

Abstract
Although dysregulated substance P (SP) has been implicated in the pathophysiology of Parkinson's disease (PD), how SP affects the survival of dopaminergic neurons remains unclear. Here, we found that mice lacking endogenous SP (TAC1−/−), but not those deficient in the SP receptor (neurokinin-1 receptor, NK1R), were more resistant to lipopolysaccharide (LPS)- and 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced nigral dopaminergic neurodegeneration than wild-type controls, suggesting a NK1R-independent toxic action of SP. In vitro dose–response studies revealed that exogenous SP enhanced LPS- and 1-methyl-4-phenylpyridinium (MPP+)-induced dopaminergic neurodegeneration in a bimodal manner, peaking at submicromolar and subpicomolar concentrations, but was substantially less effective at intermediate concentrations. Mechanistically, the actions of submicromolar levels of SP were NK1R-dependent, whereas subpicomolar SP-elicited actions required microglial NADPH oxidase (NOX2), the key superoxide-producing enzyme, but not NK1R. Subpicomolar concentrations of SP activated NOX2 by binding to the catalytic subunit gp91phox and inducing membrane translocation of the cytosolic subunits p47phox and p67phox. The importance of NOX2 was further corroborated by showing that inhibition or disruption of NOX2 blocked subpicomolar SP-exacerbated neurotoxicity. Together, our findings revealed a critical role of microglial NOX2 in mediating the neuroinflammatory and dopaminergic neurodegenerative effects of SP, which may provide new insights into the pathogenesis of PD.
Keywords: GPCR independence, NADPH oxidase, neuroinflammation, Parkinson's disease, substance P
Introduction
Chronic neuroinflammation mediated by microglia, the resident immune cells of the brain, has been implicated as a key event driving the delayed and progressive loss of dopaminergic neurons in idiopathic Parkinson's disease (PD) (Block et al., 2007; Stone et al., 2009). Postmortem analyses of PD patients have revealed the activation of microglia and an accumulation of proinflammatory factors in the substantia nigra (SN) (McGeer et al., 1988). Similarly, Gram-negative bacterial endotoxin lipopolysaccharide (LPS) induces microglia-mediated neuroinflammation in the brains of laboratory animals and recapitulates the delayed and progressive nature of nigral dopaminergic degeneration (Qin et al., 2007). Previous reports have shown that humans exposed to LPS also develop parkinsonian syndromes (Niehaus and Lange, 2003). However, how acute microglial activation becomes chronic and toxic to dopaminergic neurons is poorly understood.
Recent findings have indicated that neurogenic signals can modulate microglial activity and function (Biber et al., 2007; Domercq et al., 2013). Substance P (SP), produced in striatonigral-projecting neurons, has been shown to amplify proinflammatory responses in the CNS (Marriott, 2004). This proinflammatory peptide has been implicated in the pathogenesis of PD because of its ability to stimulate dopamine (DA) release and modulate motor behaviors (Graybiel, 1986; Reid et al., 1990). However, the functional role of SP in the regulation of neuroinflammation and dopaminergic neuron survival remains elusive. Dopaminergic neurons in the SN are more susceptible to oxidative and inflammatory insults than other types of neurons (Hastings et al., 1996; Hastings and Zigmond, 1997; Mosley et al., 2006; Reynolds et al., 2007). We (Block et al., 2006) and others (Rasley et al., 2004; Chauhan et al., 2008) have previously reported that SP is capable of stimulating or augmenting microglial activation to produce superoxide and proinflammatory cytokines in vitro. Given that SP is most concentrated in the SN (Brownstein et al., 1976), which contains a disproportionately high density of microglia (∼4.5 times that of other regions) (Kim et al., 2000), we hypothesized that SP may regulate microglial activity in vivo, thereby exacerbating dopaminergic neurodegeneration in neuroinflammatory conditions.
To test our hypothesis, we treated mice or various primary cell cultures with two commonly used toxins, LPS- or 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP, in animal)/1-methyl-4-phenylpyridinium (MPP+, in cell culture), which cause dopaminergic neurodegeneration. Several strains of mice deficient in SP (TAC1−/−), the SP receptor (NK1R−/−) or NADPH oxidase (NOX2; gp91phox−/−, the functional catalytic subunit of NOX2) were used. We found that SP potentiated LPS and MPTP-induced toxic effects on nigrostriatal dopaminergic neurons by enhancing microglia-mediated neuroinflammation. Importantly, we discovered that the activation of microglial NOX2 by ultra-low concentrations of SP was a novel, NK1R-independent signaling pathway responsible for the synergistic neurotoxicity induced by SP with either LPS or MPTP. These findings strongly suggest that SP facilitates neuroinflammation induced by toxins and plays a key role in damaging dopaminergic neurons via a novel NK1R-independent mechanism.
Materials and Methods
Animal treatments.
A single systemic LPS (Escherichia coli 0111:B4, L-3012, Sigma, catalog #L3012) injection (15 × 106 EU/kg, i.p.) or a repeated MPTP regimen (15 mg/kg, s.c. for 6 consecutive days; Sigma, catalog #M0896) was administered to male wild-type (C57BL/6J, The Jackson Laboratory, stock #000664), TAC1−/− (B6.Cg-Tac1tm1Bbm/J, The Jackson Laboratory, stock #004103), and NK1R−/− mice. The dosages of LPS (Qin et al., 2007) and MPTP (Zhang et al., 2004) were chosen based on our previous studies. Control mice received an equal volume of 0.9% saline. Ten months after the LPS injection or 3 weeks after the last MPTP treatment, the mice were killed, and their brains were collected. Housing, breeding, and experimental use of the animals were performed in strict accordance with the National Institutes of Health guidelines.
Primary cultures.
Mesencephalic neuron-glia, microglia-depleted, and microglia-enriched cultures were prepared according to a previously published protocol (Chen et al., 2013).
Immunohistochemistry.
Mouse brains were fixed in 4% PFA and processed for immunostaining as described previously (Zhang et al., 2004). Dopaminergic neurons were detected with a polyclonal antibody against tyrosine hydroxylase (TH, 1:2000, Millipore, catalog #AB152, RRID: AB_390204). Microglia were stained with rabbit anti-ionized calcium-binding adaptor molecule 1 (Iba-1, 1: 5000, Wako Chemicals, catalog #019-19741, RRID: AB_839504) or rat anti-mouse CD11b (1: 400, ABD Serotec, catalog #MCA711GT, RRID: AB_1100616) antibodies. Immunostaining was visualized using a Vectastain ABC Kit (Vector Laboratories) and diaminobenzidine substrate. Nigral densities of Iba-1 and CD11b immunostaining were measured using ImageJ software (National Institutes of Health, nif-0000-30467) (Wang et al., 2012b).
Cell counting.
The total number of THir neurons in the substantia nigra pars compacta (SNpc) was estimated stereologically using the optical fractionator method (MBF Science, MicroBrightfield ID: nif-0000-00110, Version #281824). Systematic random samples of sites with an unbiased counting frame (100 μm × 100 μm) within defined boundaries of the SN were processed. An 11 μm dissector height was used, and the guard zone was set at 2 μm. Counts were performed with an Olympus BX50 microscope using a 60 × 1.4 NA oil-immersion objective, and coefficient of error values were <0.1. Manual counts were also performed as described previously (Zhang et al., 2004), and the results were consistent with those of the stereological method (data not shown).
Real-time PCR.
Total RNA was extracted with the RNeasy Mini kit (QIAGEN) and reverse transcribed with an oligo dT primer. The real-time PCR amplification was performed using SYBR Green PCR Master Mix (Applied Biosystems) and Applied Biosystems 7900HT Fast Real-Time PCR System according to the manufacturer's protocol. The following primers and related concentrations were used: tumor necrosis factor α (TNFα), forward, GACCCTCACACTCAGATCATCTTCT (300 nm) and reverse, CCTCCACTTGGTGGTTTGCT (900 nm); inducible nitric oxide synthase (iNOS), CTGCCCCCCTGCTCACTC (300 nm) and reverse, TGGGAGGGGTCGTAATGTCC (300 nm); monocyte chemotactic protein-1 (MCP-1), forward, TTTGAATGTGAAGTTGACCCG (300 nm) and reverse, GAAGTGCTTGAGGTGGTTGTG (300 nm); and GAPDH, forward, TTCAACGGCACAGTCAAGGC (300 nm) and reverse, GACTCCACGACATACTCAGCACC (300 nm). The PCR conditions were 95°C for 10 s, 55°C for 30 s and 72°C for 30 s for 40 cycles. The data were normalized to GAPDH.
[3H]-DA uptake.
Uptake assays were performed by incubating the cultures for 20 min at 37°C with 1 μm [3H]-DA (PerkinElmer Life Sciences) as described previously (Gao et al., 2008). Nonspecific uptake was determined in the presence of 10 μm mazindol.
Measurement of superoxide, intracellular reactive oxygen species, nitrite and TNF-α.
The production of superoxide and intracellular reactive oxygen species (iROS) were determined by measuring the superoxide dismutase-inhibitable reduction of WST-1 and increase of CM-H2-DCFDA, respectively, as described previously (Qin et al., 2004; Wang et al., 2012a).
The production of nitrite was determined using the Griess reagent.
The levels of TNFα in the supernatant were measured using commercial ELISA kits (R&D Systems).
Isolation of macrophages from the peritoneal cavity.
Peritoneal macrophages were obtained from the peritoneal cavity of wild-type, gp91phox−/− and NK1R−/− mice using a modified method described previously (Ray and Dittel, 2010). The peritoneal cavities of animals were flushed with 5 ml of ice-cold PBS (containing 0.1% EDTA) after 4 d of thioglycollate medium injection. All peritoneal fluids were transferred to a 50 ml centrifuge tube and centrifuged at 1500 rpm for 8 min. The pellets were washed three times using PBS and collected for use.
[125I]-labeled SP binding.
Binding experiments were performed using both whole cells and plasma membrane fractions (Wang et al., 2012a). Briefly, whole cells and membrane pellets were suspended in binding buffer (50 mm Tris-HCl, pH 8.0, 100 mm NaCl, 1% BSA, and 1 mm PMSF). [125I]-SP (10 μCi, Phoenix Pharmaceuticals) was added into the whole-cell or membrane aliquots (0.3 mg) at a final concentration of 2, 3, 4, 6, 8, or 10 × 10−13 m. Nonspecific binding was determined in the presence of an additional 0.1 μm SP. The samples were incubated on a rotator for 2 h at 4°C. The binding assay was terminated by filtration through glass microfiber filters (GF/C, Whatman). The filters were immediately washed and counted using a γ-counter.
Immunoblotting.
Equal amounts of samples were separated by electrophoresis through a 4%–12% Bis-Tris Nu-PAGE gel and transferred to PVDF membranes. The membranes were blocked with 5% nonfat milk and incubated with primary antibodies (1:1000) against p47phox (Millipore catalog #07-500, RRID: AB_310668), p67phox (BD Biosciences, catalog #61092, RRID: AB_398229), gp91phox (BD Biosciences, catalog #611414, RRID: AB_398936), phospho-ERK (Cell Signaling Technology, catalog #9101S, RRID: AB_331646), ERK (Cell Signaling Technology, catalog #9102S, RRID: AB_10695746), phospho-p38 (Cell Signaling Technology, catalog #9215S, RRID: AB_331762), p38 (Cell Signaling Technology, catalog #9212, RRID: AB_330713), phospho-JNK (Cell Signaling Technology, catalog #9251S, RRID: AB_331659), JNK (Cell Signaling Technology, catalog #9252, RRID: AB_2250373), phospho-p65 (Cell Signaling Technology, catalog #3031S, RRID: AB_330559), p65 (Cell Signaling Technology, catalog #4764S, RRID: AB_823578), phospho-IKKα/β (Cell Signaling Technology, catalog #2697S, RRID: AB_2079382), IKKβ (Cell Signaling Technology, catalog #2678, RRID: AB_2122301), phospho-IκBα (Cell Signaling Technology, catalog #2859S, RRID: AB_561111), IκBα (Cell Signaling Technology, catalog #4814, RRID: AB_390781), and GAPDH (1: 1000, Abcam catalog #ab8245, RRID: AB_2107448) and HRP-linked anti-rabbit or mouse IgG (1:3000) for 2 h. ECL reagents were used for detection.
Statistical analysis.
All values are expressed as mean ± SEM. The differences among means were analyzed using one- or two-way ANOVA with treatment or genotype as the independent factors. When the ANOVA showed significant differences, pairwise comparisons between means were analyzed by Tukey's post hoc testing. In all analyses, a value of p < 0.05 was considered significant.
Results
Absence of endogenous SP in TAC1 gene-knock-out mice attenuates LPS- or MPTP-induced dopaminergic neurodegeneration
Mice lacking the preprotachykinin gene (TAC1) are deficient in endogenous SP and its differential splicing products, neurokinin A, neuropeptide K, and neuropeptide γ, and were used to determine whether endogenous SP plays a role in the toxins-induced loss of dopaminergic neurons. Mutant and wild-type (WT) mice were injected with LPS or MPTP (Fig. 1A), two widely used toxins known to cause neuroinflammation via different mechanisms. LPS has been used as a tool for producing neuroinflammation-driven neurodegeneration because this endotoxin does not directly cause neuronal damage (Qin et al., 2004). We have previously reported that a single systemic injection of LPS generates peripheral inflammation, and some of the circulating proinflammatory factors gain entrance to the brain to activate microglia, initiate chronic neuroinflammation, and subsequently lead to the progressive degeneration of nigral dopaminergic neurons (Qin et al., 2007). Serum TNFα is one of the major proinflammatory cytokines responsible for transferring peripheral inflammation to the brain in LPS-treated mice (Qin et al., 2007). In contrast, MPTP directly damages nigral dopaminergic neurons (Moratalla et al., 1992). Damaged neurons secrete a variety of toxic substances to initiate reactive microgliosis, which produces proinflammatory factors to cause further dopaminergic neurodegeneration (Levesque et al., 2010). Previous studies demonstrated that after LPS and MPTP injection, the levels of serum TNFα and brain MPP+ (the active metabolite of MPTP) peaked at 1 h (Qin et al., 2007) and 90 min (Przedborski et al., 1996), respectively. An initial study was conducted to ensure that LPS-induced serum TNFα and the entry of MPTP from the blood to the brain (as shown by brain content of MPP+) after 1 h and 90 min after treatment, respectively, did not differ between WT and TAC1−/− mice (TNFα (pg/ml): 6751 ± 1321 in WT versus 7069 ± 1254 in TAC1−/− mice, two-way ANOVA, genotype, F(1,15) = 0.1162, p = 0.7379; MPP+ (ng/mg tissue): 1.29 ± 0.09 in WT versus 1.49 ± 0.05 in TAC1−/− mice, two-way ANOVA, genotype, F(1,8) = 1.672, p = 0.2321).
Figure 1.

Deletion of endogenous SP production reduces LPS- and MPTP-induced nigrostriatal dopaminergic neurodegeneration in vivo. A, Experimental designs. A single dose of LPS (15 × 106 EU/kg, i.p.) or repeated MPTP regimen (15 mg/kg, s.c. for 6 consecutive days) was administered to TAC1−/− (no endogenous production of SP) or WT mice. Ten months after LPS or 21 days after the last MPTP injection, the brains were perfused for immunostaining of dopaminergic neurons. Representative images of (B) THir neurons in SNpc and (C) axon fibers in striatum are shown. Nigral THir neurons in (D) LPS-treated and (E) MPTP-treated mice were counted stereologically. F, Striatal dopaminergic neuron fibers were quantified by TH density. The results are expressed as the percentage of THir cell loss or density reduction (mean ± SEM, WT controls were considered as 0, no damage) and were analyzed using two-way ANOVA followed by Tukey's post hoc test. **p < 0.01. ***p < 0.001. n = 4–7. Scale bar, 400 μm.
Previous studies have demonstrated consistency of dopaminergic neuron quantitation by counting THir and NeuN/Nissl-immunoreactive cells in the SNpc of PD models (Gao et al., 2011a; Baiguera et al., 2012; Tonges et al., 2012). Because THir cell count affords a more precise way of quantitation, cell count based on TH staining was presented in this study. Immunostaining analysis showed a significant decrease in nigral THir neurons (TH is a marker for dopaminergic neurons) in WT mice 10 months after LPS injection or 3 weeks after MPTP treatment. The loss of nigral dopaminergic neurons was attenuated in TAC1−/− mice (Fig. 1B). Stereological counting showed that the number of THir neurons in the SNpc of WT mice was 7265 ± 255 (set as 0%, no damage). Exposure to LPS resulted in a 30% loss of nigral dopaminergic neurons in WT mice, which was reduced to a 9% loss in TAC1−/− mice (two-way ANOVA: genotype, F(1,16) = 16.9, p = 0.0008; Fig. 1D). Similarly, MPTP treatment resulted in a 40% loss in nigral dopaminergic neurons in WT mice compared with a 14% loss in TAC1−/− mice (two-way ANOVA: genotype, F(1,18) = 22.5, p = 0.0002; Fig. 1E). Consistently, the MPTP-induced decrease in the density of striatal dopaminergic fibers (detected by TH immunohistochemistry) was attenuated in TAC1−/− mice compared with WT mice (Fig. 1C,F). These results indicate that endogenous SP exacerbates LPS and MPTP-induced toxicity in dopaminergic neurons.
Deletion of endogenous SP attenuates LPS-induced nigral microglial activation and gene expression of proinflammatory factors
To determine whether the attenuated toxicity of dopaminergic neurons in LPS-treated TAC1−/− mice may be associated with reduced neuroinflammation, we measured the mRNA expression of multiple proinflammatory factors in the brains of both TAC1−/− and WT mice. Figure 2A–C shows that 10 months after LPS injection, increases in the gene expression of TNF-α, iNOS, and MCP-1 were observed in WT mice. However, upregulation of these proinflammatory factors was significantly attenuated in TAC1−/− mice (two-way ANOVA, genotype, all p < 0.05).
Figure 2.

The absence of endogenous SP reduces neuroinflammation in vivo. A–C, A single dose of LPS (15 × 106 EU/kg, i.p.) was administered to TAC1−/− or WT mice. Ten months later, the mRNA levels of TNFα (A), iNOS (B), and MCP-1 (C) were determined in the rostral halves of the brains using RT-PCR. Representative images of (D) Iba-1 and (F) CD11b staining for microglial activation in the SN 10 months after LPS injection are shown. Activated microglia, characterized by enlarged cell bodies and high staining density, are indicated by red arrowheads. E, Microglial activation was quantified by calculating the density of expression of Iba-1 and (G) CD11b in the SN. The results are expressed as a percentage of the controls (mean ± SEM) and were analyzed using two-way ANOVA followed by Tukey's post hoc test. *p < 0.05. **p < 0.01. n = 4–7. Scale bar, 50 μm.
To further investigate whether reduced neuroinflammation was the result of diminished microglial activation in LPS-treated TAC1−/− mice, the activation of microglia in the SN was morphologically examined by immunostaining for Iba1 and CD11b, two markers of microglial activation. Ten months after LPS injection, the nigral microglia of WT mice remained activated, exhibiting a hypertrophied morphology and an elevated density of Iba-1 (121 ± 7%) and CD11b (159 ± 12%) in the SN (Fig. 2D–G). In contrast, microglial activation was greatly attenuated in LPS-treated TAC1−/− mice, which exhibited a ramified morphology and a reduced expression of Iba-1 (two-way ANOVA: genotype, F(1,16) = 6.8, p = 0.0190) and CD11b (two-way ANOVA: genotype, F(1,16) = 6.2, p = 0.0245) versus the LPS-treated WT mice. Together, these data suggest that endogenous SP potentiates LPS-induced neuroinflammation.
Exogenous SP potentiates LPS- or MPP+-induced toxicity in dopaminergic neurons by activating microglia in vitro
To dissect the mechanism underlying the reduced degeneration of nigrostriatal dopaminergic neurons in LPS- or MPTP-treated TAC1−/− mice, we further examined the effects of exogenous SP in primary cell cultures. Midbrain neuron-glia cultures were coexposed to a wide range of SP concentrations (10−14 to 10−7 m), together with either LPS (10 ng/ml) or MPP+ (0.1 μm) for 7 d. LPS and MPP+ reduced DA uptake by 32% and 25%, respectively, indicating a loss of dopaminergic neurons (Fig. 3A,B). Compared with our previous report (Block et al., 2006), which demonstrated dopaminergic neurodegeneration by SP at 10−7 and 10−13 m, but not 10−10 m, this study showed that SP alone (both at 10−7 and 10−13 m) decreased DA uptake capacity; however, this change did not reach significance, likely because of minor differences in culturing conditions. However, it was interesting to observe a bimodal dose–response to SP in enhancing LPS- and MPP+-induced dopaminergic neurodegeneration. Two-way ANOVA analysis (LPS or MPP+ and SP with different concentrations) demonstrated that SP and LPS/MPP+ had significant interactions (SP × LPS, F(6,57) = 2.9, p = 0.0150; SP × MPP+, F(6,61) = 2.3, p = 0.0363). According to Tukey's comparisons, SP cotreatment augmented LPS/MPP+-induced neurotoxicity at both submicromolar (10−8 to 10−7 m) and subpicomolar concentrations (10−14 to 10−13 m), as detected by a further significant reduction of DA uptake, but not at subnanomolar concentrations (10−11 to 10−10 m; Fig. 3A,B). Possible explanations for the inactivity of SP at subnanomolar concentrations are provided in Discussion. To offer a possible explanation for this bimodal phenomenon, we speculated that SP released from the terminals of the striatonigral projecting neurons might reach submicromolar concentrations in the synaptic regions, whereas the concentrations available to act on microglia situated outside of synapses would be substantially lower, as SP must diffuse out of the synapses and escape peptidase degradation. It is difficult to determine the exact concentrations of SP released to act on microglia in the SN region, but the reported extracellular concentration of this peptide, measured by a microdialysis device followed by a radioimmunoassay, was in the picomolar range (Warsame Afrah et al., 2004; Ebner and Singewald, 2007). Based on these earlier reports, plus the bimodal dose–response (Fig. 3A,B), we speculated that subpicomolar concentrations could be more like the physiological conditions that act on microglia; thus, 10−14 to 10−13 m of SP was used for the subsequent in vitro studies. Consistent with the decreased DA uptake capacity, immunocytochemical analysis revealed synergistic effects of combined 10−14 to 10−13 m SP and LPS or MPP+ on damage to neuronal processes and loss of THir neurons in cultures treated for 7 d (SP/LPS, F(4,14) = 134.4, p < 0.0001; SP/MPP+, F(3,12) = 24.1, p < 0.0001; Tukey's comparisons test between combined SP and LPS/MPP+ and LPS/MPP+ alone, all p < 0.01; Figure 3C–E).
Figure 3.
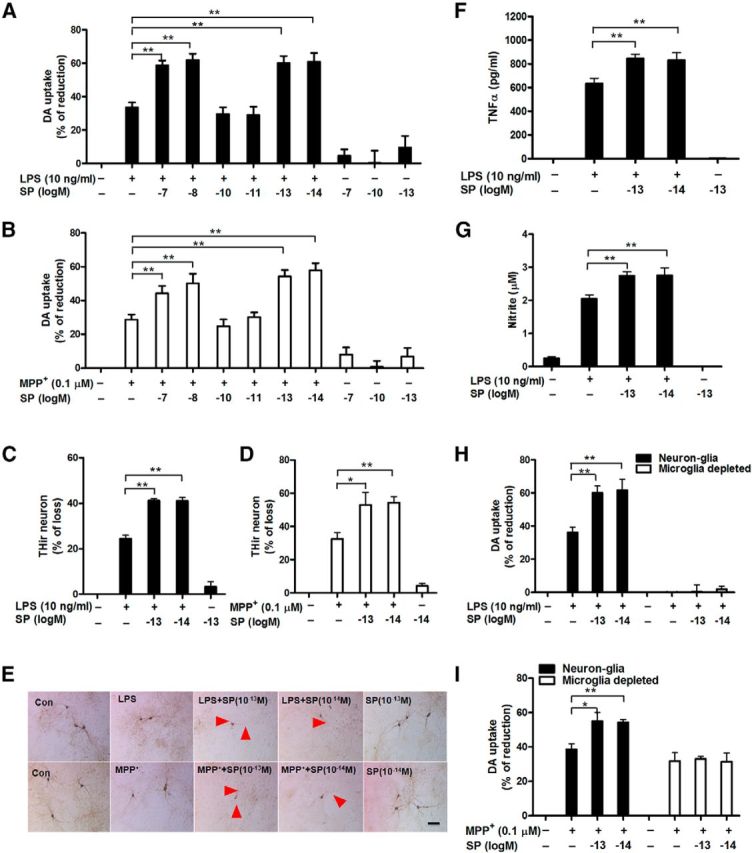
Exogenous SP potentiates dopaminergic neurodegeneration by activating microglia. A–G, Rat mesencephalic neuron-glia and microglia-depleted (H, I; microglia were removed from neuron-glia cultures by L-leucine methyl ester) cultures were treated with the indicated concentrations of SP and LPS (10 ng/ml) or MPP+ (0.1 μm). A, B, I, Seven days later, dopaminergic neurotoxicity was determined by [3H]-DA uptake assay and (C, D) concurrently quantified through immunocytochemistry for THir neuronal loss (n = 4–7) and (E) neurite atrophy (red arrowheads; n = 3). TNFα (F) and nitrite (G) levels were measured in the supernatant after 3 and 24 h of treatment, respectively. The results of [3H]-DA uptake and THir neuron counts are expressed as the percentage of reduced [3H]-DA uptake capacity and loss of THir cells (mean ± SEM, control group was considered as 0, no damage), respectively. The results of TNFα and nitrite are mean ± SEM. The data were analyzed using one- or two-way ANOVA followed by Tukey's post hoc test. *p < 0.05. **p < 0.01. Scale bar, 50 μm.
To investigate the role of microglia in subpicomolar SP-exacerbated dopaminergic neurodegeneration, we measured the production of microglia-derived proinflammatory factors, which work in concert to induce neurodegeneration (Block et al., 2007). Our results showed that 10−13 and 10−14 m SP alone failed to stimulate the release of proinflammatory factors but significantly augmented the LPS-induced production of TNFα (F(4,20) = 130.2, p < 0.0001; Fig. 3F) and nitrite (F(4,25) = 42.7, p < 0.0001; Fig. 3G). Furthermore, microglia-depleted cultures (microglia were removed from neuron-glia cultures by L-leucine methyl ester) were compared with the neuron-glia cultures for their susceptibility to neurotoxicity induced by SP and LPS or MPP+. Unlike the neuron-glia cultures, the DA uptake assay showed no synergistic toxicity to dopaminergic neurons in microglia-depleted cultures treated with combined SP and either LPS or MPP+ (two-way ANOVA, SP/LPS group: culture, F(1,30) = 125.1, p < 0.0001; SP/ MPP+ group: culture, F(1,37) = 21.8, p < 0.0001; Tukey's comparisons test between combined SP and LPS or MPP+ and LPS or MPP+ alone in microglia-depleted culture, all p > 0.05; Figure 3H,I). This finding indicates an essential role for microglia in mediating the synergistic toxic effects. The microglia-depleted cultures did not respond to LPS because microglia are required for LPS-induced toxicity (Qin et al., 2004).
NK1R deletion fails to block the synergistic effects induced by combined SP and LPS or MPTP in vivo and in vitro
To delineate whether NK1R mediates the synergistic effects of SP, we used transgenic mice with a genetic ablation of the SP receptor gene (NK1R−/−). As shown in Figure 4A, LPS treatment elicited a comparable dopaminergic neuron loss in the SN of WT and NK1R−/− mice (two-way ANOVA: genotype, F(1,16) = 0.07, p = 0.7900; treatment, F(1,16) = 57.1, p < 0.0001). Consistently, WT and NK1R−/− mice exhibited a similar sensitivity to MPTP-induced nigral DA neuron loss and fiber degeneration (Fig. 4B,C). Furthermore, NK1R deletion failed to prevent LPS-induced nigral microglial activation and the gene expression of proinflammatory factors (Fig. 4D–F), indicating that the potentiated neurotoxicity of endogenous SP was independent of NK1R.
Figure 4.

NK1R deletion fails to reduce LPS/MPTP-induced nigral dopaminergic neurodegeneration and LPS-induced neuroinflammation in vivo. A single dose of LPS (15 × 106 EU/kg, i.p.) or a repeated MPTP regimen (15 mg/kg, s.c. for 6 consecutive days) was administered to NK1R−/− or WT mice. Ten months after LPS or 21 days after the last MPTP injection, the brains were collected. A, B, Nigral THir neurons in LPS-treated (A) and MPTP-treated (B) NK1R−/− and WT mice were counted stereologically. C, Striatal dopaminergic neuron fibers were quantified by TH density. D–F, Ten months after LPS injection, the mRNA levels of TNFα (D), iNOS (E), and MCP-1 (F) were determined in brains using RT-PCR. The results are expressed as the percentage of THir cell loss or density reduction (mean ± SEM, WT controls were considered as 0, no damage) in A–C and are expressed as a percentage of the controls (mean ± SEM) in D–F. The data were analyzed using two-way ANOVA followed by Tukey's post hoc test. n = 4–7. NS, Not significant.
To further corroborate our in vivo findings, we determined whether NK1R was essential in mediating subpicomolar SP-elicited neurotoxicity in vitro. Midbrain neuron-glia cultures prepared from NK1R−/− and WT mice were treated with SP in the presence of either LPS or MPP+. We found no difference between NK1R−/− and WT groups in the subpicomolar SP-elicited synergistic neurotoxicity measured by both DA uptake and THir cell counts (Fig. 5A,B,D,E). Additional evidence supporting the NK1R-independent actions of subpicomolar SP came from the measurement of TNFα and nitrite. Subpicomolar SP alone failed to stimulate the release of TNFα and nitrite but synergistically enhanced the LPS-induced production of these two proinflammatory factors in both WT and NK1R−/− neuron-glia cultures (data not shown). It is interesting to note, in direct contrast to the results from subpicomolar SP, at submicromolar concentrations of SP (10−7 to 10−8 m), the potentiated toxicity to dopaminergic neurons with either LPS or MPP+ was completely abolished in the NK1R−/− neuron-glia cultures (Fig. 5C,F).
Figure 5.

NOX2, but not NK1R, mediates the synergistic neurotoxicity of subpicomolar concentrations of SP in combination with LPS or MPP+, although NK1R is required for submicromolar SP-potentiated toxicity. A, B, Mesencephalic neuron-glia cultures prepared from WT (C57 BL/6J), NK1R−/−, and gp91phox−/− mice were cotreated with subpicomolar concentrations of SP and LPS (10 ng/ml) or (D, E) MPP+ (0.1 μm). Seven days later, the synergistic toxicity to dopaminergic neurons was quantified by (A, D) [3H]-DA uptake and (B, E) THir neuron counts (n = 5–10). C, F, Midbrain neuron-glia cultures prepared from WT and NK1R−/− mice were cotreated with submicromolar concentrations of SP (10−8 to 10−7 m) and (C) LPS (10 ng/ml) or (F) MPP+ (0.1 μm). Seven days later, the synergistic toxicity to dopaminergic neurons was quantified by [3H]-DA uptake. The results are expressed as the percentage of reduced [3H]-DA uptake capacity or loss of THir cells (mean ± SEM, control group was considered as 0, no damage). The data were analyzed using two-way ANOVA followed by Tukey's post hoc test. *p < 0.05. **p < 0.01.
Microglial NOX2 is essential for the synergistic neurotoxicity of SP with LPS or MPP+
NOX2 is a key enzyme producing the respiratory burst in phagocytes, including microglia. The overproduction of NOX2-generated superoxide is critically linked to the secretion of a wide spectrum of proinflammatory factors from microglia, leading to chronic neuroinflammation and dopaminergic degeneration (Lambeth et al., 2008; Sorce and Krause, 2009). We previously reported that microglial NOX2 can recognize a variety of substances with a higher affinity than traditional GPCRs, including NK1R (Qin et al., 2005). To determine the role of NOX2 in the potentiating effects of subpicomolar SP, dopaminergic neurodegeneration in neuron-glia cultures prepared from either WT or NOX2-deficient (gp91phox−/−) mice was compared in the presence of SP combined with LPS or MPP+. As shown in Figure 5A, B, D, E, subpicomolar SP and LPS or MPP+ synergistically damaged dopaminergic neurons in neuron-glia cultures prepared from WT mice. LPS or MPP+ alone produced less degeneration of dopaminergic neurons in cultures prepared from gp91phox−/− mice. Importantly, subpicomolar levels of SP failed to show any potentiation of the toxic effects induced by LPS or MPP+ (two-way ANOVA, SP/LPS: genotype, DA uptake, F(1,50) = 50.0, p < 0.0001, THir counts, F(1,44) = 102.1, p < 0.0001; SP/MPP+: DA uptake, F(1,41) = 29.9, p < 0.0001, THir counts, F(1,44) = 74.4, p < 0.0001; Tukey's comparisons test between combined SP and LPS or MPP+ and LPS or MPP+ alone in DA uptake and THir neuron counts in gp91phox−/− cultures, all p > 0.05). The critical role of NOX2 in SP-potentiated toxicity was further verified by lack of synergistic TNFα and nitrite production in gp91phox−/− neuron-glia cultures (data not shown). Therefore, the presence of NOX2 was required for the synergistic toxicity induced by subpicomolar SP and LPS or MPP+.
Subpicomolar concentrations of SP enhance NOX2-generated superoxide in an NK1R-independent manner
A series of experiments were performed to determine whether the subpicomolar SP-elicited activation of microglial NOX2 occurs through direct action on this enzyme or indirectly through the activation of NK1R. We first showed that 10−13 and 10−14 m SP alone increased the production of superoxide (F(5,24) = 13.3, p < 0.0001) and iROS (F(5,28) = 17.1, p < 0.0001) and that production was further enhanced when subpicomolar SP was combined with LPS (Fig. 6A,B). Further studies have provided much evidence that clearly demonstrates that subpicomolar SP directly activated microglial NOX2 in a NK1R-independent fashion. (1) SP, at 10−14 and 10−13 m, stimulated superoxide production (F(3,21) = 9.2, p = 0.0004) as potently as 10 nm PMA (a commonly used stimulator of NOX2-generated superoxide) in COS-7-NOX2 cells, which contain all the subunits of NOX2 in COS-7 cells (Wang et al., 2012a) (Fig. 6C). Because there is no detectable NK1R expression in COS-7 cells (Fong et al., 1992), this result strongly suggested a direct activation of NOX2 by subpicomolar SP. (2) The deletion of NOX2 (gp91phox−/−), but not NK1R, abolished subpicomolar SP-induced superoxide production, further supporting the possibility that subpicomolar SP directly activates NOX2 in an NK1R-independent manner (two-way ANOVA: genotype, F(2,48) = 27.6, p < 0.0001; Tukey's comparisons test between SP and vehicle control in gp91phox−/− culture, all p < 0.01; in NK1R−/− culture, all p > 0.05; Fig. 6D). It is important to note that NK1R was necessary for submicromolar SP-induced NOX2 activation, as 10−7 and 10−8 m SP-induced NOX2-generated superoxide was greatly reduced in cultures prepared from NK1R−/− mice (data not shown).
Figure 6.

Subpicomolar SP concentrations activate NOX2 through binding to the membrane subunit, gp91phox. A, Midbrain neuron-glia cultures were treated with subpicomolar concentrations of SP with or without LPS. Superoxide and (B) iROS production was measured (n = 5–7). C, SP-elicited superoxide production was determined in COS-7-NOX2 cells (COS-7 cells were transfected with gp91phox, p22phox, and cytosolic subunits of NOX2) and (D) neuron-glia cultures prepared from WT (C57BL/6J), NK1R−/−, and gp91phox−/− mice (n = 6 or 7). PMA was used as a positive control. E, [125I]-SP binding was performed in membranes of COS-7 and (F) COS-cytochrome b558 (transfected with membrane bound subunits, gp91phox and p22phox) cells. The total binding was measured by the inclusion of [125I]-labeled SP at concentrations ranging from 2 to 10 × 10−13 m, whereas nonspecific binding was determined in the presence of an additional 0.1 μm SP. Specific binding was calculated by the difference between total binding and nonspecific binding (n = 3). G, H, The binding capacity of subpicomolar levels of SP with gp91phox was determined by using [125I]-SP in membranes or whole cell of different transfected COS-7 cells (COS-7: transfected with empty vector; COS-7-cytochrome b558: transfected with gp91phox and p22phox; COS-7-NOX2: transfected with both membrane and cytosolic subunits) and (I) macrophages derived from wild-type, gp91phox−/−and NK1R−/− mice (n = 4–7). Specific binding was calculated and expressed as a percentage of WT or empty vector-transfected COS-7 cells. J, SP-induced superoxide was measured in COS-7, COS-7-cytochrome b558, and COS-7-NOX2 cells (n = 3–5). K, The membrane translocation of cytosolic subunits p47phox and p67phox induced by SP and/or LPS was measured using Western blot, and (L) the density of the blot was quantified (n = 5 or 6). Gp91phox, an abundant membrane protein, was used as an internal control. Our previous reports indicated that gp91phox could serve as a reliable internal membrane control (Hu et al., 2008; Qian et al., 2008; Gao et al., 2011b). The results are expressed as the percentage of controls (mean ± SEM) and were analyzed using one or two-way ANOVA by Tukey's post hoc test. *p < 0.05. **p < 0.01.
Subpicomolar SP targets the membrane-bound subunit of NOX2 gp91phox
Because subpicomolar SP-stimulated NOX2 activation is NK1R independent, and SP is membrane-impermeable (because of being positively charged at physiological pH), we speculated that membrane subunit gp91phox might be the potential target of subpicomolar SP. To test this hypothesis, a series of ligand-binding experiments were performed. To characterize the binding profile, different concentrations of [125I]-SP were incubated with membranes prepared from COS-7 (transfected with an empty vector) and COS-7-cytochrome b558 cells (COS-7 cells that were stably transfected with gp91phox and p22phox plasmids). The reason for the cotransfection of p22phox with gp91phox is that p22phox facilitates the stable expression of gp91phox in the membrane (Yu et al., 1997). In the empty vector-transfected COS-7 cells, no significant specific binding was observed, as the total binding and the nonspecific binding displayed very similar linear curves (Fig. 6E). In contrast, specific binding was clearly demonstrated in COS-7-cytochrome b558 cells (Fig. 6F). Quantitative analysis revealed that the binding affinity was in the low subpicomolar range (Kd value was 3 × 10−13 m). At 3 × 10−13 m [125I]-SP, membranes from COS-7-cytochrome b558 cells had a twofold to threefold higher binding capacity for the radiolabeled ligand than did the membranes from the empty vector-transfected COS-7 cells (t(12) = 5.8, p < 0.001; Fig. 6G). Similarly, a more than threefold increase of [125I]-SP binding was observed in COS-7-NOX2 (COS-7 cells that were stably transfected with all of NOX2 subunits) compared with empty vector-transfected COS-7 cells, which do not express gp91phox (t(6) = 3.6, p = 0.0112; Fig. 6H). To further demonstrate the interaction of SP and gp91phox, [125I]-SP binding assays were performed in macrophages derived from WT and gp91phox−/− mice. We used peritoneal macrophages instead of microglia for the following reasons: (1) it is difficult to harvest a sufficient amount of enriched primary microglia for binding experiments; (2) microglia are considered to be the resident macrophages in the brain, and NOX2 is expressed in both cell types; and (3) macrophages display higher levels of NOX2 activity than do microglia. As shown in Figure 6I, gp91phox knock-out, but not NK1R knock-out, significantly decreased the binding of SP to macrophage membranes (F(2,12) = 10.1, p = 0.0027, Tukey's comparisons test between WT and NK1R−/−, p = 0.9914), indicating that SP bound to gp91phox independently of NK1R. These results suggest that subpicomolar concentrations of SP interact with flavocytochrome b558, and especially the gp91phox subunit.
Cytosolic subunits are necessary for subpicomolar SP-induced NOX2-generated superoxide
The phosphorylation of the cytosolic components p47phox and p67phox and their subsequent translocation to the plasma membrane are required for NOX2 activation. To investigate whether SP-induced NOX2 activation requires the cytosolic subunits, SP-produced superoxide was measured in different transfected COS-7 cells: (1) COS-7 cells (transfected with empty vector); (2) COS-7-cytochromeb558 cells (transfected with membrane subunits); and (3) COS-7-NOX2 cells (transfected with both membrane and cytosolic subunits). SP-stimulated superoxide was observed in COS-7-NOX2 but not in COS-7-cytochrome b558 cells (Fig. 6J), suggesting that the cytosolic subunits were necessary for SP-elicited NOX2 activation. Furthermore, Western blot analysis revealed membrane translocation of p47phox (F(5,34) = 6.9, p = 0.0002) and p67phox (F(5,24) = 10.7, p < 0.0001) in microglia treated with 10−13 and 10−14 m SP (Fig. 6K,L). In the presence of LPS, SP induced a high degree of p47phox and p67phox membrane translocation, indicating enhanced NOX2 activation. Collectively, our data suggest that subpicomolar SP activates NOX2 by binding to gp91phox and inducing the translocation of cytosolic subunits to plasma membranes.
Subpicomolar concentrations of SP potentiate LPS-induced activation of microglial MAPK and nuclear factor-κB (NF-κB) in a NOX2-dependent manner
NOX2-generated superoxide is known to increase iROS, which in turn affects downstream signaling pathways, such as mitogen-activated protein kinases (MAPK) and NF-κB, enhancing the production of proinflammatory factors (Block et al., 2007). Given that SP alone failed to produce proinflammatory cytokines but potentiated the effects of LPS (Fig. 3F,G), we hypothesized that subpicomolar SP could potentiate the LPS-induced activation of MAPK and NF-κB through NOX2 activation. As shown in Figure 7A, B, LPS significantly activated ERK1/2, p38 and JNK in microglia compared with vehicle-treated controls. SP alone at 10−13 and 10−14 m failed to activate MAPK but markedly enhanced LPS-induced phosphorylation of ERK (F(5,35) = 14.3, p < 0.0001), p38 (F(5,35) = 14.1, p < 0.0001) and JNK (F(5,33) = 13.5, p < 0.0001). A similar synergistic NF-κB activation was observed in SP and LPS-treated microglia. After LPS treatment, microglia exhibited increased phosphorylation of IKKα/β, IκBα, and p65 and reduced levels of total IκBα compared with the controls, indicating activation of the NF-κB pathway. More pronounced NF-κB activation was observed in the presence of subpicomolar SP (Fig. 7A,C).
Figure 7.

Subpicomolar SP potentiates LPS-induced activation of MAPK and NF-κB in microglia in a NOX2-dependent manner. A, Microglia-enriched cultures were treated with SP and/or LPS for 15 min. Levels of phosphorylated and nonphosphorylated MAPK (ERK1/2, p38, and JNK1/2) and NF-κB (IKKα/β, IκBα, and p65) were determined by Western blot using specific antibodies against phosphorylated or total MAPK and nuclear factor-κB, respectively. B, C, MAPK and NF-κB activation on the gel from each group were quantified by analysis of the blot density (n = 5). D, Microglia-enriched cultures were pretreated with 10−14 m DPI for 30 min before the addition of SP and/or LPS. Fifteen minutes later, the activation of MAPK and NF-κB was assessed using Western blot. E, F, The density of the blot was quantified (n = 5). The results are expressed as a percentage of the controls (mean ± SEM) and were analyzed using one-way ANOVA, followed by Tukey's post hoc test. *p < 0.05. **p < 0.01.
To test whether the synergistic activation of MAPK and NF-κB induced by subpicomolar SP and LPS was dependent on NOX2, DPI (10−14 m) was used to block NOX2 activation. As expected, NOX2 inhibition attenuated 10−14 m SP-potentiated MAPK and NF-κB activation in microglia (Fig. 7D–F). Thus, it is likely that subpicomolar levels of SP activated NOX2 to increase iROS concentrations and subsequently synergistically activated the MAPK and NF-κB pathway, contributing to the potentiated proinflammatory cytokine production.
Inhibition of NOX2-generated superoxide protects dopaminergic neuron against SP/LPS- or SP/MPP+-induced synergistic toxicity
The central role of NOX2 in the synergistic neurotoxicity of subpicomolar SP with LPS or MPP+ prompted us to test whether inhibition of NOX2-generated superoxide was capable of protecting dopaminergic neurons. Neuron-glia cultures were pretreated with DPI (10−14 m) to inhibit activation of NOX2. Pretreatment with 10−14 m DPI significantly reduced superoxide production induced by either SP alone (376 ± 59% for SP alone vs 86 ± 29% of DPI/SP group) or combined with LPS (675 ± 111% for SP/LPS vs 215 ± 108% of DPI/SP/LPS group; Fig. 8A). Similar inhibitory effects of DPI on iROS were observed (Fig. 8B). Importantly, NOX2 inhibition by DPI completely abolished the synergistic effect of SP on LPS- or MPP+- induced dopaminergic toxicities (Fig. 8C–E). Analysis of [3H]-DA uptake and THir neuronal counts revealed that DPI reduced the dopaminergic neuron loss from 40%–50% to 10%–20% in both LPS/SP- and MPP+/SP-treated cultures (DA uptake: SP/LPS, F(4,17) = 36.6, p < 0.0001; SP/MPP+, F(3,16) = 27.3, p < 0.0001; THir neuron counts: SP/LPS, F(4,28) = 23.7, p < 0.0001; SP/MPP+, F(3,22) = 27.2, p < 0.0001; Tukey's comparisons test between combined SP and LPS or MPP+ and LPS or MPP+ alone in DA uptake and THir neuron counts, all p < 0.01).
Figure 8.

Inhibition of NOX2-generated ROS protects dopaminergic neurons from SP/LPS- or SP/MPP+-induced synergistic neurotoxicity. Midbrain neuron-glia cultures were pretreated with 10−14 m DPI for 30 min before the addition of SP (10−14 m) and LPS or MPP+. Production of superoxide (A) and iROS (B) was accessed by measuring the SOD-inhibitable reduction of tetrazolium salt, WST-1 and increase of CM-H2-DCFDA, respectively (n = 5). The results of superoxide and iROS are expressed as a percentage of controls and above the control (mean ± SEM), respectively. The inhibitory effect of 10−14 m DPI on the synergistic dopaminergic neurodegeneration induced by SP and LPS (C) or MPP+ (D) was determined at 7 d of post-treatment by performing [3H]-DA uptake assay and THir neuron counts. E, Representative images confirmed the neuroprotection of dopaminergic neurons by DPI as shown by more THir neurons with longer and more abundant neurites compared with combined SP and LPS/MPP+ group (red arrowheads; n = 3). The results are expressed as a percentage of reduction of [3H]-DA uptake capacity or loss of THir cells (mean ± SEM, control group was considered as 0, no damage). Data were analyzed using one-way ANOVA, followed by Tukey's post hoc test. **p < 0.01. Scale bar, 50 μm.
Discussion
Using in vivo and in vitro dopaminergic neurodegenerative models, the current study demonstrated a novel role for SP in inflammation-related neurodegeneration and elucidated its underlying mechanisms. Our in vivo experiments found that both LPS- and MPTP-induced microglia-mediated neuroinflammation and dopaminergic neurodegeneration in the SN were greatly reduced in mice lacking endogenous SP (Figs. 1 and 2) but not in mice lacking the SP receptor, NK1R (Fig. 4). This is the first report documenting that a novel, NK1R-independent signaling pathway mediates the neurotoxic actions of endogenous SP. A series of in vitro studies revealed that microglial NOX2-generated superoxide was required for the novel NK1R-independent signaling pathway capable of modulating the proinflammatory/neurotoxic effects of SP (Fig. 9). The results from our study provide insights into the pathophysiological role of SP in dopaminergic neurodegeneration. Furthermore, our bimodal dose–response studies of SP help to reconcile the discrepancy between previous reports of its NK1R dependency.
Figure 9.

Proposed model showing how SP exacerbates inflammatory responses and dopaminergic degeneration in the presence of LPS or MPTP in an NK1R-independent manner. Left, SP release from the nerve terminals projected from the striatonigral pathway. The synaptic concentrations of SP can reach submicromolar levels and act on NK1R to mediate acute synaptic transmission. Middle, Extrasynaptic actions of SP. Some of the released SP may escape the enzyme degradation and diffuse outside the synapses to activate the surrounding microglia. Our data show that subpicomolar concentrations of SP increase superoxide production and enhance iROS through the activation of microglial NOX2. The NK1R-independent activation of NOX2 by subpicomolar levels of SP could be a key pathway mediating the potentiated effects of LPS-induced activation of both MAPK and NF-κB and leading to an enhanced production of proinflammatory factors. Right, The mechanism by which the enhanced neuroinflammation produced by subpicomolar SP and neurotoxins causes dopaminergic neurodegeneration. Although LPS and MPTP exert neurotoxic effects through different mechanisms, the activation of microglia is directly involved in the action of LPS, whereas reactive microgliosis is indirectly involved in part of MPTP-induced neurotoxicity. The common pathway underlying the potentiating effect of subpicomolar SP on these two toxins is mediated by microglial NOX2. This schematic only shows the non–NK-1R-mediated neuroinflammatory and neurodegenerative effects of SP. In all likelihood, this neuropeptide can modulate microglial function via both NK-1R dependent and independent mechanisms, depending on the local SP concentrations.
Previous studies delineating the role of NK1R in SP-mediated proinflammatory/neurotoxic effects have generated contrasting results. Exogenous SP has been shown to augment proinflammatory responses elicited by bacterial stimulation and these effects were reversed with NK1R ablation or treatment with NK1R antagonists (Chauhan et al., 2008, 2011). Similarly, SP injected into the lateral ventricle, potentiated microglial activation, and dopaminergic neurodegeneration caused by 6-hydroxydopamine, which was attenuated by NK1R antagonists (Thornton and Vink, 2012). In contrast, several studies reported the NK1R-independent actions of SP. NK1R deficiency elevated the LPS-induced proinflammatory cytokine production in a murine airway inflammation model, yet reduced cytokine production in LPS-treated TAC1−/− and TAC1/NK1R double-deficient mice (Helyes et al., 2010). Additionally, NK1R-independent inflammation of SP was also observed in brain of a mouse model of post-treatment reactive encephalopathy, in which the SP-derived neuroinflammatory response was increased in mice deficient in NK1R (Kennedy et al., 2003). Furthermore, Chu et al. (2011) reported that treatment of NK1R agonist septide [(Pyr6, Pro9)-SP (6–11)] was found to ameliorate dopaminergic neurodegenerarion and motor deficit in 6-hydroxydopamine-lesioned rats. Our in vivo data, indicating that ablation of endogenous SP, but not NK1R, decreased microglia-mediated neuroinflammation and dopaminergic neurodegeneration caused by either LPS or MPTP, supported the idea of NK1R-independent actions of SP (Figs. 1, 2, and 4). We speculated that the opposite conclusions from previous reports might be resulted from different experimental designs: the pharmacological approach by studying the effects after addition of exogenous SP versus the physiological approach by studying the consequences after deletion of either endogenous SP or the NK1R gene. We theorized that the difference in the concentrations of exogenous SP versus physiological in situ conditions could be a critical factor underlying these discrepancies. Therefore, we hypothesized that different concentrations of SP could use different signaling pathways to produce proinflammatory and neurotoxic effects.
To test our hypothesis, we performed a series of in vitro experiments using primary neuron-glia cultures. These cultures were prepared from different genetically manipulated mice and exposed to SP in the presence of either LPS or MPP+. A wide range of SP concentrations (10−14 to 10−7 m) was used. Our in vitro findings show an interesting bimodal dose–response curve for the potentiating effects of SP in the toxin-induced loss of dopaminergic neurons (Fig. 3): SP is effective at submicromolar (10−8 to 10−7 m) and subpicomolar (10−14 to 10−13 m) concentrations, but not at the intermediate concentrations (10−11 to 10−10 m). Interestingly, we found that, although the enhanced neurotoxicity was NK1R-dependent at submicromolar concentrations of SP, NK1R was not necessary for the actions of SP at subpicomolar concentrations (Fig. 5). These differences provide critical insights into the pathophysiological role of SP in regulating dopaminergic neuron survival, which will be further discussed below.
Notably, a bimodal dose–response curve was also observed in the SP-elicited production of superoxide from microglial NOX2, which is nearly identical to that observed for its neurotoxicity in dopaminergic neurons. Our subsequent experiments revealed different signaling pathways affecting superoxide production. The generation of superoxide by submicromolar SP (10−8 to 10−7 m) required NK1R (data not shown). In contrast, SP at 10−13 to 10−14 m generated as much superoxide as at 10−7 to 10−8 m, but its effect was NK1R-independent. The similar patterns of SP-induced superoxide production and dopaminergic neurotoxicity suggest a critical role for superoxide in potentiating dopaminergic neurotoxicity. Together, our results may explain the different conclusions regarding the necessity of NK1R for the proinflammatory and neurodegenerative actions of SP. On one end of the bimodal dose–response (submicromolar concentrations), our findings showed NK1R-dependent actions of SP. In contrast, subpicomolar SP-elicited actions were NK1R independent. Thus, with regard to NK1R dependency, our in vitro subpicomolar, but not submicromolar, SP data correlated well with our in vivo findings. We propose that the in vitro results, at submicromolar concentrations, represent pharmacological actions and may not reflect physiological conditions. It is likely that SP released from nerve terminals can reach submicromolar concentrations in synaptic junctions and transmits signals between nerves or nerve/effector organs through binding to NK1R (Jalkanen et al., 2011). In contrast, we believe that the extrasynaptic concentrations of SP that act on microglia to modulate their immune function are likely below the submicromolar range and the regulatory effects occur in an NK1R-independent manner. Unfortunately, there are currently no detection methods with sufficient sensitivity to detect SP at these concentrations. To support our hypothesis, we found that the synergistic toxicity elicited by subpicomolar SP was abolished in microglia-depleted cultures (Fig. 3H,I). These findings have extended our knowledge regarding the pathophysiological role of SP in neuroinflammation beyond its conventional actions on NK1R.
There are additional interesting questions related to the bimodal dose–response of SP-induced superoxide production worthy of discussion. The first question is the site of action of ultra-low SP concentrations. We have identified microglial NOX2 as a potential target site mediating the subpicomolar SP-elicited potentiation of LPS- and MPP+-induced neuroinflammation and neurodegeneration. Ligand binding studies further indicated that SP bound to the membrane catalytic subunit gp91phox of NOX2 with extremely high-affinity (Kd, ∼10−13 m) to activate this enzyme by inducing the translocation of cytosolic subunits (p47phox, p67phox) to the plasma membrane to form an active complex that generates superoxide (Fig. 6). The second question is whether submicromolar SP levels can directly act on microglial NOX2. Our findings indicate that an indirect action through NK1R is more likely. Although submicromolar SP-elicited effects were dependent on the presence of both NK1R and NOX2, submicromolar levels of SP failed to produce superoxide in NK1R-deficient microglia. The NK1R-dependent activation of NOX2 was further supported by decreasing SP-stimulated superoxide through inhibition of the downstream GPCR signals. Pharmacological inhibition of PKC decreases SP-induced superoxide generation in microglia, whereas the genetic ablation of β-arrestin 1 increases this generation (Q.S.W. et al., unpublished observation). These findings led to the third question: why do subnanomolar concentrations of SP fail to increase NOX2-generated superoxide? In addition, why don't submicromolar SP concentrations directly promote NOX2 activity? Although the answers to these questions remain unclear, we speculate that the direct action of SP on gp91phox can only stimulate this enzyme to produce superoxide at subpicomolar concentrations and may be inhibited at higher concentrations due to actions on unidentified allosteric site(s), which interferes with NOX2 activation.
In conclusion, our studies reveal a new immunomodulatory function for SP and a role for SP in the long-term survival of dopaminergic neurons in the SN. We also identified microglial NOX2/superoxide as a potential novel signaling pathway that mediates the proinflammatory and neurotoxic effects of SP. These findings help to expand our understanding beyond the conventional knowledge paradigm that peptides act exclusively on their GPCRs. These findings on the novel actions of SP could also apply to other peptides. For example, we have previously reported the bacterial chemoattractant peptide, formyl-methionyl-leucyl-phenylalanine enhanced the production of superoxide by activating microglial NOX2 (Gao et al., 2008). In contrast, several anti-inflammatory peptides, such as dynorphin (Qin et al., 2005) or pituitary adenylate cyclase-activating peptide (Yang et al., 2006), can inhibit activated microglial NOX2 at similar ultra-low concentrations. It is particularly relevant to mention that dynorphin, one of endogenous opioid peptides, colocalizes with SP in striatonigral-projecting neurons and could act as a counterbalance in maintaining immunohomeostasis and modulating the inflammatory response and subsequent dopaminergic neuron survival in pathological states.
Footnotes
This work was supported by the Intramural Research Program of the National Institutes of Health, National Institute of Environmental Health Sciences. We thank Anthony Lockhart for assistance with animal colony management and maintenance of the timed pregnant mice.
The authors declare no competing financial interests.
References
- Baiguera C, Alghisi M, Pinna A, Bellucci A, De Luca MA, Frau L, Morelli M, Ingrassia R, Benarese M, Porrini V, Pellitteri M, Bertini G, Fabene PF, Sigala S, Spillantini MG, Liou HC, Spano PF, Pizzi M. Late-onset Parkinsonism in NFkappaB/c-Rel-deficient mice. Brain. 2012;135:2750–2765. doi: 10.1093/brain/aws193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biber K, Neumann H, Inoue K, Boddeke HW. Neuronal ‘On’ and ‘Off’ signals control microglia. Trends Neurosci. 2007;30:596–602. doi: 10.1016/j.tins.2007.08.007. [DOI] [PubMed] [Google Scholar]
- Block ML, Li G, Qin L, Wu X, Pei Z, Wang T, Wilson B, Yang J, Hong JS. Potent regulation of microglia-derived oxidative stress and dopaminergic neuron survival: substance P vs dynorphin. FASEB J. 2006;20:251–258. doi: 10.1096/fj.05-4553com. [DOI] [PubMed] [Google Scholar]
- Block ML, Zecca L, Hong JS. Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nat Rev Neurosci. 2007;8:57–69. doi: 10.1038/nrn2038. [DOI] [PubMed] [Google Scholar]
- Brownstein MJ, Mroz EA, Kizer JS, Palkovits M, Leeman SE. Regional distribution of substance P in the brain of the rat. Brain Res. 1976;116:299–305. doi: 10.1016/0006-8993(76)90907-0. [DOI] [PubMed] [Google Scholar]
- Chauhan VS, Sterka DG, Jr, Gray DL, Bost KL, Marriott I. Neurogenic exacerbation of microglial and astrocyte responses to Neisseria meningitidis and Borrelia burgdorferi. J Immunol. 2008;180:8241–8249. doi: 10.4049/jimmunol.180.12.8241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chauhan VS, Kluttz JM, Bost KL, Marriott I. Prophylactic and therapeutic targeting of the neurokinin-1 receptor limits neuroinflammation in a murine model of pneumococcal meningitis. J Immunol. 2011;186:7255–7263. doi: 10.4049/jimmunol.1100721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen SH, Oyarzabal EA, Hong JS. Preparation of rodent primary cultures for neuron-glia, mixed glia, enriched microglia, and reconstituted cultures with microglia. Methods Mol Biol. 2013;1041:231–240. doi: 10.1007/978-1-62703-520-0_21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu JM, Chen LW, Chan YS, Yung KK. Neuroprotective effects of neurokinin receptor one in dopaminergic neurons are mediated through Akt/PKB cell signaling pathway. Neuropharmacology. 2011;61:1389–1398. doi: 10.1016/j.neuropharm.2011.08.027. [DOI] [PubMed] [Google Scholar]
- Domercq M, Vázquez-Villoldo N, Matute C. Neurotransmitter signaling in the pathophysiology of microglia. Front Cell Neurosci. 2013;7:49. doi: 10.3389/fncel.2013.00049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebner K, Singewald N. Stress-induced release of substance P in the locus coeruleus modulates cortical noradrenaline release. Naunyn Schmiedebergs Arch Pharmacol. 2007;376:73–82. doi: 10.1007/s00210-007-0185-3. [DOI] [PubMed] [Google Scholar]
- Fong TM, Anderson SA, Yu H, Huang RR, Strader CD. Differential activation of intracellular effector by two isoforms of human neurokinin-1 receptor. Mol Pharmacol. 1992;41:24–30. [PubMed] [Google Scholar]
- Gao HM, Zhang F, Zhou H, Kam W, Wilson B, Hong JS. Neuroinflammation and alpha-synuclein dysfunction potentiate each other, driving chronic progression of neurodegeneration in a mouse model of Parkinson's disease. Environ Health Perspect. 2011a;119:807–814. doi: 10.1289/ehp.1003013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao HM, Zhou H, Zhang F, Wilson BC, Kam W, Hong JS. HMGB1 acts on microglia Mac1 to mediate chronic neuroinflammation that drives progressive neurodegeneration. J Neurosci. 2011b;31:1081–1092. doi: 10.1523/JNEUROSCI.3732-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao X, Hu X, Qian L, Yang S, Zhang W, Zhang D, Wu X, Fraser A, Wilson B, Flood PM, Block M, Hong JS. Formyl-methionyl-leucyl-phenylalanine-induced dopaminergic neurotoxicity via microglial activation: a mediator between peripheral infection and neurodegeneration? Environ Health Perspect. 2008;116:593–598. doi: 10.1289/ehp.11031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graybiel AM. Neuropeptides in the basal ganglia. Res Publ Assoc Res Nerv Ment Dis. 1986;64:135–161. [PubMed] [Google Scholar]
- Hastings TG, Zigmond MJ. Loss of dopaminergic neurons in parkinsonism: possible role of reactive dopamine metabolites. J Neural Transm Suppl. 1997;49:103–110. doi: 10.1007/978-3-7091-6844-8_11. [DOI] [PubMed] [Google Scholar]
- Hastings TG, Lewis DA, Zigmond MJ. Reactive dopamine metabolites and neurotoxicity: implications for Parkinson's disease. Adv Exp Med Biol. 1996;387:97–106. doi: 10.1007/978-1-4757-9480-9_13. [DOI] [PubMed] [Google Scholar]
- Helyes Z, Elekes K, Sándor K, Szitter I, Kereskai L, Pintér E, Kemény A, Szolcsányi J, McLaughlin L, Vasiliou S, Kipar A, Zimmer A, Hunt SP, Stewart JP, Quinn JP. Involvement of preprotachykinin A gene-encoded peptides and the neurokinin 1 receptor in endotoxin-induced murine airway inflammation. Neuropeptides. 2010;44:399–406. doi: 10.1016/j.npep.2010.05.004. [DOI] [PubMed] [Google Scholar]
- Hu X, Zhang D, Pang H, Caudle WM, Li Y, Gao H, Liu Y, Qian L, Wilson B, Di Monte DA, Ali SF, Zhang J, Block ML, Hong JS. Macrophage antigen complex-1 mediates reactive microgliosis and progressive dopaminergic neurodegeneration in the MPTP model of Parkinson's disease. J Immunol. 2008;181:7194–7204. doi: 10.4049/jimmunol.181.10.7194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jalkanen AJ, Savolainen K, Forsberg MM. Inhibition of prolyl oligopeptidase by KYP-2047 fails to increase the extracellular neurotensin and substance P levels in rat striatum. Neurosci Lett. 2011;502:107–111. doi: 10.1016/j.neulet.2011.07.033. [DOI] [PubMed] [Google Scholar]
- Kennedy PG, Rodgers J, Bradley B, Hunt SP, Gettinby G, Leeman SE, de Felipe C, Murray M. Clinical and neuroinflammatory responses to meningoencephalitis in substance P receptor knockout mice. Brain. 2003;126:1683–1690. doi: 10.1093/brain/awg160. [DOI] [PubMed] [Google Scholar]
- Kim WG, Mohney RP, Wilson B, Jeohn GH, Liu B, Hong JS. Regional difference in susceptibility to lipopolysaccharide-induced neurotoxicity in the rat brain: role of microglia. J Neurosci. 2000;20:6309–6316. doi: 10.1523/JNEUROSCI.20-16-06309.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambeth JD, Krause KH, Clark RA. NOX enzymes as novel targets for drug development. Semin Immunopathol. 2008;30:339–363. doi: 10.1007/s00281-008-0123-6. [DOI] [PubMed] [Google Scholar]
- Levesque S, Wilson B, Gregoria V, Thorpe LB, Dallas S, Polikov VS, Hong JS, Block ML. Reactive microgliosis: extracellular micro-calpain and microglia-mediated dopaminergic neurotoxicity. Brain. 2010;133:808–821. doi: 10.1093/brain/awp333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marriott I. The role of tachykinins in central nervous system inflammatory responses. Front Biosci. 2004;9:2153–2165. doi: 10.2741/1377. [DOI] [PubMed] [Google Scholar]
- McGeer PL, Itagaki S, Boyes BE, McGeer EG. Reactive microglia are positive for HLA-DR in the substantia nigra of Parkinson's and Alzheimer's disease brains. Neurology. 1988;38:1285–1291. doi: 10.1212/WNL.38.8.1285. [DOI] [PubMed] [Google Scholar]
- Moratalla R, Quinn B, DeLanney LE, Irwin I, Langston JW, Graybiel AM. Differential vulnerability of primate caudate-putamen and striosome-matrix dopamine systems to the neurotoxic effects of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine. Proc Natl Acad Sci U S A. 1992;89:3859–3863. doi: 10.1073/pnas.89.9.3859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosley RL, Benner EJ, Kadiu I, Thomas M, Boska MD, Hasan K, Laurie C, Gendelman HE. Neuroinflammation, oxidative stress and the pathogenesis of Parkinson's disease. Clin Neurosci Res. 2006;6:261–281. doi: 10.1016/j.cnr.2006.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niehaus I, Lange JH. Endotoxin: is it an environmental factor in the cause of Parkinson's disease? Occup Environ Med. 2003;60:378. doi: 10.1136/oem.60.5.378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Przedborski S, Jackson-Lewis V, Yokoyama R, Shibata T, Dawson VL, Dawson TM. Role of neuronal nitric oxide in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced dopaminergic neurotoxicity. Proc Natl Acad Sci U S A. 1996;93:4565–4571. doi: 10.1073/pnas.93.10.4565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian L, Wei SJ, Zhang D, Hu X, Xu Z, Wilson B, El-Benna J, Hong JS, Flood PM. Potent anti-inflammatory and neuroprotective effects of TGF-beta1 are mediated through the inhibition of ERK and p47phox-Ser345 phosphorylation and translocation in microglia. J Immunol. 2008;181:660–668. doi: 10.4049/jimmunol.181.1.660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin L, Liu Y, Wang T, Wei SJ, Block ML, Wilson B, Liu B, Hong JS. NADPH oxidase mediates lipopolysaccharide-induced neurotoxicity and proinflammatory gene expression in activated microglia. J Biol Chem. 2004;279:1415–1421. doi: 10.1074/jbc.M307657200. [DOI] [PubMed] [Google Scholar]
- Qin L, Block ML, Liu Y, Bienstock RJ, Pei Z, Zhang W, Wu X, Wilson B, Burka T, Hong JS. Microglial NADPH oxidase is a novel target for femtomolar neuroprotection against oxidative stress. FASEB J. 2005;19:550–557. doi: 10.1096/fj.04-2857com. [DOI] [PubMed] [Google Scholar]
- Qin L, Wu X, Block ML, Liu Y, Breese GR, Hong JS, Knapp DJ, Crews FT. Systemic LPS causes chronic neuroinflammation and progressive neurodegeneration. Glia. 2007;55:453–462. doi: 10.1002/glia.20467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasley A, Marriott I, Halberstadt CR, Bost KL, Anguita J. Substance P augments Borrelia burgdorferi-induced prostaglandin E2 production by murine microglia. J Immunol. 2004;172:5707–5713. doi: 10.4049/jimmunol.172.9.5707. [DOI] [PubMed] [Google Scholar]
- Ray A, Dittel BN. Isolation of mouse peritoneal cavity cells. J Vis Exp. 2010;35 doi: 10.3791/1488. pii1488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid MS, Herrera-Marschitz M, Hökfelt T, Lindefors N, Persson H, Ungerstedt U. Striatonigral GABA, dynorphin, substance P and neurokinin A modulation of nigrostriatal dopamine release: evidence for direct regulatory mechanisms. Exp Brain Res. 1990;82:293–303. doi: 10.1007/BF00231249. [DOI] [PubMed] [Google Scholar]
- Reynolds A, Laurie C, Mosley RL, Gendelman HE. Oxidative stress and the pathogenesis of neurodegenerative disorders. Int Rev Neurobiol. 2007;82:297–325. doi: 10.1016/S0074-7742(07)82016-2. [DOI] [PubMed] [Google Scholar]
- Sorce S, Krause KH. NOX enzymes in the central nervous system: from signaling to disease. Antioxid Redox Signal. 2009;11:2481–2504. doi: 10.1089/ars.2009.2578. [DOI] [PubMed] [Google Scholar]
- Stone DK, Reynolds AD, Mosley RL, Gendelman HE. Innate and adaptive immunity for the pathobiology of Parkinson's disease. Antioxid Redox Signal. 2009;11:2151–2166. doi: 10.1089/ars.2009.2460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thornton E, Vink R. Treatment with a substance P receptor antagonist is neuroprotective in the intrastriatal 6-hydroxydopamine model of early Parkinson's disease. PLoS One. 2012;7:e34138. doi: 10.1371/journal.pone.0034138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tönges L, Frank T, Tatenhorst L, Saal KA, Koch JC, Szego ÉM, Bähr M, Weishaupt JH, Lingor P. Inhibition of ρ kinase enhances survival of dopaminergic neurons and attenuates axonal loss in a mouse model of Parkinson's disease. Brain. 2012;135:3355–3370. doi: 10.1093/brain/aws254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Zhou H, Gao H, Chen SH, Chu CH, Wilson B, Hong JS. Naloxone inhibits immune cell function by suppressing superoxide production through a direct interaction with gp91phox subunit of NADPH oxidase. J Neuroinflammation. 2012a;9:32. doi: 10.1186/1742-2094-9-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Shin EJ, Nguyen XK, Li Q, Bach JH, Bing G, Kim WK, Kim HC, Hong JS. Endogenous dynorphin protects against neurotoxin-elicited nigrostriatal dopaminergic neuron damage and motor deficits in mice. J Neuroinflammation. 2012b;9:124. doi: 10.1186/1742-2094-9-124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warsame Afrah A, Gustafsson H, Olgart L, Brodin E, Stiller CO, Taylor BK. Capsaicin-evoked substance P release in rat dorsal horn increases after peripheral inflammation: a microdialysis study. Neurosci Lett. 2004;368:226–230. doi: 10.1016/j.neulet.2004.07.041. [DOI] [PubMed] [Google Scholar]
- Yang S, Yang J, Yang Z, Chen P, Fraser A, Zhang W, Pang H, Gao X, Wilson B, Hong JS, Block ML. Pituitary adenylate cyclase-activating polypeptide (PACAP) 38 and PACAP4–6 are neuroprotective through inhibition of NADPH oxidase: potent regulators of microglia-mediated oxidative stress. J Pharmacol Exp Ther. 2006;319:595–603. doi: 10.1124/jpet.106.102236. [DOI] [PubMed] [Google Scholar]
- Yu L, Zhen L, Dinauer MC. Biosynthesis of the phagocyte NADPH oxidase cytochrome b558: role of heme incorporation and heterodimer formation in maturation and stability of gp91phox and p22phox subunits. J Biol Chem. 1997;272:27288–27294. doi: 10.1074/jbc.272.43.27288. [DOI] [PubMed] [Google Scholar]
- Zhang W, Wang T, Qin L, Gao HM, Wilson B, Ali SF, Zhang W, Hong JS, Liu B. Neuroprotective effect of dextromethorphan in the MPTP Parkinson's disease model: role of NADPH oxidase. FASEB J. 2004;18:589–591. doi: 10.1096/fj.03-0983fje. [DOI] [PubMed] [Google Scholar]