

Abstract
Background
Heterozygous mutations in the chromatin remodeling gene CHD7 cause CHARGE syndrome, a developmental disorder with variable craniofacial dysmorphisms and respiratory difficulties. The molecular etiologies of these malformations are not well understood. Homozygous Chd7 null mice die by E11, whereas Chd7Gt/+ heterozygous null mice are a viable and excellent model of CHARGE. We explored skeletal phenotypes in Chd7Gt/+ and Chd7 conditional knockout mice, using Foxg1-Cre to delete Chd7 (Foxg1-CKO) in the developing eye, ear, nose, pharyngeal pouch, forebrain, and gut and Wnt1-Cre (Wnt1-CKO) to delete Chd7 in migrating neural crest cells.
Results
Foxg1-CKO mice exhibited postnatal respiratory distress and death, dysplasia of the eye, concha, and frontal bone, hypoplastic maxillary shelves and nasal epithelia, and reduced tracheal rings. Wnt1-CKO mice exhibited frontal and occipital bone dysplasia, hypoplasia of the maxillary shelves and mandible, and cleft palate. In contrast, heterozygous Chd7Gt/+ mice had apparently normal skeletal development.
Conclusions
Conditional deletion of Chd7 in ectodermal and endodermal derivatives (Foxg1-Cre) or migrating neural crest cells (Wnt1-Cre) results in varied and more severe craniofacial defects than in Chd7Gt/+ mice. These studies indicate that CHD7 has an important, dosage-dependent role in development of several different craniofacial tissues.
Keywords: cleft palate, craniofacial disorders, skeletal dysplasia, trachea
Introduction
Heterozygous mutations in the gene encoding CHD7, a chromatin remodeling protein, are found in the majority of individuals diagnosed with CHARGE syndrome, a multiple anomaly disorder defined by ocular coloboma, heart defects, atresia of the choanae, retarded growth and development, genital and/or urinary abnormalities, and ear abnormalities (Janssen et al., 2012) (Janssen et al., 2012; Husu et al., 2013). Craniofacial dysmorphisms are common in CHARGE syndrome, and include square-shaped facies, facial asymmetry, external ear anomalies, cleft lip and/or palate, cranial nerve palsies, and torticollis (Zentner et al., 2010; Hughes et al., 2014). In addition, less penetrant phenotypes are associated with CHARGE and include vertebral and limb anomalies, renal anomalies, and tracheal hypoplasia or tracheo-esophageal fistula (Zentner et al., 2010; Janssen et al., 2012; Legendre et al., 2012).
Understanding the role of CHD7 during organogenesis is critical for the development of effective therapies. We have generated mouse models of CHARGE syndrome to examine molecular mechanisms of Chd7 deficiency, including Chd7 loss of function (Chd7Gt) and Chd7 conditional deletion (Chd7flox) alleles (Hurd et al., 2007; Hurd et al., 2010). Previous studies indicate that heterozygous Chd7 mutant mice exhibit many phenotypes similar to human CHARGE, including vestibular disorders, deafness, reduced growth, pubertal delay, olfactory dysfunction, and brain abnormalities (Bosman et al., 2005; Adams et al., 2007; Hurd et al., 2007; Layman et al., 2009; Hurd et al., 2010; Hurd et al., 2011; Layman et al., 2011; Hurd et al., 2012; Yu et al., 2013). Despite the myriad of skeletal abnormalities reported in humans with CHARGE (Prasad et al., 1997; Wright et al., 2009), there is no information about skeletal features of Chd7 mutant mice. Because homozygous Chd7Gt/Gt mice do not survive beyond E11 (Bosman et al., 2005; Hurd et al., 2007), we chose to focus on Chd7Gt/+ and Chd7 conditional knockout mice using Foxg1-Cre (Hebert and McConnell, 2000) and Wnt1-Cre (Danielian et al., 1998) mice. The complementary expression profiles of these two different mouse lines in neural crest (Wnt1-Cre) and various ectodermal/endodermal derivatives (Foxg1-Cre) provided an opportunity to delineate the effects of loss of Chd7 in development of various craniofacial structures.
Results
Chd7 deficient mice have craniofacial defects
The developmental origins of CHARGE-related birth defects are not fully understood. Neural crest-derived tissues are commonly affected by CHD7 deficiency (Bajpai et al., 2010), yet ectodermal structures including the brain are also affected (Legendre et al., 2012; Yu et al., 2013). In order to explore the relative contributions of CHD7 in these tissue derivatives to organ development, we generated mice with loss of Chd7 using Foxg1-Cre, which is expressed by E7.5 in the pharyngeal arches, facial and head ectoderm, otic, optic, nasal and oral epithelium, pharyngeal arches, forebrain, and foregut (Hebert and McConnell, 2000). We also generated mice with Chd7 deletion using Wnt1-Cre, which is expressed by E8.5 in developing pharyngeal arches, head mesenchyme, inner ear, neural crest, midbrain, spinal cord and nasal neural crest and mesenchyme (Danielian et al., 1998). Mice with deletion of both Chd7 alleles by Foxg1-Cre or Wnt1-Cre are hereafter referred to as Foxg1-CKO or Wnt1-CKO, respectively.
While Chd7Gt/+ mice survive to adulthood (Bosman et al., 2005; Hurd et al., 2007), both Foxg1-CKO and Wnt1-CKO mice die shortly after birth, likely from respiratory failure (neither Foxg1-CKO nor Wnt1-CKO pups had milk in their stomachs). We therefore focused our phenotypic analysis of craniofacial and other tissues on postnatal day 1 pups. Chd7 wild type, Chd7flox/+, and Chd7Gt/+ mice had no apparent abnormalities of the skull or craniofacial regions (Fig. 1A-B). In contrast, Foxg1-CKO mice had severely hypoplastic eyes (N=7/7 mice) and shortened faces (Fig. 1C) and Wnt1-CKO mice had shortened snouts (Fig. 1D). We performed comprehensive measurements of craniofacial distances on skeletal preparations of postnatal day 1 mice (Figs. 1E-H, Fig. 2 and Table 1). We observed no differences in the degree of ossification in the skulls or in craniofacial measurements (occipital bone, intersquamosal, interocular, mandible, or rostral-caudal lengths) between Chd7Gt/+ mice and wild type or Chd7flox/+ littermate controls (Table 1), suggesting that loss of one Chd7 allele does not cause major or highly penetrant skeletal defects in mice. We did note mild hypoplasia of the ethmoid bone of Chd7Gt/+ mice relative to controls (Figure 1F).
Figure 1.
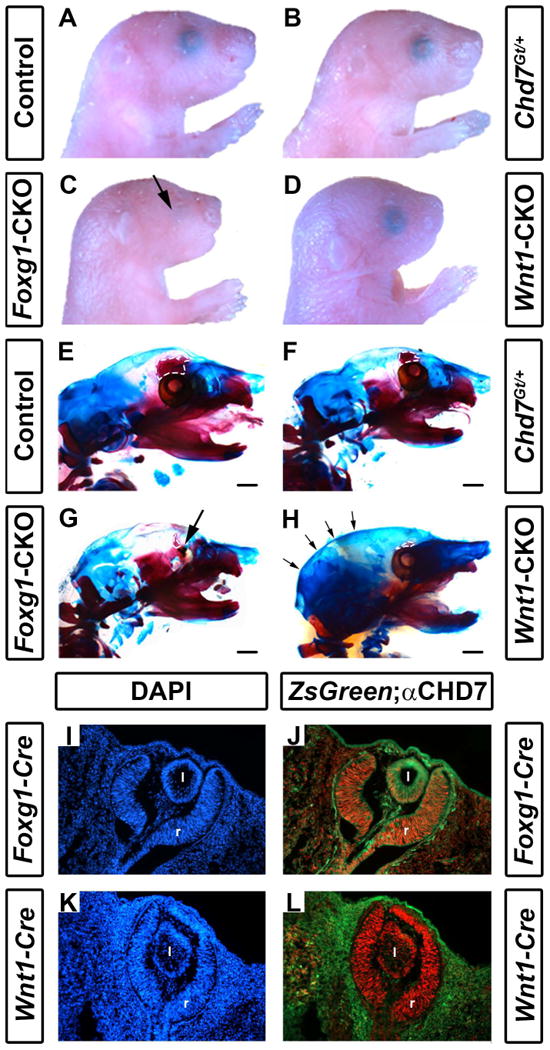
Conditional deletion of Chd7 results in ocular hypoplasia and cranial bone dysplasia. Brightfield images (A-D) and skeletal preparations (E-H) of postnatal day 1 control (Cre-negative, Chd7+/f or Chd7f/f) (A,E), Chd7Gt/+ (B,F), Foxg1-CKO (C,G) and Wnt1-CKO (D,H) pups. Chd7Gt/+ mice appear similar to controls, whereas both Foxg1-CKO (C) and Wnt1-CKO (D) mice have shortened snouts, and Foxg1-CKO mice have ocular hypoplasia (arrows in C and G). There are no abnormalities in ossification, overall morphology or size of major craniofacial bones in Chd7Gt/+ mice (B, F). In E-H, alizarin red and alcian blue staining reveal bone and cartilage, respectively. Ethmoid bones are hypoplastic in Chd7Gt/+ (F), Foxg1-CKO (G) and Wnt1-CKO (H) mice (white dashed lines in E-H). Wnt1-CKO mice (H) have a more rounded posterior cranium (indicated by arrows) than mice of other genotypes. Cre expression in Foxg1-Cre and Wnt1-Cre mice was detected using ZsGreen reporter mice and compared to CHD7 protein levels in the developing eye (I-L). Shown are transverse sections of Cre;ZsGreen positive E11.5 embryos stained with DAPI (labels cellular nuclei) or anti-CHD7 (red). Foxg1-Cre is active in CHD7-positive cells in the retina (r) and posterior lens (l) of the developing eye (J), whereas Wnt1-Cre is restricted to the surrounding periocular mesenchyme and is absent in the retina and lens proper (L). Scale bars in E-H are 1 mm.
Figure 2.
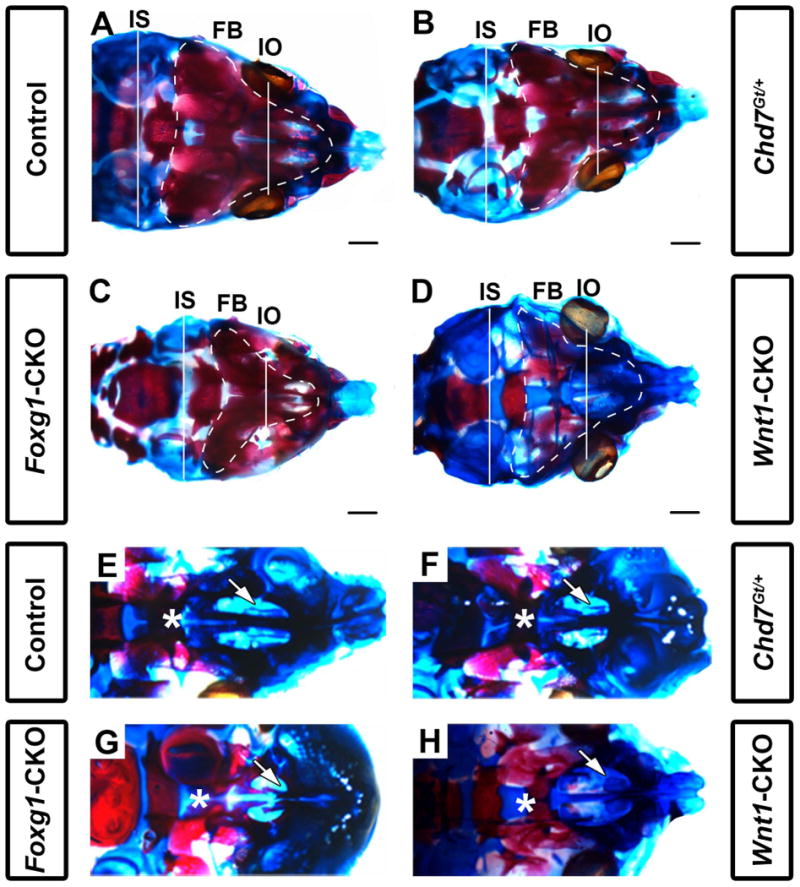
Chd7 conditional mutants have shortened intraocular and intersquamosal distances, hypoplasia of the maxillary shelves, and dysplasia of nasal conchae. Skeletal preparations of postnatal day 1 control (Cre-negative, Chd7Gt/+ or Chd7f/f) (A,E), Chd7Gt/+ (B,F), Foxg1-CKO (C,G), and Wnt1-CKO (D,H) mice. Alizarin red and alcian blue staining show bone and cartilage, respectively. (A-D) are representative dorsal images of the calvaria while (E-H) are representative dorsal images of the developing maxillary shelves (with mandibles removed). Foxg1-CKO (C) mice exhibit shorter interocular (white line, IO; p<0.001, N=7) and intersquamosal (white line, IS; p<0.001, N=7) distances compared to control (A), Chd7Gt/+ (B), and Wnt1-CKO (D) mice. Foxg1-CKO (C) mice exhibit a visibly smaller frontal bone (FB) surface area (white, dotted line) relative to control (A), Chd7Gt/+ (B), and Wnt1-CKO (D) mice. Wnt1-CKO (D) mice lack frontal bone ossification (as demonstrated by alizarin red staining) relative to control (A), Chd7Gt/+ (B), and Foxg1-CKO (C) mice. In control (E) and Chd7Gt/+ (F) mice, appropriately formed palatal shelves (asterisk) and ventral nasal conchae (white arrow) are present. Foxg1-CKO (G) mice have hypoplastic palatal shelves which approximate the midline (asterisk) and absent ventral conchae (white arrow), whereas Wnt1-CKO (H) mice have severe palatal shelf hypoplasia (asterisk) and dysplastic ventral conchae (white arrow). Scale bars in A-D are 1 mm.
Table 1.
Foxg1-Cre or Wnt1-Cre conditional loss of Chd7 in mice results in multiple skeletal abnormalities.
| Region | Feature | Foxg1-Cre Genotype | Wnt1-Cre Genotype | |||
|---|---|---|---|---|---|---|
|
Chd7+/flox (N=9) |
Chd7Gt/flox (N=6) |
CKO (N=7) |
Control (N=6) |
CKO (N=7) |
||
| Cranium | Interocular distance | 4.66 | 4.34 | 3.72 * | 5.12 | 5.01 |
| Frontal bone dysplasia† | 3 | 4 | 7 * | 0 | 6 * | |
| Eye dysplasia† | 0 | 0 | 7 * | 0 | 0 | |
| Intersquamosal distance | 6.58 | 6.38 | 5.90 * | 6.23 | 6.24 | |
| Parietal/interparietal bone dysplasia† | 0 | 0 | 0 | 0 | 0 | |
| Occipital bone length | 3.92 | 4.10 | 3.86 | 3.58 | 3.47 | |
| Occipital bone dysplasia† | 0 | 0 | 0 | 1 | 7 * | |
| Rostral-caudal length | 9.54 | 9.27 | 8.99 | 9.75 | 9.46 | |
| Ventral conchae dysplasia† | 0 | 4 * | 7 * | 1 | 7 * | |
| Dorsal conchae dysplasia† | 0 | 5 * | 7 * | 1 | 7 * | |
| Palatal shelf dysplasia† | 0 | 0 | 7* | 1 | 7 * | |
| Cleft palate† | 0 | 0 | 0 | 0 | 3 * | |
| Mandible length | 5.29 | 5.28 | 5.05 | 5.55 | 4.95 * | |
| Cervical spine | Atlas (C1) dysplasia† | 0 | 0 | 0 | 0 | 0 |
| Axis (C2) dysplasia† | 0 | 0 | 0 | 0 | 2 | |
| Thoracic spine | Rib count | 13.0 | 13.0 | 12.9 | 12.9 | 12.9 |
| Rib dysplasia (bifid)† | 1 | 3 | 1 | 2 | 4 | |
| Thorax | Clavicle length | 2.49 | 2.49 | 2.48 | 2.60 | 2.56 |
| Tracheal ring count | 17.8 | 19.3 | 11.0 * | 16.7 | 18.9 | |
All measurements are in millimeters.
These observations were scored in a binomial fashion (e.g. dysplasia of the frontal bone was identified as “present” or “absent”). As such, the number of mice affected is scored across each genotype for each observation.
Significance indicated at p < 0.05 relative to Chd7+/flox (for Foxg1-Cre) or control(Chd7+/flox or Chd7flox/flox for Wnt1-Cre) by MANOVA with post hoc Tukey's HSD test.
In contrast, we observed significant frontal bone dysplasia (either reduced ossification or hypoplasia) in both Foxg1-CKO (N=7, p<0.05) and Wnt1-CKO (N=7, p<0.05) mice compared with littermate controls (Figs. 1 E-H and 2 A-D). Interestingly, there was no dysplasia of the atlas (C1), axis (C2) or parietal/interparietal bones, or change in rostral-caudal lengths of Chd7Gt/+, Foxg1-CKO or Wnt1-CKO mice compared to controls. Some mice had dysplastic ribs, but this was infrequent, also occurred in controls, and did not correlate with changes in rib number or vary significantly across genotypes. Clavicles were also normal in mice of all genotypes. Notably, the ethmoid bones were hypoplastic in both Foxg1-CKO and Wnt1-CKO mice, and the occipital region appeared dysplastic in Wnt1-CKO mice (Figs. 1 G, H). Foxg1-CKO mice exhibited significantly shortened interocular distances (mean= 3.72 vs. 4.66 mm, N=7, p<0.05) and intersquamosal distances (mean= 5.90 vs. 6.58 mm, N=7, p<0.05) compared to controls (Fig. 2C vs. A and Table 1), whereas Wnt1-CKO mice exhibited normal interocular or intersquamosal distances, occipital bone dysplasia and reduced mandibular length (mean=4.95 vs. 5.55 mm, N=7, p<0.05) (Fig. 2D vs. A and Table 1). Intersquamosal and interocular distances were similar in Chd7Gt/+ mice (Fig. 2B) and controls (Fig. 2A). Thus, whereas Chd7Gt/+ mice appear normal, both Foxg1-CKO and Wnt1-CKO mice displayed severe but distinct craniofacial anomalies, suggesting that proper Chd7 expression within the facial ectoderm (Foxg1) and neural crest (Wnt1) is required to ensure normal craniofacial development.
Cre transgene and Chd7 expression in developing retina correlate with ocular defects
Chd7 is expressed in mouse embryonic stem cells (Schnetz et al., 2009) and broadly in early (E7.5-E8.5) developing mouse embryos (Randall et al., 2009), then by mid-gestation (E12.5) becomes restricted to specific organs and tissues such as the brain, pituitary, ear, heart, and craniofacial structures (Hurd et al., 2007). To characterize Chd7 expression relative to Foxg1-Cre and Wnt1-Cre transgene expression, we performed immunofluorescence for CHD7 on E11.5 transgenic embryos that also contained ZsGreen reporter (Madisen et al., 2010) alleles (Fig. 1 I-L). CHD7 was present in the retinae of both Foxg1-Cre;ZsGreen+ and Wnt1-Cre;ZsGreen+ E11.5 embryos. Consistent with published results, Foxg1-Cre, but not Wnt1-Cre expression was observed in the E11.5 mouse retina (Fig. 1 J vs L) (Danielian et al., 1998; Hebert and McConnell, 2000). Thus, deletion of Chd7 in Foxg1-CKO retina (with resultant hypoplastic eyes) and preservation of Chd7 (with normal eyes) in Wnt1-CKO mice is expected given these expression patterns.
Foxg1-CKO mice have upper and lower airway abnormalities including conchae dysplasia and hypoplastic nasal epithelia
Individuals with CHARGE syndrome have highly penetrant respiratory and olfactory deficits, including choanal atresia, tracheoesophageal fistula, and hyposmia or anosmia (Zentner et al., 2010; Bergman et al., 2011; Layman et al., 2011). Chd7 mutant mice are known to have severely reduced olfactory abilities and olfactory bulb hypoplasia, as well as choanal defects (Bosman et al., 2005; Layman et al., 2009; Bergman et al., 2010), but to date there is no detailed information about upper and lower airway structures. We asked whether Chd7 heterozygous and conditional mutant mice also exhibit airway dysplasia by analyzing the structure of the oral/nasal cavity in skeletal preparations of postnatal day 1 pups. The majority of Chd7Gt/+ mice (N=4/6) and all Foxg1-CKO (N=7/7) and Wnt1-CKO (N=7/7) mice displayed ventral conchae dysplasia (Fig. 2 and Table 1). Similarly, the majority of Chd7Gt/+ mice (N=5/6) and all Foxg1-CKO (N=7/7) and Wnt1-CKO (N=7/7) pups displayed dorsal conchae dysplasia (Fig. 2 and Table 1). Analysis of histologically stained sections revealed normal nasal epithelial tissues in Chd7Gt/+ and Wnt1-CKO mice, despite hypoplasia of the dorsal and ventral conchae in the latter (Fig. 3B and 3C). In contrast, the nasal epithelium in Foxg1-CKO mice was severely disorganized and hypoplastic (Fig. 3D). Comparison of X-gal staining in Chd7Gt/+ and Foxg1-CKO mice (both of which contain the Chd7Gt allele that expresses β-galactosidase under the control of the Chd7 promoter, (Hurd et al., 2007)) showed that Chd7 promoter activity is preserved in the nasal epithelium of both mutants, and that unlike Chd7Gt/+ mice which exhibit normal nasal epithelia, the Foxg1-CKO nasal epithelium is severely disrupted (Fig. 3E vs. 3F). Together, these data suggest that dysplasia of the nasal epithelium in mice with reduced Chd7 may result primarily from disruption of normal Chd7 expression in the ectoderm rather than a secondary defect in neural crest cell migration.
Figure 3.
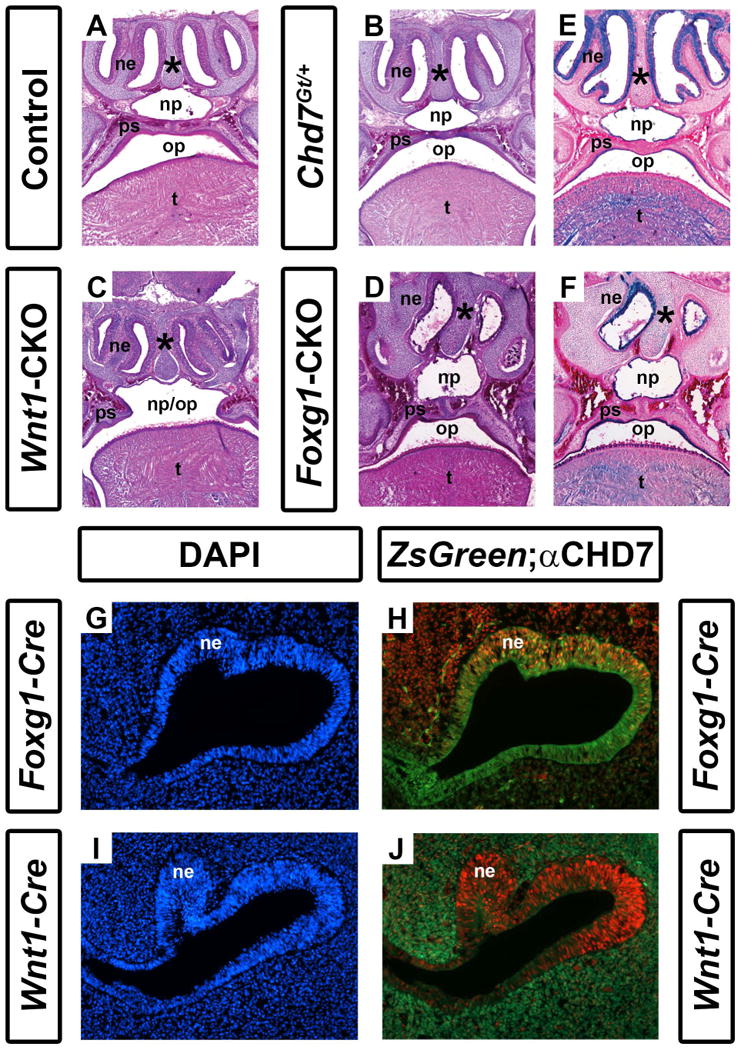
Conditional Chd7 deletion results in palatal and nasal epithelial dysplasia. Transverse sections of postnatal day 1 control (Cre-negative, Chd7+/f or Chd7f/f) (A), Chd7Gt/+ (B), Wnt1-CKO (C), and Foxg1-CKO (D) mice stained with hematoxylin and eosin. Control (A) and Chd7Gt/+ (B) mice exhibit fully fused palatal shelves (ps) with separation of the nasopharynx (np) from the oropharynx (op), whereas Wnt1-CKO(C) mice have severely hypoplastic palatal shelves (ps) resulting in a cleft palate and single pharyngeal cavity (np/op). Foxg1-CKO mice (D) have fused palatal shelves (ps) resulting in a highly-arched, intact palate , a thickened nasal septum (asterisk), and severely hypoplastic nasal epithelia (ne). (E) and (F) show X-gal/eosin stained sections of postnatal day 1 Chd7Gt mice, which express β-galactosidase under the control of the Chd7 promoter. X-gal staining is present in the nasal epithelium (ne), tongue (t) and epithelial cells lining the palatal shelves and combined oral-nasal pharynx (op/np) of Chd7Gt/flox (E) and Foxg1-CKO (F) mice. Panels (G-J) are transverse sections of Foxg1-Cre;ZsGreen (G, H) or Wnt1-Cre;ZsGreen (I, J) E11.5 embryos stained with DAPI (blue, marking cellular nuclei) or anti-CHD7 (red). Green fluorescence (ZsGreen reporter) marks Cre expressing cells and is overlaid with anti-CHD7 (red) in (H) and (J). Foxg1-Cre and Chd7 are both highly expressed in the developing nasal epithelium (ne) while Wnt1-Cre expression is restricted to the surrounding nasal mesenchyme (m) and is absent from the nasal epithelium proper.
Foxg1-CKO and Wnt1-CKO mice have palatal defects
Clefting of the lip and/or palate is reported in 33-48% of individuals with CHARGE syndrome (Zentner et al., 2010; Bergman et al., 2011). In addition, approximately one-third of Chd7whi/+ heterozygous mutant mice have cleft palates (Bosman et al., 2005). By gross morphological analysis, we found no hard or soft palatal defects in postnatal day 1 Chd7Gt/+pups (Figs. 2F and 3B). Foxg1-CKO mice exhibit mild palatal shelf hypoplasia (N=7/7); however do not exhibit cleft palates (N=0/7; Table 1, Figs. 2G and 3D). By contrast, all Wnt1-CKO mice (N=7) exhibited severe palatal shelf hypoplasia and some (N=3/7) Wnt1-CKO mice displayed complete clefting of the palate resulting in an open oral/nasal pharynx (Figs. 2D, 3C and Table 1). X-gal stained sections from Chd7Gt/+ and Foxg1-CKO mice, showed β-galactosidase activity (Chd7 promoter activity) in epithelial cells lining the palatal shelves (Fig. 3E, F). Together, these data provide evidence that Chd7 is highly expressed in the nasal/oral epithelium and may contribute to disrupted palatal formation.
Absence of long bone, vertebral, clavicular, and pelvic defects in Chd7 mutant mice
It has been proposed that CHARGE may be considered a skeletal dysplasia, with clinical reports of limb anomalies (Van de Laar et al., 2007; Wright et al., 2009), vertebral defects, and basiooccipital abnormalities (Fujita et al., 2009). We identified no visible skeletal defects in postnatal day 1 control, Foxg1-Cre conditional heterozygous (Foxg1-Cre;Chd7+/flox), or Wnt1-Cre conditional heterozygous (Wnt1-Cre;Chd7+/flox) mice. Likewise, lengths of bones in the upper extremity (humerus, ulna, or radius), pelvis (intercrest, interspinous, or crest-symphysis) and lower extremity (femur, tibia, or fibula) were similar in control, Chd7 heterozygous null (Chd7Gt/flox), Foxg1-CKO and Wnt1-CKO mice (data not shown). Bones from the right and left sides of the body of all mice were also similar across all genotypes, suggesting that loss of Chd7 does not affect axial symmetry. We observed no vertebral anomalies suggestive of spinal curvature (data not shown). Analysis of the cervical spine, thoracic spine (rib count or dysplasia) and clavicle revealed no apparent differences between Chd7Gt/+, Foxg1-CKO, Wnt1-CKO, and littermate control mice (Table 1). Thus, at the level of resolution possible by analysis of skeletal postnatal day 1 preparations, we found no defects in the long bones, vertebrae, clavicles, or pelvis of Chd7 mutant mice.
Normal development of neural crest derived cardiac structures in Wnt1-Cre conditional mutant mice
Cardiac defects including abnormal development and septation of the conotruncus and the development of the great vessels, are present in 75-77% of individuals with CHARGE syndrome (Zentner et al., 2010; Bergman et al., 2011; Janssen et al., 2012). In mice, some Chd7whi/+ embryos (heterozygous for a Chd7 null allele) have been reported to have interventricular septal defects, body edema and cardiac insufficiency (Bosman et al., 2005). In Wnt1-Cre mice, Cre-positive neural crest cells (NCCs) migrate to the conotruncus or early cardiac outflow tract, where they participate in septation of this tract into the pulmonary and aortic outflow tracts (Jiang et al., 2000). Additionally, NCCs migrate from the posterior hindbrain into the third, fourth and sixth pharyngeal arches where they participate in the asymmetric remodeling of the pharyngeal arch arteries (PAA) into the great vessels (Hutson and Kirby, 2007). We therefore asked whether Wnt1-CKO mutant mice also exhibit heart defects similar to or more severe than those previously reported. In order to demonstrate whether NCC-mediated conotruncal development is impacted by Chd7 deletion, we assessed cardiac outflow tract development in Wnt1-CKO embryos. At E16.5, a time point at which cardiac outflow tract septation is complete, we observed grossly normal aortic and pulmonary septation as well as normal formation of the major branches off the aorta (N=4 Conditional Heterozygous (CHet) (Fig. 4A), N=5 Wnt1-CKO (Fig. 4B)). Histologic analysis with H&E staining of E16.5 Wnt1-CHet (N=7; Fig. 4C) and Wnt1-CKO (N=6; Fig. 4D) hearts confirmed normal aorto-pulmonary and right-left outflow tract septation. To determine whether neural crest-mediated PAA formation was impacted by Chd7 deletion, we assessed PAA formation at E10.5 by India ink injection in Wnt1-Conditional heterozygous (Fig. 4E) and Wnt1-CKO (Fig. 4F) mice. Third, fourth, and sixth PAA formation in Wnt1-Cre expressing neural crest cells was unaffected by Chd7 deletion (N=5 CHet, N=6 Wnt1-CKO). Thus, we found no evidence for cardiac abnormalities in mice with Wnt1-Cre mediated conditional deletion of one or both copies of CHD7.
Figure 4.
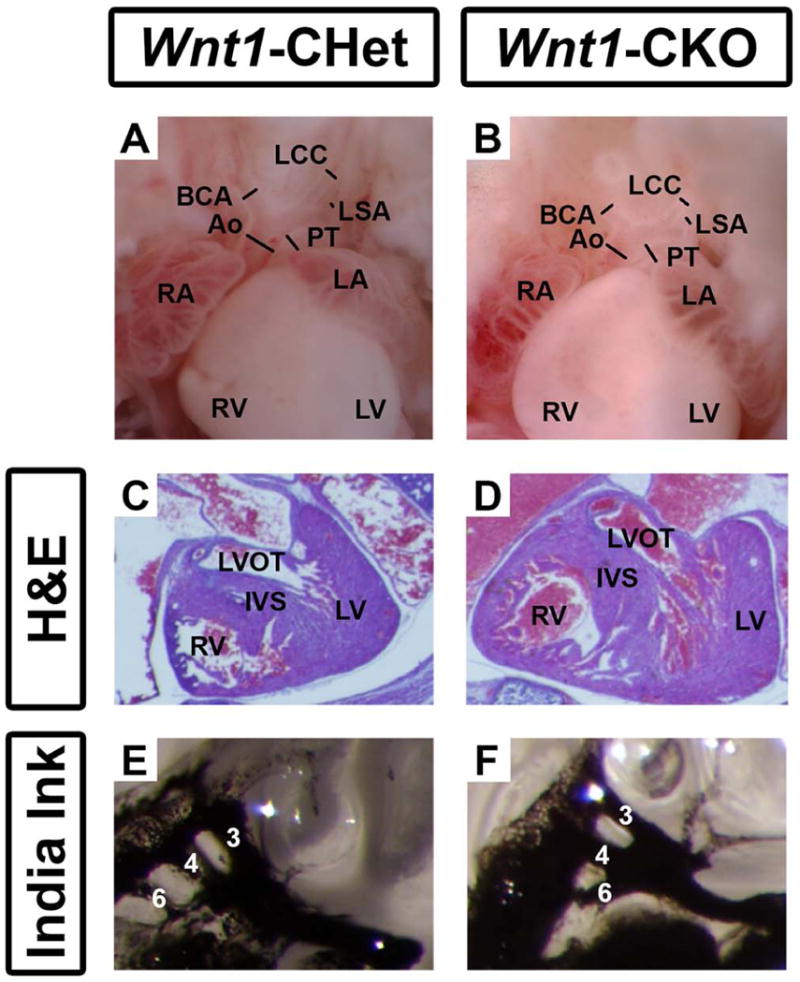
Neural crest-derived cardiac structures develop normally in Wnt1-CHet and Wnt1-CKO mice. (A) E16.5 embryonic hearts from Wnt1-CHet and Wnt1-CKO embryos exhibited normal septation of the aorta (Ao) and pulmonary trunk (PT) with normal extension of the brachiocephalic (BCA), left common carotid (LCC), and left subclavian (LSA) arteries from the aortic arch. H&E staining of sectioned E16.5 embryonic hearts from Wnt1-Cre conditional heterozygous (B) and Wnt1-CKO (C) mice revealed normal development of the left ventricular outflow tract (LVOT) and inter ventricular septum (IVS). India ink injections of E10.5 Wnt1-Cre conditional heterozygous (CHet) (D) and Wnt1-CKO (E) mice demonstrate normal development of the 3rd, 4th, and 6th pharyngeal arch arteries. Other abbreviations: RA (right atrium); LV (left ventricle); RA (right atrium); LA (left atrium). Lines span lumina of arteries.
Foxg1-Cre Chd7 conditional mutants have fewer tracheal rings
Other mediastinal abnormalities, including tracheoesophageal fistula and tracheal agenesis have also been reported at relatively high penetrance (19-29%) in CHARGE syndrome (Zentner et al., 2010; Bergman et al., 2011). Tracheal cartilage was readily visible in the skeletal preparations of postnatal day 1 pups, allowing us to characterize the mice for tracheal abnormalities. Interestingly, there were, on average, 17-18 tracheal rings in control mice (N=16), 19 in Chd7Gt/+ mice (N=6), and 19 in Wnt1-CKO mice (N=7) (Fig. 5 and Table 1). In contrast, Foxg1-CKO mice had, on average, only 11 tracheal rings (p<0.001, N=7) (Fig. 5 and Table 1). The severe reduction in tracheal rings may represent a developmental delay or arrest of tracheal development, and could contribute to their early postnatal respiratory distress and death.
Figure 5.
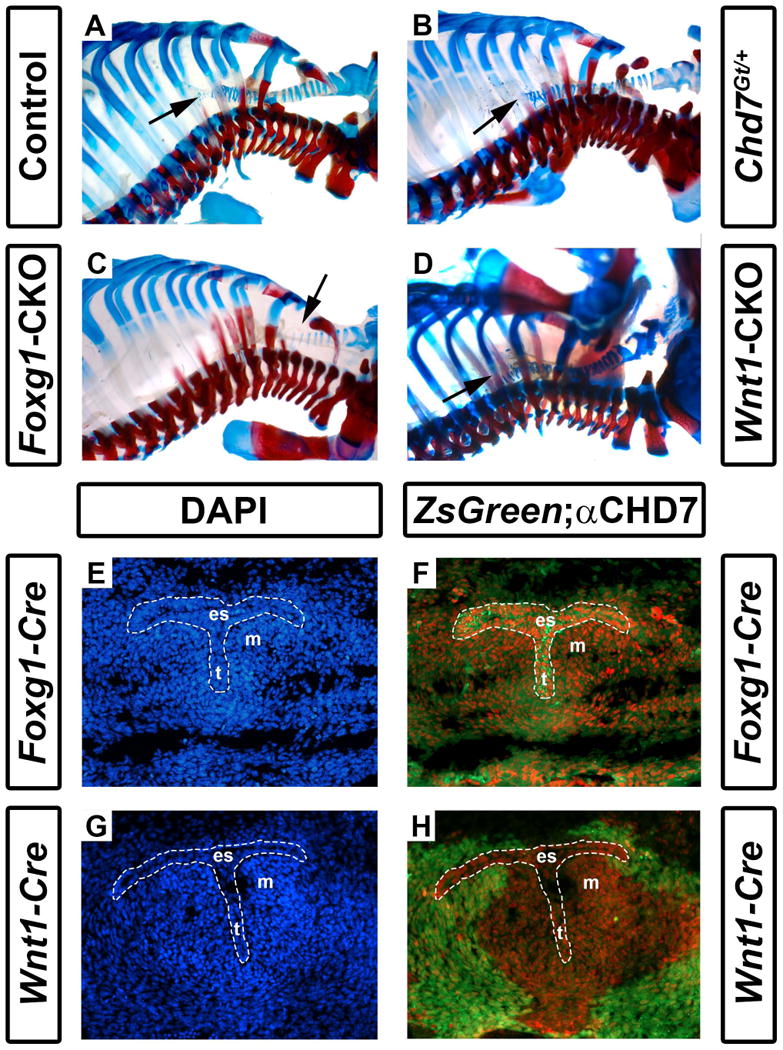
Foxg1-CKO mice have reduced numbers of cartilaginous tracheal rings. Shown are skeletal preparations of postnatal day 1 control (Cre-negative, Chd7+/f or Chd7f/f) (A), Chd7Gt/+ (B), Foxg1-CKO (C), and Wnt1-CKO (D) mice. Alizarin red and alcian blue staining reveal bone and cartilage, respectively. Normal numbers of tracheal rings (17-19) (arrows point to distal trachea) are present in control, Chd7Gt/+, and Wnt1-CKO mice, whereas Foxg1-CKO mice have fewer (11) tracheal rings. Panels E-H are transverse sections of Foxg1-Cre;ZsGreen (E, F) or Wnt1-Cre;ZsGreen (G, H) E11.5 embryos at the level of the laryngotracheal groove (indicated by the area within the dashed white lines) stained with DAPI (blue, labeling cellular nuclei) or anti-CHD7 (red). Green fluorescence (ZsGreen reporter) marks Cre-expressing cells and is overlaid with anti-CHD7 (red) in (F) and (H). Chd7 and Foxg1-Cre are highly co-expressed in the developing esophagus (es), trachea (t), and the surrounding mesenchyme (m), whereas Wnt1-Cre activity is restricted to the surrounding mesenchymal tissues and does not overlap with Chd7 expression in the presumptive esophagus or trachea.
We then asked whether Chd7 and Wnt1-Cre/Foxg1-Cre transgenes are expressed in the embryonic trachea proper or in surrounding mesenchymal tissues (Woo et al., 2011). As in Figs. 1 and 3, we stained cross-sections through E11.5 Foxg1-Cre;ZsGreen+ and Wnt1-Cre;ZsGreen+ embryos at the level of the mediastinum for immunofluorescence with anti-CHD7 antibody (Fig. 5E-H). We observed abundant CHD7 staining in the presumptive esophagus and trachea, with less bright label in the surrounding mesenchyme of Foxg1-Cre;ZsGreen+ embryos. Interestingly, Foxg1-Cre transgene activity was widespread in both the tracheal epithelium and surrounding mesenchymal tissues (Fig. 5E, F) whereas Wnt1-Cre transgene activity was restricted to the peri-tracheal mesenchyme (Fig. 5G, H). These data suggest that CHD7 is concentrated in the tracheal primordium, and is also expressed in adjacent mesenchyme, and that loss of CHD7 in the tracheal primordium leads to severely disrupted tracheal development.
Discussion
In this study, we present data showing that Chd7 is required for certain aspects of murine craniofacial and mediastinal development; specifically, loss of Chd7 disrupts formation of the orbit and multiple regions of the respiratory tract. By using mice expressing Cre recombinase in ectoderm- and endoderm-derived cells of the developing telencephalon and pharyngeal arches (Foxg1-Cre) and in migratory neural crest cells (Wnt1-Cre), we identified components of the upper and lower respiratory tracts that likely contribute to acute respiratory failure and perinatal death of Chd7-conditional mutant mice. Interestingly, Chd7 is required for proper development of multiple components along the upper and lower respiratory tracts, including the nose, hard palate, and trachea. Complex malformations of each of these organs have been noted in humans with CHARGE syndrome, and postnatal respiratory distress is a common cause for early morbidity and mortality.
Upper and lower airway malformations with Chd7 deficiency
Nasal conchae, or turbinates, are critical for air filtration and proper formation and maintenance of the olfactory epithelium. Specifically, the conchae provide increased nasal epithelial surface area while humidifying the air, which may help protect olfactory nerve axons traveling through the cribriform plate to synapse in the olfactory bulb (Kim et al., 2003; Kastl et al., 2009). These structures develop, in part, from the ethmoid bone, a complex structure which forms the caudal nasal cavity (Jacob and Chole, 2006). We found that Chd7Gt/+ mice exhibit mild hypoplasia of the dorsal and ventral conchae as well as the ethmoid bone, whereas conditional deletion of Chd7 in Foxg1-Cre-expressing mice resulted in complete loss of the nasal choanae and ethmoid bone. Interestingly, Wnt1-CKO mice also exhibited nasal conchae and ethmoid dysplasia, with little disruption to the nasal passageways. Given the importance of the conchae in olfactory development, their disruption could worsen olfactory neuron dysfunction in Chd7 mutant mice and contribute to previously reported hyposmia/anosmia phenotypes (Layman et al., 2009). In addition to disruption of the nasal conchae, we observed marked disruption of the nasal passageways in Foxg1-CKO mice, reminiscent of the posterior nasal aperture blockage or choanal atresia reported in some Chd7whi/+ mice (Bosman et al., 2005). The nose, like most other craniofacial structures, is composed of facial ectoderm and migrating cranial neural crest cells, and Chd7 may be necessary for proper integration of these cell types. Interestingly, external naris occlusion impairs nasal turbinate development in the mouse (Coppola and Craven, 2013), evidence that development of the conchae is dependent on proper airflow. We postulate that some of the nasal abnormalities observed in Chd7 mutant mice reflect underlying contributions of Chd7 to development of cells derived from neural crest, ectoderm, and underlying mesenchyme.
In addition to choanal atresia, other airway abnormalities such as sinus blockage, cleft palate, and laryngomalacia, are common in CHARGE syndrome (Zentner et al., 2010; Janssen et al., 2012; Hughes et al., 2014). We observed thickening and vaulting of the palatal shelves in Foxg1-CKO mice and complete cleft of the palate in Wnt1-CKO mice. In humans, highly-arched palates indicate disruptions in maxillary shelf development, are often reported following cleft palate repair, and are commonly associated with dental crowding and speech difficulties (Eppley et al., 2005; Chapman et al., 2008; Wang et al., 2009). The occurrence of highly-arched palates has been documented in individuals with CHARGE who have intact palates or following surgical repair of a cleft palate (Akisu et al., 1998). Both Foxg1-Cre and Wnt1-Cre are expressed in cells critical for maxillary development; indeed, deletions of other genes including β-catenin and Bmpr1a using these Cre recombinases results in mild and more severe maxillary malformations including complete aplasia (Li et al., 2011; Wang et al., 2011). Interestingly, we did not observe palatal vaulting or clefting in our Chd7Gt/+ mice. This could be a result of low sample size or effects of genetic background. Cleft palate occurs in 25% of Chd7whi/+ mice (Bosman et al., 2005) and 33-48% of humans with CHARGE syndrome (Zentner et al., 2010; Janssen et al., 2012). Together, these results suggest that Chd7 plays critical roles in Wnt1-Cre derived cranial neural crest cells that contribute to osteogenesis and to the overlying Foxg1-Cre derived oral epithelium, both of which are critical for proper maxillary development (Bush and Jiang, 2012).
In addition to malformations of the upper airway, we observed hypoplasia of tracheal cartilaginous rings in Foxg1-CKO mice. This is consistent with high levels of Cre expression in the tracheal primordium of Foxg1-Cre mice. Tracheal cartilage is critical for maintaining patency of the trachea upon expiration (Doshi and Krawiec, 2007). Thus Chd7 may play an intrinsic role in tracheal development, consistent with the tracheal abnormalities (malacia, dysgenesis, and agenesis) observed in individuals with CHARGE. The proximity of foregut derivatives to the trachea and the high incidence of tracheoesophageal fistula in CHARGE suggest that CHD7 may be important for gut development (Perl et al., 2002). Further, we observed significant, colocalized Chd7 and Foxg1-Cre activity in the presumptive esophagus suggesting that development of the larygotracheal groove is critically dependent on proper expression of Chd7. More detailed analysis mice will be necessary to determine the extent to which Chd7 contributes to foregut development.
Potential dual roles for Chd7 in the eye
Development of the eye involves a dual process whereby neuroectoderm forms a proto-retinal outpouching from the rostral telencephalon (Sinn and Wittbrodt, 2013). Upon contact with growing retinal tissue, the overlying surface ectoderm induces optic cup formation. The lens is the principal structure derived from the overlying surface ectoderm. Recent reports suggest that spatiotemporal localization of lens and retinal tissue is driven by neural crest cells, which themselves differentiate into the sclera separating the developing lens from the retina (Grocott et al., 2011). These neural crest cells help direct the generation of the optic cup, which involves a complex folding process resulting in formation of the choroid fissure. In the mature eye, the choroidal fissure is almost completely closed, providing continuity to the inferior iris and retina. Defects in choroidal fissure closure present as colobomata.
Ocular colobomata of the retina and/or iris, are sometimes associated with microphthalmia, occur in 75-81% of individuals with CHARGE syndrome (Zentner et al., 2010; Bergman et al., 2011). Previous studies of Chd7whi/+ mice showed keratoconjunctivitis sicca (dry eye) (Bosman et al., 2005), but there are no reports in Chd7 mutant mice of ocular coloboma or other major eye defects commonly seen in CHARGE syndrome. One of our most striking findings was severe hypoplasia of the eye in Foxg1-CKO mice. Our immunofluorescence results from E11.5 mice also demonstrate high levels of both Foxg1-Cre and CHD7 in retinal tissue. Notably, in addition to the high CHD7 in retina, we detected abundant CHD7 in the developing lens. We observe no obvious defects in eye development in Wnt1-CKO mice, suggesting that Chd7 may have subtle or no significant functions in neural crest cells that contribute to ocular development. Recent data from zebrafish (D. rerio) have implicated chd7 in the regulation of retinal cell specification (Patten et al., 2012); our data provide the first evidence that loss of Chd7 within the retina contributes to ocular malformations in mammals. Future studies of Chd7 function in the developing eye should help determine the mechanistic relationship between ocular hypoplasia and colobomata.
Lack of evidence for Chd7 function in cardiac neural crest cells
Fourth pharyngeal arch artery (PAA) hypoplasia and malformations of the transverse aorta and right subclavian artery (fourth PAA derivatives) have been reported in Chd7whi/+ mice (Randall et al., 2009). Importantly, attempted rescue of these malformations using Wnt1-Cre, expressed in neural crest cells, and a conditional Chd7 wild-type allele was unsuccessful, suggesting minimal contributions of Chd7 in Wnt1-Cre lineages to heart formation (Randall et al., 2009). Given these observations, we further explored the role of cardiac neural crest by examining conditional deletion of Chd7 using Wnt1-Cre. Neither Wnt1-conditional heterozygous nor Wnt1-CKO mice exhibited any deformity in PAA development at E10.5 or in arterial branching at E16.5, consistent with minimal or no Chd7 function in cardiac neural crest cells. Alternatively, Wnt1-Cre activity may be mosaic or inefficient in the cardiac neural crest. Additional studies are necessary to distinguish among these possibilities.
Development of craniofacial structures requires correct spatiotemporal Chd7 expression
Our results demonstrate that proper expression of Chd7 is necessary for development of the craniofacial structures contributing to the upper respiratory tract and the cartilaginous rings of the trachea. Two complementary themes emerge from our data: (1) Chd7 expression is required in the surface ectoderm, endoderm, and migrating neural crest cells which all contribute to the development of facial structures and the eye and (2) Chd7 likely has a significant role in the development of cartilage and bone. Foxg1-Cre is highly expressed by E7.5 in surface ectoderm and endoderm which populate the pharyngeal arch and pouch derivatives, respectively (Hebert and McConnell, 2000). Components of the first pharyngeal arch contribute to many of the facial bones affected in Chd7 deficient mice and humans, including the ethmoid, maxilla, and mandible (Sperber, 1989). Migrating neural crest cell populations expressing Wnt1-Cre invade pharyngeal arch structures and are critical for their proper specification. Loss of Chd7 in the embryonic frog (X. laevis) has been reported to result in failed neural crest cell migration from the dorsal neural tube (Bajpai et al., 2010). We found that disruptions to Chd7 in Wnt1-Cre expressing neural crest cells results in developmental abnormalities in the nose and palate, but not the heart. Interestingly, we observed phenotypic differences in the nose and palate between Foxg1-CKO and Wnt1-CKO mice that may reflect differences in sites or timing of Cre activity. In Foxg1-Cre mice, Cre is induced around E7.5, which may affect certain populations of pre-migratory neural crest cells (Nichols, 1986), whereas Wnt1-Cre is activated slightly later, at E8.5, in migratory neural crest (Danielian et al., 1998). Much remains to be clarified regarding how CHD7 regulates proper gene expression in both the facial ectoderm and neural crest cells that contribute to craniofacial development.
Our results highlight the potential role that CHD7 has in regulating development of bone. Interestingly, a previous report suggests that CHD7 is required for inducing Wnt5a-dependent osteoblastogenesis in mesenchymal stem cells (Takada et al., 2007). Indeed, these authors determined that reduction in Chd7 mRNA levels by RNAi caused a cell fate switch leading to the accumulation of adipocytes. While this research was performed using mesoderm-derived cell populations, CHD7 may function similarly in craniofacial osteoblasts derived from cranial neural crest cells (Minoux and Rijli, 2010). Further investigation is necessary to determine the specific function(s) of CHD7 in development of craniofacial and tracheal structures affected in CHARGE syndrome.
Experimental Procedures
Mice
Foxg1-Cre;Chd7Gt/+ mice and Chd7flox/flox mice were generated and genotyped as previously described (Hurd et al., 2007; Hurd et al., 2010). Specifically, these mice were maintained on a mixed C57BL/6;129S1/SvImJ background and inbred to N=6-8. Mice of the following genotypes were analyzed: control (Chd7+/flox ), Chd7 heterozygous mutant (Chd7Gt/flox or Chd7Gt/+), Foxg1 conditional heterozygous mutant (Foxg1-Cre;Chd7+/flox) and Foxg1conditional knockout mutant (Foxg1-Cre;Chd7Gt/flox or Foxg1-CKO). Wnt1-Cre;Chd7+/flox mice were crossed with Chd7flox/flox mice, and the following genotypes were analyzed: control (Chd7+/flox or Chd7flox/flox), Wnt1-Cre conditional heterozygous mutant (Wnt1-Cre;Chd7+/flox) and Wnt1conditional knockout mutant (Wnt1-Cre;Chd7flox/flox or Wnt1-CKO). ZsGreen Cre reporter mice (Madisen et al., 2010) (JAX#007906) were crossed with Foxg1-Cre or Wnt1-Cre mice to identify sites of Cre positive cells. The University of Michigan University Committee on the Use and Care of Animals (UCUCA) approved all procedures.
Skeletal Preparations
Postnatal day 1 mice were terminally anaesthetized, eviscerated, and fixed in 95% ethanol overnight and incubated in acetone for three days. Preparations were placed in dye solution (0.06 mg/ml Alizarin Red (Sigma, St. Louis, MO) and 0.18 mg/mL Alcian Blue (Sigma) in 42 mL of 72% ethanol and 3 mL of glacial acetic acid) and agitated gently on a rotary shaker for four days. Skeletons were washed in ddH2O for two hours to rinse excess dye then placed in 1% KOH clearing solution for two days. Samples were then immersed in increasing concentrations of glycerol (1:4 glycerol / 1% KOH; 1:1 glycerol / 1% KOH; 8:2 glycerol / 1% KOH) for five to seven days per immersion. Finally, skeletal preparations were stored in 100% glycerol, photographed using a Leica DMRB microscope and measured using ImageJ (National Institutes of Health, Bethesda, MD). Images were processed using Adobe Photoshop CS6 (Adobe, San Jose, CA).
Histology and X-gal staining
Postnatal day 1 mice were collected, decapitated and heads fixed in 4% paraformaldehyde overnight at 4°C. After washing in PBS, heads were incubated in 30% sucrose, embedded in OCT medium (Tissue-Tek, Torrance, CA), frozen, and sectioned at 12-15 μm. Sections were either stained with hematoxylin and eosin (H&E) (Sigma) for histological analysis, or X-gal for expression analysis as previously described (Hurd et al., 2007). Sections were photographed by light microscopy on a Leica upright DMRB microscope and processed in Photoshop CS2 (Adobe, San Jose, CA).
Immunofluorescence and Cre lineage tracing
For Cre lineage tracing and anti-CHD7 immunofluorescence, E11.5 Foxg1-Cre;ZsGreen;Chd7+/+ and Wnt1-Cre;ZsGreen;Chd7+/+ embryos were harvested, dissected, and embedded in OCT cryosectioning media (Tissue-Tek, Torrance, CA). Embryos were cryosectioned at 14 μm, stained with 1:7500 anti-CHD7 rabbit IgG monoclonal antibody (in TSA block, #6505 Cell Signaling Technology (Danvers, MA) and counterstained with 1:200 biotinylated anti-rabbit IgG goat secondary antibody (in TSA block, BA-1000, Vector Laboratories (Burlingame, CA)). Strepavidin-HRP (1:100 in TSA block) was added to slides and Alexa 555 tyramide (1:100 in Amplification Buffer, Molecular Probes (Grand Island, NY)) was applied to generate fluorescence. DAPI (5 μg/mL) was added and slides mounted for imaging using a Leica DMRB microscope. Final image processing was performed using Adobe Photoshop CS6 (Adobe, San Jose, CA).
Cardiac Evaluation
For gross inspection, E16.5 embryos were harvested and dissected. For the hematoxylin/eosin stained sections, embryos were fixed in 4% paraformaldehyde for 12–18 hours at 4°C, dehydrated and embedded in Leica Histowax according to standard procedures. After embedding, sections were cut at 5–7 μm. Images were analyzed by brightfield microscopy. For India ink injections, E10.5 embryos were dissected and placed in ice cold PBS. Using a pulled glass pipette, India ink was injected into the ventricles until the ink penetrated the small vessels. Embryos were post-fixed in 10% buffered formalin for 12 hours, dehydrated and cleared in benzylbenzoate: benzyl alcohol (2:1v/v).
Statistical analysis
Measurements of anatomical regions were performed on control (N=16), Chd7 heterozygous mutant (N=6), Foxg1 conditional heterozygous mutant (N=7), Wnt1 conditional heterozygous mutant (N=2), Foxg1 conditional knockout mutant (N=7) and Wnt1 conditional knockout mutant (N=7) mice using stereoscope-standardized pictures analyzed with ImageJ (National Institutes of Health, Bethesda, MD). Discrete observations (i.e. presence or absence of dysplasia) were scored in a binomial fashion. Bone lengths were measured using ImageJ (National Institutes of Health, Bethesda, MD). All measurements were analyzed in SPSS (v.19.0) by MANOVA with a post hoc Tukey's honest significant difference (HSD) test; significance was limited to p ≤ 0.05.
Acknowledgments
We thank John L. Zeller, M.D., Ph.D. (Division of Anatomical Sciences, University of Michigan) for assistance with interpretation of skeletal anatomy. This work was supported by the CHARGE Syndrome Foundation (EAH and ABS), Elizabeth E. Kennedy Research Award (EAH), Hearing Health Foundation (EAH), and NIH grants 5T32GM007863-33 (EDS), 5T35GL007690-30 (ENR), and R01-DC009410 (DMM).
Grant Sponsor: NIH R01-DC009410.
References
- Adams ME, Hurd EA, Beyer LA, Swiderski DL, Raphael Y, Martin DM. Defects in vestibular sensory epithelia and innervation in mice with loss of Chd7 function: implications for human CHARGE syndrome. J Comp Neurol. 2007;504:519–532. doi: 10.1002/cne.21460. [DOI] [PubMed] [Google Scholar]
- Akisu M, Ozkinay F, Ozyurek R, Kucuktas A, Kultursay N. The CHARGE association in a newborn infant. Turk J Pediatr. 1998;40:283–287. [PubMed] [Google Scholar]
- Bajpai R, Chen DA, Rada-Iglesias A, Zhang J, Xiong Y, Helms J, Chang CP, Zhao Y, Swigut T, Wysocka J. CHD7 cooperates with PBAF to control multipotent neural crest formation. Nature. 2010;463:958–962. doi: 10.1038/nature08733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergman JEH, Bosman EA, van Ravenswaaij-Arts CMA, Steel KP. Study of smell and reproductive organs in a mouse model for CHARGE syndrome. European Journal of Human Genetics. 2010;18:171–177. doi: 10.1038/ejhg.2009.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergman JEH, Janssen N, Hoefsloot LH, Jongmans MCJ, Hofstra RMW, van Ravenswaaij-Arts CMA. CHD7 mutations and CHARGE syndrome: the clinical implications of an expanding phenotype. Journal of Medical Genetics. 2011;48:334–342. doi: 10.1136/jmg.2010.087106. [DOI] [PubMed] [Google Scholar]
- Bosman EA, Penn AC, Ambrose JC, Kettleborough R, Stemple DL, Steel KP. Multiple mutations in mouse Chd7 provide models for CHARGE syndrome. Hum Mol Genet. 2005;14:3463–3476. doi: 10.1093/hmg/ddi375. [DOI] [PubMed] [Google Scholar]
- Bush JO, Jiang R. Palatogenesis: morphogenetic and molecular mechanisms of secondary palate development. Development. 2012;139:231–243. doi: 10.1242/dev.067082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman KL, Hardin-Jones MA, Goldstein JA, Halter KA, Havlik RJ, Schulte J. Timing of palatal surgery and speech outcome. Cleft Palate Craniofac J. 2008;45:297–308. doi: 10.1597/06-244. [DOI] [PubMed] [Google Scholar]
- Coppola D, Craven B. The effects of naris occlusion on mouse nasal turbinate development. J Exp Biol. 2013 doi: 10.1242/jeb.092940. [DOI] [PubMed] [Google Scholar]
- Danielian PS, Muccino D, Rowitch DH, Michael SK, McMahon AP. Modification of gene activity in mouse embryos in utero by a tamoxifen-inducible form of Cre recombinase. Curr Biol. 1998;8:1323–1326. doi: 10.1016/s0960-9822(07)00562-3. [DOI] [PubMed] [Google Scholar]
- Doshi J, Krawiec ME. Clinical manifestations of airway malacia in young children. J Allergy Clin Immunol. 2007;120:1276–1278. doi: 10.1016/j.jaci.2007.09.048. [DOI] [PubMed] [Google Scholar]
- Eppley BL, van Aalst JA, Robey A, Havlik RJ, Sadove AM. The spectrum of orofacial clefting. Plast Reconstr Surg. 2005;115:101e–114e. doi: 10.1097/01.prs.0000164494.45986.91. [DOI] [PubMed] [Google Scholar]
- Fujita K, Aida N, Asakura Y, Kurosawa K, Niwa T, Muroya K, Adachi M, Nishimura G, Inoue T. Abnormal basiocciput development in CHARGE syndrome. AJNR Am J Neuroradiol. 2009;30:629–634. doi: 10.3174/ajnr.A1380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grocott T, Johnson S, Bailey AP, Streit A. Neural crest cells organize the eye via TGF-beta and canonical Wnt signalling. Nat Commun. 2011;2:265. doi: 10.1038/ncomms1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hebert JM, McConnell SK. Targeting of cre to the Foxg1 (BF-1) locus mediates loxP recombination in the telencephalon and other developing head structures. Dev Biol. 2000;222:296–306. doi: 10.1006/dbio.2000.9732. [DOI] [PubMed] [Google Scholar]
- Hughes SS, Welsh HI, Safina NP, Bejaoui K, Ardinger HH. Family history and clefting as major criteria for CHARGE syndrome. Am J Med Genet A. 2014;164A:48–53. doi: 10.1002/ajmg.a.36192. [DOI] [PubMed] [Google Scholar]
- Hurd EA, Adams ME, Layman WS, Swiderski DL, Beyer LA, Halsey KE, Benson JM, Gong TW, Dolan DF, Raphael Y, Martin DM. Mature middle and inner ears express Chd7 and exhibit distinctive pathologies in a mouse model of CHARGE syndrome. Hear Res. 2011 doi: 10.1016/j.heares.2011.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurd EA, Capers PL, Blauwkamp MN, Adams ME, Raphael Y, Poucher HK, Martin DM. Loss of Chd7 function in gene-trapped reporter mice is embryonic lethal and associated with severe defects in multiple developing tissues. Mamm Genome. 2007;18:94–104. doi: 10.1007/s00335-006-0107-6. [DOI] [PubMed] [Google Scholar]
- Hurd EA, Micucci JA, Reamer EN, Martin DM. Delayed fusion and altered gene expression contribute to semicircular canal defects in Chd7 deficient mice. Mech Dev. 2012 doi: 10.1016/j.mod.2012.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurd EA, Poucher HK, Cheng K, Raphael Y, Martin DM. The ATP-dependent chromatin remodeling enzyme CHD7 regulates pro-neural gene expression and neurogenesis in the inner ear. Development. 2010;137:3139–3150. doi: 10.1242/dev.047894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Husu E, Hove HD, Farholt S, Bille M, Tranebjaerg L, Vogel I, Kreiborg S. Phenotype in 18 Danish subjects with genetically verified CHARGE syndrome. Clin Genet. 2013;83:125–134. doi: 10.1111/j.1399-0004.2012.01884.x. [DOI] [PubMed] [Google Scholar]
- Hutson MR, Kirby ML. Model systems for the study of heart development and disease. Cardiac neural crest and conotruncal malformations. Semin Cell Dev Biol. 2007;18:101–110. doi: 10.1016/j.semcdb.2006.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacob A, Chole RA. Survey anatomy of the paranasal sinuses in the normal mouse. Laryngoscope. 2006;116:558–563. doi: 10.1097/01.MLG.0000202085.23454.2F. [DOI] [PubMed] [Google Scholar]
- Janssen N, Bergman JE, Swertz MA, Tranebjaerg L, Lodahl M, Schoots J, Hofstra RM, van Ravenswaaij-Arts CM, Hoefsloot LH. Mutation update on the CHD7 gene involved in CHARGE syndrome. Hum Mutat. 2012;33:1149–1160. doi: 10.1002/humu.22086. [DOI] [PubMed] [Google Scholar]
- Jiang X, Rowitch DH, Soriano P, McMahon AP, Sucov HM. Fate of the mammalian cardiac neural crest. Development. 2000;127:1607–1616. doi: 10.1242/dev.127.8.1607. [DOI] [PubMed] [Google Scholar]
- Kastl KG, Rettinger G, Keck T. The impact of nasal surgery on air-conditioning of the nasal airways. Rhinology. 2009;47:237–241. doi: 10.4193/Rhin08.014. [DOI] [PubMed] [Google Scholar]
- Kim KS, Choi YS, Kim HJ, Yoon JH. The risk of olfactory disturbance from conchal plate injury during ethmoidectomy. Am J Rhinol. 2003;17:307–310. [PubMed] [Google Scholar]
- Layman WS, Hurd EA, Martin DM. Reproductive dysfunction and decreased GnRH neurogenesis in a mouse model of CHARGE syndrome. Hum Mol Genet. 2011;20:3138–3150. doi: 10.1093/hmg/ddr216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Layman WS, McEwen DP, Beyer LA, Lalani SR, Fernbach SD, Oh E, Swaroop A, Hegg CC, Raphael Y, Martens JR, Martin DM. Defects in neural stem cell proliferation and olfaction in Chd7 deficient mice indicate a mechanism for hyposmia in human CHARGE syndrome. Hum Mol Genet. 2009;18:1909–1923. doi: 10.1093/hmg/ddp112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Legendre M, Gonzales M, Goudefroye G, Bilan F, Parisot P, Perez MJ, Bonniere M, Bessieres B, Martinovic J, Delezoide AL, Jossic F, Fallet-Bianco C, Bucourt M, Tantau J, Loget P, Loeuillet L, Laurent N, Leroy B, Salhi H, Bigi N, Rouleau C, Guimiot F, Quelin C, Bazin A, Alby C, Ichkou A, Gesny R, Kitzis A, Ville Y, Lyonnet S, Razavi F, Gilbert-Dussardier B, Vekemans M, Attie-Bitach T. Antenatal spectrum of CHARGE syndrome in 40 fetuses with CHD7 mutations. J Med Genet. 2012 doi: 10.1136/jmedgenet-2012-100926. [DOI] [PubMed] [Google Scholar]
- Li L, Lin M, Wang Y, Cserjesi P, Chen Z, Chen Y. BmprIa is required in mesenchymal tissue and has limited redundant function with BmprIb in tooth and palate development. Dev Biol. 2011;349:451–461. doi: 10.1016/j.ydbio.2010.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madisen L, Zwingman TA, Sunkin SM, Oh SW, Zariwala HA, Gu H, Ng LL, Palmiter RD, Hawrylycz MJ, Jones AR, Lein ES, Zeng H. A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat Neurosci. 2010;13:133–140. doi: 10.1038/nn.2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minoux M, Rijli FM. Molecular mechanisms of cranial neural crest cell migration and patterning in craniofacial development. Development. 2010;137:2605–2621. doi: 10.1242/dev.040048. [DOI] [PubMed] [Google Scholar]
- Nichols DH. Formation and distribution of neural crest mesenchyme to the first pharyngeal arch region of the mouse embryo. Am J Anat. 1986;176:221–231. doi: 10.1002/aja.1001760210. [DOI] [PubMed] [Google Scholar]
- Patten SA, Jacobs-McDaniels NL, Zaouter C, Drapeau P, Albertson RC, Moldovan F. Role of Chd7 in zebrafish: a model for CHARGE syndrome. PLoS One. 2012;7:e31650. doi: 10.1371/journal.pone.0031650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perl AK, Wert SE, Nagy A, Lobe CG, Whitsett JA. Early restriction of peripheral and proximal cell lineages during formation of the lung. Proc Natl Acad Sci U S A. 2002;99:10482–10487. doi: 10.1073/pnas.152238499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasad C, Quackenbush EJ, Whiteman D, Korf B. Limb anomalies in DiGeorge and CHARGE syndromes. Am J Med Genet. 1997;68:179–181. doi: 10.1002/(sici)1096-8628(19970120)68:2<179::aid-ajmg11>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- Randall V, McCue K, Roberts C, Kyriakopoulou V, Beddow S, Barrett AN, Vitelli F, Prescott K, Shaw-Smith C, Devriendt K, Bosman E, Steffes G, Steel KP, Simrick S, Basson MA, Illingworth E, Scambler PJ. Great vessel development requires biallelic expression of Chd7 and Tbx1 in pharyngeal ectoderm in mice. J Clin Invest. 2009;119:3301–3310. doi: 10.1172/JCI37561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnetz MP, Bartels CF, Shastri K, Balasubramanian D, Zentner GE, Balaji R, Zhang X, Song L, Wang Z, Laframboise T, Crawford GE, Scacheri PC. Genomic distribution of CHD7 on chromatin tracks H3K4 methylation patterns. Genome Res. 2009;19:590–601. doi: 10.1101/gr.086983.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinn R, Wittbrodt J. An eye on eye development. Mech Dev. 2013;130:347–358. doi: 10.1016/j.mod.2013.05.001. [DOI] [PubMed] [Google Scholar]
- Sperber GH. Craniofacial Embryology. Great Britain: Wright; 1989. [Google Scholar]
- Takada I, Mihara M, Suzawa M, Ohtake F, Kobayashi S, Igarashi M, Youn MY, Takeyama K, Nakamura T, Mezaki Y, Takezawa S, Yogiashi Y, Kitagawa H, Yamada G, Takada S, Minami Y, Shibuya H, Matsumoto K, Kato S. A histone lysine methyltransferase activated by non-canonical Wnt signalling suppresses PPAR-gamma transactivation. Nat Cell Biol. 2007;9:1273–1285. doi: 10.1038/ncb1647. [DOI] [PubMed] [Google Scholar]
- Van de Laar I, Dooijes D, Hoefsloot L, Simon M, Hoogeboom J, Devriendt K. Limb anomalies in patients with CHARGE syndrome: an expansion of the phenotype. Am J Med Genet A. 2007;143A:2712–2715. doi: 10.1002/ajmg.a.32008. [DOI] [PubMed] [Google Scholar]
- Wang XX, Wang X, Li ZL, Yi B, Liang C, Jia YL, Zou BS. Anterior maxillary segmental distraction for correction of maxillary hypoplasia and dental crowding in cleft palate patients: a preliminary report. Int J Oral Maxillofac Surg. 2009;38:1237–1243. doi: 10.1016/j.ijom.2009.06.028. [DOI] [PubMed] [Google Scholar]
- Wang Y, Song L, Zhou CJ. The canonical Wnt/beta-catenin signaling pathway regulates Fgf signaling for early facial development. Dev Biol. 2011;349:250–260. doi: 10.1016/j.ydbio.2010.11.004. [DOI] [PubMed] [Google Scholar]
- Woo J, Miletich I, Kim BM, Sharpe PT, Shivdasani RA. Barx1-mediated inhibition of Wnt signaling in the mouse thoracic foregut controls tracheo-esophageal septation and epithelial differentiation. PLoS One. 2011;6:e22493. doi: 10.1371/journal.pone.0022493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright EM, O'Connor R, Kerr BA. Radial aplasia in CHARGE syndrome: a new association. Eur J Med Genet. 2009;52:239–241. doi: 10.1016/j.ejmg.2009.03.017. [DOI] [PubMed] [Google Scholar]
- Yu T, Meiners LC, Danielsen K, Wong MT, Bowler T, Reinberg D, Scambler PJ, van Ravenswaaij-Arts CM, Basson MA. Deregulated FGF and homeotic gene expression underlies cerebellar vermis hypoplasia in CHARGE syndrome. Elife. 2013;2:e01305. doi: 10.7554/eLife.01305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zentner GE, Layman WS, Martin DM, Scacheri PC. Molecular and phenotypic aspects of CHD7 mutation in CHARGE syndrome. Am J Med Genet A. 2010;152A:674–686. doi: 10.1002/ajmg.a.33323. [DOI] [PMC free article] [PubMed] [Google Scholar]