

Abstract
Recent studies have demonstrated that Notch-1 expression is increased in the hippocampus of Alzheimer's disease patients. We speculate that Notch-1 signaling may be involved in PC12 cell apoptosis induced by amyloid beta-peptide (25–35) (Aβ25–35). In the present study, PC12 cells were cultured with different doses (0, 0.1, 1.0, 10 and 100 nmol/L) of N-[N-(3,5-Difluorophenacetyl)-L-alanyl]-S-phenylglycine t-butyl ester, a Notch-1 signaling pathway inhibitor, for 30 minutes. Then cultured cells were induced with Aβ25–35 for 48 hours. Pretreatment of PC12 cells with high doses of N-[N-(3,5-Difluorophenacetyl)-L-alanyl]-S-phenylglycine t-butyl ester (> 10 nmol/L) prolonged the survival of PC12 cells after Aβ25–35 induction, decreased the expression of apoptosis-related proteins caspase-3, -8, -9, increased the activity of oxidative stress-related superoxide dismutase and catalase, inhibited the production of active oxygen, and reduced nuclear factor kappa B expression. This study indicates that the Notch-1 signaling pathway plays a pivotal role in Aβ25–35-induced PC12 apoptosis.
Keywords: nerve regeneration, Alzheimer's disease, amyloid beta-peptide (25–35), Notch-1, PC12 cells, apoptosis, oxidative stress, nuclear factor kappa B, neural regeneration
Introduction
Alzheimer's disease is one of the most common neural degenerative diseases in humans and is characterized by memory impairment (Glenner and Wong, 1984; Hardy and Higgins, 1992; Tomita, 2011; Drachman, 2014). Studies indicated that synaptic changes and β-amyloid (Aβ), a 39- to 43-amino acid β-sheet peptide derived from proteolytic processing at the N-terminus of the amyloid precursor protein, are characteristic histopathological features of Alzheimer's disease patients (Selkoe, 1991; Levine, 1993; Selkoe, 1994; Hardy, 1997; Crump et al., 2013). From a physiological point of view, Aβ25–35, a derivative of Aβ1–40 and Aβ1–42, has been demonstrated to be the shortest fragment that exhibits biological activity and retains toxicity of the full-length peptide(s) (Shearman et al., 1994; Terzi et al., 1994; Fuller et al., 1995; Iversen et al., 1995; Pike et al., 1995).
Notch-1 signaling is an important signaling pathway and has an important role in individual developmental processes, cell proliferation, differentiation and cell fate decisions by interacting with transcriptional regulators (Yu et al., 2000; Selkoe, 2001; Sisodia and St George-Hyslop, 2002; Harper et al., 2003; Ahmed et al., 2014; Liao et al., 2014). Recently, some studies demonstrated that Notch-1 was also expressed in the hippocampus of adult human brains, indicating Notch-1 may have a specific function in neural developmental. Notch-1 expression was significantly increased in the hippocampus of Alzheimer's disease patients compared with normal subjects (Berezovska et al., 1999; Mitani et al., 2014; Wagner et al., 2014). It is well known that the hippocampus relates to the generation and formation of new memories. Notch-1 potentially influences neurogenesis and neuronal plasticity in the hippocampus (Albensi and Mattson, 2000; Wang et al., 2004; Oikawa et al., 2012). To date, whether the Notch signaling pathway is involved in Aβ-induced neuronal cell apoptosis and the underlying molecular mechanism are unknown.
The present study demonstrated an effect of N-[N-(3,5-Di-fluorophenacetyl)-L-alanyl]-S-phenylglycine t-butyl ester (DAPT), a Notch-1 signaling pathway inhibitor, on PC cell apoptosis induced by Aβ25–35 and oxidative stress, in a broad attempt to explore the prevention and treatment of Alzheimer's disease.
Materials and Methods
PC12 cell culture and intervention
PC12 cells (American Type Culture Collection, Manassas, VA, USA) were cultured with complete RPMI-1640 medium (Hyclone, Logan, Utah, USA) supplemented with 5% fetal calf serum (Hyclone), 10% horse serum (Hyclone), 100 U/mL penicillin, and 100 mg/mL streptomycin at 37°C in a 5% CO2 incubator. Logarithmic growth phase cells were digested and seeded at appropriate densities on poly-L-lysine-coated plates or chambers. PC12 cells were pre-incubated with different concentrations of DAPT (0, 0.1, 1.0, 10 and 100 nmol/L), a γ-secretase inhibitor and indirect inhibitor of Notch-1 signaling (Xiao et al., 2014) (Gene Operation, Ann Arbor, MI, USA) for 30 minutes. Subsequently, the cells were treated with 10 μmol/L Aβ25–35 (Sigma-Aldrich, St. Louis, MO, USA) for 48 hours. Concentrations of 0, 1.0 or 10 nmol/L were used to study the mechanisms of DAPT in PC12 cell apoptosis.
PC12 cell viability detected by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay
MTT assay was used to detect cell viability by measuring levels of formazan produced. PC12 cells at a density of 1 × 104 were plated in 96-well plates with 100 μL medium in every well. After 24 hours, the cells were incubated with 10 μmol/L Aβ25–35 for 48 hours pretreated with various concentrations of DAPT (0.1–100 nmol/L) for 30 minutes. After incubation, cells were treated with 20 μL MTT solution (5 mg/mL; Beyotime Institute of Biotechnology, Shanghai, China) for an additional 4 hours. Then the medium was removed and 200 μL dimethylsulfoxide was added to every well. Absorbance was determined with a microplate reader (Becton Dickenson, San Francisco, CA, USA) at 570 nm. Cell viability was normalized as a percentage of the absorbance values compared to the controls, which were not exposed to DAPT or Aβ25–35.
Measurement of intracellular reactive oxygen species generation in PC12 cells detected by flow cytometry
The level of intracellular reactive oxygen species was determined by a change in fluorescence resulting from intracellular esterases to non-fluorescent 2′,7′-dichlorofluorescin diacetate (DCFH), which was performed using a Becton Dickenson FACScan™ flow cytometer (Becton Dickenson) with a reactive oxygen species-sensitive dye, hydroethidine. PC12 cells were plated at a density of 3 × 105 cells per 6-well dish. Twenty-four hours later, PC12 cells were pre-incubated for 30 minutes with DAPT, and then incubated with 10 μmol/L Aβ25–35. The cells were then placed in 10 μmol/L DCFH-DA for 20 minutes at 37°C, and washed three times with DMEM. Reactive oxygen species levels were detected by flow cytometry. A total of 10,000 events were recorded for each analysis and the value for each treatment group was shown as a percentage of the control value.
Morphology of apoptotic PC12 cells observed by Hoechst 33342/propidium iodide double staining
Hoechst 33342/propidium iodide double staining was used for detection of morphological changes of apoptotic cells. PC12 cells at a density of 1 × 106 were plated in 6-well plates with 2 mL of medium in every well, and were treated as previously described. After treatment, cells were stained with the DNA dye Hoechst 33342/propidium iodide (Beyotime Institute of Biotechnology) for 15 minutes, followed by fixing with 4% formaldehyde in PBS for 5 minutes at 4°C. After being washed with PBS three times, the cells were visualized under a fluorescence microscope (Olympus, Tokyo, Japan).
Superoxide dismutase activity in PC12 cells detected by microplate reader
Superoxide dismutase activity was estimated according to the previously described method (Beauchamp and Fridovich, 1971; Marcus et al., 1998) by assaying the auto-oxidation and illumination of pyrogallol at 440 nm. This method employs xanthine and xanthine oxidase to generate superoxide radicals, which react with 2-(4-iodophenyl)-3-(4-nitrophenol)-5-phenyltetrazolium chloride to form a red formazan dye. Superoxide dismutase activity is then measured by the degree of inhibition of this reaction. Superoxide dismutase inhibits the reaction by converting the superoxide radical to oxygen. The absorbance at 505 nm was measured by spectrophotometer (Shimadzu UV-1700, Tokyo, Japan) and used to calculate superoxide dismutase activity.
Catalase activity in PC12 cells detected by microplate reader
Catalase activity was measured according to the instructions of the Catalase Assay Kit (Cayman Chemical, Ann Arbor, MI, USA), based on the reaction of catalase with methanol in the presence of an optimal concentration of H2O2. The cells were treated as previously, and equal amounts of total proteins were used for detection as described in the manufacturer's instructions. The absorbance at 450 nm was measured by spectrophotometer and used to calculate catalase activity.
Expression of caspase-9, caspase-8, caspase-3, Notch-1, nuclear factor kappa B, catalase, superoxide dismutase in PC12 cells detected by western blot analysis
PC12 cells were subcultured and treated as previously described. After pretreatment with DAPT for 30 minutes and Aβ25–35 for 48 hours, the cells were collected and lysed in RIPA buffer (including 1% Triton, 0.1% sodium dodecylsulfate, 0.5% deoxycholate, ethylenediaminetetraacetic acid 1 mmol/L, Tris 20 mmol/L (pH 7.4), NaCl 150 mmol/L, and NaF 10 mmol/L). Insoluble material was removed by centrifugation at 12,000 r/min for 20 minutes at 4°C. A bovine serum albumin kit was used for quantifying protein concentrations. The samples were equalized for protein concentration. Total proteins were separated by 12% SDS-PAGE, and transferred to polyvinyl difluoride membranes. The membranes were blocked with 5% non-fat milk in PBST buffer for 1 hour at room temperature prior to incubation with rabbit anti-rat caspase-9 (pro-form), caspase-8 (pro-form), caspase-3 (activated form), Notch-1, nuclear factor kappa B, catalase, and superoxide dismutase monoclonal antibodies (Santa Cruz Biotechnology, Santa Cruz, CA, USA) overnight at 4°C, followed by goat anti-rabbit IgG conjugated to HRP (1:1,000, Santa Cruz Biotechnology). The results were scanned and analyzed with ImageJ software (http://rsbweb.nih.gov/ij/download.html). The expression level was corrected to β-actin. The results are shown as relative absorbance detected by spectrophotometer (BioTek, Winooski, VT, USA).
Statistical analysis
SPSS 11.0 software (SPSS, Chicago, IL, USA) was used for statistical analysis. All data were expressed as mean ± SD. Statistical analysis was performed using the two sample independent t-test for comparison of two groups and differences of P < 0.05 were considered statistically significant. All experiments were repeated at least three times.
Results
Notch-1 signaling inhibitor inhibited Aβ25–35-induced reduction of PC12 cell viability
MTT assay indicated that the viability of PC12 cells was reduced significantly after Aβ25–35 treatment, which decreased to 40.22% of the control group (P < 0.05).
The viability of PC12 cells incubated with Aβ25–35 was significantly increased after pretreatment with different concentrations of DAPT (1–100 nmol/L) (P < 0.05 or P < 0.01). Cell viability increased slightly by treatment with 0.1 nmol/L DAPT, but there was no statistically significant difference compared with the Aβ25–35 treatment group (P > 0.05; Figure 1A).
Figure 1.
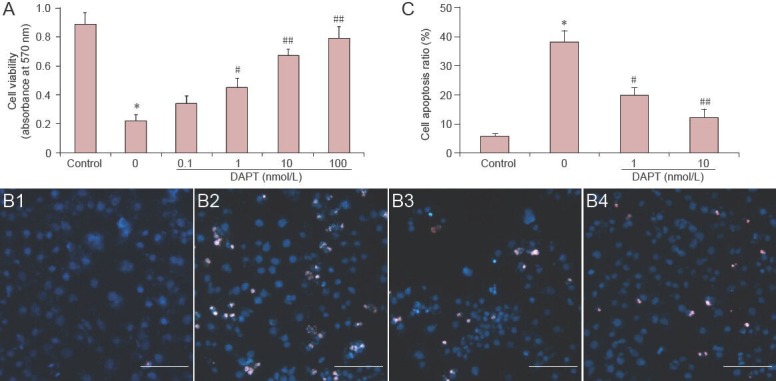
Effect of Notch-1 signaling on PC12 cell viability, apoptosis, and morphology induced by amyloid beta-peptide (25–35) (Aβ25–35) treatment.
(A) Effect of Notch-1 signaling on PC12 cell viability induced by Aβ25–35 treatment. PC12 cells were pretreated with 0.1–100 nmol/L DAPT for 30 minutes, followed by Aβ25–35 for 48 hours. Cell viability was detected by MTT assay. (B) Cell morphology was monitored by Hoechst/propidium iodide double staining. (B1) Control group; (B2) model group (0): PC12 cells treated with Aβ25–35 for indicated times without DAPT incubation; (B3, 4) 1, 10 nmol/L DAPT groups (1, 10 nmol/L). Scale bars: 100 μm. (C) The apoptosis ratio of PC12 cells was determined by Hoechst/propidium iodide double staining. (A, C) Data were expressed as mean ± SD (n = 3). Statistical analysis was performed by two sample independent t-test for comparison between two groups. All experiments were repeated at least three times. *P < 0.05, vs. control group; #P < 0.05, ##P < 0.01, vs. model group (0).
Notch-1 signaling inhibitor reduced PC12 cell apoptosis induced by Aβ25–35
The morphological changes of apoptotic cells were confirmed by Hoechst 33342/propidium iodide double staining. PC12 cells treated with Aβ25–35 alone appeared to undergo cellular nuclear condensation, contraction and fragmentation, suggesting that Aβ25–35 induced apoptosis in PC12 cells. The number of Hoechst 33342/propidium iodide positive cells was decreased upon pretreatment with 1 and 10 nmol/L DAPT (P < 0.05; Figure 1B, C). We also examined the expression of apoptotic proteins by western blot analysis. Caspase-3, caspase-8, and caspase-9 expression was significantly increased in PC12 cells in response to treatment with Aβ25–35 (P < 0.05, Aβ25–35 vs. control). However, the expression of these proteins significantly decreased in groups pretreated with 1 or 10 nmol/L DAPT (P < 0.05; Figure 2).
Figure 2.
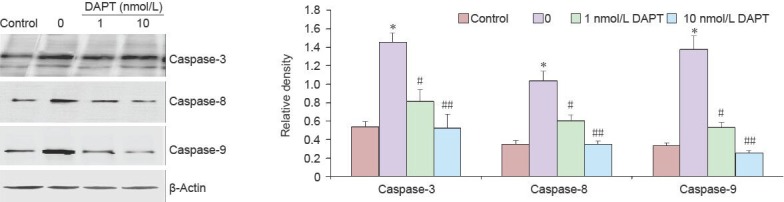
Role of Notch-1 signaling on the expression of apoptotic proteins after amyloid beta-peptide (25–35) (Aβ25–35) treatment.
Levels of apoptosis related proteins were detected by western blot analysis. Values presented are absorbance ratios of caspase-3, -8, and -9 to β-actin, which was used as an equal protein loading marker. Results are presented as mean ± SD (n = 5). Statistical analysis was performed by two sample independent t-test for comparison between two groups. All experiments were repeated at least three times. *P < 0.05, vs. control group; #P < 0.05, ##P < 0.01, vs. model group (0). DAPT: N-[N-(3,5-Difluorophenacetyl)-L-alanyl]-S-phenylglycine t-butyl ester.
Notch-1 signaling inhibitor attenuated oxidative stress in PC12 cells induced by Aβ25–35
After PC12 cells were pretreated with Aβ25–35, the activity of superoxide dismutase and catalase in cells was significantly decreased, while the production of intracellular reactive oxygen species was significantly increased (P < 0.05). Furthermore, the activity of superoxide dismutase and catalase in cells was significantly increased after DAPT treatment, and the levels of reactive oxygen species were reduced (P < 0.05; Figure 3). Western blot analysis showed that Aβ25–35 treatment increased the levels of Notch-1, nuclear factor kappa B, superoxide dismutase and catalase proteins in PC12 cells (P < 0.05). Notch-1 and nuclear factor kappa B expression was reduced, while superoxide dismutase and catalase protein levels were increased by treatment with 1–10 nmol/L of DAPT (P < 0.05; Figure 4).
Figure 3.
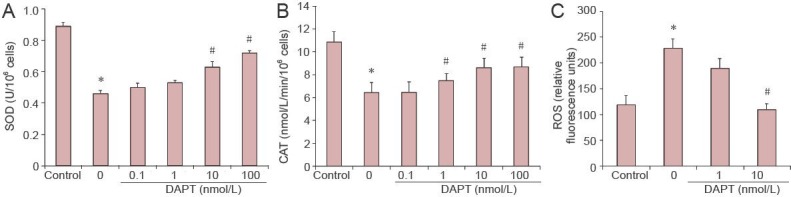
Role of Notch-1 signaling on oxidative stress in PC12 cells after amyloid beta-peptide (25–35) (Aβ25–35) treatment.
(A) Superoxide dismutase (SOD); (B) catalase (CAT); (C) reactive oxygen species (ROS). Data are expressed as mean ± SD (n = 3). Statistical analysis was performed by two sample independent t-test for comparison of two groups. All experiments were repeated at least three times. *P < 0.05, vs. control group; #P < 0.05, vs. model group (0). DAPT: N-[N-(3,5-Difluorophenacetyl)-L-alanyl]-S-phenylglycine t-butyl ester.
Figure 4.
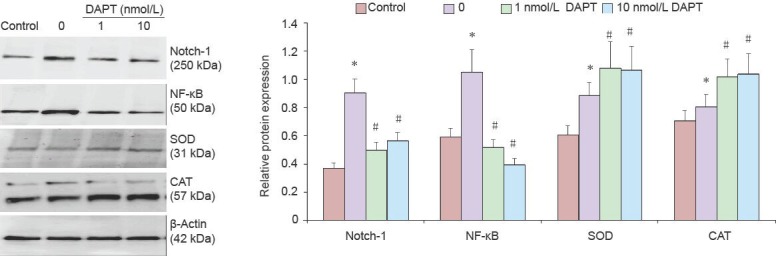
Role of Notch-1 signaling on cellular redox regulation after amyloid beta-peptide (25–35) (Aβ25–35) treatment.
The protein level of Notch-1, nuclear factor kappa B (NF-κB), superoxide dismutase (SOD), and catalase (CAT) were detected by western blot assay. Values presented are absorbance ratios of Notch-1, NF-κB, SOD, and CAT to β-actin, which was used as an equal protein loading marker. Results are presented as mean ± SD (n = 5). Statistical analysis was performed by two sample independent t-test for comparison between two groups. All experiments were repeated at least three times. *P < 0.05, vs. control group; #P < 0.05, vs. model group (0). DAPT: N-[N-(3,5-Difluorophenacetyl)-L-alanyl]-S-phenylglycine t-butyl ester.
Discussion
The PC12 cell line is usually used as a cellular model to study neurodegenerative diseases (Vaudry et al., 2002; Yan et al., 2013). Previous studies have shown that Aβ25–35 not only induced cytotoxicity, but also elicited excessive reactive oxygen species production, apoptosis and cell death in PC12 cells (Xiao et al., 2002; Ge et al., 2008; Chen et al., 2013; Dimitrov et al., 2013; Grimm et al., 2013; Prox et al., 2013). However, to date, the role of Notch signaling in the regulation of apoptosis induced by Aβ25–35 remains unknown. Therefore, the present study explored whether DAPT has a protective role against Aβ25–35-induced apoptosis in PC12 cells. This study showed that PC12 cells treated with Aβ25–35 underwent apoptotic cell death in accordance with previous studies. A significant cytotoxic effect of Aβ25–35 on PC12 cells was detected by MTT assay and Hoechst 33342/propidium iodide double staining. Apoptosis induced by Aβ25–35 was confirmed to be the activation of caspase-3 and high levels of caspase-8 and caspase-9. We also demonstrated that the cytotoxicity of Aβ25–35 was associated with oxidative stress. The level of intracellular reactive oxygen species in PC12 cells increased and the activities of superoxide dismutase and catalase decreased when PC12 cells were treated with Aβ25–35.
Notch signaling is an important pathway that is widely expressed in many tissues (Hansson et al., 2004; Lasky and Wu, 2005; Bonini et al., 2013; Newman et al., 2014). Recent research demonstrated that Notch is highly expressed and has high activity in the brain, particularly in Alzheimer's disease patients, suggesting Notch signaling might play an important role in neuron development (Redmond and Ghosh, 2001; Gaiano and Fishell, 2002; Woo et al., 2009; Dimitrov et al., 2013; Shen, 2013; Singh et al., 2013). Studies also demonstrated that overexpression of Notch and exogenous Notch had a role in neuronal cell protection to oxidative and ischemic insults, and exogenous Notch reduced blood-brain barrier permeability and preserved tissue against injury (Deane and Zlokovic, 2007; Li et al., 2013; McKee et al., 2013). However, the molecular mechanisms by which Notch is involved in neuronal impairment remain unclear. We speculated that a Notch inhibitor might have a protective role in the neurodegenerative process in diseases such as Alzheimer's disease by decreasing the oxidative stress induced by Aβ.
Previous research suggested that Notch signaling was involved in the regulation of cell apoptosis through the nuclear factor kappa B signaling pathway (Wang et al., 2008; Abdallah and Kassem, 2012; Xie et al., 2012; García-Escudero et al., 2013). Many studies have shown that Aβ-induced neurotoxicity is mediated by free radicals in vitro (Butterfield et al., 2001; Cai et al., 2011; Alberi et al., 2013). Consistent with these findings, results confirmed that Aβ stimulated reactive oxygen species production associated with nuclear factor kappa B signaling pathway. Furthermore, Aβ25–35 treatment decreased survival and increased apoptosis of PC12 cells associated with reactive oxygen species overproduction. However, the effects were reversed significantly when PC12 cells were pretreated with DAPT before the addition of Aβ25–35. In addition, elevated reactive oxygen species levels by Aβ25–35 were decreased after treatment with DAPT.
To explore the molecular mechanism of Notch involvement in protection of PC12 cells against apoptosis induced by Aβ25–35, the generation of reactive oxygen species was detected. Administration of a Notch inhibitor reduced reactive oxygen species production by elevating superoxide dismutase and catalase levels. The expression of activated caspase-3 was significantly increased, indicating apoptosis initiation. Administration of the Notch inhibitor also significantly decreased the Aβ-induced expression of activated caspase-3, suggesting it exerts protective effects against Aβ25–35-induced apoptosis.
In summary, the present study demonstrated that Notch signaling is involved in the regulation of PC12 cell apoptosis induced by Aβ treatment. The use of Notch inhibitors might be useful in cellular defense against oxidative stress during the neurodegenerative process in Alzheimer's disease.
Footnotes
Conflicts of interest: None declared.
Copyedited by Croxford L, Norman C, Yu J, Yang Y, Li CH, Song LP, Zhao M
References
- Abdallah BM, Kassem M. New factors controlling the balance between osteoblastogenesis and adipogenesis. Bone. 2012;50:540–545. doi: 10.1016/j.bone.2011.06.030. [DOI] [PubMed] [Google Scholar]
- Ahmed MM, Dhanasekaran AR, Block A, Tong S, Costa AC, Gardiner KJ. Protein profiles associated with context fear conditioning and their modulation by memantine. Mol Cell Proteomics. 2014;13:919–937. doi: 10.1074/mcp.M113.035568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albensi BC, Mattson MP. Evidence for the involvement of TNF and NF-κB in hippocampal synaptic plasticity. Synapse. 2000;35:151–159. doi: 10.1002/(SICI)1098-2396(200002)35:2<151::AID-SYN8>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- Alberi L, Hoey SE, Brai E, Scotti AL, Marathe S. Notch signaling in the brain: in good and bad times. Ageing Res Rev. 2013;12:801–814. doi: 10.1016/j.arr.2013.03.004. [DOI] [PubMed] [Google Scholar]
- Beauchamp C, Fridovich I. Superoxide dismutase: improved assays and an assay applicable to acrylamide gels. Anal Biochem. 1971;44:276–287. doi: 10.1016/0003-2697(71)90370-8. [DOI] [PubMed] [Google Scholar]
- Berezovska O, Frosch M, McLean P, Knowles R, Koo E, Kang D, Shen J, Lu FM, Lux SE, Tonegawa S, Hyman BT. The Alzheimer-related gene presenilin 1 facilitates notch 1 in primary mammalian neurons. Brain Res Mol Brain Res. 1999;69:273–280. doi: 10.1016/s0169-328x(99)00119-9. [DOI] [PubMed] [Google Scholar]
- Bonini SA, Ferrari-Toninelli G, Montinaro M, Memo M. Notch signalling in adult neurons: a potential target for microtubule stabilization. Ther Adv Neurol Disord. 2013;6:375–385. doi: 10.1177/1756285613490051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butterfield DA, Drake J, Pocernich C, Castegna A. Evidence of oxidative damage in Alzheimer's disease brain: central role for amyloid β-peptide. Trends Mol Med. 2001;7:548–554. doi: 10.1016/s1471-4914(01)02173-6. [DOI] [PubMed] [Google Scholar]
- Cai Z, Zhao B, Ratka A. Oxidative stress and β-amyloid protein in Alzheimer's disease. Neuromolecular Med. 2011;13:223–250. doi: 10.1007/s12017-011-8155-9. [DOI] [PubMed] [Google Scholar]
- Chen DL, Zhang P, Lin L, Shuai O, Zhang HM, Liu SH, Wang JY. Protective effect of bajijiasu against β-amyloid-induced neurotoxicity in PC12 cells. Cell Mol Neurobiol. 2013;33:837–850. doi: 10.1007/s10571-013-9950-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crump CJ, Johnson DS, Li YM. Development and mechanism of γ-secretase modulators for Alzheimer's disease. Biochemistry. 2013;52:3197–3216. doi: 10.1021/bi400377p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deane R, Zlokovic BV. Role of the blood-brain barrier in the pathogenesis of Alzheimers disease. Curr Alzheimer Res. 2007;4:191–197. doi: 10.2174/156720507780362245. [DOI] [PubMed] [Google Scholar]
- Dimitrov M, Alattia JR, Lemmin T, Lehal R, Fligier A, Houacine J, Hussain I, Radtke F, Dal Peraro M, Beher D, Fraering PC. Alzheimer's disease mutations in APP but not γ-secretase modulators affect epsilon-cleavage-dependent AICD production. Nat Commun. 2013;4:2246. doi: 10.1038/ncomms3246. [DOI] [PubMed] [Google Scholar]
- Drachman DA. The amyloid hypothesis, time to move on: amyloid is the downstream result, not cause, of Alzheimer's disease. Alzheimers Dement. 2014;10:372–380. doi: 10.1016/j.jalz.2013.11.003. [DOI] [PubMed] [Google Scholar]
- Fuller SJ, Storey E, Li QX, Smith AI, Beyreuther K, Masters CL. Intracellular production of beta A4 amyloid of Alzheimer's disease: modulation by phosphoramidon and lack of coupling to the secretion of the amyloid precursor protein. Biochemistry. 1995;34:8091–8098. doi: 10.1021/bi00025a015. [DOI] [PubMed] [Google Scholar]
- Gaiano N, Fishell G. The role of notch in promoting glial and neural stem cell fates. Annu Rev Neurosci. 2002;25:471–490. doi: 10.1146/annurev.neuro.25.030702.130823. [DOI] [PubMed] [Google Scholar]
- García-Escudero V, Martín-Maestro P, Perry G, Avila J. Deconstructing mitochondrial dysfunction in Alzheimer disease. Oxid Med Cell Longev. 2013:162152. doi: 10.1155/2013/162152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge J, Yu Y, Chui DH. Protective effect of Xylocoside G on Aβ25-35-induced neurotoxicity in PC12 cells. Zhongguo Yaoxue Zazhi. 2008;18:21–26. [Google Scholar]
- Glenner GG, Wong CW. Alzheimer's disease: initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem Biophys Res Commun. 1984;120:885–890. doi: 10.1016/s0006-291x(84)80190-4. [DOI] [PubMed] [Google Scholar]
- Grimm MO, Mett J, Stahlmann CP, Haupenthal VJ, Zimmer VC, Hartmann T. Neprilysin and Aβ clearance: impact of the APP intracellular domain in NEP regulation and implications in Alzheimer's disease. Front Aging Neurosci. 2013;5:98. doi: 10.3389/fnagi.2013.00098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansson EM, Lendahl U, Chapman G. Notch signaling in development and disease. Semin Cancer Biol. 2004;14:320–328. doi: 10.1016/j.semcancer.2004.04.011. [DOI] [PubMed] [Google Scholar]
- Hardy J. Amyloid, the presenilins and Alzheimer's disease. Trends Neurosci. 1997;20:154–159. doi: 10.1016/s0166-2236(96)01030-2. [DOI] [PubMed] [Google Scholar]
- Hardy JA, Higgins GA. Alzheimer's disease: the amyloid cascade hypothesis. Science. 1992;256:184–185. doi: 10.1126/science.1566067. [DOI] [PubMed] [Google Scholar]
- Harper JA, Yuan JS, Tan JB, Visan I, Guidos CJ. Notch signaling in development and disease. Clin Genet. 2003;64:461–472. doi: 10.1046/j.1399-0004.2003.00194.x. [DOI] [PubMed] [Google Scholar]
- Iversen LL, Mortishire-Smith RJ, Pollack SJ, Shearman MS. The toxicity in vitro of beta-amyloid protein. Biochem J. 1995;311:1–16. doi: 10.1042/bj3110001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lasky JL, Wu H. Notch signaling, brain development, and human disease. Pediatr Res. 2005;57:104R–109R. doi: 10.1203/01.PDR.0000159632.70510.3D. [DOI] [PubMed] [Google Scholar]
- Levine H. Thioflavine T interaction with synthetic Alzheimer's disease β-amyloid peptides: Detection of amyloid aggregation in solution. Protein Sci. 1993;2:404–410. doi: 10.1002/pro.5560020312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Yu S, Wu J, Zou Y, Zhao Y. Sulfiredoxin-1 protects PC12 cells against oxidative stress induced by hydrogen peroxide. J Neurosci Res. 2013;91:861–870. doi: 10.1002/jnr.23218. [DOI] [PubMed] [Google Scholar]
- Liao YF, Tang YC, Chang MY, Wang BJ, Hu MK. Discovery of small molecular (d)-leucinamides as potent, Notch-sparing γ-secretase modulators. Eur J Med Chem. 2014;79:143–151. doi: 10.1016/j.ejmech.2014.04.006. [DOI] [PubMed] [Google Scholar]
- Marcus DL, Thomas C, Rodriguez C, Simberkoff K, Tsai JS, Strafaci JA, Freedman ML. Increased peroxidation and reduced antioxidant enzyme activity in Alzheimer's disease. Exp Neurol. 1998;150:40–44. doi: 10.1006/exnr.1997.6750. [DOI] [PubMed] [Google Scholar]
- McKee TD, Loureiro R, Dumin JA, Zarayskiy V, Tate B. An improved cell-based method for determining the γ-secretase enzyme activity against both Notch and APP substrates. J Neurosci Methods. 2013;213:14–21. doi: 10.1016/j.jneumeth.2012.11.011. [DOI] [PubMed] [Google Scholar]
- Mitani Y, Akashiba H, Saita K, Yarimizu J, Uchino H, Okabe M, Asai M, Yamasaki S, Nozawa T, Ishikawa N, Shitaka Y, Ni K, Matsuoka N. Pharmacological characterization of the novel γ-secretase modulator AS2715348, a potential therapy for Alzheimer's disease, in rodents and nonhuman primates. Neuropharmacology. 2014;79:412–419. doi: 10.1016/j.neuropharm.2013.12.013. [DOI] [PubMed] [Google Scholar]
- Newman M, Wilson L, Verdile G, Lim A, Khan I, Nik SHM, Pursglove S, Chapman G, Martins RN, Lardelli M. Differential, dominant activation and inhibition of Notch signalling and APP cleavage by truncations of PSEN1 in human disease. Hum Mol Genet. 2014;23:602–617. doi: 10.1093/hmg/ddt448. [DOI] [PubMed] [Google Scholar]
- Oikawa N, Goto M, Ikeda K, Taguchi R, Yanagisawa K. The γ-secretase inhibitor DAPT increases the levels of gangliosides at neuritic terminals of differentiating PC12 cells. Neurosci Lett. 2012;525:49–53. doi: 10.1016/j.neulet.2012.07.027. [DOI] [PubMed] [Google Scholar]
- Pike CJ, Walencewicz-Wasserman AJ, Kosmoski J, Cribbs DH, Glabe CG, Cotman CW. Structure-activity analyses of β-amyloid peptides: contributions of the β25–35 region to aggregation and neurotoxicity. J Neurochem. 1995;64:253–265. doi: 10.1046/j.1471-4159.1995.64010253.x. [DOI] [PubMed] [Google Scholar]
- Prox J, Bernreuther C, Altmeppen H, Grendel J, Glatzel M, D’Hooge R, Stroobants S, Ahmed T, Balschun D, Willem M, Lammich S, Isbrandt D, Schweizer M, Horré K, De Strooper B, Saftig P. Postnatal disruption of the disintegrin/metalloproteinase ADAM10 in brain causes epileptic seizures, learning deficits, altered spine morphology, and defective synaptic functions. J Neurosci. 2013;33:12915–12928. doi: 10.1523/JNEUROSCI.5910-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redmond L, Ghosh A. The role of Notch and Rho GTPase signaling in the control of dendritic development. Curr Opin Neurobiol. 2001;11:111–117. doi: 10.1016/s0959-4388(00)00181-1. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ. The molecular pathology of Alzheimer's disease. Neuron. 1991;6:487–498. doi: 10.1016/0896-6273(91)90052-2. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ. Normal and abnormal biology of the beta-amyloid precursor protein. Annu Rev Neurosci. 1994;17:489–517. doi: 10.1146/annurev.ne.17.030194.002421. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ. Presenilin, Notch, and the genesis and treatment of Alzheimer's disease. Proc Natl Acad Sci U S A. 2001;98:11039–11041. doi: 10.1073/pnas.211352598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shearman MS, Ragan CI, Iversen LL. Inhibition of PC12 cell redox activity is a specific, early indicator of the mechanism of beta-amyloid-mediated cell death. Proc Natl Acad Sci U S A. 1994;91:1470–1474. doi: 10.1073/pnas.91.4.1470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen J. Function and dysfunction of presenilin. Neurodegener Dis. 2013;13:61–63. doi: 10.1159/000354971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh A, Zapata MC, Choi YS, Yoon SO. GSI promotes vincristine-induced apoptosis by enhancing multi-polar spindle formation. Cell Cycle. 2013;13:157–166. doi: 10.4161/cc.26951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sisodia SS, St George-Hyslop PH. gamma-Secretase, Notch, Abeta and Alzheimer's disease: where do the presenilins fit in? Nat Rev Neurosci. 2002;3:281–290. doi: 10.1038/nrn785. [DOI] [PubMed] [Google Scholar]
- Terzi E, Hoelzemann G, Seelig J. Reversible random coil-beta-sheet transition of the Alzheimer beta-amyloid fragment (25-35) Biochemistry. 1994;33:1345–1350. doi: 10.1021/bi00172a009. [DOI] [PubMed] [Google Scholar]
- Tomita T. Development of Alzheimer's disease treatment based on the molecular mechanism of γ-secretase activity. Rinsho Shinkeigaku. 2011;52:1165–1167. doi: 10.5692/clinicalneurol.52.1165. [DOI] [PubMed] [Google Scholar]
- Vaudry D, Stork PJ, Lazarovici P, Eiden LE. Signaling pathways for PC12 cell differentiation: making the right connections. Science. 2002;296:1648–1649. doi: 10.1126/science.1071552. [DOI] [PubMed] [Google Scholar]
- Wagner SL, Zhang C, Cheng S, Nguyen P, Zhang X, Rynearson KD, Wang R, Li Y, Sisodia SS, Mobley WC, Tanzi RE. Soluble γ-secretase modulators selectively inhibit the production of the 42-amino acid amyloid β peptide variant and augment the production of multiple carboxy-truncated amyloid β species. Biochemistry. 2014;53:702–713. doi: 10.1021/bi401537v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Chan SL, Miele L, Yao PJ, Mackes J, Ingram DK, Mattson MP, Furukawa K. Involvement of Notch signaling in hippocampal synaptic plasticity. Proc Natl Acad Sci U S A. 2004;101:9458–9462. doi: 10.1073/pnas.0308126101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang YL, Cai ZY, Luo Y, Gong JM. Influence of edaravone on Notch1 and nuclear factor-kappaB in rats with cerebral ischemia/reperfusion injury. Neural Regen Res. 2008;3:1342–1347. [Google Scholar]
- Woo HN, Park JS, Gwon A, Arumugam TV, Jo DG. Alzheimer's disease and Notch signaling. Biochem Biophys Res Commun. 2009;390:1093–1097. doi: 10.1016/j.bbrc.2009.10.093. [DOI] [PubMed] [Google Scholar]
- Xiao XQ, Zhang HY, Tang XC. Huperzine A attenuates amyloid beta-peptide fragment 25-35-induced apoptosis in rat cortical neurons via inhibiting reactive oxygen species formation and caspase-3 activation. J Neurosci Res. 2002;67:30–36. doi: 10.1002/jnr.10075. [DOI] [PubMed] [Google Scholar]
- Xiao YG, Wang W, Gong D, Mao ZF. γ-Secretase inhibitor DAPT attenuates intimal hyperplasia of vein grafts by inhibition of Notch1 signaling. Lab Invest. 2014;94:654–662. doi: 10.1038/labinvest.2014.58. [DOI] [PubMed] [Google Scholar]
- Xie Z, Dong Y, Maeda U, Xia W, Tanzi RE. RNAi-mediated knock-down of Dab and Numb attenuate Aβ levels via γ-secretase mediated APP processing. Transl Neurodegener. 2012;1:8. doi: 10.1186/2047-9158-1-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan FL, Han GL, Wu GJ. Cytotoxic role of advanced glycation end-products in PC12 cells treated with β-amyloid peptide. Mol Med Rep. 2013;8:367–372. doi: 10.3892/mmr.2013.1545. [DOI] [PubMed] [Google Scholar]
- Yu G, Nishimura M, Arawaka S, Levitan D, Zhang L, Tandon A, Song YQ, Rogaeva E, Chen F, Kawarai T, Supala A, Levesque L, Yu H, Yang DS, Holmes E, Milman P, Liang Y, Zhang DM, Xu DH, Sato C, et al. Nicastrin modulates presenilin-mediated notch/glp-1 signal transduction and betaAPP processing. Nature. 2000;407:48–54. doi: 10.1038/35024009. [DOI] [PubMed] [Google Scholar]