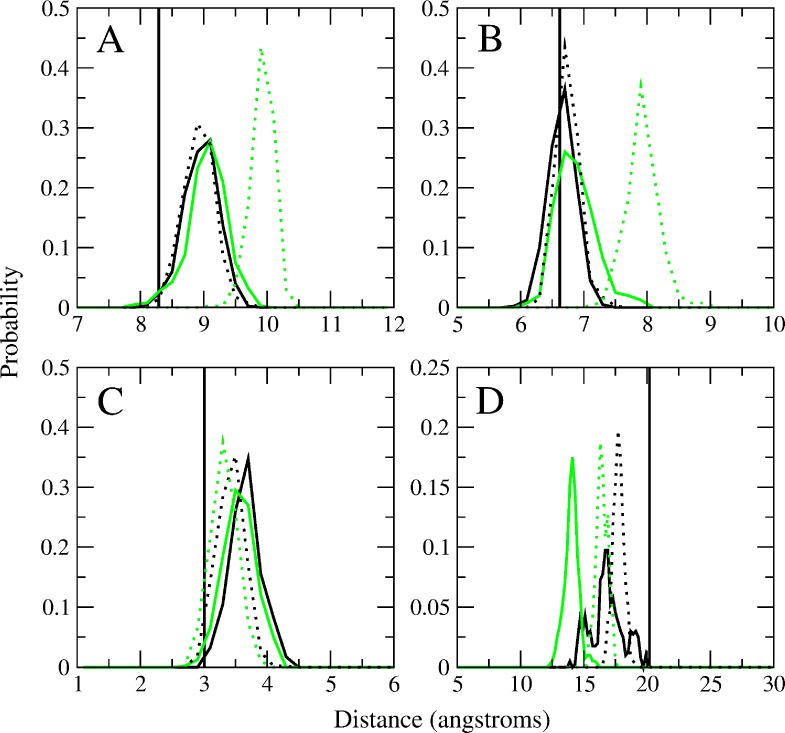
CSP probability distributions for selected atom pairs in 2 and 6 in the WT E. coli ribosome (2, solid black; 6, solid green) and in A2058G mutant (2, dashed black; 6, dashed green). The vertical line corresponds to the crystallographic distances from PDB 3OAT. Atom pairs are shown in Figure 3
The biological evaluation of 4-desmethyl telithromycin (6) by means of MIC assays (Table 1) does not provide direct evidence that it is in fact binding (and inhibiting) the bacterial ribosome in analogy to telithromycin (2). As our hypothesis is predicated on this particular mechanism of action, we sought to determine whether 6 would in fact bind the bacterial ribosome. Yan and co-workers had demonstrated that a fluorescence polarization-based competitive binding assay utilizing boron-dipyrromethene (BODIPY)-labeled erythromycin and isolated E. coli ribosomes was a reliable method for determining the dissociation constants (Kd) of various macrolide antibiotics.33 Telithromycin (2) bound E. coli ribosomes with a Kd of 6.4–11 nM. We applied the same method to 6 and found that it bound with a Kd of 21.25 ± 6.4 nM, which is close to the reported value for 2 and establishes that desmethyl analogue 6 is exerting its bioactivity by binding the macrolide binding site of the bacterial ribosome.
In conclusion, 4-desmethyl telithromycin (6) was prepared via total synthesis using an intermolecular NHK/Yamaguchi macrolactonization approach in 36 steps (26 steps in the longest linear sequence) from commercially available materials. It is worthy to note that the strategy employed for each desmethyl congener was unique, which underscores the complexity of these targets. Evaluation of the biological activity of 6 via MIC assays revealed it was equipotent with comparator telithromycin (2) against an E. coli WT strain but was 4-fold less potent against the A2058G mutant. MD simulations were employed to help rationalize these results and revealed 6 adopts a slightly different conformation once bound to the A2058G ribosome, thus impacting noncovalent interactions reflected in a lower MIC value. Altogether, these data do not support the hypothesis that strategic desmethylation of ketolide antibiotics can directly address resistance arising from the pathogenically relevant A2058G mutation. The sequential addition of methyl residues (i.e., 3 → 6) corresponded with an increase in bioactivity, thus revealing the critical nature of these in biasing macrolactone conformation as well as participating in VDW interactions with the macrolide binding site in the 50S subunit of the bacterial ribosome. Finally, we showed that 6 binds the bacterial ribosome with nanomolar affinity using an established fluorescence polarization assay.
Acknowledgments
We thank Prof. Shura Mankin (University of Illinois at Chicago) for helpful suggestions. We thank Dr. Barry S. Cooperman (University of Pennsylvania) for providing 70S E. coli ribosomes and for helpful discussions. Finally, we dedicate this work to the memory of Dr. Tapas Paul.
Glossary
Abbreviations
- BODIPY
boron-dipyrromethene
- CDI
carbonyldiimidazole
- CSA
camphorsulfonic acid
- CSP
conformationally sampled pharmacophore
- DDQ
2,3-dichloro-5,6-dicyano-1,4-benzoquinone
- DMAD
N6-dimethyladenine
- DMAP
4-dimethylaminopyridine
- DMF
N,N-dimethylformamide
- DMP
Dess-Martin Periodinane
- DMSO
dimethyl sulfoxide
- DTBMP
2,6-di-tert-butyl-4-methylpyridine
- Hm
Haloarcula marismortui
- HREX MD
Hamiltonian replica exchange dynamics
- LDA
lithium diisopropylamide
- MAD
N6-methyladenine
- MD
molecular dynamics
- MIC
minimum inhibitory concentration
- NCS
N-chlorosuccinimide
- NHK
Nozaki–Hiyama–Kishi
- PMB
para-methoxybenzyl
- Pyr
pyridine
- TAS-F
tris(dimethylamino)sulfonium difluorotrimethylsilicate
- TBAF
tetrabutylammonium fluoride
- TBS
tert-butyldimethylsilyl
- TEL
telithromycin
- TES
triethylsilyl
- THF
tetrahydrofuran
- TMS
trimethylsilyl
- Tf
triflyl
- VDW
van der Waals
- WT
wild-type
Supporting Information Available
General experimental protocols, computational methods, fluorescence polarization experiments with BODIPY-labeled erythromycin, full structural assignment of 21, and characterization of all new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
This work was supported by the NIH (AI080968 and GM070855) and the University of Maryland Computer-Aided Drug Design Center.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Laxminarayan R.; Duse A.; Wattal C.; Zaidi A. K. M.; Wertheim H. F. L.; Sumpradit N.; Vlieghe E.; Hara G. L.; Gould I. M.; Goossens H.; Greko C.; So A. D.; Bigdeli M.; Tomson G.; Woodhouse W.; Ombaka E.; Peralta A. Q.; Qamar F. N.; Mir F.; Kariuki S.; Bhutta Z. A.; Coates A.; Bergstrom R.; Wright G. D.; Brown E. D.; Cars O. Antibiotic resistance-the need for global solutions. Lancet Infect. Dis. 2013, 13, 1057–1098. [DOI] [PubMed] [Google Scholar]
- Fox J. L. The business of developing antibacterials. Nat. Biotechnol. 2006, 24, 1521–1528. [DOI] [PubMed] [Google Scholar]
- Denis A.; Agouridas C.; Auger J. M.; Benedetti Y.; Bonnefoy A.; Bretin F.; Chantot J. F.; Dussarat A.; Fromentin C.; D’Ambrieres S. G.; Lachaud S.; Laurin P.; Le Martret O.; Loyau V.; Tessot N.; Pejac J. M.; Perron S. Synthesis and antibacterial activity of HMR 3647 a new ketolide highly potent against erythromycin-resistant and susceptible pathogens. Bioorg. Med. Chem. Lett. 1999, 9, 3075–3080. [DOI] [PubMed] [Google Scholar]
- Velvadapu V.; Paul T.; Wagh B.; Klepacki D.; Guvench O.; MacKerell A.; Andrade R. B. Desmethyl macrolides: synthesis and evaluation of 4,8,10-tridesmethyl telithromycin. ACS Med. Chem. Lett. 2011, 2, 68–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velvadapu V.; Paul T.; Wagh B.; Glassford I.; DeBrosse C.; Andrade R. B. Total synthesis of (−)-4,8,10-tridesmethyl telithromycin. J. Org. Chem. 2011, 76, 7516–7527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velvadapu V.; Glassford I.; Lee M.; Paul T.; DeBrosse C.; Klepacki D.; Small M. C.; MacKerell A. D. Jr.; Andrade R. B. Desmethyl macrolides: synthesis and evaluation of 4,10-didesmethyl telithromycin. ACS Med. Chem. Lett. 2012, 3, 211–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagh B.; Paul T.; Glassford I.; DeBrosse C.; Klepacki D.; Small M. C.; MacKerell A. D. Jr.; Andrade R. B. Desmethyl macrolides: synthesis and evaluation of 4,8-didesmethyl telithromycin. ACS Med. Chem. Lett. 2012, 3, 1013–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mankin A. S. Macrolide myths. Curr. Opin. Microbiol. 2008, 11, 414–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spahn C. M. T.; Prescott C. D. Throwing a spanner in the works: antibiotics and the translation apparatus. J. Mol. Med. 1996, 74, 423–439. [DOI] [PubMed] [Google Scholar]
- Tu D.; Blaha G.; Moore P. B.; Steitz T. A. Structures of MLSBK antibiotics bound to mutated large ribosomal subunits provide a structural explanation for resistance. Cell 2005, 121, 257–270. [DOI] [PubMed] [Google Scholar]
- Dunkle J. A.; Xiong L.; Mankin A. S.; Cate J. H. Structures of the Escherichia coli ribosome with antibiotics bound near the peptidyl transferase center explain spectra of drug action. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 17152–17157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphrey W.; Dalke A.; Schulten K. VMD: Visual molecular dynamics. J. Mol. Graphics 1996, 14, 33–38. [DOI] [PubMed] [Google Scholar]
- Reller L. B.; Weinstein M.; Jorgensen J. H.; Ferraro M. J. Antimicrobial susceptibility testing: a review of general principles and contemporary practices. Clin. Infect. Dis. 2009, 49, 1749–1755. [DOI] [PubMed] [Google Scholar]
- Wagh B.; Paul T.; DeBrosse C.; Klepacki D.; Small M. C.; MacKerell A. D.; Andrade R. B. Desmethyl macrolides: synthesis and evaluation of 4,8,10-tridesmethyl cethromycin. ACS Med. Chem. Lett. 2013, 4, 1114–1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fürstner A. Carbon–carbon bond formations involving organochromium(III) reagents. Chem. Rev. 1999, 99, 991–1045. [DOI] [PubMed] [Google Scholar]
- Oikawa Y.; Tanaka T.; Horita K.; Yonemitsu O. Selective hydrogenolysis of the benzyl protecting group for hydroxy function with raney nickel in the presence of the MPM (4-methoxybenzyl) and DMPM (3,4-dimethoxybenzyl) protecting groups. Tetrahedron Lett. 1984, 25, 5397–5400. [Google Scholar]
- Mancuso A. J.; Swern D. Activated dimethyl sulfoxide: useful reagents for synthesis. Synthesis 1981, 165–185. [Google Scholar]
- Evans D. A.; Bartroli J.; Shih T. L. Enantioselective aldol condensations. 2. Erythro-selective chiral aldol condensations via boron enolates. J. Am. Chem. Soc. 1981, 103, 2127–2129. [Google Scholar]
- Nelson T. D.; Crouch R. D. Selective deprotection of silyl ethers. Synthesis 1996, 1031–1069. [Google Scholar]
- Dess D. B.; Martin J. C. A useful 12-I-5 triacetoxyperiodinane (the Dess–Martin periodinane) for the selective oxidation of primary or secondary alcohols and a variety of related 12-I-5 species. J. Am. Chem. Soc. 1991, 113, 7277–7287. [Google Scholar]
- Parenty A.; Moreau X.; Campagne J. M. Macrolactonizations in the total synthesis of natural products. Chem. Rev. 2006, 106, 911–939. [DOI] [PubMed] [Google Scholar]
- Inanaga J.; Hirata K.; Saeki H.; Katsuki T.; Yamaguchi M. A rapid esterification by means of mixed anhydride and its application to large-ring lactonization. Bull. Chem. Soc. Jpn. 1979, 52, 1989–1993. [Google Scholar]
- Velvadapu V.; Andrade R. B. Concise syntheses of d-desosamine, 2-thiopyrimidinyl desosamine donors, and methyl desosaminide analogues from d-glucose. Carbohydr. Res. 2008, 343, 145–150. [DOI] [PubMed] [Google Scholar]
- Luche J. L.; Rodriguezhahn L.; Crabbe P. Reduction of natural enones in the presence of cerium trichloride. Chem. Commun. 1978, 601–602. [Google Scholar]
- Baker W. R.; Clark J. D.; Stephens R. L.; Kim K. H. Modification of macrolide antibiotics. Synthesis of 11-deoxy-11-(carboxyamino)-6-O-methylerythromycin A 11,12-(cyclic esters) via an intramolecular Michael reaction of O-carbamates with an alpha,beta-unsaturated ketone. J. Org. Chem. 1988, 53, 2340–2345. [Google Scholar]
- Grant E. B., III.Preparation of macrolide 9-alkyl-and 9-alkylidenyl-6-O-alkyl-11, 12-carbamate ketolide clarithromycin derivatives as anti-bacterial agents. WO patent 2006047167A2, 2006.
- Scheidt K. A.; Chen H.; Follows B. C.; Chemler S. R.; Coffey D. S.; Roush W. R. Tris(dimethylamino)sulfonium difluorotrimethylsilicate, a mild reagent for the removal of silicon protecting groups. J. Org. Chem. 1998, 63, 6436–6437. [Google Scholar]
- Corey E. J.; Kim C. U. New and highly effective method for oxidation of primary and secondary alcohols to carbonyl-compounds. J. Am. Chem. Soc. 1972, 94, 7586–7587. [Google Scholar]
- Jiang W.; Hodoscek M.; Roux B. Computation of absolute hydration and binding free energy with free energy perturbation distributed replica-exchange molecular dynamics (FEP/REMD). J. Chem. Theory Comput. 2009, 5, 2583–2588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kannan S.; Zacharias M. Enhanced sampling of peptide and protein conformations using replica exchange simulations with a peptide backbone biasing potential. Proteins 2007, 66, 697–706. [DOI] [PubMed] [Google Scholar]
- Zuckerman D. M.; Lyman E. A second look at canonical sampling of biomolecules using replica exchange simulation. J. Chem. Theory Comput. 2006, 2, 1200–1202. [DOI] [PubMed] [Google Scholar]
- Small M. C.; Lopes P.; Andrade R. B.; Mackerell A. D. Jr. Impact of ribosomal modification on the binding of the antibiotic telithromycin using a combined grand canonical monte carlo/molecular dynamics simulation approach. PLoS Comput. Biol. 2013, 9, e1003113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan K.; Hunt E.; Berge J.; May E.; Copeland R. A.; Gontarek R. R. Fluorescence polarization method to characterize macrolide-ribosome interactions. Antimicrob. Agents Chemother. 2005, 49, 3367–3372. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.