

Abstract
A series of NO-donor praziquantel hybrid compounds was obtained by combining praziquantel (PZQ) and furoxan moieties in a single entity. NO-donor properties of the furoxan derivatives were evaluated by detecting nitrite after incubation of the products in 7.4 pH buffered solution in the presence of L-cysteine. Structurally-related furazans, devoid of NO release capacity, were also synthesized for control purposes. All products were studied for their ability to inhibit recombinant Schistosoma mansoni thioredoxin glutathione reductase (TGR). Mobility and death of adult Schistosoma mansoni worms cultured in the presence of the products were evaluated versus PZQ. Analysis of the results showed that some products were endowed with both PZQ and NO-dependent antiparasitic properties. Compounds 6, 7, 18, and 24 emerged as the most interesting balanced hybrids, worthy of additional study on PZQ-resistant parasites.
Keywords: furoxans, praziquantel, schistosomiasis, thioredoxin glutathione reductase, NO-donor praziquantel
Introduction
Schistosomiasis, also known as Bilharzia, is one of the most prominent neglected tropical diseases (NTDs). It is a parasitosis caused by blood-dwelling flatworms of the genus Schistosoma; the principal species parasitizing humans are S. mansoni, S. japonicum, and S. haematobium.1 Estimates indicate that 600 million people are at risk of parasitosis and that more than 200 million people suffer from schistosomiasis, resulting in 200,000 deaths each year. The highest incidence of schistosomiasis is in sub-Saharan Africa, where the principal species responsible are S. mansoni and S. haematobium.2–4 Praziquantel (PZQ) is the drug of choice for the treatment of schistosomiasis.4,5 The structure (2-(cyclohexylcarbonyl)-1,2,3,6,7,11b-hexahydro-4H-pyrazino[2,1-a]isoquinolin-4-one) (PZQ, Chart 1) assigned to the drug was confirmed by X-ray analysis.6 It is a fairly lipophilic product, and is consequently endowed with low water solubility.7 PZQ has a stereogenic center and can thus exist as two optical stereoisomers: the levo isomer ((−)PZQ) is more active than the dextro isomer ((+)PZQ), but since the dextro form has no side effects, for economic reasons the drug is used as an orally-administered racemic mixture. PZQ is active against the adult forms of all schistosome species, but not against the juvenile forms;8 its mechanism of action is still unclear. Calcium accumulation, alteration of schistosomal membrane fluidity, reduction of glutathione concentration, destruction of the tegument following binding to schistosomal actin, and interference with components of the parasite’s aerobic metabolism have all been proposed as possible action mechanisms.4,9 A number of structural modifications of PZQ have been introduced with the goal of improving its antihelmintic action.7,10 Most of the resulting products are devoid of substantial activity; others show only moderate effects, and none is better than the lead.
Chart 1.

PZQ, Praziquantel; A, general structure of the furoxan hybrids 5–7, and of the related furazans 8–10; B, general structure of the furoxan hybrids 17, 18, 24, and of the related furazans 20, 21, 26; 1, R=R’=H, 1,2,5-oxadiazole 2-oxide (furoxan); 2, R=C6H5, R’=CN, 4-phenylfuroxan-3-carbonitrile.
One problem associated with the extensive use of PZQ is the risk of the parasite’s developing resistance.11,12 To date, there is no clear evidence for large-scale clinical resistance of schistosomes to treatment with PZQ. Conversely, laboratory studies clearly indicate that resistance to PZQ can be selected. In particular it has been found that a schistosomal homologue of the mammalian P-glycoprotein (P-gp) is upregulated in PZQ-treated worms and in juvenile worms, which are resistant to PZQ activity.13 For this reason there is an urgent need to develop new chemical classes of drugs for the treatment of schistosomiasis.
Recently, through a quantitative high-throughput screen, 1,2,5,-oxadiazole 2-oxides (furoxans) have been identified as a new chemical class for the control of schistosomiasis.14,15 Furoxan (1, Chart 1) is an old heterocyclic system, well known to chemists because of arguments over its structure.16 In the recent past, renewed interest has surrounded furoxan derivatives due to the discovery that they can release nitric oxide (NO) under the action of thiol cofactors.17,18 The presence of electron withdrawing groups at the ring, in particular at the 3-position, generally increases this capacity.
It has been shown that furoxan derivatives are able to inhibit thioredoxin glutathione reductase (TGR), a multifunctional parasite protein that reduces both thioredoxin and glutathione disulfide and provides deglutathionylation (glutaredoxin) activity in worms.19 Specific reaction of furoxans with TGR results in localized NO production and subsequent S- (or Se-) nitrosation and inactivation of TGR. The exact nature of the resulting TGR modifications is not known.20 Furoxan-3-carbonitrile derivatives are the most interesting compounds studied thus far. The prototype of this series is 4-phenylfuroxan-3-carbonitrile (2, Chart 1). The product was found to be an irreversible inhibitor of TGR (IC50 = 6.3 µM). It was active against all stages of S. mansoni and against cultured adult S. japonicum, and S. haematobium worms. Intraperitoneal injection of 2 at 10 mg/Kg in S. mansoni infected mice led to a marked reduction in worm burden when treatment occurred 1 day after infection (skin-stage parasites), 23 days after infection (juvenile, liver stage parasites), or 37 days after infection (adult, egg-laying parasites).14,20 More recently, a number of phenylsulfonyl substituted furoxans have been found to be endowed with potent antischistosomal activity.21
This paper describes a new series of compounds with potential dual antischistosomal action obtained by combining, in a single entity, PZQ and NO-donor furoxan derivatives bearing at the 3- position CN, CONH2, COOMe, or SO2C6H5 moieties. In the first group of hybrids, the furoxan substructures were substituted for the cyclohexyl group of PZQ (Chart 1, general structure A; Scheme 1, der.s 5–7). In the second group of hybrids the furoxan moiety was linked to the 10-position of PZQ through appropriate bridges (Chart 1, general structure B; Scheme 2, der.s 17, 18, 24). The synthesis, structural characterization, and preliminary in vitro and ex vivo pharmacological profiles of all these products are reported and discussed. Related furazan (1,2,5-oxadiazoles) derivatives, (Chart 1, general structure A; Scheme 1, der.s 8–10; Chart 1, general structure B; Scheme 2, der.s 20, 21, 26) were also considered for comparison, since their structures are closely related to those of the corresponding furoxans, but they do not release NO (des-NO furazans). Therefore, if a given biological activity of a furoxan derivative is similar to that of its furazan analogue, it will be assumed that NO is not involved in that activity. The approach to compare the pharmacological profile of a NO donor with that of its analogue des-NO, is frequently used in the study of hybrid NO-donor drugs.22,a,b,c
Scheme 1.

Reaction conditions: (a) N-hydroxysuccinimide, EDC˙HCl, cat. DMAP, CH2Cl2/THF; (b) CH3OH(NH3); (c) TFAA, pyridine, THF, 0°C; (d) (CH3O)3P reflux.
Scheme 2.

Reaction conditions: (a) NaOH/H2O, TEBAC, CHCl3, CH2Cl2, reflux; (b) paraformaldehyde, 2,2-dimethoxyethylamine, cyclohexyl carboxylic acid, CH3OH; (c) methanesulfonic acid, 0 °C then 70 °C; (d) BBr3, CH2Cl2; (e) NaH 60% mineral oil disp., THF, reflux; (f) TFAA, pyridine, THF, 0°C; (g) 3-bromopropan-1-ol, K2CO3, CH3CN, reflux; (h) DBU, CH2Cl2.
Results and Discussion
Chemistry
Synthesis
The first series of NO-donor PZQ hybrids (5–7) and of the related des-NO furazans (8–10) was obtained through the synthetic pathway reported in Scheme 1. The known hexahydro-4H-pyrazinoisoquinoline derivative 3 was coupled with 3-methoxycarbonylfuroxan-4-carboxylic acid (4) in CH2Cl2/THF solution, in the presence of N-hydroxysuccinimide, 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC), and a catalytic amount of 4-(dimethylamino)pyridine (DMAP), to give the furoxan ester 5. Treatment of this ester with methanol saturated with ammonia afforded the related amide 6. Dehydration of this latter product with trifluoroacetic anhydride in THF solution, in the presence of pyridine, gave rise to the corresponding nitrile 7. The furazan target product 8 was obtained by reduction with trimethylphosphite of the related furoxan 5. Furazans 9 and 10 were prepared from 8, following the same procedures used to obtain the corresponding furoxans 6 and 7 from 5.
The second series of NO-donor PZQ hybrids (17, 18, 24) and of the related des-NO furazans (20, 21, 26) was obtained through the synthetic pathway reported in Scheme 2. The commercial 2-(4-methoxyphenyl)ethanamine 11 was transformed into the related isocyanide 12 by treatment with chloroform under basic conditions, in the presence of a catalytic amount of triethylbenzylammonium chloride (TEBAC). The 10-methoxy-substituted PZQ 14, the common intermediate in preparing all target compounds, was obtained from 12 by the same stepwise Ugi four-component reaction and Pictect–Spengler reaction used by Cao et al. to prepare PZQ from 2-phenylethanamine.23 Briefly, a mixture of paraformaldehyde, dimethoxyethylamine and cyclohexylcarboxylic acid in methanol was treated with 12 to give 13 (Ugi reaction). This intermediate was added to methanesulfonic acid to afford 17 (Pictect-Spengler reaction). Cleavage with BBr3 of the methoxy group present in 14 gave rise to the phenol 15. Reaction of 15 with the 4-(bromomethyl)furoxan-3-carboxamide (16), or with the related furazan amide 19, in the presence of NaH yielded the expected final compounds 17 and 20, respectively. Dehydration of these latter compounds with trifluoroacetic anhydride in pyridine produced the final cyano-substituted related hybrids 18, 21. To prepare the phenylsulfonyl-substituted target structures 24, 26, the phenol derivative 15 was coupled with 3-bromopropan-1-ol in CH3CN in the presence of K2CO3, to give the alcohol 22. This intermediate, treated either with 3,4-bisphenylsulfonylfuroxan (23) or with the related furazan 25, gave the desired final products.
NO-release
The capacity of the final furoxan hybrids to release NO was evaluated on the basis of the amount of nitrite produced following incubation in buffered pH 7.4 solution in the presence of a 5:1 molar excess of L-cysteine. Nitrite was detected by the Griess reaction. The results expressed as percentages of NO2− are reported in Table 1. NO production ranks the series 7 > 18 > 24 > 6 ≈ 17 > 5. This sequence should parallel the different capacities of the products to release NO under the action of free cellular thiols (‘nonspecific’ release). Potentially, the NO formed can diffuse into the worm resulting in antiparasitic action, following interaction with a variety of cellular targets.24
Table 1.
Activity of compounds against TGR and cultured adult Schistosoma mansoni worms and NO donation ability expressed as % of NO2−.
| Compound | TGR IC50 (µM) |
Worm contraction | Worm killinga | %NO2− (mol/mol) ± SEb |
|||
|---|---|---|---|---|---|---|---|
| 50 µM | 10 µM | 50 µM | 10 µM | ||||
| PZQ | n.i. | Yes | Yes | 40%c | 15%c | --- | |
| 2d | 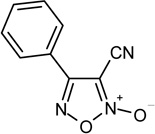 |
5.0 | No | No | 24 hrs | 96 hrs | 28.9 ± 0.3 |
| 5 | 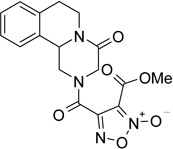 |
0.148 | Yes | Yese | 50%c | 23% | <1 |
| 6 | 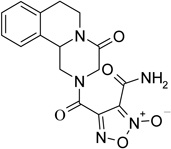 |
0.01 | Yes | Yese | 80% | 30% | 1.8 ± 0.1 |
| 7 | 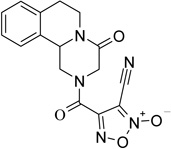 |
0.316 | Yes | ♀ only | 72 hrs | 30%c | 56.2 ± 0.3 |
| 8 | 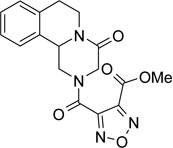 |
>50 | Yese | Yese | 18% | 15% | --- |
| 9 | 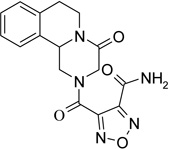 |
>50 | Yese | Weake | 23% | 23% | --- |
| 10 | 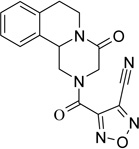 |
>50 | Yese | Yese | 25% | 20% | --- |
| 17 | 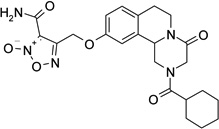 |
0.083 | No | No | No | No | 1.1 ± 0.2 |
| 18 | 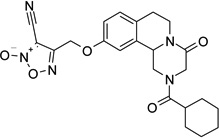 |
0.35 | Yesf | No | 24 hrs | Noc | 33.8 ± 0.1 |
| 20 |  |
> 50 | No | No | No | No | --- |
| 21 | 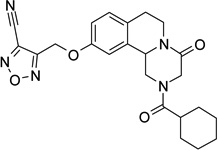 |
> 50 | Yesf | No | No | No | --- |
| 24 | 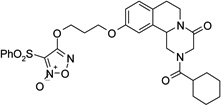 |
8.5 | ♂only | Some, not all | 24 hrs | 48 hrs | 15.7 ±0.1 |
| 26 | 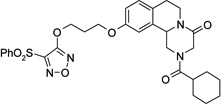 |
> 50 | Yes | Yese | Noc | No | --- |
n.i. – no inhibition; n.d. – not determined;
time (hrs) to 100% worm death or % dead worms at 144 hrs;
nitrite was measured (Griess reaction) after 1 hr incubation at 37 °C of the product in buffered solution (pH = 7.4) containing 5:1 excess of L-cysteine;
living worms moved very slowly;
Rai et al. (reference 20);
worms contracted immediately upon addition of compound, then relaxed over 24–48 hrs;
worms contracted immediately upon addition of compound, then relaxed after < 10 min.
Biology
Inhibition of TGR
The inhibitory activity of all products described in this paper was evaluated against recombinant S. mansoni TGR; the results, expressed as IC50, are reported in Table 1. All the hybrid furoxan products acted as potent TGR inhibitors; their inhibitory potency follows the series 6 > 17 > 5 > 7 ≈ 18 > 24. This sequence should parallel the different capacities of the compounds to release NO under action of the enzyme. This ‘specific’ NO release is dependent upon both binding affinity and appropriately aligned reactivity of each product, and is a measure of each product’s capacity to trigger antiparasitic action following its ability to inactivate TGR. As expected, the des-NO furazan analogues did not display inhibitory activity when tested up to 50 µM concentration.
Action of compounds against ex vivo parasites
Adult S. mansoni worms were cultured in the presence of two different concentrations of furoxan and furazan derivatives: 10 and 50 µM, in DMSO. The mobility and death of the parasites were monitored over 144 hrs. In Table 1, the results are compared with those for PZQ and 2, taken as references. Contraction and paralysis of the worms are the first observable effects of PZQ (Figure 1). After overnight culture with PZQ, these effects are reversible and the worms return to normal length over several days (>2 days). At the concentrations of PZQ tested, worms begin to die 4–5 days after PZQ is removed from the culture media. The furoxan 2 displayed no effect on worm length (i.e., it did not cause contractile paralysis), but quickly killed 100% of worms at both the concentrations tested. This indicates that NO is not involved in worm contraction, but is responsible for the rapid death of the parasites.
Figure 1.

Images of adult Schistosoma mansoni worms 24 hrs after addition of compounds.
Worm morphology was monitored after treatment with hybrid compounds to indirectly assess their PZQ-like activity. Worms were contracted and mobility impaired immediately after exposure to the first group of products 5–10. In the case of furazans 8–10, the subsequent recovery after their removal from the medium was faster than that observed for PZQ, at both concentrations tested. The hybrid furoxans 5–7 retained PZQ’s full worm-contraction ability at the higher concentration tested, but mobility recovered faster at the 10 µM concentration. The hybrid furoxans were more effective worm killers than the furazan analogues (compare 5/8, 6/9, 7/10), in particular at the 50 µM concentration, indicating an involvement of NO in this action, in keeping with what was observed with 2. The furoxans 6 and 7 emerge as the most interesting products, because of their balance between PZQ and NO-dependent activities. Findings concerning TGR IC50 and cysteine-induced NO release suggest that, in the case of compound 7 (high NO release under the action of cysteine, high TGR-inhibitory potency) the worm-killing activity might be induced by a combination of both specific and nonspecific NO production provided that the compound’s pharmacokinetic properties (e.g. worm uptake, metabolic fate, increased efflux) do not limit its access to the TGR target. In the case of 6 (low nonspecific release, high TGR inhibitory potency) specific NO release and TGR inhibition appear to be principally involved. The modestly higher worm-killing activity of 5 compared to 8 is in keeping with the low cysteine-induced NO release of product but not with its high TGR inhibitory potency. This suggests that pharmacokinetic properties of 5 limit its access to this target.
The results obtained with the second group of compounds, 17, 18, 20, 21, 24, and 26, indicate that, although at the higher concentration tested several products induced worm contraction, this effect was of a much shorter duration than that of PZQ (less than 10 minutes to ~24 hrs, depending on the compound), suggesting that these compounds’ PZQ-like activity was limited (Figure 1). This is in line with findings that modification of the PZQ aromatic ring significantly reduces activity.7,10 This might be due to decreased interaction with parasite target protein(s), or to increased efflux of the compounds. Furoxans 18 and 24 have better worm killing activity than the related furazans 21 and 26 suggesting that NO is involved in this action. Since both furoxan derivatives are quite efficient NO-donors under the action of cysteine, and quite potent TGR inhibitors, we can infer that both specific (if no critical structural modifications of the products occur during their random walk to the target) and nonspecific production of NO might be implicated in the killing activity. Furoxan 17 and the related furazan 20 did not kill worms. This inability is surprising in the case of the furoxan derivative since it is a quite potent TGR inhibitor, although it is a weak NO-donor under the action of cysteine. Metabolic fate, increased efflux, and lack of worm uptake could be responsible for its inactivity. In light of these results, the specific NO release following the direct interaction of furoxan derivatives with TGR does not directly correlate with the ex-vivo killing activity due to the complexity of the biological system involved. In conclusion, the only products of a certain interest among those belonging to this group are the hybrids 18 and 24, which retain some PZQ-like activity resulting in worm paralysis and also display NO-dependent worm-killing capacity.
We recently demonstrated that furoxans can inhibit the activity of P-gp, MRP1 and MRP3 transporters in different kinds of cells.25,26 Since multidrug resistance transporters (MDR) are considered potentially attractive targets for the development of new antischistosomal drugs,13 selected members of this new class of PZQ/furoxan hybrid products are worthy of additional investigation, in view of their potential activity against PZQ-resistant schistosomes.
Conclusions
A new class of NO-donor PZQ hybrids was developed by joining NO-donor furoxan moieties to different areas of the PZQ structure. The inhibitory activity of these products, and that of the related des-NO furazan derivatives, was evaluated against recombinant S. mansoni TGR; their antiparasitic action against ex vivo adult S. mansoni worms was likewise evaluated. Products 6, 7, 18, and 24 emerged as potent antischistosomal agents, endowed with both PZQ-like and NO-dependent antiparasitic activity. These compounds are worthy of additional studies in view of their potential activity against PZQ-resistant schistosomes.
Experimental section
Chemistry
1H and 13C NMR spectra were recorded on a Bruker Avance 300 at 300 and 75 MHz, respectively, using SiMe4 as internal standard. Low resolution mass spectra were recorded with a Finnigan-Mat TSQ-700. Melting points were determined with a capillary apparatus (Büchi 540). Flash column chromatography was performed on silica gel (Merck Kieselgel 60, 230–400 mesh ASTM). The progress of the reactions was monitored by thin layer chromatography (TLC) on 5 cm × 20 cm plates with a layer thickness of 0.2 mm. Organic solvents were removed under vacuum at 30 °C. Elemental analyses (C, H, N) of the target compounds were performed by Section de Pharmacie, Service de Microanalyse (Geneva), and the results are within 0.4% of the theoretical values, unless otherwise stated. Target compounds were prepared, as assessed using the aforementioned standard spectroscopic techniques and elemental analyses, in ≥95% purity. Compounds 3;27 4;28 16;29 19;30 2331 and 2532 were obtained as described elsewhere.
Methyl 4-[(4-oxo-1,3,4,6,7,11b-hexahydro-2H-pyrazino[2,1-a]isoquinolin-2-yl)carbonyl]furoxan-3-carboxylate (5)
3-methoxycarbonyl-4-furoxancarboxylic acid (300 mg, 1.59 mmol) was dissolved in anhydrous THF (7 ml) and CH2Cl2 (10 ml) and the solution obtained was cooled in an ice bath. N-hydroxysuccinimide (1.1 eq.) and EDC˙HCl (1.5 eq.) were added to the solution, which was kept under stirring at room temperature for one hour. A solution of 1,2,3,6,7,11b-hexahydro-pyrazino[2,1-a]isoquinolin-4-one (1 eq.) in anhydrous CH2Cl2 (10 ml) was then added and the resulting mixture was kept under stirring at room temperature for 20 minutes. The solvent was removed under reduced pressure, the residue was taken up with 30 ml of CH2Cl2 and washed with water (2 × 20 ml), brine, dried over Na2SO4 and the solvent was evaporated under reduced pressure. The residue was purified by flash chromatography on silica gel (eluent CH2Cl2/EtOAc 90/10) to give the title product as a white solid, yield 42%, mp 150–152 °C dec (MeOH).
1H-NMR (CDCl3): δ 2.78–3.06 (3H, m, H-6, 2 × H-7), 3.16 (0.5H, dd, J = 2.4, 13.2 Hz, H-1), 3.43 (0.5H, dd, J = 3.0, 13.8 Hz, H-1), 3.95 (3H, d, J = 6.0 Hz, -COOCH3), 4.05–4.41 (2H, m, 2 × H-3), 4.76–4.92 (1H, m, H-1), 4.98–5.22 (2H, m, H-6 and H-11b), 7.22–7.34 (4H, m, aromatic protons).
13C-NMR (CDCl3): δ 28.7, 39.0, 46.1, 50.0, 53.9, 55.0, 106.5, 125.3, 127.1, 127.9, 129.6, 131.6, 135.0, 150.3, 155.2, 155.9, 163.1.
MS CI (isobutane) (m/z): 372 [MH+].
Anal. (C17H16N4O6) C, H, N.
4-[(4-oxo-1,3,4,6,7,11b-hexahydro-2H-pyrazino[2,1-a]isoquinolin-2-yl)carbonyl]furoxan-3-carboxamide (6)
A solution of 5 (390 mg, 1.05 mmol) in MeOH saturated with NH3 (15 ml) was kept under stirring at room temperature for 10 minutes. A white solid precipitated and was recovered by filtration. The solid obtained was recrystallized from MeOH, to give the title product, yield 53%, mp 206–208 °C dec.
1H-NMR (DMSO-d6): δ 2.74–2.95 (3H, m, H-6, 2 × H-7), 3.26 (0.5H, dd, J = 2.1, 13.9 Hz, H-1), 3.45 (0.5H, dd, J = 3.0, 13.7 Hz, H-1), 3.96–4.15 (1H, m, H-1), 4.46–4.68 (3H, m, 2 × H-3 and H-6), 4.91–4.99 (1H, m, H-11b), 7.18–7.39 (4H, m, aromatic protons), 7.86 (1H, d, J = 13.5 Hz, –NH2), 8.50 (1H, d, J = 3.9 Hz, –NH2).
13C-NMR (DMSO-d6): δ 28.0, 45.2, 48.6, 53.9, 110.3, 125.6, 126.3, 126.6, 127.1, 128.9, 132.7, 134.9, 151.9, 154.8, 155.7, 163.0.
MS CI (isobutane) (m/z): 358 [MH+].
Anal. (C16H15N5O5) C, H, N.
4-[(4-oxo-1,3,4,6,7,11b-hexahydro-2H-pyrazino[2,1-a]isoquinolin-2-yl)carbonyl]furoxan-3-carbonitrile (7)
A solution of 6 (130 mg, 0.36 mmol) in anhydrous THF (20 ml) was cooled in an ice bath; anhydrous pyridine (4 eq.) and TFAA (4 eq.) were added to the solution. The mixture was kept under stirring at 0°C for 30 minutes, then the solvent was removed under reduced pressure. The residue was taken up with 20 ml of CH2Cl2 and washed with H2SO4 0.5 M (2 × 10 ml), water (2 × 10 ml), brine, dried over Na2SO4, filtered and evaporated under reduced pressure. The yellow solid obtained was purified by flash chromatography on silica gel (eluent CH2Cl2/EtOAc 90/10) to give the title product as white solid yield 83%, mp 183–185 °C dec. (CHCl3/n-Hex).
1H-NMR (CDCl3): δ 2.80–3.02 (3H, m, H-6, 2 × H-7), 3.18 (0.5H, dd, J = 2.7, 13.5 Hz, H-1), 3.47 (0.5H, dd, J = 3.0, 13.7 Hz, H-1), 4.09 (0.5H, d, J = 18.6 Hz, H-1), 4.43 (0.5H, d, J = 18.3 Hz, H-1), 4.82–5.22 (4H, m, 2 × H-3, H-6 and H-11b), 7.22–7.32 (4H, m, aromatic protons).
13C-NMR (CDCl3): δ 28.7, 39.0, 47.0, 50.0, 54.3, 55.7, 104.5, 125.4, 127.2, 128.0, 129.7, 131.3, 135.0, 150.4, 153.4, 163.0.
MS CI (isobutane) (m/z): 340 [MH+].
Anal. (C16H13N5O4) C, H, N.
Methyl 4-[(4-oxo-1,3,4,6,7,11b-hexahydro-2H-pyrazino[2,1-a]isoquinolin-2-yl)carbonyl]furazan-3-carboxylate (8)
A solution of 5 (330 mg, 0.89 mmol) in (CH3O)3P (5 ml) was refluxed for 4 hours. The mixture was then poured into 20 ml of 2.5 M H2SO4 and cooled in an ice bath. A white solid precipitated and was collected by filtration. The filtrate was extracted with CH2Cl2 (2 × 10 ml), then the organic phase was washed with H2SO4 2.5 M (3 × 10 ml), NaHCO3 saturated solution (3 × 10 ml), brine, dried over Na2SO4, filtered and evaporated under reduced pressure. The residue was combined with the filtered-out white solid and crystallized from MeOH/H2O, to give the title product as a white solid, yield 47%, mp 141–142 °C.
1H-NMR (CDCl3): δ 2.78–3.01 (3H, m, H-6, 2 × H-7), 3.16 (0.5H, dd, J = 2.7, 13.4 Hz, H-1), 3.39 (0.5H, dd, J = 3.0, 13.7 Hz, H-1), 4.03 (3H, d, J = 3.9 Hz, -COOCH3), 4.14 (1H, s, H-3), 4.27 (1H, m, H-3), 4.79–5.06 (2H, m, H-1 and H-6), 5.23–5.28 (1H, m, H-11b), 7.21–7.34 (4H, m, aromatic protons).
13C-NMR (CDCl3): δ 28.7, 39.0, 46.2, 50.2, 54.0, 54.5, 55.6, 125.3, 127.5, 129.5, 131.6, 135.0, 147.0, 148.8, 155.7, 157.6, 163.1.
MS CI (isobutane) (m/z): 357 [MH+].
Anal. (C17H16N4O5) C, H, N.
4-[(4-oxo-1,3,4,6,7,11b-hexahydro-2H-pyrazino[2,1-a]isoquinolin-2-yl)carbonyl]furazan-3-carboxamide (9)
A solution of 8 (240 mg, 0.67 mmol) in MeOH saturated with NH3 (9 ml) was stirred at room temperature for 10 minutes. A white solid precipitated and was recovered by filtration. The solid obtained was purified by flash chromatography on silica gel (CH2Cl2/acetone 97/3) to give the product as a white solid, yield 83%, mp 138–139 °C (iPrOH).
1H-NMR (CDCl3): δ 2.79–3.04 (3H, m, H-6, 2 × H-7), 3.15 (0.5H, dd, J = 2.4, 13.4 Hz, H-1), 3.39 (0.5H, dd, J = 2.4, 13.5 Hz, H-1), 4.14 (1H, s, H-3), 4.23–4.29 (1H, m, H-3), 4.74–5.13 (2H, m, H-1 and H-6), 5.22–5.28 (1H, m, H-11b), 6.61 (1H, br. s, - NH2), 6.76 (0.5H, br. s, -NH2), 7.18–7.35 (4H, m, aromatic protons), 7.53 (0.5H, br. s, -NH2).
13C-NMR (CDCl3): δ 28.7, 39.0, 46.3, 49.6, 54.9, 125.4, 127.1, 127.8, 129.5, 131.6, 135.0, 147.8, 148.3, 156.4, 157.0, 163.5.
MS CI (isobutane) (m/z): 342 [MH+].
Anal. calc. for C16H15N5O4˙0.7H2O: C, H, N 19.79%; found: C, H, N 19.19%.
4-[(4-oxo-1,3,4,6,7,11b-hexahydro-2H-pyrazino[2,1-a]isoquinolin-2-yl)carbonyl]furazan-3-carbonitrile (10)
A solution of 9, (170 mg 0.50 mmol) in anhydrous THF (20 ml) was cooled in an ice bath; anhydrous pyridine (4 eq.) and TFAA (4 eq.) were added to the solution. The mixture was stirred at 0°C for 30 minutes, then the solvent was removed under reduced pressure. The residue was taken up with 20 ml of CH2Cl2 and washed with H2SO4 0.5 M (2 × 10 ml), water (2 × 10 ml), brine, dried over Na2SO4, filtered and evaporated under reduced pressure. The resulting yellow solid was purified by flash chromatography on silica gel (eluent CH2Cl2/EtOAc 95/5) and then crystallized from CHCl3/n-hexane to give the product as a white solid, yield 83%, mp 149–150 °C.
1H-NMR (CDCl3): δ 2.80–3.02 (3H, m, H-6, 2 × H-7), 3.19 (0.5H, dd, J = 2.4, 13.2 Hz, H-1), 3.49 (0.5H, dd, J = 2.7, 14.3 Hz, H-1), 4.11 (0.5H, d, J = 18.3 Hz, H-1), 4.42 (0.5H, d, J = 17.7 Hz, H-1), 4.79–5.25 (4H, m, 2 × H-3, H-6 and H-11b), 7.18–7.34 (4H, m, aromatic protons).
13C-NMR (CDCl3): δ 28.7, 39.0, 46.9, 50.3, 54.3, 55.7, 106.2, 125.4, 127.6, 129.7, 131.4, 134.3, 135.0, 149.8, 153.5, 163.0.
MS CI (isobutane) (m/z): 324 [MH+].
Anal.(C16H13N5O3) C, H, N.
1-(2-isocyanoethyl)-4-methoxybenzene (12)
A solution of 2-(4-methoxyphenyl)ethanamine 11 (15 ml, 100 mmol), TEBAC (0.01 eq.) and CHCl3 (1 eq.) in CH2Cl2 (32 ml) was added dropwise over 10 minutes to an aqueous solution of NaOH (8 eq., 32 ml). The mixture was refluxed for 4 hours, and then a mixture of ice and water (50 ml) was added. The organic phase was collected and the aqueous phase was extracted with CH2Cl2 (2 × 50 ml). The organic phases were then washed with H2SO4 0.5 M (3 × 100 ml), water (100 ml), brine, dried over Na2SO4 and evaporated under reduced pressure. The residue was purified by flash chromatography on silica gel (eluent CH2Cl2) to give the product as a yellow oil, yield 57%.
1H-NMR (CDCl3): δ 2.91 (2H, t, J = 6.9 Hz, -CH2CH2-NC), 3.55 (2H, t, J = 6.9 Hz, - CH2CH2-NC), 3.79 (3H, s, -OCH3), 6.87 (2H, d, J = 8.4 Hz, H-2 and H-6 aromatic protons), 7.14 (2H, d, J = 8.4 Hz, H-3 and H-5 aromatic protons).
13C-NMR (CDCl3): δ 34.8, 43.2, 52.3, 114.2, 128.7, 129.4, 129.7, 156.4.
MS CI (isobutane) (m/z): 162 [MH+].
N-(2,2-dimethoxyethyl)-N-(2-oxo-2-(2-(4-methoxy)phenethylamino)-ethyl)cyclohexanecarboxamide (13)
1-(2-isocyanoethyl)-4-methoxybenzene 12 (9.2 g, 0.057 mol) was added portion-wise to a suspension of paraformaldehyde (1 eq.), 2,2-dimethoxyethylamine (1 eq.), cyclohexanecarboxylic acid (1 eq.) in MeOH (60 ml) cooled in an ice bath, and the mixture was stirred at room temperature for 48 hours. The solvent was then evaporated under reduced pressure and the residue was dissolved in diethyl ether (150 ml). The organic phase was washed with H2SO4 0.5 M (50 ml), NaOH 0.5 M (50 ml), water, brine, dried over Na2SO4 and evaporated under reduced pressure. The yellow oil solidifies to give the product as a pale yellow solid pure enough for the next step, yield 89%.
1H-NMR (CDCl3): δ 1.22–1.25 (3H, m, cyclohexane protons), 1.45 (2H, m, cyclohexane protons), 1.60–1.76 (5H, m, cyclohexane protons), 2.24 (0.5H, t, J = 11.1 Hz, cyclohexane protons), 2.59 (0.5H, t, J = 11.4 Hz, cyclohexane protons), 2.70–2.78 (2H, m, -Ph-CH2CH2NH-), 3.35 (3H, s, (CH3O)2CHCH2-), 3.38 (3H, s, (CH3O)2CHCH2-), 3.43 (2H, t, J = 5.1 Hz, (CH3O)2CHCH2-), 3.48–3.55 (2H, m, -Ph-CH2CH2NH-), 3.78 (3H, s, CH3O-Ph), 3.99 (2H, d, J = 5.1 Hz, -COCH2-), 4.40 (0.5H, t, J = 4.8 Hz, (CH3O)2CHCH2-), 4.58 (0.5H, t, J = 4.8 Hz, (CH3O)2CHCH2-), 6.50 (0.5H, br. s, -NH), 6.83 (2H, d, J = 8.4 Hz, H-2 and H-6 aromatic protons), 6.98 (0.5H, br. s, -NH), 7.10 (2H, d, J = 8.1 Hz, H-3 and H-5 aromatic protons).
13C-NMR (CDCl3): δ 25.5, 29.2, 34.7, 40.3, 41.2, 42.8, 50.3, 51.3, 52.3, 52.7, 54.9, 56.4, 102.9, 103.8, 114.0, 129.6, 130.6, 131.3, 158.5, 169.2, 169.6, 178.2.
MS CI (isobutane) (m/z): 377 [MH+].
2-(cyclohexylcarbonyl)-10-methoxy-1,2,3,6,7,11b-hexahydro-4H-pyrazino[2,1-a]isoquinolin-4-one (14)
13 (19.0 g, 0.051 mol) was added portion-wise to methanesulphonic acid (20 eq.) cooled in an ice bath. The mixture was heated to 70°C for 5 hours, then poured into an ice-water mixture. The mixture was alkalinized to pH 8 with a 20% solution of NaOH, then extracted with CH2Cl2 (4 × 50 ml). The organic phase was washed with water (2 × 30 ml), brine, dried over Na2SO4 and evaporated under reduced pressure. The resulting brown oil was purified by flash chromatography on silica gel (eluent CH2Cl2/MeOH 98/2) to give a yellow solid, which was crystallized from EtOAc/n-hexane 1/1 to give the product as a white solid, yield 40%, mp 145–146 °C.
1H-NMR (CDCl3): δ 1.26–1.32 (3H, m, cyclohexane protons), 1.49–1.61 (2H, m, cyclohexane protons), 1.73–1.81 (5H, m, cyclohexane protons), 2.47 (1H, t, J = 11.1 Hz, cyclohexane proton), 2.69–2.91 (4H, m, 2 × H-1 and 2 × H-7), 3.79 (3H, s, - OCH3), 4.08 (1H, d, J = 17.4 Hz, H-3), 4.47 (1H, d, J = 17.4 Hz, H-3), 4.75–4.82 (2H, m, 2 × H-6), 5.14 (1H, dd, J = 13.2 Hz, H-11b), 6.80 (2H, d, J = 9.6 Hz, H-9 and H-11 aromatic protons), 7.09 (1H, d, J = 8.1 Hz, H-8 aromatic proton).
13C-NMR (CDCl3): δ 25.7, 27.9, 29.0, 29.2, 39.4, 40.8, 45.2, 49.0, 55.1, 55.4, 110.1, 114.1, 126.7, 130.3, 133.7, 158.5, 164.4, 174.8.
MS CI (isobutane) (m/z): 343 [MH+].
2-(cyclohexylcarbonyl)-10-hydroxy-1,2,3,6,7,11b-hexahydro-4H-pyrazino[2,1-a]isoquinolin-4-one (15)
BBr3 (1 M solution in CH2Cl2) (3.5 eq.) was added dropwise to a solution of 14 (6.4 g, 0.019 mol) in CH2Cl2 (300 ml) cooled in an ice bath. The mixture was stirred at room temperature for 3 hours, and then cooled to 0°C. An aqueous solution of KHCO3 (10 eq.) was added dropwise to the reaction mixture, and the resulting suspension was stirred for 1 hour. The organic phase was collected and washed with water (100 ml), brine, dried over Na2SO4 and evaporated under reduced pressure. The residue was crystallized from CHCl3/n-hexane to give the product as a white solid, yield 81%, mp 238–239 °C.
1H-NMR (DMSO-d6): δ 1.18–1.39 (5H, m, cyclohexane protons), 1.64–1.72 (5H, m, cyclohexane protons), 2.65–2.86 (5H, m, 1 cyclohexane proton, 2 × H-1 and 2 × H-7), 3.72 (0.5H, d, J = 17.7 Hz, H-3), 4.08 (0.5H, d, J = 17.1 Hz, H-3), 4.39 (1H, s, H-3), 4.45–4.51 (2H, m, 2 × H-6), 4.66–4.78 (1H, m, H-11b), 6.64 (2H, d, J = 9.3 Hz, H-9 and H-11 aromatic protons), 6.99 (1H, d, J = 8.1 Hz, H-8 aromatic proton), 9.38 (1H, br. s, -OH).
13C-NMR (DMSO-d6): δ, 24.9, 25.5, 27.3, 28.9, 30.9, 45.5, 48.1, 48.3, 54.7, 112.1, 114.4, 124.9, 129.8, 134.0, 155.8, 164.6, 173.6.
MS CI (isobutane) (m/z): 329 [MH+].
4-({[2-(cyclohexylcarbonyl)-4-oxo-1,3,4,6,7,11b-hexahydro-2H-pyrazino[2,1-a]isoquinolin-10-yl]oxy}methyl)furoxan-3-carboxamide (17)
NaH (60% suspension in mineral oil, 4 eq.) was added to a suspension of 15 (1.0 g, 3.1 mmol) in anhydrous THF (100 ml) cooled in an ice bath. The mixture was refluxed for 1.5 hours, and then cooled to room temperature. 4-Bromomethylfuroxan-3-carboxamide (16) (1 eq.) was added to the mixture, which was then stirred at 60°C for 2 hours. The mixture was then cooled in an ice bath and water was added. THF was evaporated under reduced pressure and the aqueous phase was extracted with EtOAc (5 × 20 ml). The organic phase was washed with water (20 ml), brine, dried over Na2SO4 and evaporated under reduced pressure. The residue was purified by flash chromatography on silica gel (eluent CH2Cl2/MeOH 98/2) to give a yellow solid which was crystallized from iPrOH, to give the product as a white solid, yield 42%, mp 200–202 °C (dec.).
1H-NMR (DMSO-d6): δ 1.03 (6H, d, J = 6 Hz, 2 × CH3 iPrOH) 1.25–1.31 (5H, m, cyclohexane protons), 1.52–1.80 (5H, m, cyclohexane protons), 2.38–1.61 (1H, m, cyclohexane proton), 2.71–2.89 (4H, m, 2 × H-1 and 2 × H-7), 4.06–4.12 (2H, m, CH iPrOH, H-3), 4.47 (1H, d, J = 17.4 Hz, H-3), 4.75–4.82 (2H, m, 2 × H-6), 5.08 (1H, m, H-11b), 5.44 (2H, s,-CH2O-), 6.4 (1H, br. s, -CONH2), 6.90–6.96 (2H, m, H-9 and H-11 aromatic protons), 7.12 (1H, d, J = 8.4 Hz, H-8 aromatic proton), 7.55 (1H, br. s, -CONH2).
13C-NMR (DMSO-d6): δ 22.7, 25.4, 25.7, 27.95, 28.01, 39.4, 40.8, 45.0, 49.0, 54.9, 61.3, 64.9 110.4, 111.8, 114.9, 126.0, 128.4, 130.4, 134.1, 154.7, 156.6, 164.4, 174.9.
MS CI (isobutane) (m/z): 470 [MH+].
Anal. calc. for C23H27N5O6˙iPrOH: C, H, N.
4-({[2-(cyclohexylcarbonyl)-4-oxo-1,3,4,6,7,11b-hexahydro-2H-pyrazino[2,1-a]isoquinolin-10-yl]oxy}methyl)furoxan-3-carbonitrile (18)
Anhydrous pyridine (4 eq.) and TFAA (4 eq.) were added to a solution of 17 (200 mg, 0.43 mmol) in anhydrous THF (25 ml). The mixture was stirred at room temperature for 45 minutes. The solvent was then evaporated under reduced pressure and the residue was dissolved in CH2Cl2 (50 ml). The organic phase was washed with H2SO4 0.5 M (2 × 30 ml), water, brine, dried over Na2SO4, filtered and evaporated under reduced pressure. The resulting yellow solid was purified by flash chromatography on silica gel (eluent CH2Cl2/EtOAc 95/5) to give the product as a white solid, yield 41%, mp 95–97 °C (iPr2O/EtOAc).
1H-NMR (CDCl3): δ 1.26–1.31 (5H, m, cyclohexane protons), 1.48–1.81 (5H, m, cyclohexane protons), 2.46 (1H, m, cyclohexane proton), 2.73–2.98 (4H, m, 2 × H-1 and 2 × H-7), 4.10 (1H, d, J = 17.7 Hz, H-3), 4.46 (1H, d, J = 17.7 Hz, H-3), 4.76–4.83 (2H, m, 2 × H-6), 5.04 (1H, dd, J = 2.4, 10.8 Hz, H-11b), 5.24 (2H, s, -CH2O-), 6.91–6.94 (2H, m, H-9 and H-11 aromatic protons), 7.16 (1H, d, J = 8.3 Hz, H-8 aromatic proton).
13C-NMR (CDCl3): δ 25.7, 27.9, 29.0, 29.2, 39.3, 40.8, 44.8, 49.1, 53.5, 60.9, 96.0, 104.9, 109.7, 114.9, 129.3, 130.8, 134.5, 153.4, 155.8, 164.4, 174.8.
MS CI (isobutane) (m/z): 452 [MH+].
Anal. (C23H25N5O5˙0.5H2O) C, H, N.
4-({[2-(cyclohexylcarbonyl)-4-oxo-1,3,4,6,7,11b-hexahydro-2H-pyrazino[2,1-a]isoquinolin-10-yl]oxy}methyl)furazan-3-carboxamide (20)
NaH (60% suspension in mineral oil, 4 eq.) was added to a suspension of 15 (1.0 g, 3.1 mmol) in anhydrous THF (100 ml) cooled in an ice bath. The mixture was refluxed for 1.5 hours, and then cooled to room temperature. 4-Bromomethylfurazan-3-carboxamide (19) (1 eq.) was added to the mixture which was stirred at 60°C for 2 hours. The mixture was then cooled in an ice bath and water was added. THF was evaporated under reduced pressure and the aqueous phase was extracted with EtOAc (4 × 20 ml). The organic phase was washed with water (2 × 20 ml), brine, dried over Na2SO4 and evaporated under reduced pressure. The residue was purified by flash chromatography on silica gel (eluent CH2Cl2/MeOH 98/2) to give a white solid which was crystallized from EtOAc, yield 42%, mp 203–204 °C.
1H-NMR (DMF-d7): δ 1.11–1.43 (5H, m, cyclohexane protons), 1.64–1.79 (5H, m, cyclohexane protons), 2.74–2.92 (5H, m, 1 cyclohexane proton, 2 × H-1 and 2 × H-7), 4.16–4.21 (1H, m, H-3), 4.47–4.55 (1H, m, H-3), 4.71–4.92 (2H, m, 2 × H-6), 4.96–5.04 (1H, m, H-11b), 5.66 (2H, s, -CH2O-), 7.03 (2H, d, J= 7.8 Hz, H-9 and H-11 aromatic protons), 7.21 (1H, d, J= 8.4 Hz, H-8 aromatic proton), 8.30 (1H, br. s, -CONH2), 8.67 (1H, br. s, -CONH2).
13C-NMR (DMF-d7): δ 25.9, 26.5, 28.3, 30.0, 39.3, 39.8, 46.5, 49.0, 56.0, 60.4, 113.1, 114.5, 129.2, 130.8, 149.7, 153.2, 157.4, 159.2, 162.9, 165.7, 174.9.
MS CI (isobutane) (m/z): 454 [MH+].
Anal. (C23H27N5O5) C, H, N.
4-({[2-(cyclohexylcarbonyl)-4-oxo-1,3,4,6,7,11b-hexahydro-2H-pyrazino[2,1-a]isoquinolin-10-yl]oxy}methyl)furazan-3-carbonitrile (21)
Anhydrous pyridine (4 eq.) and TFAA(4 eq.) were added to a solution of 20 (550 mg, 1.21 mmol) in anhydrous THF (70 ml). The mixture was stirred at room temperature for 45 minutes. The solvent was then evaporated under reduced pressure, and the residue was dissolved in CH2Cl2 (50 ml). The organic phase was washed with H2SO4 0.5 M (2 × 30 ml), water, brine, dried over Na2SO4, filtered and evaporated under reduced pressure. The resulting yellow solid was purified by flash chromatography on silica gel (eluent CH2Cl2/MeOH 98/2), to give the product as a white solid, yield 28%, mp 69–70 °C (iPr2O/EtOAc).
1H-NMR (CDCl3): δ 1.27–1.57 (5H, m, cyclohexane protons), 1.72–1.81 (5H, m, cyclohexane protons), 2.46 (1H, m, cyclohexane proton), 2.73–2.96 (4H, m, 2 × H-1 and 2 × H-7), 4.10 (1H, d, J = 17.4 Hz, H-3), 4.46 (1H, d, J = 17.7 Hz, H-3), 4.76–4.83 (2H, m, 2 × H-6), 5.04–5.09 (1H, m, H-11b), 5.40 (2H, s, -CH2O-), 6.92 (2H, d, J = 6.6 Hz, H-9 and H-11 aromatic protons), 7.16 (1H, d, J = 8.7 Hz, H-8 aromatic proton).
13C-NMR (CDCl3): δ, 21.0, 25.7, 27.9, 29.1, 39.3, 40.8, 44.9, 49.0, 55.0, 60.4, 111.3, 114.8, 129.2, 130.8, 132.5, 134.4, 153.0, 155.9, 164.4, 171.2, 174.8.
MS CI (isobutane) (m/z): 436 [MH+].
Anal. (C23H25N5O4 H2O) C, H, N.
2-(cyclohexylcarbonyl)-10-(3-hydroxypropoxy)-1,2,3,6,7,11b-hexahydro-4H-pyrazino[2,1-a]isoquinolin-4-one (22)
3-Bromopropan-1-ol (1 eq.) and K2CO3 (2 eq.) were added to a suspension of 15 (1.0 g, 3.2 mmol) in CH3CN. The mixture was refluxed for 24 hours, and then cooled to room temperature. The solvent was evaporated under reduced pressure; the residue was dissolved in EtOAc (50 ml) and washed with 0.5 M NaOH solution (4 × 30 ml), water (30 ml), brine, dried over Na2SO4 and evaporated under reduced pressure.
The yellow solid was purified by flash chromatography on silica gel (eluent CH2Cl2/MeOH 98/2) to give the product as a white solid, yield 66%, mp 187–188 °C (MeOH/H2O).
1H-NMR (CDCl3): δ 1.18–1.39 (3H, m, cyclohexane protons), 1.52–1.60 (2H, m, cyclohexane protons), 1.73–1.80 (5H, m, cyclohexane protons), 2.02–2.17 (2H, m, cyclohexane proton and H-1), 2.43–2.50 (1H, m, H-1), 2.69–2.76 (2H, m, 2 × H-7), 2.81–2.95 (2H, m, HO-CH2CH2CH2O-), 3.84–3.87 (2H, m, 2 × H-3), 4.05–4.25 (3H, m, -OH, HO-CH2CH2CH2O-), 4.28–4.36 (1H, m, H-6), 4.43–4.49 (1H, m, H-6), 4.73–4.82 (2H, m, HO-CH2CH2CH2O-), 5.06–5.10 (1H, m, H-11b), 6.81 (2H, d, J = 8.1 Hz, H-9 and H-11 aromatic protons), 7.08 (1H, d, J = 7.8 Hz, H-8 aromatic protons).
13C-NMR (CDCl3): δ 25.7, 28.6, 29.0, 29.5, 31.0, 39.4, 40.8, 45.2, 49.0, 55.1, 60.0, 65.8, 110.8, 114.6, 126.9, 130.2, 133.7, 157.7, 164.4, 174.9.
MS CI (isobutane) (m/z): 387 [MH+].
2-(cyclohexylcarbonyl)-10-(3-{[3-(phenylsulfonyl)furoxan-4-yl]oxy}propoxy)-1,2,3,6,7,11b-hexahydro-4H-pyrazino[2,1-a]isoquinolin-4-one (24)
DBU (2 eq.) was added to a solution of 23 (1.2 eq.) in CH2Cl2 (20 ml). The solution was cooled in an ice bath and a solution of 22 (140 mg, 0.36 mmol) in CH2Cl2 (10 ml) was added. The mixture was stirred at room temperature for 1 hour, washed with a 0.5 M H2SO4 solution (2 × 20 ml), water (2 × 20 ml), brine, dried over Na2SO4 and evaporated under reduced pressure. The resulting yellow oil was partially purified by flash chromatography on silica gel (eluent CH2Cl2/MeOH 98/2) and then purified by HPLC (eluent CH3CN/H2O 0.1% CF3COOH 70/30), to give the product as a white solid, yield 14%, mp 82–84 °C (iPrOH).
1H-NMR (CDCl3): δ 1.26–1.32 (3H, m, cyclohexane protons), 1.49–1.57 (2H, m, cyclohexane protons), 1.72–1,80 (5H, m, cyclohexane protons), 2.34–2.47 (3H, m, cyclohexane proton and 2 × H-1), 2.71–2.92 (4H, m, 2 × H-7 and HO-CH2CH2CH2O-), 4.06–4.50 (4H, m, 2 × H-3 and HO-CH2CH2CH2O-), 4.62–4.66 (2H, m, 2 × H-6), 4.75–4.83 (2H, m, HO-CH2CH2CH2O-), 5.08–5.17 (1H, m, H-11b), 6.83 (2H, d, J = 7.5 Hz, H-9 and H-11 aromatic protons), 7.11 (1H, d, J = 7.8 Hz, H-8 aromatic protons), 7.54–7.59 (2H, m, aromatic protons), 7.74 (1H, t, J = 7.5 Hz, aromatic protons), 8.02 (2H, d, J = 7.5 Hz, aromatic protons).
13C-NMR (CDCl3): δ 25.7, 27.9, 28.5, 29.0, 29.2, 39.4, 40.8, 45.2, 49.0, 55.0, 63.7, 68.1, 110.5, 110.8, 111.2, 114.3, 127.2, 128.5, 129.6, 130.4, 133.9, 135.6, 138.0, 157.5, 158.9, 164.4, 174.8, 177.2.
MS CI (isobutane) (m/z): 611 [MH+].
Anal. (C30H34N4O8S): C, H, N.
2-(cyclohexylcarbonyl)-10-(3-{[4-(phenylsulfonyl)furazan-3-yl]oxy}propoxy)-1,2,3,6,7,11b-hexahydro-4H-pyrazino[2,1-a]isoquinolin-4-one (26)
DBU (2 eq.) was added to a solution of 25 (1.2 eq.) in CH2Cl2 (20 ml). The solution was cooled in an ice bath and a solution of 22 (150 mg, 0.39 mmol) in CH2Cl2 (10 ml) was added to it. The mixture was stirred at room temperature for 1 hour, washed with H2SO4 0.5 M solution (2 × 20 ml), water (2 × 20 ml), brine, dried over Na2SO4 and evaporated under reduced pressure. The resulting yellow oil was partially purified by flash chromatography on silica gel (eluent CH2Cl2/MeOH 98/2) and then purified by HPLC (eluent CH3CN/H2O 0.1% CF3COOH 75/25), to give the product as a white solid, yield 35%, mp 70–71 °C (iPrOH).
1H-NMR (CDCl3): δ 1.28–1.31 (3H, m, cyclohexane protons), 1.49–1.57 (2H, m, cyclohexane protons), 1.72–1,80 (5H, m, cyclohexane protons), 2.20–2.34 (3H, m, cyclohexane proton and 2 × H-1), 2.71–2.95 (4H, m, 2 × H-7 and HO-CH2CH2CH2O-), 4.06–4.50 (4H, m, 2 × H-3 and HO-CH2CH2CH2O-), 4.57–4.61 (2H, m, 2 × H-6), 4.74–4.83 (2H, m, HO-CH2CH2CH2O-), 5.09–5.14 (1H, m, H-11b), 6.73–6.81 (2H, m, H-9 and H-11 aromatic protons), 7.10 (1H, d, J = 8.7 Hz, H-8 aromatic protons), 7.56–7.66 (2H, m, aromatic protons), 7.73 (1H, t, J = 7.2 Hz, aromatic protons), 8.07 (2H, d, J = 7.8 Hz, aromatic protons).
13C-NMR (CDCl3): δ 25.7, 28.0, 28.6, 29.2, 29.6, 39.4, 40.8, 45.2, 49.0, 55.0, 63.7, 70.5, 111.1, 114.3, 127.2, 129.0, 129.6, 129.7, 130.4, 133.8, 135.4, 137.9, 148.8, 157.4, 161.2, 164.1, 164.4, 174.8.
MS CI (isobutane) (m/z): 595 [MH+].
Anal. (C30H34N4O7S): C, H, N.
Biology
Female Swiss-Webster mice infected with S. mansoni cercariae by percutaneous tail exposure were obtained from the Biomedical Research Institute, Rockville, MD. Parasites were obtained from mice 7 weeks after infection by perfusion with RPMI 1640 medium (Cellgro, Fisher).33 Worms were washed thoroughly several times in RPMI 1640 in a laminar flow hood; they were then washed several times in complete RPMI 1640 (cRPMI) containing 25 mM HEPES (Cellgro, Fisher), 150 units/mL penicillin, 125 µg/mL streptomycin, and 10% fetal bovine serum (Atlanta Biologicals). Worms were then distributed into 6-well tissue culture plates with 15–20 worms per well and cultured overnight in 5 mL cRPMI in 5% CO2 at 37°C. The following day, media were removed from each well and replaced with 5 mL fresh cRPMI. Test compounds were dissolved in DMSO at 10 mM and added to the wells at the indicated concentrations. The same volume of DMSO was added to each well. Negative control worms were treated with an equal volume of DMSO alone. After overnight culture, the media with compounds were removed and replaced with fresh media alone. Worm mobility and survival were observed under a stereomicroscope (Zeiss Stemi 2000-C) for 10 seconds per worm at the indicated time points. Media were removed and replaced with fresh media every 48 hours. This study was approved by the Institutional Animal Care and Use Committee at Rush University Medical Center (IACUC number 08–058; DHHS animal welfare assurance number A3120-01).
TGR preparation and IC50 determination were performed as described.34 Briefly, assays were run in 96-well plates in 200 µL with 20 nM enzyme and 100 µM NADPH in 0.1 M potassium phosphate (pH 7.4), 10 mM EDTA. Test compounds were dissolved in DMSO. The enzyme was incubated for 10 minutes at room temperature with NADPH and compounds at 50 µM, 20 µM, 10 µM, 1 µM, 500 nM, 100 nM, 10 nM, or 5 nM before the addition of 3 mM 5, 5'-dithio-bis(2-nitrobenzoic acid). Enzyme activity was monitored by observing the change in A412 due to the formation of 5-thio-2-nitrobenzoic acid during the first 3 minutes. Residual TGR activity was compared to controls, incubated with equal volumes of DMSO. All assays were done in triplicate.
Highlights.
New NO-donor derivatives of praziquantel (PZQ) have been synthesized and studied.
The compounds are potential therapeutic tools against schistosomiasis.
Several compounds showed interesting TGR inhibiting and anti-parasitic activities.
The compounds could be useful in case of infection by PZQ-resistant schistosomes.
Acknowledgements
We would like to thank Dr. Matthew S. Tucker, Biomedical Research Institute, Rockville, MD, for parasite material through NIH-NIAID contract HHSN272201000005I. This work was supported in part by the NIH/National Institute of Allergy and Infectious Diseases (NIAID) grant R01AI065622 (DLW) and by MIUR grant (PRIN 2009 prot. 20097FJHPZ_002).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Gryseels B. Schistosomiasis. Infect. Dis. Clin. of N. Am. 2012;26:383–397. doi: 10.1016/j.idc.2012.03.004. [DOI] [PubMed] [Google Scholar]
- 2.Steinmann P, Keiser J, Bos R, Tanner M, Utzinger J. Schistosomiasis and water resources development: systematic review, meta-analysis, and estimates of people at risk. Lancet Infect. Dis. 2006;6:411–425. doi: 10.1016/S1473-3099(06)70521-7. [DOI] [PubMed] [Google Scholar]
- 3.Van der Werf MJ, de Vlas SJ, Brooker S, Looman CW, Nagelkerke NJ, Habbema JD, Engels D. Quantification of clinical morbidity associated with schistosoma infection in sub-Saharan Africa. Acta Trop. 2003;86:125–139. doi: 10.1016/s0001-706x(03)00029-9. [DOI] [PubMed] [Google Scholar]
- 4.Thetiot-Laurent SAL, Boissier J, Robert A, Meunier B. Schistosomiasis Chemotherapy. Angewandte Chemie-International Edition. 2013;53:17936–17956. doi: 10.1002/anie.201208390. [DOI] [PubMed] [Google Scholar]
- 5.Gonnert R, Andrews P. Praziquantel, a new broad-spectrum antischistosomal agent. Z. Parasitenk. 1977;52:129–150. doi: 10.1007/BF00389899. [DOI] [PubMed] [Google Scholar]
- 6.Liu Y, Wang X, Wang JK, Ching CB. Investigation of the phase diagrams of chiral praziquantel. Chirality. 2006;18:259–264. doi: 10.1002/chir.20251. [DOI] [PubMed] [Google Scholar]
- 7. Doemling A, Khoury K. Praziquantel and Schistosomiasis. ChemMedChem. 2010;5:1420–1434. doi: 10.1002/cmdc.201000202. and references therein.
- 8.Fenwick A, Webster JP. Schistosomiasis: challenges for control, treatment and drug resistance. Curr. Opin. Infect. Dis. 2006;19:577–582. doi: 10.1097/01.qco.0000247591.13671.6a. [DOI] [PubMed] [Google Scholar]
- 9.Aragon AD, Imani RA, Blackburn VR, Cupit PM, Melman SD, Goronga T, Webb T, Loker ES, Cunningham C. Towards an understanding of the mechanism of action of praziquantel. Mol. Biochem. Parasit. 2009;164:57–65. doi: 10.1016/j.molbiopara.2008.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sadhu PS, Kumar SN, Chandrasekharam M, Pica-Mattoccia L, Cioli D, Rao VJ. Synthesis of new praziquantel analogues: Potential candidates for the treatment of schistosomiasis. Bioorg. Med. Chem. Lett. 2012;22:1103–1106. doi: 10.1016/j.bmcl.2011.11.108. [DOI] [PubMed] [Google Scholar]
- 11.van den Enden E. Pharmacotherapy of helminth infection. Expert Opinion on Pharmacotherapy. 2009;10:435–451. doi: 10.1517/14656560902722463. [DOI] [PubMed] [Google Scholar]
- 12.Wang W, Wang L, Liang YS. Susceptibility or Resistance of Praziquantel in Human Schistosomiasis: A Review. Parasitol. Res. 2011;111:1871–1877. doi: 10.1007/s00436-012-3151-z. [DOI] [PubMed] [Google Scholar]
- 13.Kasinathan RS, Morgan WM, Greenberg RM. Schistosoma mansoni express higher levels of multidrug resistance-associated protein 1 (SmMRP1) juvenile worms and in response to praziquantel. Molecular & Biochemical Parasitology. 2010;173:25–31. doi: 10.1016/j.molbiopara.2010.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sayed AA, Simeonov A, Thomas CJ, Inglese J, Austin CP, Williams DL. Identification of oxadiazoles as new drug leads for the control of schistosomiasis. Nat. Med. 2008;14:407–412. doi: 10.1038/nm1737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rai G, Thomas CJ, Leister W, Maloney DJ. Synthesis of oxadiazole-2-oxide analogues as potential antischistosomal agents. Tetrahedron Lett. 2009;50:1710–1713. doi: 10.1016/j.tetlet.2009.01.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gasco A, Boulton AJ. Furoxans and Benzofuroxans. Adv. Heterocycl. Chem. 1981;29:251. [Google Scholar]
- 17.Feelisch M, Schonafinger K, Noack E. Thiol-Mediated Generation of Nitric-Oxide Accounts For The Vasodilator Action of Furoxans. Biochem. Pharmacol. 1992;44:1149–1157. doi: 10.1016/0006-2952(92)90379-w. [DOI] [PubMed] [Google Scholar]
- 18.Gasco A, Schoenafinger K. The NO-releasing Heterocycles. In: Wang PG, Cai TB, Taniguchi N, editors. Nitric Oxide Donors. WILEY-VCH Verlag GmbH & Co KGaA Weinheim; 2005. pp. 131–175. [Google Scholar]
- 19.Alger HM, Sayed AA, Stadecker MS, Williams DL. Molecular and enzimatic characterization of Schistosoma mansoni thioredoxin. Int. J. Parasitol. 2002;32:1285–1292. doi: 10.1016/s0020-7519(02)00108-x. [DOI] [PubMed] [Google Scholar]
- 20.Rai G, Sayed AA, Lea WA, Luecke HF, Chakrapani H, Prast-Nielsen S, Jadhav A, Leister W, Shen M, Inglese J, Austin CP, Keefer L, Arner ESJ, Simeonov A, Maloney DJ, Williams DL, Thomas CJ. Structure Mechanism Insights and the Role of Nitric Oxide Donation Guide the Development of Oxadiazole-2-Oxides as Therapeutic Agents against Schistosomiasis. J. Med. Chem. 2009;52:6474–6483. doi: 10.1021/jm901021k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.To be submitted for publication.
- 22.a) Fruttero R, Boschi D, Di Stilo A, Gasco A. The Furoxan System as a Useful Tool for Balancing "Hybrids" with Mixed alpha-1-Antagonist and NO-737 Like Vasodilator Activities". J. Med. Chem. 1995;38:4944. doi: 10.1021/jm00025a012. [DOI] [PubMed] [Google Scholar]; b) Buonsanti MF, Bertinaria M, Di Stilo A, Cena C, Fruttero R, Gasco A. Nitric Oxide Donor beta(2)-Agonists: Furoxan Derivatives Containing the Fenoterol Moiety and Related Furazans. J. Med. Chem. 2007;50:5003–5011. doi: 10.1021/jm0704595. [DOI] [PubMed] [Google Scholar]; c) Dunlap T, Abdul-Hay SO, Chandrasena REP, Hagos GK, Sinha V, Wang Z, Wang H, Thatcher GRJ. Nitrates and NO-NSAIDs in cancer chemoprevention and therapy: In vitro evidence querying the NO donor functionality. Nitric Oxide-Biology and Chemistry. 2008;19:115–124. doi: 10.1016/j.niox.2008.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cao H, Liu H, Doemling A. Efficient Multicomponent Reaction Synthesis of the Schistosomiasis Drug Praziquantel. Chem. Eur. J. 2010;16:12296–12298. doi: 10.1002/chem.201002046. [DOI] [PubMed] [Google Scholar]
- 24.Colasanti M, Gradoni L, Mattu M, Persichini T, Salvati L, Venturini G, Ascenzi P. Molecular bases for the anti-parasitic effect of NO (Review) International Journal of Molecular Medicine. 2002;9:131–134. [PubMed] [Google Scholar]
- 25.Fruttero R, Crosetti M, Chegaev K, Guglielmo S, Gasco A, Berardi F, Niso M, Perrone R, Panaro MA, Colabufo NA. Phenylsulfonylfuroxans as modulators of multidrug-resistance associated protein-1 and P-glycoprotein. J. Med. Chem. 2010;53:5467–5475. doi: 10.1021/jm100066y. [DOI] [PubMed] [Google Scholar]
- 26.Chegaev K, Riganti C, Lazzarato L, Rolando B, Guglielmo S, Campia I, Fruttero R, Bosia A, Gasco A. Nitric Oxide Donor Doxorubicins Accumulate into Doxorubicin-Resistant Human Colon Cancer Cells Inducing Cytotoxicity. Medicinal Chemistry Letters. 2011;2:494–497. doi: 10.1021/ml100302t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kim JH, Lee YS, Park H, Kim CS. Formation of Pyrazinoisoquinoline Ring System by the Tandem Amidoalkylation and N-Acyliminium ion Cyclization: An Efficient Synthesis of Praziquantel. Tetrahedron. 1998;54:7395–7400. [Google Scholar]
- 28.Grundmann C, Nickel GW, Bansal RK. Das Tetramere der Knallsaure (Isocyanilsaure) und seine Derivate (Nitrile oxides. XVIII. Tetramer of fulminic acid (isocyanilic acid) and its derivatives) Liebigs Ann. Chem. 1976:1029–1050. [Google Scholar]
- 29.Di Stilo A, Visentin S, Cena C, Gasco AM, Ermondi G, Gasco A. New 1,4-Dihydropyridines Conjugated to Furoxanyl Moieties, Endowed with Both Nitric Oxide-like and Calcium Channel Antagonist Vasodilator Activities. J. Med. Chem. 1998;41:5393–5401. doi: 10.1021/jm9803267. [DOI] [PubMed] [Google Scholar]
- 30.Bertinaria M, Galli U, Sorba G, Fruttero R, Gasco A, Brenciaglia MI, Scaltrito MM, Dubini F. Synthesis and anti-Helicobacter pylori properties of NO-donor/metronidazole hybrids and related compounds. Drug. Dev. Res. 2003;60:225–239. [Google Scholar]
- 31.Farrar WV. The 3,4-bisarenesulphonylfuroxans. J. Chem. Soc. 1964:904–906. [Google Scholar]
- 32.Boschi D, Di Stilo A, Cena C, Lolli M, Fruttero R, Gasco A. Studies on agents with mixed NO-dependent vasodilating and beta-blocking activities. Pharm. Res. 1997;14:1750–1758. doi: 10.1023/a:1012136030849. [DOI] [PubMed] [Google Scholar]
- 33.Lewis F. Schistosomiasis. In: Coligan JE, Kruisbeek AM, Margulies DH, Shevach EM, Strober W, editors. Current Protocols in Immunology. New York: John Wiley & Sons; 1998. pp. 19.11.11–19.11.28. Edition edn. [Google Scholar]
- 34.Kuntz AN, Davioud-Charvet E, Sayed AA, Califf LL, Dessolin J, Arnér ESJ, Williams DL. Thioredoxin glutathione reductase from Schistosoma mansoni: an essential parasite enzyme and a key drug target. PLoS Med. 2007;4:1071–1086. doi: 10.1371/journal.pmed.0040206. [DOI] [PMC free article] [PubMed] [Google Scholar]