

Abstract
Little is known about the temporal progression and regulation of the mechanisms underlying memory consolidation. Brain-derived-neurotrophic-factor (BDNF) has been shown to mediate the maintenance of memory consolidation, but the mechanisms of this regulation remain unclear. Using inhibitory avoidance (IA) in rats, here we show that a hippocampal BDNF-positive autoregulatory feedback loop via CCAAT-enhancer binding protein β (C/EBPβ) is necessary to mediate memory consolidation. At training, a very rapid, learning-induced requirement of BDNF accompanied by rapid de novo translation controls the induction of a persistent activation of cAMP-response element binding-protein (CREB) and C/EBPβ expression. The latter, in turn, controls an increase in expression of bdnf exon IV transcripts and BDNF protein, both of which are necessary and, together with the initial BDNF requirement, mediate memory consolidation. The autoregulatory loop terminates by 48 h after training with decreased C/EBPβ and pCREB and increased methyl-CpG binding protein-2, histone-deacetylase-2, and switch-independent-3a binding at the bdnf exon IV promoter.
Keywords: BDNF, C/EBP, consolidation, hippocampus, memory, memory persistence
Introduction
Long-term memories are initially labile, but over time become insensitive to disruption through a process known as consolidation (Davis and Squire, 1984; Dudai, 2004; Alberini, 2009). Widely studied pharmacological compounds that disrupt consolidation are protein and RNA synthesis inhibitors, which have provided evidence that de novo transcription and translation are evolutionarily conserved mechanisms of memory consolidation. Numerous investigations, especially based on genetic mutations or deletions, have thus focused on the identification of which genes and proteins mediate long-term memory formation. Although the nature of several genes and proteins necessary for memory formation have been established, the temporal progression of the learning-dependent de novo protein synthesis and of molecular events that accompany the progression of memory consolidation remains poorly characterized. This characterization is critical for understanding the progression of the functional mechanisms of memory consolidation and storage, which provides important information for how/when to intervene to strengthen or weaken memories.
Most early studies on memory consolidation led to the belief that de novo gene expression is required only for a very brief temporal window, on the order of 1–2 h (h) after training. However, in the last decade several reports have indicated that training activates hippocampal molecular changes whose progression is critical for a day or longer. These changes include redistribution of glutamate receptors, activation of protein kinases, and gene expression regulation (Taubenfeld et al., 2001a,b; Katche et al., 2013). In particular, in the hippocampus of rats trained with inhibitory avoidance (IA), the critical role of the evolutionarily conserved CREB-C/EBP pathway begins immediately after training and progresses for >20 h. In fact, IA memory formation is accompanied by a significant increase in CREB activation (phosphorylation of Ser133, pCREB; Taubenfeld et al., 2001a,b), which lasts for >20 h after training and requires the expression of the CREB-downstream target gene C/EBPβ for >24 h post training (Taubenfeld et al., 2001a). These data imply that de novo protein synthesis critical for memory consolidation must still take place 24 h after IA training (Alberini, 2009). In agreement, anisomycin injected into the hippocampus 12 h after step-down IA training leaves memory intact at 2 d after training, but this memory decays at 7 d after training, indicating that de novo protein synthesis ∼12 h after training is essential for memory persistence (Bekinschtein et al., 2007a). Furthermore, the expression of hippocampal BDNF is increased at 12 h after IA training, and blocking hippocampal BDNF 12 h after training blocks memory persistence. Finally, coinjection of recombinant BDNF into the hippocampus rescues the memory deficit caused by anisomycin injection at 12 h after training (Bekinschtein et al., 2008), indicating that BDNF is critical for memory persistence (Medina et al., 2008). Despite this knowledge, the molecular mechanisms that mediate this temporally extended requirement of protein synthesis and BDNF on memory persistence and its regulation are not known. Furthermore, it is not known for how long the fragile phase of memory consolidation continues and which mechanisms end the process. In this study, we asked precisely these questions: Are the extended gene expression and BDNF requirements linear processes? Are they connected? If so, how? And, how are they terminated? We used rat IA to elucidate the temporal evolution of gene expression and BDNF-dependent mechanisms required for induction, progression, and completion of memory consolidation.
Materials and Methods
Animals.
Adult male, Long–Evans rats weighing between 200 and 250 g served as subjects in all experiments. Animals were all singly housed and maintained on a 12 h light/dark cycle. Experiments were performed during the light cycle. All rats were allowed ad libitum access to food and water and were handled for 3 min per day for 5 d before any procedure. All protocols complied with the NIH Guide for the Care and Use of Laboratory Animals and were approved by the Mt. Sinai School of Medicine and New York University Animal Care Committees.
Cannulae implants and hippocampal injections.
Rats were anesthetized with ketamine (65 mg/kg, i.p.) and xylazine (7.5 mg/kg, i.p.), and stainless steel guide cannulae (22 gauge) were stereotactically implanted to bilaterally target the hippocampus (4.0 mm posterior to the bregma, 2.6 mm lateral from midline, and 2.0 mm ventral; Taubenfeld et al., 2001a). The rats were returned to their home cages and allowed to recover from surgery for 7 d. At the indicated time points before or after training or retrieval, rats received bilateral injections of compounds as specified. All injections are indicated by arrows in the experimental schedule. All hippocampal injections were performed in 1 μl per side. Hippocampal injections used a 28 gauge needle that extended 1.5 mm beyond the tip of the guide cannula and connected via polyethylene tubing to a Hamilton syringe. The infusions were delivered at a rate of 0.33 μl/min using an infusion pump. The injection needle was left in place for 2 min after the injection to allow complete dispersion of the solution. To verify proper placement of cannula implants, at the end of the behavioral experiments, rats were killed and their brains were postfixed with 10% buffered formalin. Forty micrometer coronal sections were cut through the hippocampus and examined under a light microscope. Rats with incorrect cannula placement (<2%) were discarded from the study.
IA.
IA was performed as previously described (Taubenfeld et al., 2001a). The IA chamber (Med Associates) consisted of a rectangular Perspex box divided into a safe compartment and a shock compartment. The safe compartment was white and illuminated and the shock compartment was black and dark. Footshocks were delivered to the grid floor of the shock chamber via a constant current scrambler circuit. The apparatus was located in a sound-attenuated, nonilluminated room. During training sessions, each rat was placed in the safe compartment with its head facing away from the door. After 10 s, the door separating the compartments was automatically opened, allowing the rat access to the shock compartment; the rats usually enter the shock (dark) compartment within 10–20 s of the door opening. The door closed 1 s after the rat entered the shock compartment, and a 2 s footshock was administered. All initial behavioral experiments involving anti-BDNF, TrkB-Fc, anisomycin, and rapamycin were performed using a 0.6 mA footshock. Subsequent biochemistry and the behavioral experiments were performed using a 0.9 mA footshock, because higher shock intensity produces stronger molecular changes. To ensure that the behavioral results were confirmed with the higher footshock, we repeated both pretraining TrkB-Fc and post-training anti-BDNF injections using a 0.9 mA footshock and obtained comparable results (Fig. 5; Chen et al., 2012). Latency to enter the shock compartment was taken in seconds as acquisition. The rat was then returned to its home cage and tested for memory retention at the designated time point(s). Retention tests were done by placing the rat back in the safe compartment and measuring its latency to enter the shock compartment. Footshocks were not administered on the retention tests, and testing was terminated at 540 s or 900 s, as detailed. In some experiments, as indicated, we also used, as a control, an unpaired IA paradigm. In this paradigm, rats were exposed to the context and 1 h later they were placed on the footshock grid and a footshock was immediately delivered. Hence, this protocol temporally dissociates, within subject, context and footshock exposure; this unpaired protocol does not produce IA memory retention (Chen et al., 2011). All behavioral tests were done blind to the experimental groups. For biochemical studies, rats were not tested for memory retention.
Figure 5.

BDNF recruits C/EBPβ in an autoregulatory-positive feedback loop. A, qRT-PCR analysis of bdnf exon IV on dorsal hippocampal cDNAs of trained rats killed at the indicated time points (normalized to gapdh). Compared with naive, training significantly increased bdnf exon IV levels at 12 and 20 h post training (n = 4–8/group; 12 h, p < 0.05; 20 h, p < 0.01; one-way ANOVA followed by Dunnett's multiple-comparison post hoc test). No increase in bdnf exon IV levels occurred in the hippocampi of rats at 12 h after exposure to both context and shock separated by 1 h (unpaired protocol). Data are expressed as mean fold change ± SEM. B, Bdnf exon IV AS, but not SCR ODN, disrupted memory consolidation. Experimental schedule shown above the graph. ODNs were injected (arrow) 6 h after IA training. Rats were tested 2 d (Test 1) and 7 d (Test 2) after training. Compared with SCR, AS significantly decreased memory retention at both Tests 1 and 2 (n = 9–10/group; AS vs SCR, p < 0.001, two-way ANOVA followed by Bonferroni post hoc tests; treatment effect F(1,51) = 29.14). No significant reinstatement was observed following a reminder footshock (p > 0.05). Data are expressed as mean latency ± SEM. ***p < 0.001. C, Representative PCR fragments in agarose gel stained with ethidium bromide and graphs showing qRT-PCRs of bdnf exon IV promoter obtained following ChIP with an anti-C/EBPβ. These fragments were obtained from dorsal hippocampi of trained rats killed at 30 min or 12 h or 48 h after training. Training resulted in a significant increase in C/EBPβ binding to the bdnf exon IV promoter 12 h after training (n = 4/group; naive vs trained, 12 h, p < 0.05, one-way ANOVA followed by Dunnett's multiple-comparison post hoc test; F(3,15) = 3.998, p = 0.0346). Data are presented as mean percentage input enrichment ± SEM. D, qRT-PCR analysis of bdnf exon IV in hippocampi of naive and trained rats injected (arrow) with either SCR-ODN or β-ODN 5 h after training and killed 12 h after training (normalized to gapdh). Training significantly increased bdnf exon IV levels at 12 h; this increase was blocked by β-ODN (n = 6–8/group; naive-SCR-ODN vs trained SCR-ODN p < 0.01, trained-SCR-ODN vs trained β-ODN p < 0.05, one-way ANOVA followed by Newman–Keuls multiple-comparison post hoc tests; F(2,21) = 8.136, p = 0.0028). Data are expressed as mean fold change ± SEM. E, Western blot examples and densitometric quantitative Western blot analyses of BDNF protein levels in dorsal hippocampal extracts from naive and trained rats injected (arrow) with either SCR-ODN or β-ODN 5 h after training and killed 20 h after training (normalized to actin). Training significantly increased BDNF levels at 20 h; this increase was blocked by β-ODN (n = 6–8/group; naive-SCR-ODN vs trained SCR-ODN and trained-SCR-ODN vs trained β-ODN p < 0.05, one-way ANOVA followed by Newman–Keuls multiple-comparison post hoc tests; F(2,20) = 5.568, p = 0.0131). Data are expressed as mean percentage ± SEM naive controls injected with SCR-ODN. F, qRT-PCR analysis of bdnf exon IV cDNA obtained from dorsal hippocampi of naive and trained rats injected (arrow) with either IgG or anti-BDNF 15 min before training and killed 12 h after training (normalized to gapdh). Anti-BDNF blocked the training-dependent significant increase in bdnf exon IV (n = 8–12/group; naive-IgG vs trained-IgG and trained-IgG vs trained-anti-BDNF p < 0.05, one-way ANOVA followed by Newman–Keuls multiple-comparison post hoc tests; F(2,27) = 4.949, p = 0.0155). Data are expressed as mean fold change ± SEM; *p < 0.05, **p < 0.01.
Drug and oligodeoxynucleotide injections.
The sheep anti-BDNF antibody was purchased from Millipore and dissolved in 1× PBS. Anti-BDNF antibody was injected at 0.5 μg per injection per side. Recombinant human TrkB-Fc chimera was purchased from R&D Systems and was dissolved in PBS. TrkB-Fc was injected at 0.5 μg per injection per side. At these dosages, anti-BDNF and TrkB-Fc have been used to disrupt long-term memory consolidation when injected into the hippocampus (Alonso et al., 2002). Control sheep IgG was purchased from Sigma-Aldrich and dissolved in 1× PBS and injected at 0.5 μg per injection per side. Recombinant BDNF and NT-3 were purchased from PeproTech and dissolved in PBS and injected at 0.25 μg per injection per side. Recombinant IGF-II was purchased from R&D Systems and dissolved in 0.1% BSA in 1× PBS. Anisomycin (Sigma-Aldrich) was dissolved in 0.9% saline, pH 7.4, and injected at 125 μg/μl. This dose blocks >80% of protein synthesis in the dorsal hippocampus for up to 6 h (Milekic et al., 2006). Rapamycin was purchased from LC Laboratories, dissolved in DMSO, and diluted in 1× PBS to a final concentration of 1 ng/μl per side. The dose of rapamycin used was previously shown to block S6 kinase phosphorylation and spatial memory in rats (Dash et al., 2006). Antisense oligodeoxynucleotides (ODNs) and relative scrambled sequences (SCR-ODNs) were injected at 2 nmol/μl in all antisense experiments. Sequences were as follows: C/EBPβ antisense (β-ODN: 5′-CCAGCAGGCGGTGCATGAAC-3′), C/EBPβ scrambled (SCR-ODN: 5′-TCGGAGACTAAGCGCGGCAC-3′), bdnf exon IV antisense (5′-CAGTCACTACTTGTCAAAGTA-3′), and bdnf exon IV scrambled (5′-ATTAACTACAAGCGTTCGACT-3′). The respective SCR-ODNs, which served as controls, contained the same base composition but in a random order and show no homology to sequences in the GenBank database. All ODNs were phosphorothioated on the three terminal bases of both 5′ and 3′ ends to produce increased stability. ODNs were reverse-phased-cartridge-purified and obtained from Gene Link.
Tissue preparation and Western blot analysis.
Dorsal hippocampi were dissected in ice-cold cortical dissection buffer and snap frozen on dry ice. The tissues were homogenized using a Polytron homogenizer in lysis buffer (0.2 m NaCl, 5 mm EDTA, 10% glycerol, 100 mm HEPES, 2 mm sodium phosphate) containing a protease inhibitor cocktail (Sigma; used as recommended by the manufacturer), 0.5 mm phenylmethylsulfonyl fluoride, 2 mm dithiothreitol, the phosphatase inhibitor cocktail (Sigma; used as recommended by the manufacturer), 2 mm NaF, 1 μm microcystin LR, and 1 mm sodium orthovanadate. Protein concentration was determined by using the Bio-Rad protein assay (Bio-Rad Laboratories). Five to 20 μg of protein was resolved on 7.5%, 10%, or 15% polyacrylamide gels, according to each marker's molecular weight, and then transferred to Immobilon-P membranes (Millipore). Membranes were blocked and incubated with primary antibodies according to manufacturers' recommendation. After the primary antibody incubation overnight at 4°C, the membranes were washed and treated with a secondary horseradish peroxidase (HRP)-labeled goat anti-rabbit (1:4000) or goat-anti-mouse (1:4000), as required, for 1 h at room temperature. Actin was used to account for loading variation. Membranes were washed and incubated with enhanced chemiluminescence detection reagents (GE Healthcare) and exposed to HyBlot CL Autoradiography Film (Denville Scientific). Quantitative densitometric analysis was performed using NIH Image software. Antibodies are as follows: anti-pCREB (1/1000) and anti-CaMKIIα (1/2000; Millipore); anti-CREB (1/1000), anti-cofilin, and anti-pCaMKIIα (1/5000; Cell Signaling Technology); anti-pCofilin (1:1000; Abcam); anti-C/EBPβ C-19 (1/1000); anti-BDNF (1:1000); and anti-actin-HRP (1/4000; Santa Cruz Biotechnology).
Chromatin immunoprecipitation.
Chromatin immunoprecipitation (ChIP) was performed as described previously (Tsankova et al., 2004). Rat hippocampi were dissected and minced into ∼1 mm pieces, and immediately cross-linked in 1% formaldehyde for 17 min at room temperature with rotation. The cross-linking reaction was stopped by adding glycine to a final concentration of 0.125 m and incubated for 7 min. The tissue was washed five times in cold PBS containing protease inhibitor (Roche Applied Sciences) and then frozen on dry ice. The chromatin was solubilized and extracted by adding 500 μl of lysis buffer (1% SDS, 50 mm Tris-HCl, pH 8.1, and 10 mm EDTA) followed by sonication. The homogenate was diluted in 1.1 ml ChIP dilution buffer (1.1% Triton X-100, 167 mm NaCl, 16.7 mm Tris-HCl, pH 8.1, 1.2 mm EDTA, and 0.01% SDS). The homogenate was then used for ChIP. Thirty microliters of Magnetic Protein A beads (EZ-Magna ChIP A kit; Millipore) and 7.5 μg of C/EBPβ antibody (Abcam), MeCP2 antibody (Abcam), Sin3a antibody (Abcam), HDAC2 antibody (Abcam), or pCREB antibody (Millipore) were added to the homogenate. The mixture was incubated rotating overnight at 4°C. The wash, elution, and reverse cross-link to free DNA were all performed according to the manufacturer's protocol (EZ-Magna ChIP A/G kit). Specific primers were designed to amplify the proximal promoter region of ∼200 bp 5′ of exon IV of rat bdnf, which contains a putative C/EBP binding site. Putative C/EBP binding site was predicted using on-line programs AliBaba 2.1 and Matinspector. Similar C/EBP binding sites have been identified in other species (van Dijk et al., 1992). Primer sequences used were as follows: forward 5′GGC TTC TGT GTG CGT GAG TTC GC 3′; reverse 5′AAA GTG GGT GGG AGT CCA CGA G 3′ based on previously published results (Martinowich et al., 2003). A standard 35 cycle PCR was performed as follows: denature at 95°C for 30 s, anneal at 58°C for 30 s and extend for 30 s at 72°C. The PCR was resolved on a 2% agarose gel and sequenced. Sequencing confirmed the identity of the fragment. DNA sequencing was performed by W.M. Keck Facility at Yale University, New Haven, CT. For ChIP time course studies, quantitative Real-Time PCR analysis was performed as described below.
Real-time quantitative RT-PCR.
Hippocampal total RNA was extracted with TRIzol (Invitrogen) and reverse transcribed using SuperScript II RNase H minus RT (Invitrogen) or Bio-Rad iScript first-strand synthesis kit (Bio-Rad). Real-time PCR was done with an ABI Prism 7900HT (Applied Biosystems) or Bio-Rad My-IQ single-color RT-PCR machine; 500 pg of the first-strand cDNA was subjected to PCR amplification using a QuantiTect SYBR Green PCR kit (Qiagen) or Bio-Rad iQ SYBR Green Supermix (Bio-Rad). Forty cycles of PCR amplification were performed as follows: denature at 95°C for 30 s, anneal at 55°C for 30 s, and extend for 30 s at 72°C. Three PCR assays with triplicates were performed for each cDNA sample. Bdnf exon IV (Forward, 5′ CCCAGTCTCTGCCTAGATCAAATGG 3′, Reverse, 5′ ACTCGCACGCCTTCAGTGAGAA 3′), Gapdh (Forward, 5′GAACATCATCCCTGCATCCA 3′, Reverse 5′CCAGTGAGCTTCCCGTTCA 3′) was used as internal control. Data were analyzed with Sequence Detector System version 2.0 software (Applied Biosystems). The cycle threshold method (CT; see Applied Biosystems User Bulletin Number 2, P/N 4303859), ddCT, was chosen to determine the relative quantification of gene expression in trained and control rats.
Statistical analyses.
One-way ANOVA followed by Newman–Keuls post hoc tests was performed when comparing groups where a pairwise post hoc analysis of each group was required (Figs. 4, 5C–E). One-way ANOVA followed by Dunnett's post hoc tests was used when each group was compared with a single control group (Naive; Figs. 5A,B, 7). Two-way ANOVAs followed by Bonferroni post hoc tests were used when two factors were compared (e.g., treatment and testing; Figs. 1–3, 5, 6). Student's t test was used when two groups were compared (e.g., reminder shock).
Figure 4.

Hippocampal BDNF at training is required for the increase of pCREB, pCofilin, pCaMKIIα, and C/EBPβ. A, Examples and densitometric quantitative Western blot analyses of dorsal hippocampal extracts of naive and trained rats given hippocampal injections of IgG or anti-BDNF 15 min before training and killed 12 h after training. The expression of pCREB, pCofilin, pCaMKIIα, and C/EBPβ is significantly increased after training and this increase is significantly blocked by anti-BDNF injections (n = 6–12/group, naive vs trained-IgG and naive vs trained-anti-BDNF, one-way ANOVA followed by Newman–Keuls multiple-comparison post hoc tests; pCREB: F(2,30) = 7.608, p = 0.0023; pCofilin: F(2,20) = 5.796, p = 0.0114; pCaMKIIα: F(2,32) = 8.621, p = 0.0011; C/EBPβ: F(2,23) = 5.308, p = 0.0136). No significant changes were observed in levels of total CREB, total CaMKIIα, or total Cofilin (p > 0.05, data not shown). Data are expressed as mean percentage ± SEM naive controls injected with IgG; *p < 0.05, **p < 0.01, and ***p < 0.001. B, This figure summarizes the temporal profile of training-dependent increases in pCREB and pCaMKIIα in dorsal hippocampal extracts obtained from rats injected with either anti-BDNF antibody or control IgG antibody 15 min before IA training (n = 5–8/group) and killed at 30 min or 12 h or 20 h after training. The graphs include previously reported data at 30 min and 20 h (Chen et al., 2012) in addition to the 12 h time point investigated here. Densitometric quantitative Western blot analyses data expressed as mean percentage ± SEM naive, IgG-injected controls killed at matched time points. All values were normalized against actin. Compared with naive controls injected with IgG (light gray circles), rats trained in IA showed a significant induction of hippocampal pCREB and pCaMKIIα at 30 min and 12 and 20 h after training (black squares). The induction of pCREB at all three time points and the induction of pCaMKIIα at 12 and 20 h but not 30 min are blocked by an intrahippocampal injection of anti-BDNF 15 min before training (dark gray triangles; one-way ANOVA followed by Newman–Keuls multiple-comparison post hoc tests). *Indicates significance levels comparing Naive-IgG to Trained-IgG groups, *p < 0.05, **p < 0.01, and ***p < 0.001. #Indicates significance levels comparing Trained-IgG to Trained-Anti-BDNF groups, #p < 0.05, ##p < 0.01, and ###p < 0.001.
Figure 7.
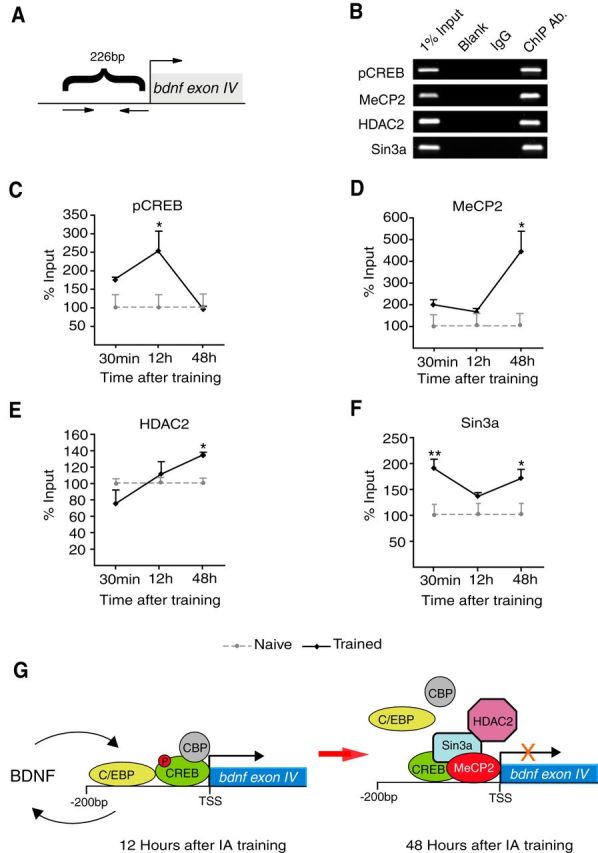
Increased ratio of repressor/activator binding at the bdnf exon IV promoter correlates with the termination of the feedback loop. A, Scheme of the 226bp upstream region of bdnf exon IV promoter analyzed with ChIP. B, Representative PCR fragments in agarose gel stained with ethidium bromide and (C–F) qRT-PCR analysis of bdnf exon IV promoter fragment following ChIP using an antibody against pCREB, MeCP2, HDAC2, or Sin3a. The qRT-PCRs were obtained from dorsal hippocampi of trained rats killed at 30 min or 12 h or 48 h after training. Data are presented as mean percentage input enrichment ± SEM (n = 4–7/group; p < 0.05, p < 0.01, one-way ANOVA followed by Dunnett's multiple-comparison post hoc test; pCREB: F(3,15) = 5.437, p = 0.0136; MeCP2: F(3,15) = 7.069, p = 0.0054; HDAC2: F(3,19) = 5.960, p = 0.0063; Sin3a: F(3,15) = 5.842, p = 0.0104). G, Schematic model of transcriptional regulation of the bdnf exon IV promoter following IA training. BDNF at training leads to an increase in pCREB and C/EBPβ binding at the bdnf exon IV promoter at 12 h after IA training, which correlates with increased transcription of bdnf exon IV as well as BDNF protein (Fig. 5). At 48 h after training, the binding of pCREB and C/EBPβ at the bdnf exon IV promoter returns to baseline, while the binding of the inhibitors Sin3a, MeCP2, and HDAC2 increases correlating with return to baseline of BDNF level. *p < 0.05, **p < 0.01.
Figure 1.
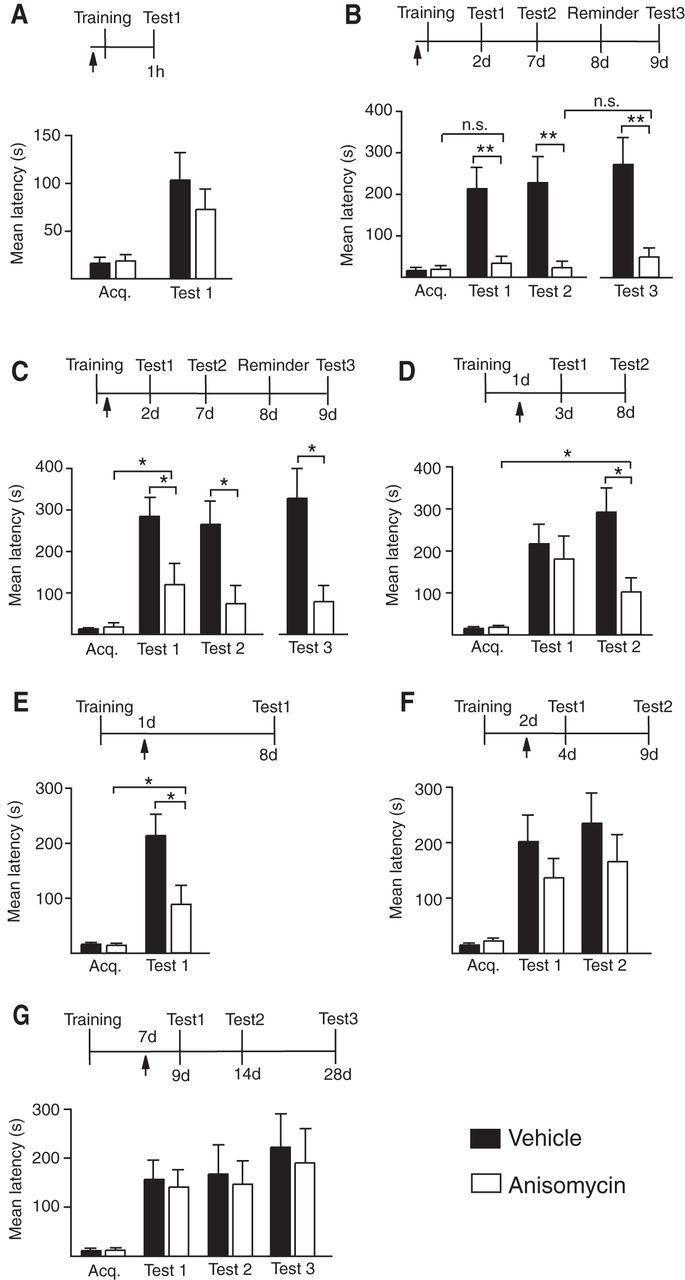
Temporal profile of the hippocampal protein synthesis requirement during memory consolidation. Experimental schedule is shown above each figure. A–G, Acquisition (Acq.) and retention are expressed as mean latency ± SEM (in seconds) of rats given bilateral hippocampal injections (arrow) of vehicle or anisomycin at designated time points. A, Hippocampal injections of anisomycin 15 min before IA training had no effect on short-term memory tested at 1 h after training (n = 8/group, Test 1, vehicle vs anisomycin, p > 0.05, Student's t test). B, Hippocampal injections of anisomycin 15 min before IA training disrupted memory tested 2 and 7 d after training (n = 8–9/group, Test 1 and Test 2, vehicle vs anisomycin, p < 0.01, two-way ANOVA followed by Bonferroni post hoc tests; treatment effect F(1,28) = 22.37, p < 0.0001) as well as 1 d after a reminder footshock (Test 3, vehicle vs anisomycin, p < 0.01, Student's t test). Memory retention at Test 1, Test 2, or Test 3 was not significantly different from acquisition latency (p > 0.05, Student's t test). C, Hippocampal injections of anisomycin immediately after IA training disrupted memory tested 2 and 7 d after training (n = 8/group, Test 1 and Test 2, vehicle vs anisomycin, p < 0.05, two-way ANOVA followed by Bonferroni post hoc tests; treatment effect F(1,28) = 13.79, p = 0.0009) as well as 1 d after a reminder footshock (Test 3, vehicle vs anisomycin, p < 0.05, Student's t test). Memory retention at Test 1 was significantly different from acquisition latency (p < 0.05, Student's t test). D, Hippocampal injections of anisomycin 1 d after IA training had no effect on memory tested 2 d after the injection (n = 10/group, Test 1, vehicle vs anisomycin, p > 0.05, two-way ANOVA followed by Bonferroni post hoc tests), but significantly disrupted memory retested at 7 d after the injection (Test 2, p < 0.05, two-way ANOVA followed by Bonferroni post hoc tests; treatment effect F(1,36) = 5.277, p = 0.0275). Student's t test comparing Test 2 of the anisomycin group with its relative acquisition latency shows a statistically significant difference (Student's t test, p = 0.0210). E, Hippocampal injections of anisomycin 1 d after IA training significantly disrupted memory tested at 7 d after the injection (n = 8/group, Test 1, vehicle vs anisomycin, p < 0.05, Student's t test). Student's t test comparing Test 1 of the anisomycin group with its relative acquisition latency shows a statistically significant difference (Student's t test, p = 0.0412). F, Hippocampal injections of anisomycin 2 d after training had no effect on memory tested 2 d (Test 1) or 7 d (Test 2) after the injection (n = 12–14/group, Test 1 and Test 2, vehicle vs anisomycin, p > 0.05, two-way ANOVA followed by Bonferroni post hoc tests for both experiments). G, Hippocampal injections of anisomycin 7 d after training had no effect on memory tested 2 d (Test 1), 7 d (Test 2), or 21 d (Test 3) after the injection (n = 8–9/group, Test 1, Test2, and Test 3, vehicle vs anisomycin, p > 0.05, two-way ANOVA followed by Bonferroni post hoc tests); *p < 0.05, **p < 0.01.
Figure 2.
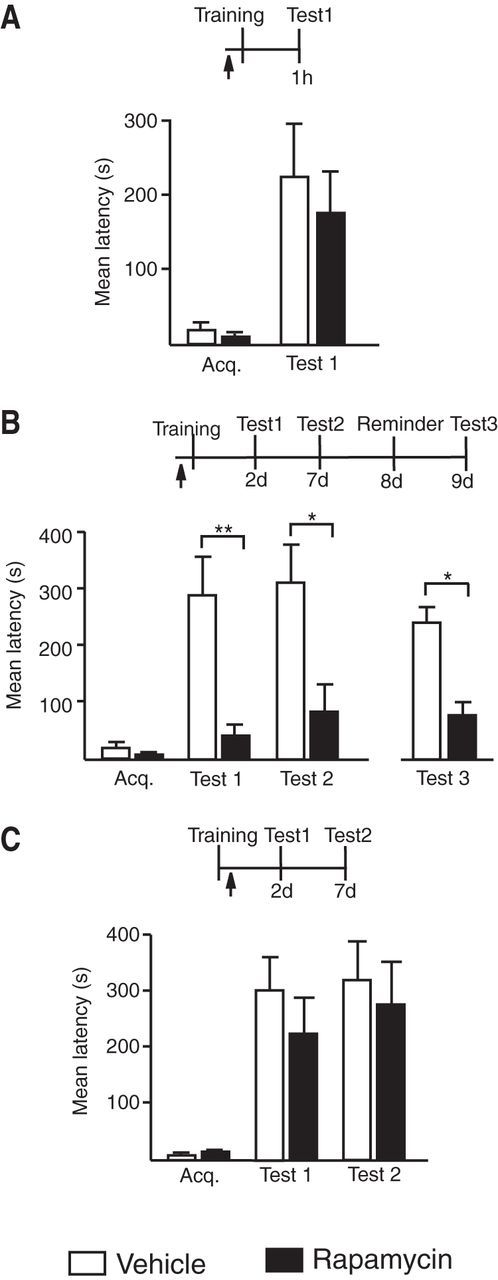
A rapid rapamycin-sensitive phase is required for long-term memory consolidation. Experimental schedule above each figure (A–C). Acquisition (Acq.) and retention are expressed as mean latency ± SEM (in seconds). Arrow indicates a bilateral hippocampal injection of vehicle or rapamycin 15 min before or immediately after IA training. Compared with vehicle, rapamycin injected 15 min before training (A) did not affect short-term memory (n = 7/group, vehicle vs rapamycin, p > 0.05, Student's t test), but (B) completely disrupted memory retention at both Test 1 and Test 2 (n = 7/group, Test 1, vehicle vs rapamycin, p < 0.01rapamycin; Test 2, vehicle vs rapamycin, p < 0.05, two-way ANOVA followed by Bonferroni post hoc tests; treatment effect F(1,24) = 19.77, p = 0.0002, each rapamycin group compared with Acq., p > 0.05 Student's t test). Memory did not reinstate following the reminder shock (Test 3, vehicle vs rapamycin, p < 0.05, Student's t test). C, Rapamycin injected immediately after training had no effect on memory retention (n = 9–10/group, Test 1 and Test 2, vehicle vs rapamycin, p > 0.05, two-way ANOVA followed by Bonferroni post hoc tests); *p < 0.05, **p < 0.01.
Figure 3.
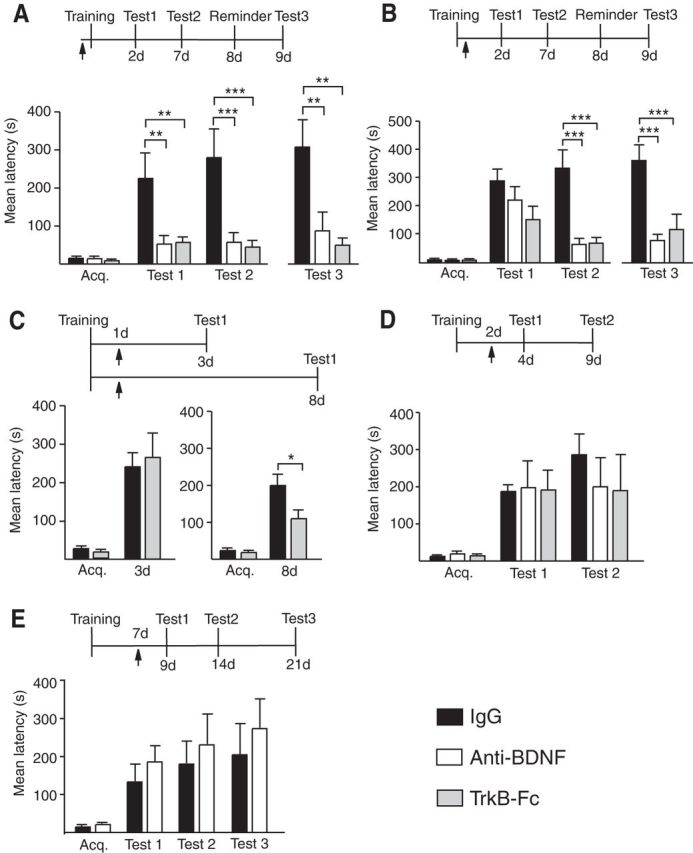
Temporal profile of hippocampal BDNF requirement during long-term memory consolidation. Experimental schedule above each figure. Acquisition (Acq.) and retention are expressed as mean latency ± SEM (in seconds) of rats given a bilateral hippocampal injection (arrow) of IgG, anti-BDNF, or TrkB-Fc at designated time points (A–E). A, Hippocampal injections of anti-BDNF or TrkB-Fc 15 min before IA training significantly disrupted memory tested at 2 and 7 d after training (n = 8–9/group, IgG vs anti-BDNF or vs TrkB-Fc Test 1, p < 0.01 and Test 2, p < 0.001, two-way ANOVA followed by Bonferroni post hoc tests; treatment effect F(2,46) = 17.08, p < 0.0001) as well as 1 d after a reminder footshock (Test 3, IgG vs anti-BDNF or vs TrkB-Fc, both treatments p < 0.01, one-way ANOVA with Newman–Keuls multiple-comparison post hoc tests; treatment effect F(2,25) = 8.174, p = 0.0021). B, Hippocampal injections of anti-BDNF or TrkB-Fc immediately after training had no significant effect on memory tested 2 d after the injection (n = 8–10/group, Test 1, IgG vs anti-BDNF or vs TrkB-Fc p > 0.05, two-way ANOVA followed by Bonferroni post hoc tests) but significantly disrupted memory retested 7 d after the injection (Test 2, both anti-BDNF and TrkB-Fc, p < 0.001, two-way ANOVA followed by Bonferroni post hoc tests; treatment effect F(2,48) = 12.93, p < 0.0001). The significant impairment persisted after a reminder shock (Test 3, both treatments p < 0.001, one-way ANOVA followed by Newman–Keuls multiple-comparison post hoc tests; treatment effect F(2,26) = 9.958, p = 0.0007). C, Hippocampal injections of TrkB-Fc 1 d after IA training had no effect on memory tested 2 d after the injection (n = 8–9/group, Test 1, 3 d, IgG vs TrkB-Fc p > 0.05, Student's t test) but significantly disrupted memory tested 7 d after the injection (n = 7–8 /group, Test 1, 8 d, IgG vs TrkB-Fc p < 0.05, Student's t test). D, Hippocampal injections of anti-BDNF or TrkB-Fc 2 d after IA training had no effect on memory tested at 2 d (Test 1) or 7 d (Test 2) after the injection (n = 6–9/group, IgG vs anti-BDNF or vs TrkB-Fc p > 0.05; two-way ANOVA followed by Bonferroni post hoc tests). E, Hippocampal injections of anti-BDNF 7 d after IA training had no effect on memory tested at 2 d (Test 1), 7 d (Test 2), or 14 d (Test 3) after the injection (n = 7/group, IgG vs anti-BDNF p > 0.05, two-way ANOVA followed by Bonferroni post hoc tests); *p < 0.05, **p < 0.01, and ***p < 0.001.
Figure 6.
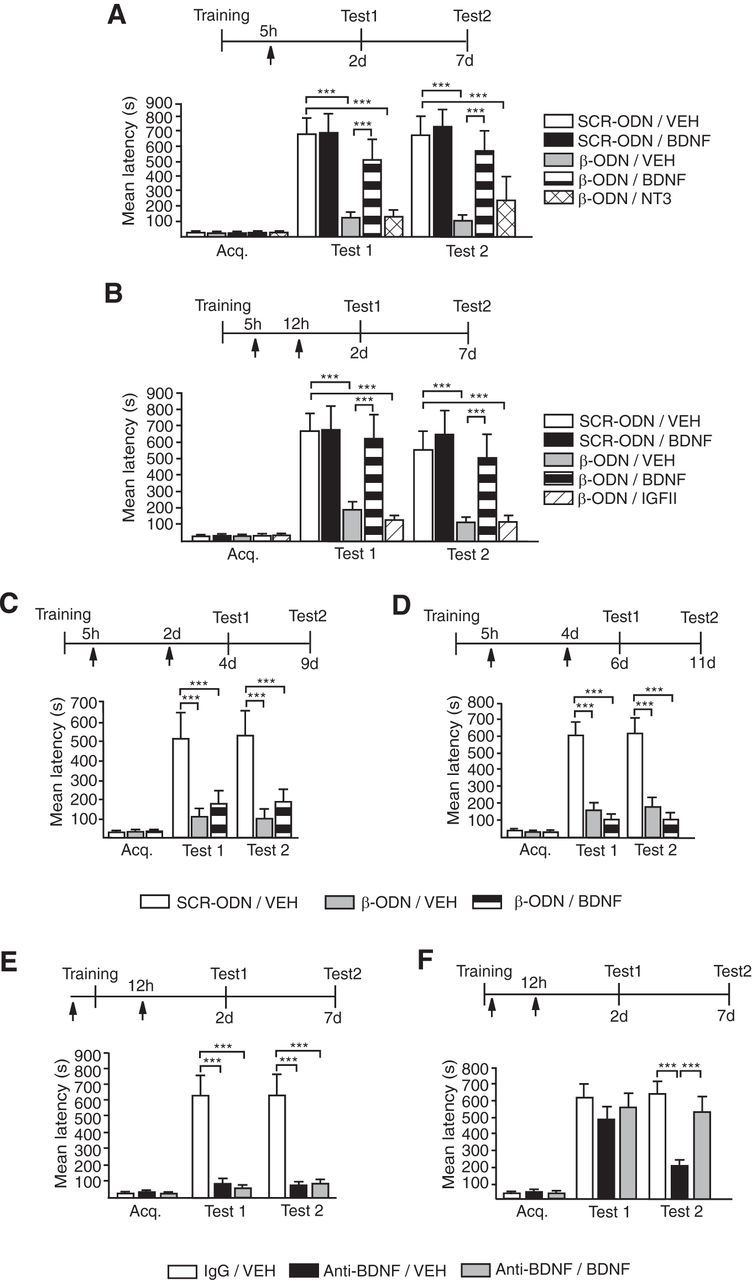
The memory impairment due to C/EBPβ or BDNF blockade after training is rescued by BDNF. Data are expressed as mean latency ± SEM. Experimental schedule is shown above each figure (A–F). A, Rec-BDNF but not rec-NT3 injected 5 h after IA training rescued the memory impairment caused by blocking C/EBPβ expression. β-ODN or SCR-ODN was injected (arrow) 5 h after IA training along with rec-BDNF, Rec-NT3, or Vehicle. Rats were tested 2 d (Test 1) or 7 d (Test2) after the injection. Compared with SCR-ODN, β-ODN significantly impaired memory retention at both 2 and 7 d after training (n = 6–8/group; SCR-ODN/VEH vs β-ODN/VEH, p < 0.001, two-way ANOVA followed by Bonferroni post hoc tests; treatment effect F(4,58) = 13.73, p < 0.0001). BDNF, but not NT3, significantly and completely rescued this amnesia and the rescuing effect persisted (β-ODN/VEH vs β-ODN/BDNF, p < 0.001; β-ODN/VEH vs β-ODN/NT-3, p > 0.05; SCR-ODN/VEH vs β-ODN/BDNF, p > 0.05, two-way ANOVA followed by Bonferroni post hoc tests). B, Rec-BDNF but not rec-IGF-II injected at 12 h after IA training significantly and completely rescued the memory impairment caused by β-ODN injected 5 h after training (n = 6–11/group, β-ODN/VEH vs β-ODN/BDNF, p < 0.001; β-ODN/VEH vs β-ODN/IGFII, p > 0.05; SCR-ODN/VEH vs β-ODN/BDNF p > 0.05, two-way ANOVA followed by Bonferroni post hoc tests; treatment effect F(2,34) = 13.51, p < 0.0001). Rats were tested 2 d (Test 1) or 7 d (Test 2) after the injection. C, D, Rec-BDNF injected 2 d (C) or 4 d (D) after training did not rescue the memory impairment caused by blocking C/EBPβ expression. β-ODN or SCR-ODN was injected 5 h after IA training. Rats were tested 2 d (Test 1) or 7 d (Test 2) after the injection (n = 6–8/group; β-ODN/VEH vs β-ODN/BDNF, p > 0.05, two-way ANOVA followed by Bonferroni post hoc tests; treatment effect: for C, F(2,34) = 13.51, p < 0.0001; for D, F(2,40) = 46.42, p < 0.0001), E, Rec-BDNF injected 12 h after IA training does not rescue the memory impairment caused by pretraining anti-BDNF. IgG or anti-BDNF antibody was injected (arrow) 15 min before training. Rec-BDNF or vehicle was injected (arrow) 12 h after training. Rats were tested 2 d (Test 1) or 7 d (Test 2) after the injection (n = 6–7/group; anti-BDNF/VEH vs anti-BDNF/BDNF, p > 0.05, two-way ANOVA followed by Bonferroni post hoc tests; treatment effect F(2,32) = 41.39, p < 0.0001). F, Rec-BDNF injected 12 h after IA training rescues the memory decay caused by post-training anti-BDNF injection. IgG or anti-BDNF antibody was injected (arrow) immediately after training. Rec-BDNF or Vehicle was injected (arrow) 12 h after training. Rats were tested 2 d (Test 1) or 7 d (Test 2) after the injection (n = 9–10/group; anti-BDNF/VEH vs anti-BDNF/BDNF, Test 2, p < 0.001, two-way ANOVA followed by Bonferroni post hoc tests; treatment effect F(2,50) = 8.36, p = 0.0007); ***p < 0.001.
Results
Temporal evolution of hippocampal de novo protein-synthesis requirement during memory consolidation
To define the temporal requirement of the dorsal hippocampal de novo protein synthesis during long-term IA memory consolidation, we bilaterally injected 125 μg of the translation inhibitor anisomycin into the dorsal hippocampi of rats at 15 min before, immediately after, or 24 h, 48 h, or 7 d after training. This dose blocks >80% of protein synthesis in the dorsal hippocampus for up to 6 h (Milekic et al., 2006). Memory retention was tested at 2 d (Test 1) and 7 d (Test 2) after the injection, or, in specific cases, only at the indicated time.
Compared with vehicle, anisomycin injected 15 min before training completely disrupted memory retention at Tests 1 and 2 without affecting short-term memory tested at 1 h after training (Fig. 1A,B). At both Tests 1 and 2 the retention of the anisomycin-injected rats was not significantly different from acquisition, indicating that memory was completely disrupted. A reminder footshock of intensity equal to that of training in a different context 24 h after Test 2 did not reinstate the memory tested 24 h later (Test 3), indicating that the memory loss was not due to temporary retention/retrieval inhibition. Such a footshock rescues IA extinction (Inda et al., 2011).
Anisomycin injected immediately after training, while still causing significant memory impairment compared with vehicle (Tests 1 and 2), produced a significantly higher retention relative to acquisition, indicating that a few minutes of de novo protein synthesis following training is sufficient to produce some memory retention that lasts for 2 d. A reminder shock 1 d after Test 2 failed to rescue the memory at Test 3 (Fig. 1C).
Rats injected with anisomycin 24 h after training had retentions similar to those of vehicle-injected controls 2 d later (Test 1), but 5 d later (Test 2) significantly lost their memory compared with controls (Fig. 1D). This loss of memory was not complete, as the retention was significantly higher than acquisition. To test whether Test 1 contributed to the effect seen at Test 2 (Inda et al., 2011), rats were given the same treatments and were tested only at 7 d after training (Fig. 1E). Compared with vehicle, anisomycin significantly impaired memory retention, indicating that translation at 24 h after training is required for memory persistence.
In contrast, anisomycin injected either at 2 d or 7 d after training had no effect on memory retention tested at 2 d, 7d, or 21 d after injection (Fig. 1F,G), indicating that the hippocampal de novo protein synthesis requirement for IA memory consolidation is completed by 2 d after training.
Finally, rapamycin, which is known to inhibit the mTOR pathway and target the translation of a subset of proteins in dendrites (Richter and Klann, 2009), injected 15 min before training completely disrupted memory retention, whereas the same treatment after training had no effect, indicating that a very brief phase of hippocampal, rapamycin-dependent mechanisms are required for IA memory consolidation (Fig. 2).
Thus, a rapid phase of de novo protein synthesis is required to initiate memory consolidation. This requirement persists for >24 h and terminates by 48 h after training. Interfering with translation hours or a day after training leads to the formation of a memory, which, however, only persists for a few days, but then rapidly decays.
The temporal evolution of BDNF requirement in the hippocampus during memory consolidation parallels that of de novo protein synthesis
Based on previous studies showing that (1) BDNF leads to mTOR activation and protein synthesis, which are required for LTP and long-term memory formation (Tang et al., 2002; Slipczuk et al., 2009); (2) hippocampal blockade of BDNF or TrkB 15 min before training disrupts memory at 2 d after training without affecting short-term memory (Chen et al., 2012); (3) hippocampal BDNF is required at 12 h post training for the persistence but not formation of step-down IA memory (Bekinschtein et al., 2007a); and (4) BDNF rescues the memory decay caused by protein synthesis inhibitors injected 12 h after training in a step-down IA (Bekinschtein et al., 2008), we set out to determine whether BDNF is both a fast upstream mediator of the rapid phase of translation required at training to initiate consolidation and a critical persistent mediator of the progression of the entire consolidation process. Hence, we tested whether the temporal progression of BDNF requirement parallels that of the entire protein synthesis-dependent phase.
Pretraining bilateral injections into the dorsal hippocampus of either anti-BDNF or its receptor scavenger TrkB-Fc completely disrupted memory retention at both 2 d (Test 1; Fig. 3A) and 7 d after training compared with control IgG (Test 2). No reinstatement was found following a reminder footshock (Test 3; Fig. 3A).
The same treatments given immediately post training caused a nonsignificant trend toward memory decrease at Test 1 (Fig. 3B). This memory retention significantly decayed by Test 2. No memory reinstatement was found following the reminder footshock (Test 3), indicating that the memory decline from Test 1 to Test 2 was not a result of extinction, but of consolidation impairment (Fig. 3B).
Further, similar to what was found with anisomycin, injection of TrkB-Fc 1 d after training did not affect retention 2 d later (Test 1; Fig. 3D) but significantly impaired memory tested only at 7 d after the injection (Fig. 3C, test 1, 8 d). Again similar to that of de novo protein synthesis, the requirement for BDNF signaling in the hippocampus was completed by 2 d after training. In fact, injecting anti-BDNF or TrkB-Fc either 2 d or 7 d after training had no effect on retention when tested 2, 7, and 21 d after the injection (Tests 1–3; Fig. 3D,E).
Together, these data indicate that both BDNF signaling and de novo protein synthesis in the hippocampus follow a similar temporal progression of requirement during memory consolidation. They also indicate that a rapid BDNF-dependent activation is critical for initiating memory consolidation and that a sustained or cycling activation of the BDNF-dependent pathway during the first 1–2 d after training is required for the completion of hippocampal memory consolidation.
Initial activation of BDNF signaling is required for initiating and maintaining a sustained increase in pCREB, C/EBPβ, pCaMKIIα, and pCofilin
Because consolidation requires gene expression, we then hypothesized that an autoregulatory-positive feedback loop of BDNF-dependent increase in BDNF expression is a critical mechanism for memory consolidation.
We first examined the effect of blocking BDNF on post-training temporal profiles of gene expression known to underlie IA memory formation. Previous results from Chen et al. (2012) showed that blocking BDNF in the dorsal hippocampus before training completely blocks the rapid (at 30 min) IA training-dependent induction of pCREB. This blockade persists at 20 h after training together with the blockade of phosphorylation of calcium pCaMKIIα and Synapsin1.
Here we extended the investigation of molecular changes by testing the effect of pretraining anti-BDNF on the expression levels of the same and additional markers at 12 h after training. We chose this time point because it reflects the progression phase of the gene expression initiated at training. It also reports the changes occurring midway between the immediate phase after training and the 24 h time point at which protein synthesis and BDNF are still required. Quantitative Western blot analysis revealed that pretraining anti-BDNF treatment significantly blocks the training-dependent increase in hippocampal pCREB, phosphorylation of the actin severing protein cofilin (at Ser-9; pCofilin), and pCaMKIIα as well as the induction of C/EBPβ (Fig. 4A). These changes were not observed in the hippocampi of rats that underwent an unpaired context- shock protocol (UN), which is known to not induce an IA memory [Student's t test for all comparisons: 12 h, pCREB: naive = 100.0 ± 9.380 (n = 5), UN = 81.05 ± 12.47 (n = 6), p = 0.27; C/EBPβ: naive = 100.0 ± 11.96 (n = 5), UN = 81.34 ± 16.46 (n = 6), p = 0.40; pCaMKIIα: naive = 100.0 ± 16.99 (n = 5), UN = 82.98 ± 9.508 (n = 6), p = 0.38; pCofilin: naive = 100.0 ± 17.96 (n = 5), UN = 109.9 ± 13.41 (n = 6), p = 0.66; 20 h, BDNF: naive = 100.0 ± 10.31 (n = 5), UN = 93.37 ± 7.577 (n = 5), p = 0.62].
No significant changes were observed in levels of total CREB, total CaMKIIα, or total cofilin (data not shown).
A summary of the temporal progression of hippocampal molecular changes occurring in vivo downstream BDNF in response to IA learning is shown in Figure 4B. All the changes after training are shown compared with the levels of naive rats killed at matched time points. Furthermore, in the hippocampi of rats exposed to unpaired context-shock, shock alone, or context alone and killed at matched time points, the levels of pCREB, pCaMKIIα, and BDNF have been found to be similar to those of naive rats, indicating that the training-dependent changes correlate with the context-shock association (Taubenfeld et al., 2001b; Chen et al., 2012). Hence, training-elicited molecular changes are shown compared with naive conditions.
All together, these data indicate that BDNF is necessary for the activation of the pCREB-C/EBPβ-dependent cascade of events, which are sustained for >24 h. The progression and completion of this cascade of events are necessary for memory consolidation.
BDNF is regulated by C/EBPβ in the hippocampus during memory consolidation via an autoregulatory-positive feedback loop
CREB is required for BDNF transcription (Tao et al., 1998) and BDNF is known to mediate CREB activation (Finkbeiner et al., 1997; Pizzorusso et al., 2000; Alonso et al., 2002). Similarly, as shown above, the training-dependent induction of C/EBPβ in the hippocampus is also downstream BDNF and in vitro data indicate that, in addition to CREs, there are putative C/EBP binding sites on BDNF promoters (Shieh et al., 1998; Tao et al., 1998; Takeuchi et al., 2002). We therefore tested whether the sustained BDNF-dependent requirement for memory consolidation for over 24 h could be due to a positive autoregulatory feedback loop of BDNF via C/EBPβ.
Hence, we first investigated the temporal profile of bdnf exon IV mRNA, as this exon has been reported to be regulated in synaptic plasticity and memory consolidation (Hong et al., 2008; Lubin et al., 2008). Dorsal hippocampal RNA of rats killed at 40 min and 6, 12, 20, and 48 h after IA training was probed with quantitative real-time PCR (qRT-PCR) for bdnf exon IV mRNA. Results showed that, compared with naive rats, trained rats had a significant increase in bdnf exon IV mRNA level at 12 and 20 h after training but not at the other time points tested (Fig. 5A).
To determine whether bdnf exon IV expression is critical for learning, we used an antisense ODN against a sequence selective for the bdnf exon IV to knock down this transcript. Rats received bilateral hippocampal injections of either bdnf exon IV antisense (AS) or SCR ODN 6 h after IA training. AS injection significantly decreased BDNF levels at 20 h after training [AS = 64.32 ± 4.254 (n = 5), SCR = 100.0 ± 12.01 (n = 5), p = 0.0221, Student's t test], validating the knock-down effect of the AS. Compared with SCR, AS significantly impaired memory retention at both 2 and 7 d after training (Fig. 5B). Furthermore, the memory of the AS-injected rats was not reinstated by a reminder footshock (Fig. 5B), indicating that bdnf exon IV expression is in fact required for IA memory consolidation.
We then asked whether bdnf exon IV is a transcriptional target of C/EBPβ in vivo. ChIP of dorsal hippocampal extracts with an anti-C/EBPβ or a control IgG antibody (Fig. 5C) followed by PCR using primers flanking putative C/EBPβ binding sites on the bdnf exon IV promoter (Figs. 5C, 7A) showed significant binding of anti-C/EBPβ compared with control IgG antibody (Fig. 5C). Hence, we conducted ChIP qRT-PCR analysis on dorsal hippocampal extracts from rats trained and killed at 30 min or 12 h or 48 h after training. Compared with naive, the hippocampi of trained rats had a significant increase of C/EBPβ binding to the exon IV promoter at 12 h but not at 30 min or 48 h after training when it returned to control levels (Fig. 5C). This profile correlates with that of bdnf exon IV mRNA as well as that of the training-induced C/EBPβ expression (Taubenfeld et al., 2001b), thus suggesting that bdnf is likely a direct transcriptional target of C/EBPβ in the rat hippocampus in vivo.
To further define whether bdnf is a functional target of C/EBPβ, we determined whether blocking the training-induced upregulation of C/EBPβ affects the induction of bdnf exon IV mRNA. The antisense-mediated knockdown of C/EBPβ with the oligodeoxynucleotide β-ODN at 5 h after training, a protocol that has been shown to block the induction of C/EBPβ 12 h after training (Taubenfeld et al., 2001a), completely blocked the increase of bdnf exon IV mRNA measured by qRT-PCR analysis (Fig. 5D). Furthermore, the training-dependent induction of BDNF protein at 20 h after training was blocked by β-ODN injected 5 h after training, confirming that the training-evoked BDNF increase is downstream C/EBPβ (Fig. 5E).
Since the induction of C/EBPβ requires BDNF at training (Fig. 4), we tested whether the bdnf exon IV mRNA induction at 12 h would also be blocked by pretraining injections of an anti-BDNF antibody. qRT-PCR analysis revealed that compared with IgG, anti-BDNF antibodies injected 15 min before training blocked the induction of bdnf exon IV at 12 h after training (Fig. 5F).
Together, these data indicate that a rapid BDNF signaling at training leads to the recruitment of the CREB-C/EBP pathway, which in turn via C/EBPβ upregulates the expression of BDNF. This autoregulatory-positive feedback loop is required for the successful completion of memory consolidation.
BDNF rescues the memory impairment mediated by C/EBPβ knockdown
Given that BDNF recruits the CREB-C/EBP pathway to regulate its own expression, we asked whether BDNF is sufficient to rescue the memory impairment caused by C/EBPβ knockdown 5 h after IA training. Rats received bilateral hippocampal injections of either β-ODN or SCR-ODN 5 h after IA training. The β-ODN-injected rats were further divided into three groups: one was coinjected with recombinant BDNF (Rec-BDNF), one with vehicle, and the third with another recombinant trophic factor, neurotrophin 3 (Rec-NT3). All rats were tested 2 d (Test 1) and 7 d (Test 2) after training. Confirming our previous result, compared with SCR-ODN, β-ODN significantly impaired memory retention at both 2 and 7 d after training (Fig. 6A). BDNF significantly and completely rescued this amnesia while NT3 did not. Moreover, the rescuing effect of BDNF persisted over time (Fig. 6A). Thus, BDNF selectively rescues the memory impairment mediated by C/EBPβ knockdown.
To investigate whether the rescuing effect of BDNF occurs during the temporally restricted window of the CREB-C/EBP-dependent gene expression phase, which, as shown above, regulates the endogenous increase in BDNF, we monitored the effect of rec-BDNF hippocampal injection over time. Rats received bilateral intrahippocampal injections of either β-ODN or SCR-ODN 5 h after IA training. Both groups received either vehicle or rec-BDNF at 12 h, 2 d, or 4 d after training and were tested at 2 d (Test 1) and 7 d (Test 2) after injection (Fig. 6B–D). Rec-BDNF selectively and completely rescued the memory impairment caused by β-ODN when injected at 12 h after training without changing memory retention when injected with SCR-ODN (Fig. 6B). This effect was selective, because IGF-II, another C/EBPβ target growth factor, did not rescue the memory deficit caused by C/EBPβ knockdown. Notably, BDNF no longer rescued the memory impairment if injected at 2 d or 4 d after training (Fig. 6C,D), suggesting that the effect is limited to the gene expression-dependent consolidation phase. Thus, BDNF rescues the memory impairment caused by C/EBPβ knockdown in a selective and temporally restricted manner. The temporal window within which this effect occurs is similar to that of the requirement for hippocampal de novo protein synthesis critical for memory consolidation, lending further support for our hypothesis that BDNF mediates at least part of the protein synthesis required for memory consolidation.
Last, since pretraining anti-BDNF antibody led to amnesia and blocked the induction of C/EBPβ at 12 h after training, we tested whether rec-BDNF could rescue this amnesia. Rats were given intradorsal hippocampal injections of anti-BDNF or IgG 15 min before IA training; 12 h later, while the anti-BDNF rats were injected with either rec-BDNF or vehicle, the IgG-injected controls received vehicle. All rats were tested at 2 d (Test 1) and 7 d (Test 2) after training (Fig. 6E). BDNF failed to rescue the memory deficit caused by pretraining intrahippocampal anti-BDNF injections (Fig. 6E), suggesting that pretraining blockade of BDNF completely prevents the rapid changes that initiate the consolidation process on which BDNF can act to rescue the memory. In contrast, blocking C/EBPβ induction at 5 h post training using β-ODN allows for at least 5 h of consolidation mechanisms, which leaves substrate/s at 12 h on which BDNF rescues the memory. To exclude that the anti-BDNF antibody injected 15 min before training was still present at 12 h after training, thus preventing the rescuing effect of BDNF at 12 h, anti-BDNF antibody was delivered into the dorsal hippocampi of rats immediately after training followed by injection of either vehicle or rec-BDNF 12 h after training. As shown in Figure 3B, we confirmed that the post-training injection of anti-BDNF failed to significantly change memory at 2 d after training but significantly disrupted it at 7 d after training. This decay was completely rescued by rec-BDNF injected at 12 h after training (Fig. 6F), confirming that a rapid, BDNF-dependent mechanism is required for the rescuing effect of BDNF at later times.
Hence, BDNF is sufficient to rescue the memory impairment caused by hippocampal C/EBPβ knockdown during a temporally restricted window that overlaps with that of the de novo protein-synthesis requirement for consolidation. Moreover, BDNF can rescue a memory only if the consolidation process has been initiated or is ongoing, but not beyond this temporal window.
BDNF-dependent transcriptional repression and the termination of the feedback loop correlate with binding of HDAC2, MeCP2, and Sin3A to its promoter
Which mechanism ends the temporal window of de novo protein synthesis and BDNF autoregulatory feedback loop by 48 h after training? Since bdnf mRNA levels return to baseline by 48 h after training, we hypothesized that repressing chromatin structure at the bdnf exon IV might act as a molecular brake that signals the end of the BDNF feedback loop. Moreover, the training-dependent increases in pCREB and C/EBPβ also return to control levels by 48 h. We therefore examined the binding of pCREB and C/EBPβ as well as the transcriptional repressor MeCP2, along with its corepressors HDAC2 and Sin3a at the bdnf exon IV promoter, because these factors have previously been implicated in transcriptional control of the bdnf exon IV promoter in vitro (Chen et al., 2003; Martinowich et al., 2003).
ChIPs with anti-pCREB, anti-C/EBPβ, anti-MeCP2, anti-HDAC2, and anti-Sin3a of dorsal hippocampal extracts taken from trained rats killed 30 min, 12 h, or 48 h after training followed by qRT-PCR analysis of the bdnf exon IV promoter region (Fig. 7A) revealed that pCREB and C/EBPβ binding at the bdnf exon IV promoter is significantly increased at 12 h but not 30 min after training and returns to naive control levels by 48 h (Figs. 7B,C, 5B). Conversely, MeCP2 exhibited significantly higher binding to the bdnf exon IV promoter at 48 h but not at 30 min or 12 h after training compared with naive controls (Fig. 7B,D). Similarly, HDAC2 binding was significantly increased at 48 h after training but not at earlier time points (Fig. 7E). We also observed significantly increased Sin3a binding at 30 min and 48 h but not 12 h after training, indicating that Sin3a may have different binding partners at different time points. Notably, Sin3a binding to the exon IV promoter appears anti-correlated with bdnf exon IV mRNA expression (Fig. 7F). These data imply that increased transcriptional repression at the bdnf exon IV promoter, likely mediated by MeCP2, HDAC2, and Sin3a, with consequent decrease in BDNF transcription at 48 h after training, accompanies the termination of the consolidation process.
Discussion
We have shown that, to consolidate, IA memory requires hippocampal de novo protein synthesis that begins very rapidly at training, lingers for >24 h, and ends by 48 h after training. A key mechanism paralleling this process is BDNF release and its expression regulation via a positive autoregulatory feedback loop, which initiates, sustains, and ends the gene expression-dependent phase of consolidation. Interfering with translation or BDNF function at 24 h after training compromises the progression of consolidation and leads to memory decay. C/EBPβ is critical for the positive BDNF feedback loop: the induction of C/EBPβ requires BDNF at training and in turn C/EBPβ regulates the increase of BDNF expression during the lasting phase of consolidation.
The requirements for both de novo protein synthesis and BDNF/TrkB in the hippocampus are crucial during the immediate phase after training to rapidly initiate memory consolidation, and a few minutes of BDNF/TrkB signaling and translation are sufficient to produce some memory retention that is maintained for a few days but then rapidly decays. Our anisomycin results extend the temporal window proposed in classical studies of a very rapid, initial phase of translation sufficient to promote memory consolidation (Squire and Davis, 1975). In most cases, in fact, these investigations did not continue the temporal analysis beyond the first day or two after training (Mark and Watts, 1971; Squire and Davis, 1975). Our findings also expand those of Medina's laboratory showing that anisomycin injected into the dorsal hippocampus at 12 h after step-down IA training but not at 9 h or 24 h post training disrupts memory retention tested at 7 d but not 2 d post training (Bekinschtein et al., 2007a, 2010). Moreover, similar temporal effects of translation inhibitors were reported in Aplysia californica neuronal cultures where a late sustained phase of translation was shown to be critical for the stabilization of long-term facilitation, indicating that a sustained requirement of translation is an evolutionarily conserved mechanism in long-term plasticity and memory (Miniaci et al., 2008). In summary our experiments extensively characterized the temporal profile of translation requirement occurring in the dorsal hippocampus during IA memory consolidation leading to the conclusion that de novo protein synthesis is essential for more than 1 d but less than 2 d after training.
Additionally, we showed that the requirement for translation to initiate memory consolidation follows a temporal progression similar to that of BDNF, although the difference between before and after training treatments is more dramatic with BDNF blockade than anisomycin. This difference may be due to the efficiency or concentration of blockers, but may also result from distinct target mechanisms of the anti-BDNF antibody versus those of anisomycin. Indeed, BDNF targets several signal transduction pathways, including those mediated by MAP kinase, Akt, and PLCγ, in addition to affecting translation and the mTOR pathway (Takei et al., 2004; Musumeci and Minichiello, 2011). Moreover, rapamycin, known to affect a subset of translation targeted by anisomycin as well as by BDNF, showed, in agreement with previous studies (Bekinschtein et al., 2007b), an even more dramatic difference with hippocampal pretraining versus post-training injections. This suggests that the targets of rapamycin act very rapidly to initiate memory consolidation, which then progresses via rapamycin-independent mechanisms.
Both BDNF/TrkB and translation continue to be necessary for more than a day but less than two to complete the consolidation process. These data extend those of Medina's laboratory showing that blocking translation or BDNF in the dorsal hippocampus at 12 h after step-down IA training but not at 9 h or 24 h post training disrupts memory retention tested at 7 d but not 2 d post training (Bekinschtein et al., 2007a, 2010). The discrepancy between this study and our data for the effect of BDNF blockade at 24 h on memory retention at 7 d after training might be only apparent and likely due to the effect of retesting in their experiments.
The fact that hippocampal-mediated consolidation requires one entire day to complete is intriguing, as it suggests that circadian rhythms and sleep may significantly be involved (Eckel-Mahan and Storm, 2009; Wang et al., 2011; Tononi and Cirelli, 2014).
Our results showed that the extended role of BDNF requires an upregulation of BDNF expression, which follows a temporal profile that parallels that of C/EBPβ induction, as bdnf exon IV mRNA level is significantly elevated at 12 and 20 h but not at 30 min or 6 h post training and returns to baseline by 48 h after training. This increase in mRNA is paralleled by an increase in BDNF protein. As bdnf exon IV knockdown results in decreased levels of BDNF protein and disrupts memory consolidation, we conclude that in vivo bdnf exon IV expression is necessary for long-term memory formation. Furthermore, we showed that C/EBPβ is a critical activator of BDNF expression because first, C/EBPβ binds to the promoter of the bdnf exon IV and this binding, like that of pCREB, significantly increases at 12 h after training and returns to baseline by 48 h post training when BDNF levels are also back to baseline. Second, C/EBPβ knockdown 5 h after training prevents BDNF increase and impairs long-term memory. Notably, this memory loss is fully rescued by rec-BDNF, indicating that BDNF is sufficient to rescue the C/EBPβ-dependent mechanisms.
As C/EBPβ induction occurs downstream the BDNF requirement at training, which in turn controls the induction of BDNF expression, we conclude that a BDNF-C/EBPβ-BDNF-positive autoregulatory loop in the dorsal hippocampus is necessary for initiating and sustaining the process of consolidation until completion when memory becomes fully resistant to hippocampal molecular interference. BDNF-induced BDNF synthesis regulated by CREB had been described in neuronal culture studies (Finkbeiner et al., 1997; Tao et al., 1998) and a feedback loop involving BDNF has been hypothesized to occur in the hippocampus in response to exercise (Vaynman et al., 2003).
To our knowledge our results are the first demonstration of a role of an autoregulatory-positive feedback loop of BDNF in long-term memory consolidation in vivo.
Administering BDNF after training does not rescue the amnesia caused by blocking BDNF before training, suggesting that BDNF-dependent initial changes, in addition to those downstream C/EBPβ, are necessary for memory consolidation. However, blocking BDNF immediately after training permits BDNF to rescue memory decay. These results confirm that a few minutes of BDNF-induced changes following training are essential for the initiation of memory consolidation during which BDNF is required but not sufficient, and indicate that after this initiation BDNF is sufficient to sustain the entire process. We speculate that the changes occurring during the rapid, initial phase may include activity-dependent, post-translational modifications as well as translation of critical transcripts at synaptic sites.
Finally, we identified increased binding to the bdnf exon IV promoter of transcriptional inhibitors, which are likely critical for turning off the autoregulatory-positive BDNF loop. MeCP2, a protein that is mutated in Rett syndrome and functions as a transcriptional repressor and/or global regulator of chromatin architecture (Chahrour et al., 2008; Skene et al., 2010; Cohen et al., 2011), together with Sin3a and HDAC2 significantly increase their binding to bdnf exon IV promoter at 48 h after training when BDNF mRNA is back to baseline, but not at 12 h after training when BDNF mRNA is increased as a result of training. Both HDAC2 and Sin3a have been previously shown to bind to bdnf exon IV promoter both in vitro and in vivo and to downregulate bdnf transcription in cortical neuronal cultures (Chen et al., 2003; Martinowich et al., 2003; Guan et al., 2009). Our results link and extend this knowledge to in vivo hippocampal mechanisms regulating memory consolidation, suggesting that the increased binding of MeCP2 and its corepressors Sin3a and HDAC2 at the bdnf exon IV promoter, together with the decreased binding of pCREB and C/EBPβ, are critical mechanisms by which the BDNF-positive autoregulation ends (Fig. 7G). Ending the positive autoregulation of BDNF expression is likely to be a critical step for an efficient consolidation, and its alteration may lead to functional impairments, as also suggested by the fact that overexpression of BDNF indeed leads to memory impairments (Cunha et al., 2009, 2010).
In summary, we propose that BDNF plays a persistent, critical role in the hippocampus for >24 h after training through a positive autoregulatory feedback loop mediated by C/EBPβ and terminated by transcription inhibition mediated by MeCP2, HDAC2, and Sin3a. The sustained molecular mechanisms required for consolidation may facilitate memory trace reactivation during the first 24 h after training, which is suggested to promote hippocampal–neocortical interaction, thus leading to memory storage (Frankland and Bontempi, 2005). We speculate that a similar mechanism may support long-term plasticity responses in other brain regions and functions where BDNF plays a critical role.
This knowledge is important for better understanding how memories are consolidated and stored and formulating new working hypotheses about memory deficits or loss occurring in aging and diseases. It is also important for developing new strategies to either enhance or interfere with memory consolidation, hence combating cognitive impairments or disorders linked to pathogenic memories such as post-traumatic stress disorder and addiction.
Footnotes
This work was supported by grants from the U.S. National Institutes of Health (RO1-MH065635 and R37-MH065635 to C.M.A. and F31-MH816213 to D.Y.C.). We thank Ja Wook Koo and Victoria Swiss for technical support.
The authors declare no competing financial interests.
References
- Alberini CM. Transcription factors in long-term memory and synaptic plasticity. Physiol Rev. 2009;89:121–145. doi: 10.1152/physrev.00017.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alonso M, Vianna MR, Izquierdo I, Medina JH. Signaling mechanisms mediating BDNF modulation of memory formation in vivo in the hippocampus. Cell Mol Neurobiol. 2002;22:663–674. doi: 10.1023/A:1021848706159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bekinschtein P, Cammarota M, Igaz LM, Bevilaqua LR, Izquierdo I, Medina JH. Persistence of long-term memory storage requires a late protein synthesis- and BDNF- dependent phase in the hippocampus. Neuron. 2007a;53:261–277. doi: 10.1016/j.neuron.2006.11.025. [DOI] [PubMed] [Google Scholar]
- Bekinschtein P, Katche C, Slipczuk LN, Igaz LM, Cammarota M, Izquierdo I, Medina JH. mTOR signaling in the hippocampus is necessary for memory formation. Neurobiol Learn Mem. 2007b;87:303–307. doi: 10.1016/j.nlm.2006.08.007. [DOI] [PubMed] [Google Scholar]
- Bekinschtein P, Cammarota M, Katche C, Slipczuk L, Rossato JI, Goldin A, Izquierdo I, Medina JH. BDNF is essential to promote persistence of long-term memory storage. Proc Natl Acad Sci U S A. 2008;105:2711–2716. doi: 10.1073/pnas.0711863105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bekinschtein P, Katche C, Slipczuk L, Gonzalez C, Dorman G, Cammarota M, Izquierdo I, Medina JH. Persistence of long-term memory storage: new insights into its molecular signatures in the hippocampus and related structures. Neurotox Res. 2010;18:377–385. doi: 10.1007/s12640-010-9155-5. [DOI] [PubMed] [Google Scholar]
- Chahrour M, Jung SY, Shaw C, Zhou X, Wong ST, Qin J, Zoghbi HY. MeCP2, a key contributor to neurological disease, activates and represses transcription. Science. 2008;320:1224–1229. doi: 10.1126/science.1153252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen DY, Stern SA, Garcia-Osta A, Saunier-Rebori B, Pollonini G, Bambah-Mukku D, Blitzer RD, Alberini CM. A critical role for IGF-II in memory consolidation and enhancement. Nature. 2011;469:491–497. doi: 10.1038/nature09667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen DY, Bambah-Mukku D, Pollonini G, Alberini CM. Glucocorticoid receptors recruit the CaMKIIalpha-BDNF-CREB pathways to mediate memory consolidation. Nat Neurosci. 2012;15:1707–1714. doi: 10.1038/nn.3266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen WG, Chang Q, Lin Y, Meissner A, West AE, Griffith EC, Jaenisch R, Greenberg ME. Derepression of BDNF transcription involves calcium-dependent phosphorylation of MeCP2. Science. 2003;302:885–889. doi: 10.1126/science.1086446. [DOI] [PubMed] [Google Scholar]
- Cohen S, Gabel HW, Hemberg M, Hutchinson AN, Sadacca LA, Ebert DH, Harmin DA, Greenberg RS, Verdine VK, Zhou Z, Wetsel WC, West AE, Greenberg ME. Genome-wide activity-dependent MeCP2 phosphorylation regulates nervous system development and function. Neuron. 2011;72:72–85. doi: 10.1016/j.neuron.2011.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunha C, Angelucci A, D'Antoni A, Dobrossy MD, Dunnett SB, Berardi N, Brambilla R. Brain-derived neurotrophic factor (BDNF) overexpression in the forebrain results in learning and memory impairments. Neurobiol Dis. 2009;33:358–368. doi: 10.1016/j.nbd.2008.11.004. [DOI] [PubMed] [Google Scholar]
- Cunha C, Brambilla R, Thomas KL. A simple role for BDNF in learning and memory? Front Mol Neurosci. 2010;3:1–14. doi: 10.3389/neuro.02.001.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dash PK, Orsi SA, Moore AN. Spatial memory formation and memory-enhancing effect of glucose involves activation of the tuberous sclerosis complex-Mammalian target of rapamycin pathway. J Neurosci. 2006;26:8048–8056. doi: 10.1523/JNEUROSCI.0671-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis HP, Squire LR. Protein synthesis and memory: a review. Psychol Bull. 1984;96:518–559. doi: 10.1037/0033-2909.96.3.518. [DOI] [PubMed] [Google Scholar]
- Dudai Y. The neurobiology of consolidations, or, how stable is the engram? Annu Rev Psychol. 2004;55:51–86. doi: 10.1146/annurev.psych.55.090902.142050. [DOI] [PubMed] [Google Scholar]
- Eckel-Mahan KL, Storm DR. Circadian rhythms and memory: not so simple as cogs and gears. EMBO Rep. 2009;10:584–591. doi: 10.1038/embor.2009.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finkbeiner S, Tavazoie SF, Maloratsky A, Jacobs KM, Harris KM, Greenberg ME. CREB: a major mediator of neuronal neurotrophin responses. Neuron. 1997;19:1031–1047. doi: 10.1016/S0896-6273(00)80395-5. [DOI] [PubMed] [Google Scholar]
- Frankland PW, Bontempi B. The organization of recent and remote memories. Nat Rev Neurosci. 2005;6:119–130. doi: 10.1038/nrn1607. [DOI] [PubMed] [Google Scholar]
- Guan JS, Haggarty SJ, Giacometti E, Dannenberg JH, Joseph N, Gao J, Nieland TJ, Zhou Y, Wang X, Mazitschek R, Bradner JE, DePinho RA, Jaenisch R, Tsai LH. HDAC2 negatively regulates memory formation and synaptic plasticity. Nature. 2009;459:55–60. doi: 10.1038/nature07925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong EJ, McCord AE, Greenberg ME. A biological function for the neuronal activity-dependent component of Bdnf transcription in the development of cortical inhibition. Neuron. 2008;60:610–624. doi: 10.1016/j.neuron.2008.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inda MC, Muravieva EV, Alberini CM. Memory retrieval and the passage of time: from reconsolidation and strengthening to extinction. J Neurosci. 2011;31:1635–1643. doi: 10.1523/JNEUROSCI.4736-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katche C, Cammarota M, Medina JH. Molecular signatures and mechanisms of long-lasting memory consolidation and storage. Neurobiol Learn Mem. 2013;106:40–47. doi: 10.1016/j.nlm.2013.06.018. [DOI] [PubMed] [Google Scholar]
- Lubin FD, Roth TL, Sweatt JD. Epigenetic regulation of BDNF gene transcription in the consolidation of fear memory. J Neurosci. 2008;28:10576–10586. doi: 10.1523/JNEUROSCI.1786-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mark RF, Watts ME. Drug inhibition of memory formation in chickens. I. Long-term memory. Proc R Soc Lond B Biol Sci. 1971;178:439–454. doi: 10.1098/rspb.1971.0074. [DOI] [PubMed] [Google Scholar]
- Martinowich K, Hattori D, Wu H, Fouse S, He F, Hu Y, Fan G, Sun YE. DNA methylation-related chromatin remodeling in activity-dependent BDNF gene regulation. Science. 2003;302:890–893. doi: 10.1126/science.1090842. [DOI] [PubMed] [Google Scholar]
- Medina JH, Bekinschtein P, Cammarota M, Izquierdo I. Do memories consolidate to persist or do they persist to consolidate? Behav Brain Res. 2008;192:61–69. doi: 10.1016/j.bbr.2008.02.006. [DOI] [PubMed] [Google Scholar]
- Milekic MH, Brown SD, Castellini C, Alberini CM. Persistent disruption of an established morphine conditioned place preference. J Neurosci. 2006;26:3010–3020. doi: 10.1523/JNEUROSCI.4818-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miniaci MC, Kim JH, Puthanveettil SV, Si K, Zhu H, Kandel ER, Bailey CH. Sustained CPEB-dependent local protein synthesis is required to stabilize synaptic growth for persistence of long-term facilitation in Aplysia. Neuron. 2008;59:1024–1036. doi: 10.1016/j.neuron.2008.07.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musumeci G, Minichiello L. BDNF-TrkB signalling in fear learning: from genetics to neural networks. Rev Neurosci. 2011;22:303–315. doi: 10.1515/RNS.2011.031. [DOI] [PubMed] [Google Scholar]
- Pizzorusso T, Ratto GM, Putignano E, Maffei L. Brain-derived neurotrophic factor causes cAMP response element-binding protein phosphorylation in absence of calcium increases in slices and cultured neurons from rat visual cortex. J Neurosci. 2000;20:2809–2816. doi: 10.1523/JNEUROSCI.20-08-02809.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter JD, Klann E. Making synaptic plasticity and memory last: mechanisms of translational regulation. Genes Dev. 2009;23:1–11. doi: 10.1101/gad.1735809. [DOI] [PubMed] [Google Scholar]
- Shieh PB, Hu SC, Bobb K, Timmusk T, Ghosh A. Identification of a signaling pathway involved in calcium regulation of BDNF expression. Neuron. 1998;20:727–740. doi: 10.1016/S0896-6273(00)81011-9. [DOI] [PubMed] [Google Scholar]
- Skene PJ, Illingworth RS, Webb S, Kerr AR, James KD, Turner DJ, Andrews R, Bird AP. Neuronal MeCP2 is expressed at near histone-octamer levels and globally alters the chromatin state. Mol Cell. 2010;37:457–468. doi: 10.1016/j.molcel.2010.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slipczuk L, Bekinschtein P, Katche C, Cammarota M, Izquierdo I, Medina JH. BDNF activates mTOR to regulate GluR1 expression required for memory formation. PLoS One. 2009;4:e6007. doi: 10.1371/journal.pone.0006007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Squire LR, Davis HP. Cerebral protein synthesis inhibition and discrimination training: effect of extent and duration of inhibition. Behav Biol. 1975;13:49–57. doi: 10.1016/S0091-6773(75)90778-6. [DOI] [PubMed] [Google Scholar]
- Takei N, Inamura N, Kawamura M, Namba H, Hara K, Yonezawa K, Nawa H. Brain-derived neurotrophic factor induces mammalian target of rapamycin-dependent local activation of translation machinery and protein synthesis in neuronal dendrites. J Neurosci. 2004;24:9760–9769. doi: 10.1523/JNEUROSCI.1427-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeuchi Y, Miyamoto E, Fukunaga K. Analysis on the promoter region of exon IV brain-derived neurotrophic factor in NG108–15 cells. J Neurochem. 2002;83:67–79. doi: 10.1046/j.1471-4159.2002.01096.x. [DOI] [PubMed] [Google Scholar]
- Tang SJ, Reis G, Kang H, Gingras AC, Sonenberg N, Schuman EM. A rapamycin-sensitive signaling pathway contributes to long-term synaptic plasticity in the hippocampus. Proc Natl Acad Sci U S A. 2002;99:467–472. doi: 10.1073/pnas.012605299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao X, Finkbeiner S, Arnold DB, Shaywitz AJ, Greenberg ME. Ca2+ influx regulates BDNF transcription by a CREB family transcription factor-dependent mechanism. Neuron. 1998;20:709–726. doi: 10.1016/S0896-6273(00)81010-7. [DOI] [PubMed] [Google Scholar]
- Taubenfeld SM, Milekic MH, Monti B, Alberini CM. The consolidation of new but not reactivated memory requires hippocampal C/EBPbeta. Nat Neurosci. 2001a;4:813–818. doi: 10.1038/90520. [DOI] [PubMed] [Google Scholar]
- Taubenfeld SM, Wiig KA, Monti B, Dolan B, Pollonini G, Alberini CM. Fornix-dependent induction of hippocampal CCAAT enhancer-binding protein [beta] and [delta] Co-localizes with phosphorylated cAMP response element-binding protein and accompanies long-term memory consolidation. J Neurosci. 2001b;21:84–91. doi: 10.1523/JNEUROSCI.21-01-00084.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tononi G, Cirelli C. Sleep and the price of plasticity: from synaptic and cellular homeostasis to memory consolidation and integration. Neuron. 2014;81:12–34. doi: 10.1016/j.neuron.2013.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsankova NM, Kumar A, Nestler EJ. Histone modifications at gene promoter regions in rat hippocampus after acute and chronic electroconvulsive seizures. J Neurosci. 2004;24:5603–5610. doi: 10.1523/JNEUROSCI.0589-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Dijk MA, Rodenburg RJ, Holthuizen P, Sussenbach JS. The liver-specific promoter of the human insulin-like growth factor II gene is activated by CCAAT/enhancer binding protein (C/EBP) Nucleic Acids Res. 1992;20:3099–3104. doi: 10.1093/nar/20.12.3099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaynman S, Ying Z, Gomez-Pinilla F. Interplay between brain-derived neurotrophic factor and signal transduction modulators in the regulation of the effects of exercise on synaptic-plasticity. Neuroscience. 2003;122:647–657. doi: 10.1016/j.neuroscience.2003.08.001. [DOI] [PubMed] [Google Scholar]
- Wang G, Grone B, Colas D, Appelbaum L, Mourrain P. Synaptic plasticity in sleep: learning, homeostasis and disease. Trends Neurosci. 2011;34:452–463. doi: 10.1016/j.tins.2011.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]