

Abstract
The bicyclic β-lactam/2-pyrrolidine precursor to all carbapenem antibiotics is biosynthesized by attachment of a carboxymethylene unit to C5 of L-proline followed by β-lactam ring closure. Carbapenem synthase (CarC), an Fe(II)- and 2-(oxo)glutarate-dependent (Fe/2OG) oxygenase, then inverts the C5 configuration. Here we report the structure of CarC in complex with its substrate and biophysical dissection of its reaction to reveal the stereoinversion mechanism. An Fe(IV)-oxo intermediate abstracts the hydrogen (H•) from C5, and tyrosine 165, a residue not visualized in the published structures of CarC lacking bound substrate, donates H• to the opposite face of the resultant radical. The reaction oxidizes the Fe(II) cofactor to Fe(III), limiting wild-type CarC to one turnover, but substitution of the H•-donating tyrosine disables stereoinversion and confers to CarC the capacity for catalytic substrate oxidation.
β-Lactam compounds constitute more than half of the global antibiotic drug market (1, 2). In the past decade, the increased incidence of bacterial β-lactam resistance has forced increasing reliance on a relatively new subclass of these drugs known as carbapenems (Fig. 1A) (2, 3). Defined by the bicyclic β-lactam/pyrroline nucleus (1), carbapenems are generally less susceptible than many members of the β-lactam class to hydrolytic inactivation by β-lactamases. With more than 40 naturally occurring members, the carbapenem sub-class retains activity against a broad range of both Gram-positive and Gram-negative bacteria, including Staphylococcus epidermidis, Staphylococcus aureus, and Pseudomonas aeruginosa (4). An understanding of the biosynthetic pathways to these natural carbapenems could enable the engineering of new drug variants to combat resistance, which has been emerging at an alarming rate, even to this most robust β-lactam sub-class (5).
Figure 1.
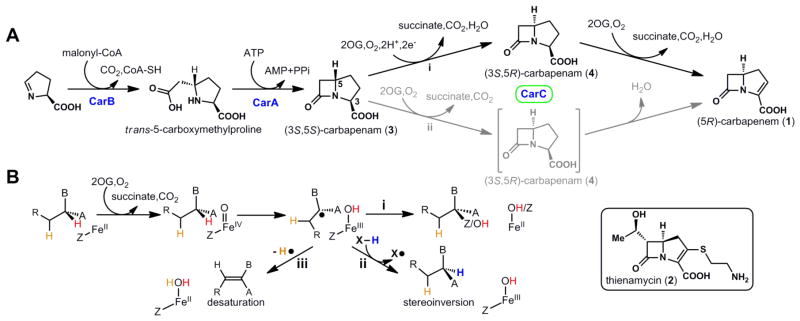
Carbapenem biosynthesis, the role of CarC, and the relationship of the CarC reactions to those of other Fe/2OG oxygenases. (A) The proposed carbapenem biosynthetic pathway. (B) Divergence of the (i) radical-group transfer, (ii) stereoinversion, and (iii) desaturation outcomes known or thought to be mediated by Fe/2OG oxygenases, including CarC.
Following the initial isolation of a carbapenem, thienamycin (2), from Streptomyces cattleya in 1976 (6), the structurally simplest carbapenem, 1, was isolated from Erwinia carotovora and other species (7). Early biochemical studies identified the car gene cluster encoding the enzymatic apparatus to produce 1 (8, 9). Later studies confirmed that this cluster is conserved in all strains that produce carbapenem and is distinct from gene clusters specifying penicillin and cephalosporin biosynthesis (2). Introduction of the carA, carB and carC genes into Escherichia coli established that the three encoded enzymes, carbapenam synthetase (CarA), carboxymethylproline synthase (CarB), and carbapenem synthase (CarC), are sufficient to produce 3, 4, and 1 and enabled in vitro dissection of the biosynthetic sequence (10) (Fig. 1A). Using malonyl-CoA as co-substrate, CarB attaches a carboxymethylene unit to 1-pyrroline-5-carboxylate (which may be supplied in carbapenem-producing microorganisms from L-proline by the carD gene product) to form trans-5-carboxymethylproline (11, 12). CarA then uses ATP hydrolysis (to AMP and pyrophosphate) to drive β-lactam ring closure, generating (3S,5S)-carbapenam (3) (13). The stereochemical configuration at C5 established by CarB is opposite to that in all natural carbapenem antibiotics isolated to date (2). Further elaboration of 3 apparently requires C5 stereoinversion by CarC, an Fe(II)- and 2-(oxo)glutarate-dependent (Fe/2OG) oxygenase, which produces (3S,5R)-carbapenam (4) as its initial product (10). It has been concluded that only after inversion of C5 can CarC then desaturate across the C2–C3 bond to produce (5R)-carbapenem (1) (14). The R configuration at C5 and the C2=C3 double bond of 1 are both crucial for optimal antibiotic activity (15, 16). To our knowledge, no CarB ortholog has yet been shown to produce the cis-5-carboxymethylproline, with C5 in the configuration found in the drug compounds (2, 17–19). Moreover, although it has been suggested that C5 epimerases structurally and mechanistically unrelated to CarC might be operant in other carbapenem biosynthetic pathways [e.g., to thienamycin (17, 20)], none has been identified to date. An understanding of the structure and function of CarC is thus central to efforts to bioengineer new carbapenems.
Mononuclear non-heme-iron oxidases and oxygenases, including those in the Fe/2OG sub-class, generally effect oxidation of their primary substrates (21). In the most well-understood cases, addition of O2 to the Fe(II) cofactor and delivery of two electrons from a co-substrate generates an Fe(IV)-oxo (ferryl) intermediate, which either abstracts a hydrogen atom (H•) from an aliphatic carbon of the substrate or adds as an electrophile to a double bond (22). In cases of H• abstraction, an iron ligand then transfers as a radical to the substrate radical, completing the oxidative substitution of hydrogen by a heteroatom (O, Cl/Br, or S; Fig. 1B, reaction i). This step regenerates the Fe(II) form of the cofactor for subsequent turnovers (21–24).
By contrast to these oxidative substitution reactions, the CarC stereoinversion reaction is redox neutral with respect to its primary substrate (3). This outcome has been rationalized by mechanisms invoking H• abstraction from C5 by the canonical ferryl species and subsequent transfer of a different H• to the opposite face of the substrate radical (Fig. 1B, reaction ii) by, presumably, an amino acid donor (X-H) (25). Computational analyses have supported such a mechanism and involvement of a dedicated H• donor (26). A fluorescence-based in vivo assay for carbapenem production was coupled with amino acid mutagenesis to obtain data suggesting that tyrosine (Y) 67, visualized near the bound N-acetyl-L-proline intended to mimic the substrate in one of two published X-ray crystal structures of CarC, is the H• donor (27, 28). The most recent of these studies employed this structure for molecular-docking analysis that identified a hypothetical substrate-binding mode with C5 in sufficient proximity to Y67 for H• transfer (28). However, because multiple segments of the protein are disordered in this structure, its use as a starting point for such an analysis is inherently speculative. Published mechanisms have further suggested that, after inverting C5, CarC then either uses the two oxidizing equivalents still present in the active site (Fig. 1A, pathway ii) or, following their reduction, undergoes a second round of O2 activation and ferryl formation (pathway i), to effect C2–C3 desaturation (28). As depicted, this second reaction is a mechanistically distinct two-electron oxidation requiring removal of an H• equivalent from each of two adjacent carbon atoms (Fig. 1B, reaction iii) (28, 29).
Studies on other Fe/2OG oxygenases and related oxidative enzymes have established that the crucial tenets of the proposed CarC stereoinversion mechanism, including H• abstraction by a ferryl intermediate and formation of a tyrosyl radical, could be definitively tested by rapid-kinetic and spectroscopic analysis of the reaction (22, 30, 31). In addition, the structure of the enzyme with its authentic substrate, 3, would be expected to reveal proximity of C5 to the iron cofactor and the H• donor, as intentionally enforced for the case of Y67 in the recent computational study (28). Instability of the substrate (7, 28, 32) and enzyme (28, 33) have repeatedly been cited as barriers to such direct in vitro mechanistic and structural analysis. To obtain the enzyme, we expressed N-terminally His6-affinity-tagged Pectobacterium carotovorum CarC in Escherichia coli at 16 °C and purified it on Ni(II)-nitrilo-tris-acetate agarose affinity resin, obtaining ~ 30 mg of > 90% pure protein per gram of wet cell mass (fig. S1). To obtain the substrate, we made use of the observation by multiple investigators that carboxylate-esterified analogs of 3 are sufficiently stable to be isolated (32, 34, 35). We prepared the methyl ester of 3 (7) and its [5-2H]-labeled isotopolog (5-d-7) by a hybrid of two published syntheses (fig. S2A and Supplementary Methods) and then rapidly, enzymatically hydrolyzed the esters to 3 and 5-d-3 just before carrying out in vitro experiments (fig. S2B and Supplementary Methods). Following this deprotection, we found 3 to have a half-life of ~ 14 h at 25 °C and pH 7.5 (in 100 mM Tris-chloride).
By this method of in situ deprotection, we overcame the longstanding barrier posed by the substrate’s instability to determine both the structure of the CarC•Fe(II) •2OG•3 quaternary complex and the mechanism of C5 stereoinversion. To enable structural characterization, crystals of the CarC•Fe(II) •2OG ternary complex obtained after a ~ 24-h incubation in the absence of O2 were soaked in freshly prepared anoxic solutions of 3 to form the quaternary complex, and the crystals were frozen before 3 could decompose (after 2 h; see Supplementary Methods). The structure, solved at a resolution of 2.1 Å by X-ray diffraction experiments (Table S1 and fig. S3), reveals three CarC molecules in the asymmetric unit (ASU). Significant electron density for carbapenam 3 is readily visualized in the active site of two of the three monomers (Fig. 2A and fig. S4), but a crystal lattice contact that distorts the structure of the active site prevents proper substrate binding in the third molecule of the ASU (fig. S5). The bridgehead C5 of 3 resides 4.2 Å from the iron site with its hydrogen directed toward the Fe cofactor in appropriate geometry for its abstraction by the ferryl intermediate (Fig. 2B and fig. S6). This binding orientation is unequivocally established by the shape of the electron density and the positioning of hydrogen-bonding residues to interact with the carboxylate and carbonyl groups of 3. The presence of the actual substrate (rather than the N-acetyl-L-proline bound in one of two published structures of CarC (36)) yields clear electron density for two previously disordered loops surrounding the active site (fig. S4), allowing their structures to be modeled in two of the three molecules in the ASU. Residues 67–78 (loop 1) and 162–172 (loop 2) close over the active site, thereby isolating the substrate from bulk solvent. Such protection would seem to be critical for control of the free-radical reaction(s) mediated by CarC, a conclusion that is consistent with the results of recent mutational-saturation studies that showed that substitutions of bulky or aromatic residues in the active site are incompatible with activity (28). In loop 1, Y67 and Y78 adopt new positions to participate with R267 and Q269 in an extended hydrogen-bonding network that anchors the carboxylate of 3 in the binding pocket. Y164 in loop 2 also interacts directly with the carboxylate. The most mechanistically significant change relative to published structures is the ordering of loop 2 to position Y165 directly above C5, opposite the iron center, with its hydroxyl group 4.7 Å from the substrate C5 atom (Fig. 2B). In this position, Y165 is ideally poised to donate H• to the C5 radical to generate the (3S,5R) epimer, 4. By contrast, Y67, the residue suggested in previous work to be the H• donor (27, 28), is positioned toward the edge of the substrate, inappropriately to serve in this capacity (Fig. 2; figs. S4 and S5).
Figure 2.

Structure of the active site in the CarC•Fe(II) •2OG•3 complex and the mechanism of stereoinversion indicated by the structure and biophysical results presented in this study. (A) 2Fo-Fc (gray mesh, 1.2σ) and Fo-Fc omit (blue mesh, 2.6σ) electron density maps for 3 (carbon atoms shown in yellow). Dashed lines illustrate hydrogen-bonding interactions involving the substrate or bonds between the Fe(II) cofactor and its ligands. (B) Surface representation of the substrate-binding pocket showing the locations of nearby aromatic amino acids including Y165, the H• donor. Black lines indicate the distances between C5 of 3 and the Fe(II) or the hydroxyl substituent on Y165. Selected amino acid side chains, 2OG, and substrate 3 are shown in stick format. The Fe(II) ion and water molecules are shown as green and red spheres, respectively. (C) Mechanism of the CarC-mediated, stoichiometric stereoinversion of 3 to 4, involving abstraction of H• from C5 by a ferryl complex and H• donation by Y165.
The structure of the CarC•Fe(II) •2OG•3 complex is consistent with key features of the published mechanisms, including abstraction of H• from C5 of 3 by the ferryl intermediate and transfer of H• to the opposite side of the C5 radical by a tyrosine, but it implies that Y165, a residue not visualized in any of the published structures and therefore never invoked in the mechanisms, is the dedicated H• donor (Fig. 2C). To test these clear functional implications, we dissected the mechanism by quantification of the reaction products (at completion and with time) and by rapid-kinetic and spectroscopic experiments on both wild-type and Y→F variant CarC proteins with both 3 and 5-d-3 substrates. All results are consistent with the chemical mechanism in Fig. 2C and the kinetic elaboration of this mechanism in fig. S7. Crucial features revealed by the analysis are that the stereoinversion reaction is, in the absence of a reductant, stoichiometric (Fig. 1A, pathway i) rather than catalytic (pathway ii) as a result of oxidation of the Fe(II) cofactor to a stable Fe(III) form, and that only one-third of the enzyme’s Fe(II) sites react, either because its quaternary structure (the trimeric ASU and overall hexamer seen in the crystal structure) enforces a “one-third-of-sites reactivity” or because the previously noted instability of the protein resulted in an inactive fraction. These features are most obvious from the production of a maximum of 0.32 equiv of the succinate co-product in the presence of excess 3, 2OG and O2 (fig. S8A, B). The reaction of only one-third of the Fe(II) sites is also reflected in the accumulation of sub-stoichiometric quantities of the intermediate states b and c (fig. S7).
The intermediacy of a high-spin ferryl complex (Fig. 2C, state b), which may be general to all Fe/2OG oxygenases (22), is implied by Mössbauer spectra of a sample prepared by mixing the CarC•Fe(II) •2OG•3 complex at 5 °C with excess O2 and freeze-quenching after 0.15 s (Fig. 3B and fig. S9). The spectrum at 4.2 K in an applied magnetic field of 53-mT (hash marks) exhibits a new feature at ~ 1 mm/s that is not present in the spectrum of the reactant complex (Fig. 3A). This feature is the high-energy line of a quadrupole doublet with isomer shift (δ) of 0.28 mm/s and quadrupole splitting (ΔEQ) of 0.87 mm/s (Fig. 3B, solid line; fig. S9), parameters very similar to those of high-spin (S = 2) ferryl complexes detected in other Fe/2OG oxygenases (37–41). Spectra collected in an 8-T applied field (fig. S10) confirm that the Fe(IV) complex has a high-spin ground state. The features of the complex account for 24% of the total intensity, equating to 0.24 equiv relative to CarC•Fe(II). As noted, the modest accumulation reflects the reaction of only one-third of the Fe(II) sites. The ferryl complex is, as expected, transient, decaying to < 3 % of its maximal intensity by a reaction time of 3 s (Fig. 3C and fig. S7). Its decay is accompanied by formation of high-spin (S = 5/2) Fe(III) species (Fig. 3C, arrows, and fig. S10), consistent with formation of states c and d in Fig. 2C. Conversion of the ferryl complex to accumulating Fe(III) species, which is also reflected in absorption and EPR spectra discussed below, contrasts with events in the catalytic oxidation reactions of other well-studied Fe/2OG oxygenases, in which the ferryl complexes decay back to Fe(II) species (37, 39, 41, 42). As noted, this difference reflects the non-oxidative and stoichiometric nature of the stereoinversion reaction.
Figure 3.

Mössbauer spectra at 4.2 K and 53 mT demonstrating accumulation of an Fe(IV) intermediate (Fig. 2C, state b) and Fe(III) species (states c and d) in the CarC reaction. (A) Spectrum of the O2-free CarC•Fe(II) •2OG•3 reactant solution. (B and C) Spectra of samples freeze-quenched 0.15 s and 3 s, respectively, after initiating the reaction by mixing with O2. The solid trace is the simulated spectrum of the high-spin Fe(IV) (presumptively ferryl) species, and the arrows mark the features of the high-spin Fe(III) species. Note the different x-scales for the spectra: the top scale applies to A and B and the bottom scale to C. The dotted lines indicate the relationship of the two scales. Reaction conditions are provided in Supplementary Methods.
Stopped-flow absorption experiments initiated by mixing the CarC•Fe(II) •2OG•3 with excess O2 confirm that decay of the ferryl complex between 0.15 s and 3 s is associated with accumulation of a tyrosyl radical (Fig. 2C, state c). A sharp absorption feature at 410 nm and broader peak at 390 nm, unmistakable hallmarks of a tyrosyl radical (Y•), reach their maximum intensity at a reaction time of ~ 3 s and then decay slowly (Fig. 4A, B). The kinetics of formation and decay of the Y• (Fig. 4B, red trace), deduced by the height of the 410-nm peak (Fig. 4A, gold markings), are simulated well according to the kinetic mechanism in fig. S7 (compare red data and black simulation traces). The absorption in the UV region of the spectrum remaining after decay of the Y• (Fig. 4A, cyan trace) reflects the conversion of the Fe(II) cofactor to Fe(III) in the stoichiometric stereoinversion, as also revealed by the Mössbauer spectra. The enzyme reacts with O2 much less rapidly, and no Y• accumulates, in analogous reactions lacking 3 in the protein reactant solution (Fig. 4B, compare red and orange traces). This activating effect of the substrate, which we have termed “substrate triggering,” is common to the Fe/2OG oxygenases (22, 39).
Figure 4.

Kinetic and spectroscopic evidence for state c (Fig. 2C), containing Y165•, Fe(III), and 4, in the CarC reaction. (A) Change in absorbance at the indicated reaction times after mixing the CarC•Fe(II) •2OG•3 complex with O2. The spectra shown were obtained by subtracting the spectrum at 5 ms from the spectra at the indicated reaction times. (B) Kinetics of Y• formation and decay, as reported by the height of the sharp peak at 410 nm, in the reactions of wild-type CarC (red), and the Y67F (black), Y164F (yellow), Y165F (green), and Y191F (cyan) variants under the same conditions as in A. The orange trace is a control reaction of the wild-type enzyme from which 3 was omitted from the protein syringe. The dark blue trace is from the reaction of the wild-type enzyme in the presence of the 5-d-3 substrate. Each trace in B is the average of three trials constructed as indicated by the gold lines, bracket, and arrow in A as A410-(A404+A416)/2 (A410 dropline). (C) X-band EPR spectra of the Y• in reaction samples freeze-quenched at the indicated times. The 100-K spectrum of the 3-s sample (gray trace) has been scaled by 0.5 for clarity. Measurement conditions: microwave frequency, 9.4 GHz; microwave power, 2 μW for 10-K spectra and 20 μW for 100-K spectrum; modulation amplitude, 1 mT for 10-K spectra and 0.2 mT for 100-K spectrum; modulation frequency, 100 kHz. (D) Chemical-quench, LC-MS analysis of the stereoinversion of 3 to 4. Negative-mode, single-ion chromatograms at m/z = 172 for acid hydrolysis of the 3 synthetic standard (3→12, red trace) and the 3.5:1 4:3 synthetic standard (4→13, blue trace) and for CarC reactions acid-quenched at the indicated times. Reaction conditions for the four experiments, the procedures used to synthesize the standards, and details of the LC-MS analysis are provided in the Supplementary Methods.
EPR spectra at 10 K of samples freeze quenched at reaction times of 0.5, 3.0, and 30 s (Fig. 4C) exhibit a signal at g = 2.0 suggestive of an organic radical. The signal develops and decays with the same kinetics (fig. S7, red circles) as the absorption features from the Y• (black trace). In the 3.0-s spectrum, the intensity of the signal reflects the accumulation of 0.25 equiv of a radical, similar to the maximum quantity of the ferryl complex. The g-value and kinetic behavior associate it with the Y•, but the signal is atypical of a magnetically isolated Y•. It has broad features well outside the envelope of a typical Y• spectrum and does not obviously exhibit the characteristic hyperfine couplings with the hydrogens on the β-carbon and phenol ring. The unusual lineshape results from magnetic interaction of the Y• with the nearby high-spin Fe(III) species in state c, as explained below.
These 10-K EPR spectra have additional resonances at g = 4.28 and 6.95 (fig. S11). The g = 4.28 signal develops slowly and is then stable, and we attribute it to the stable Fe(III) cofactor form(s) in state d of the productive reaction (Fig. 2C and fig. S7) and perhaps also generated by slow oxidation of the Fe(II) in the two-thirds of the enzyme that fails to react productively. The g = 6.95 signal is transient and develops and decays along with the Y• (fig. S11A). It arises from the ground Kramers doublet of a high-spin (S = 5/2) Fe(III) center with a positive axial zero-field-splitting parameter (D > 0) and a rhombicity (E/D) of ~0.05. Its characteristics associate it with the Fe(III) site present along with the Y• in state c (Fig. 2C). Evidence for the expected interaction between the two paramagnetic species is provided by the temperature dependencies of the g = 6.95 and g = 2.0 signals. With increasing temperature from 10 K to 100 K, the g = 6.95 signal of the Fe(III) ion becomes markedly less intense (fig. S11B). It broadens almost into the baseline at the higher temperatures as a result of more rapid spin-relaxation. Because the two species interact magnetically, the faster relaxation of the Fe(III) center impacts the g = 2.0 signal of the Y•. At 100 K, the Y• signal is sharper than at 10 K and now clearly does exhibit the characteristic hyperfine interactions with the β- and ring hydrogens (Fig. 4C, gray trace, and fig. S12). The faster spin-relaxation of the Fe(III) species at the higher temperature effectively decouples the spins of the two paramagnetic centers on the EPR timescale. The failure of the magnetic interaction with the Y• to perturb the Mössbauer spectra of the Fe(III) center is explained in Supplementary Results (fig. S13).
The results of stopped-flow experiments employing 5-d-3 in place of 3 verify that conversion of b to c involves cleavage of the C5-H bond. The features of the Y• develop more slowly and to a much lesser extent with the deuterated substrate (Fig. 4B and fig. S7, compare blue and red traces). That the Y• forms more slowly reflects the deuterium kinetic isotope effect on abstraction of the C5 hydrogen and implies that Y• formation follows cleavage of this bond. That much less Y• accumulates implies that the H•-abstracting ferryl complex can decay by at least one additional, unproductive pathway (the downward branch from b to ? in fig. S7), which contributes to a minor extent in the reaction with 3 but becomes predominant when the intermediate is challenged by the heavier hydrogen isotope in 5-d-3. The “uncoupling” of O2 activation from H• abstraction upon substrate deuteration has been seen in other Fe/2OG oxygenases (30, 42, 43). Accounting for the unproductive pathway(s) permits the kinetics of formation and decay of the Y• in the reaction with 5-d-3 (fig. S7, blue trace) to be simulated accurately (gray trace).
The results of chemical-quench kinetic experiments confirm that C5 stereoinversion also occurs in the conversion of b to c. Acid hydrolysis of 3 yields trans-5-carboxymethyl-L-proline, 12, whereas hydrolysis of 4 yields the cis diastereomer, 13 (fig. S14A). LC-MS analysis of these hydrolysis products (fig. S14B, red and blue traces) along with samples of 5-carboxymethyl-L-proline enriched by synthetic methods in either 12 (purple trace) or 13 (green trace) reveals that the two ring-opened compounds can be resolved sufficiently well by hydrophilic interaction chromatography (see Supplementary Methods) to enable the conversion of 3 to 4 in the CarC reaction to be monitored. LC-MS analysis of a sample of the CarC•Fe(II) •2OG•3 complex (prepared with nominally 5 molar equiv of CarC•Fe(II) relative to 3) denatured in formic acid without prior exposure to O2 reveals a single sharp peak at m/z = 172.0 (Figure 4D, black trace) co-eluting at 7.8 min with the acid-hydrolyzed substrate (red trace). As expected, no peak (or shoulder) is seen at 8.0 min, when the cis product from hydrolysis of 4 elutes (dark blue trace). The chromatogram for a sample quenched in formic acid 3 s after the CarC•Fe(II) •2OG•3 complex was mixed with excess O2 (cyan trace) reveals a diminution in the peak at 7.8 min elution time corresponding to 12 (trans) and development of the broad peak at 8.0 min corresponding to 13 (cis), signifying conversion of 3 to 4. The corresponding chromatogram from a sample acid-quenched after reacting for just 0.15 s (when state b predominates) has only a small shoulder for 13 (light green trace), and the trace from the 10-s sample reveals little additional conversion occurring after 3 s, the time at which state c accumulates maximally. The results confirm that state b has 3 bound to the enzyme whereas state c has 4 bound.
The clear implication of the CarC•Fe(II) •2OG•3 structure that Y165 is the H• donor and primary site of the Y• is confirmed by the results of stopped-flow experiments on four variants of CarC each having one of the four Y residues in the halo of aromatic residues that encircles the substrate (Fig. 2B) replaced by phenylalanine, which is incompetent for H• donation. The Y67F, Y164F, and Y191F variants all form the sharp 410-nm peak of the Y• upon reaction of their enzyme•Fe(II) •2OG•3 complexes with O2 (Fig 4B, black, yellow, and cyan traces, respectively). The amplitudes and kinetics are perturbed to varying extents by the substitutions, consistent with auxiliary roles for these Y residues (e.g., in binding the substrate, in ensuring the proper conformation of the H•-donating Y, or in stabilizing the Y•). By contrast, no 410-nm signal develops in the reaction of the Y165F variant of CarC (dark green trace), and no conversion of 3 to 4 is detected in LC-MS analysis of the acid-quenched reaction products (as in Figure 4D). These observations confirm that Y165 is the H• donor and primary site of the radical.
Figure 1B implies that H• donation to C5 diverts the CarC reaction from either of two possible catalytic oxidation outcomes (reactions i and iii) to the stoichiometric, redox-neutral stereoinversion (reaction ii). Consistent with this notion, the Y165F substitution defaults CarC back to a fully catalytic oxygenase. By contrast to the mediation of only 0.25–0.35 turnovers by the wild-type enzyme, the Y165F variant protein effects minimally 3 turnovers under the same conditions, as seen by quantification of both succinate produced (figs. S8A, five rear traces, and S8B, red data points) and 3 consumed (fig. S8C, five rear traces). If only one-third of the Fe(II) sites react also in the Y165F variant, as suggested by the modest amplitude of decay of the ~ 510-nm Fe(II)→2OG metal-to-ligand charge-transfer absorption band (23) upon mixing of the CarC-Y165•Fe(II) •2OG•3 complex with O2 (fig. S15), then the reactive fraction performs ~ 10 turnovers. The omission of the prior acid treatment in the LC-MS analysis of fig. S8 leaves 3 and 4 intact, and they are not resolved chromatographically. Under this analysis, the intensity of the single peak at m/z = 154 and 1.8 min elution time from the combination of 3 and 4 is not impacted by the stereoinversion outcome (fig. S8C, compare red and blue traces). The almost complete diminution of this peak in samples of the CarC-Y165F reaction (fig. S8C, rear traces) thus signifies consumption of 3 by a reaction other than stereoinversion. In fig. S8D, we posit that the altered outcome is hydroxylation of C5 according to the classical “oxygen-rebound” mechanism (44), but the actual outcome remains to be determined. Regardless, the results establish that the H•-donating Y165 residue, brought into position by the substrate-driven closure of a lid loop, is necessary and sufficient to direct the non-redox, stereoinversion outcome that distinguishes CarC from other Fe/2OG oxygenases and is required for the biosynthesis of all carbapenem antibiotics.
Supplementary Material
Acknowledgments
Atomic coordinates and structure factors have been deposited in the Protein Data Bank with accession code 4OJ8. This work was supported by grants from the National Institutes of Health (GM 058518 to A.C.R., GM 100011 to A.K.B., and GM 069657 to C.K. and J.M.B). Use of the Advanced Photon Source, an Office of Science User Facility operated for the U.S. Department of Energy (DOE) Office of Science by Argonne National Laboratory, was supported by the U.S. DOE under Contract No. DE-AC02-06CH11357. Use of the LS-CAT Sector 21 was supported by the Michigan Economic Development Corporation and the Michigan Technology Tri-Corridor (Grant 085P1000817). We thank E. Y. Tamanaha for assistance in the kinetic analysis.
Footnotes
This manuscript has been accepted for publication in Science. This version has not undergone final editing. Please refer to the complete version of record at http://www.sciencemag.org/. The manuscript may not be reproduced or used in any manner that does not fall within the fair use provisions of the Copyright Act without the prior, written permission of AAAS.
Materials and Methods
References
- 1.Elander RP. Industrial production of beta-lactam antibiotics. Appl Microbiol Biotechnol. 2003;61:385. doi: 10.1007/s00253-003-1274-y. [DOI] [PubMed] [Google Scholar]
- 2.Hamed RB, et al. The enzymes of beta-lactam biosynthesis. Nat Prod Rep. 2013;30:21. doi: 10.1039/c2np20065a. [DOI] [PubMed] [Google Scholar]
- 3.Coulthurst SJ, Barnard AM, Salmond GP. Regulation and biosynthesis of carbapenem antibiotics in bacteria. Nat Rev Microbiol. 2005;3:295. doi: 10.1038/nrmicro1128. [DOI] [PubMed] [Google Scholar]
- 4.Bradley JS, et al. Carbapenems in clinical practice: a guide to their use in serious infection. Int J Antimicrob Agents. 1999;11:93. doi: 10.1016/s0924-8579(98)00094-6. [DOI] [PubMed] [Google Scholar]
- 5.The antibiotic alarm. Nature. 2013;495:141. doi: 10.1038/495141a. [DOI] [PubMed] [Google Scholar]
- 6.Kahan JS, et al. Thienamycin, a new beta-lactam antibiotic. I. Discovery, taxonomy, isolation and physical properties. J Antibiot (Tokyo) 1979;32:1. doi: 10.7164/antibiotics.32.1. [DOI] [PubMed] [Google Scholar]
- 7.Parker WL, et al. SQ 27,860, a simple carbapenem produced by species of Serratia and Erwinia. J Antibiot (Tokyo) 1982;35:653. doi: 10.7164/antibiotics.35.653. [DOI] [PubMed] [Google Scholar]
- 8.McGowan SJ, et al. Analysis of bacterial carbapenem antibiotic production genes reveals a novel beta-lactam biosynthesis pathway. Mol Microbiol. 1996;22:415. [PubMed] [Google Scholar]
- 9.Demain AL, Elander RP. The beta-lactam antibiotics: past, present, and future. Antonie Van Leeuwenhoek. 1999;75:5. doi: 10.1023/a:1001738823146. [DOI] [PubMed] [Google Scholar]
- 10.Li RF, Stapon A, Blanchfield JT, Townsend CA. Three unusual reactions mediate carbapenem and carbapenam biosynthesis. J Am Chem Soc. 2000;122:9296. [Google Scholar]
- 11.Gerratana B, Arnett SO, Stapon A, Townsend CA. Carboxymethylproline synthase from Pectobacterium carotorova: a multifaceted member of the crotonase superfamily. Biochemistry. 2004;43:15936. doi: 10.1021/bi0483662. [DOI] [PubMed] [Google Scholar]
- 12.Hamed RB, Mecinovic J, Ducho C, Claridge TD, Schofield CJ. Carboxymethylproline synthase catalysed syntheses of functionalised N-heterocycles. Chem Commun. 2010;46:1413. doi: 10.1039/b924519g. [DOI] [PubMed] [Google Scholar]
- 13.Gerratana B, Stapon A, Townsend CA. Inhibition and alternate substrate studies on the mechanism of carbapenam synthetase from Erwinia carotovora. Biochemistry. 2003;42:7836. doi: 10.1021/bi034361d. [DOI] [PubMed] [Google Scholar]
- 14.Stapon A, Li R, Townsend CA. Carbapenem biosynthesis: confirmation of stereochemical assignments and the role of CarC in the ring stereoinversion process from L-proline. J Am Chem Soc. 2003;125:8486. doi: 10.1021/ja034248a. [DOI] [PubMed] [Google Scholar]
- 15.Woodward RB. Penems and related substances. Philos Trans R Soc Lond B Biol Sci. 1980;289:239. doi: 10.1098/rstb.1980.0042. [DOI] [PubMed] [Google Scholar]
- 16.Nangia A, Biradha K, Desiraju GR. Correlation of biological activity in beta-lactam antibiotics with Woodward and Cohen structural parameters - A Cambridge database study. J Chem Soc, Perk Trans. 1996;2:943. [Google Scholar]
- 17.Hamed RB, Batchelar ET, Mecinovic J, Claridge TD, Schofield CJ. Evidence that thienamycin biosynthesis proceeds via C-5 epimerization: ThnE catalyzes the formation of (2S,5S)-trans-carboxymethylproline. Chembiochem. 2009;10:246. doi: 10.1002/cbic.200800652. [DOI] [PubMed] [Google Scholar]
- 18.Hamed RB, et al. Stereoselective C-C bond formation catalysed by engineered carboxymethylproline synthases. Nat Chem. 2011;3:365. doi: 10.1038/nchem.1011. [DOI] [PubMed] [Google Scholar]
- 19.Hamed RB, et al. Crotonase catalysis enables flexible production of functionalized prolines and carbapenams. J Am Chem Soc. 2012;134:471. doi: 10.1021/ja208318d. [DOI] [PubMed] [Google Scholar]
- 20.Bodner MJ, Phelan RM, Freeman MF, Li R, Townsend CA. Non-heme iron oxygenases generate natural structural diversity in carbapenem antibiotics. J Am Chem Soc. 2010;132:12. doi: 10.1021/ja907320n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hausinger RP. Fe(II)/alpha-ketoglutarate-dependent hydroxylases and related enzymes. Crit Rev Biochem Mol Biol. 2004;39:21. doi: 10.1080/10409230490440541. [DOI] [PubMed] [Google Scholar]
- 22.Krebs C, Galonić Fujimori D, Walsh CT, Bollinger JM., Jr Non-heme Fe(IV)-oxo intermediates. Acc Chem Res. 2007;40:484. doi: 10.1021/ar700066p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Solomon EI, et al. Geometric and electronic structure/function correlations in non-heme iron enzymes. Chem Rev. 2000;100:235. doi: 10.1021/cr9900275. [DOI] [PubMed] [Google Scholar]
- 24.Costas M, Mehn MP, Jensen MP, Que L., Jr Dioxygen activation at mononuclear nonheme iron active sites: enzymes, models, and intermediates. Chem Rev. 2004;104:939. doi: 10.1021/cr020628n. [DOI] [PubMed] [Google Scholar]
- 25.Topf M, et al. The unusual bifunctional catalysis of epimerization and desaturation by carbapenem synthase. J Am Chem Soc. 2004;126:9932. doi: 10.1021/ja047899v. [DOI] [PubMed] [Google Scholar]
- 26.Borowski T, Broclawik E, Schofield CJ, Siegbahn PEM. Epimerization and desaturation by carbapenem synthase (CarC). A hybrid DFT study. J Comput Chem. 2006;27:740. doi: 10.1002/jcc.20384. [DOI] [PubMed] [Google Scholar]
- 27.Phelan RM, DiPardo BJ, Townsend CA. A high-throughput screen for the engineered production of beta-lactam antibiotics. ACS Chem Biol. 2012;7:835. doi: 10.1021/cb200504g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Phelan RM, Townsend CA. Mechanistic insights into the bifunctional non-heme iron oxygenase carbapenem synthase by active site saturation mutagenesis. J Am Chem Soc. 2013;135:7496. doi: 10.1021/ja311078s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhou J, et al. Spectroscopic studies of substrate interactions with clavaminate synthase 2, a multifunctional alpha-KG-dependent non-heme iron enzyme: correlation with mechanisms and reactivities. J Am Chem Soc. 2001;123:7388. doi: 10.1021/ja004025+. [DOI] [PubMed] [Google Scholar]
- 30.Bollinger JM, Jr, Krebs C. Stalking intermediates in oxygen activation by iron enzymes: motivation and method. J Inorg Biochem. 2006;100:586. doi: 10.1016/j.jinorgbio.2006.01.022. [DOI] [PubMed] [Google Scholar]
- 31.Bollinger JM, Jr, et al. Use of rapid kinetics methods to study the assembly of the diferric-tyrosyl radical cofactor of E. coli ribonucleotide reductase. Methods Enzymol. 1995;258:278. doi: 10.1016/0076-6879(95)58052-2. [DOI] [PubMed] [Google Scholar]
- 32.Pfaendler HR, Gosteli J, Woodward RB, Rihs G. Structure, reactivity, and biological activity of strained bicyclic beta-lactams. J Am Chem Soc. 1981;103:4526. [Google Scholar]
- 33.Sleeman MC, et al. Biosynthesis of carbapenem antibiotics: new carbapenam substrates for carbapenem synthase (CarC) Chembiochem. 2004;5:879. doi: 10.1002/cbic.200300908. [DOI] [PubMed] [Google Scholar]
- 34.Bycroft BW, Chhabra SR. A chiral synthesis of trans-carbapenam-3-carboxylic acid and the assignment of (3S,5S) configuration to the corresponding product from Serratia and Erwinia species. J Chem Soc, Chem Commun. 1989:423. [Google Scholar]
- 35.Avenoza A, Barriobero JI, Busto JH, Peregrina JM. Enantiopure synthesis of all four stereoisomers of carbapenam-3-carboxylic acid methyl ester. J Org Chem. 2003;68:2889. doi: 10.1021/jo026804+. [DOI] [PubMed] [Google Scholar]
- 36.Clifton IJ, et al. Crystal structure of carbapenem synthase (CarC) J Biol Chem. 2003;278:20843. doi: 10.1074/jbc.M213054200. [DOI] [PubMed] [Google Scholar]
- 37.Hoffart LM, Barr EW, Guyer RB, Bollinger JM, Jr, Krebs C. Direct spectroscopic detection of a C-H-cleaving high-spin Fe(IV) complex in a prolyl-4-hydroxylase. Proc Natl Acad Sci USA. 2006;103:14738. doi: 10.1073/pnas.0604005103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Galonić DP, Barr EW, Walsh CT, Bollinger JM, Jr, Krebs C. Two interconverting Fe(IV) intermediates in aliphatic chlorination by the halogenase CytC3. Nat Chem Biol. 2007;3:113. doi: 10.1038/nchembio856. [DOI] [PubMed] [Google Scholar]
- 39.Matthews ML, et al. Substrate-triggered formation and remarkable stability of the C-H bond-cleaving chloroferryl intermediate in the aliphatic halogenase, SyrB2. Biochemistry. 2009;48:4331. doi: 10.1021/bi900109z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Matthews ML, et al. Substrate positioning controls the partition between halogenation and hydroxylation in the aliphatic halogenase, SyrB2. Proc Natl Acad Sci USA. 2009;106:17723. doi: 10.1073/pnas.0909649106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Price JC, Barr EW, Tirupati B, Bollinger JM, Jr, Krebs C. The first direct characterization of a high-valent iron intermediate in the reaction of an alpha-ketoglutarate-dependent dioxygenase: a high-spin Fe(IV) complex in taurine/alpha-ketoglutarate dioxygenase (TauD) from Escherichia coli. Biochemistry. 2003;42:7497. doi: 10.1021/bi030011f. [DOI] [PubMed] [Google Scholar]
- 42.Bollinger JM, Jr, Price JC, Hoffart LM, Barr EW, Krebs C. Mechanism of taurine: alpha-ketoglutarate dioxygenase (TauD) from Escherichia coli. Eur J Inorg Chem. 2005:4245. doi: 10.1021/bi050227c. [DOI] [PubMed] [Google Scholar]
- 43.McCusker KP, Klinman JP. Modular behavior of TauD provides insight into the origin of specificity in alpha-ketoglutarate-dependent nonheme iron oxygenases. Proc Natl Acad Sci USA. 2009;106:19791. doi: 10.1073/pnas.0910660106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Groves JT. Key elements of the chemistry of cytochrome-P-450 - the oxygen rebound mechanism. J Chem Educ. 1985;62:928. [Google Scholar]
- 45.Pedregal C, Ezquerra J, Escribano A, Carreno MC, Ruano JLG. Highly chemoselective reduction of N-Boc protected lactams. Tetrahedron Lett. 1994;35:2053. [Google Scholar]
- 46.Collado I, Ezquerra J, Vaquero JJ, Pedregal C. Diastereoselective functionalization of 5-hydroxy prolinates by tandem Horner-Emmons-Michael reaction. Tetrahedron Lett. 1994;35:8037. [Google Scholar]
- 47.Ranatunga S, Liyanage W, Del Valle JR. Synthesis and conformational analysis of bicyclic extended dipeptide surrogates. J Org Chem. 2010;75:5113. doi: 10.1021/jo1008433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bycroft BW, Chhabra SR, Kellam B, Smith P. Convenient syntheses of (3S,5S)-carbapenam-3-carboxylates and their biosynthetic relevance. Tetrahedron Lett. 2003;44:973. [Google Scholar]
- 49.Bollinger JM, Jr, et al. Mechanism of assembly of the tyrosyl radical-diiron(III) cofactor of Escherichia coli ribonucleotide reductase .2. kinetics of the excess Fe2+ reaction by optical, EPR, and Mössbauer spectroscopies. J Am Chem Soc. 1994;116:8015. [Google Scholar]
- 50.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 51.McCoy AJ, Grosse-Kunstleve RW, Storoni LC, Read RJ. Likelihood-enhanced fast translation functions. Acta Crystallogr. 2005;D61:458. doi: 10.1107/S0907444905001617. [DOI] [PubMed] [Google Scholar]
- 52.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. 1997;D53:240. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 53.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr. 2004;D60:2126. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 54.Schüttelkopf AW, van Aalten DMF. PRODRG: a tool for high-throughput crystallography of protein-ligand complexes. Acta Crystallogr. 2004;D60:1355. doi: 10.1107/S0907444904011679. [DOI] [PubMed] [Google Scholar]
- 55.Krissinel E, Henrick K. Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Crystallogr. 2004;D60:2256. doi: 10.1107/S0907444904026460. [DOI] [PubMed] [Google Scholar]
- 56.Laskowski RA. PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Cryst. 1993;26:283. [Google Scholar]
- 57.Chen VB, et al. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. 2010;D66:12. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ten Eyck LF. Fast Fourier transform calculation of electron density maps. Methods Enzymol. 1985;115:324. doi: 10.1016/0076-6879(85)15024-x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.