

Abstract
Introduction
Heat shock protein 90 (HSP90) serves as a critical facilitator for oncogene addiction. There has been augmenting enthusiasm in pursuing HSP90 as an anticancer strategy. In fact, since the initial serendipitous discovery that geldanamycin (GM) inhibits HSP90, the field has rapidly moved from proof-of-concept clinical studies with GM derivatives to novel second-generation inhibitors.
Areas covered
The authors highlight the current status of the second-generation HSP90 inhibitors in clinical development. Herein, the authors note the lessons learned from the completed clinical trials of first- and second-generation inhibitors and describe various assays attempting to serve for a more rational implementation of these agents to cancer treatment. Finally, the authors discuss the future perspectives for this promising class of agents.
Expert opinion
The knowledge gained thus far provides perhaps only a glimpse at the potential of HSP90 for which there is still much work to be done. Lessons from the clinical trials suggest that HSP90 therapy would advance at a faster pace if patient selection and tumor pharmacokinetics of these drugs were better understood and applied to their clinical development. It is also evident that combining HSP90 inhibitors with other potent anticancer therapies holds great promise not only due to synergistic antitumor activity but also due to the potential of prolonging or preventing the development of drug resistance.
Keywords: cancer, clinical development, heat shock protein 90 inhibitors, patient selection
1. Introduction
Heat shock protein 90 (HSP90) is an ATP-dependent molecular chaperone that aids its ‘client proteins’ to fold properly. To achieve its role, HSP90 uses a complex cycle regulated by binding and hydrolysis of ATP and its various co-chaperones (HSP70, Aha1, p23, HSP-organizing protein (HOP), Cdc37). Inhibition of the HSP90 chaperone cycle leads to destabilization of these client proteins, their ubiquitination and ensuing degradation by the proteasome [1-4]. The acquired knowledge over the course of years that many of HSP90's clients are bona fide oncoproteins [e.g., human epidermal growth factor receptor (HER2), EGFR, CDK4, serine/threonine-protein kinase C-Raf (CRAF), serine/threonine-protein kinase B-Raf (BRAF), also known as Protein Kinase B (PKB) (AKT), mesenchymal epithelial transition factor (MET), BCR-ABL] has propelled this chaperone protein as a promising target for the treatment of cancers [1-4]. Importantly, HSP90 inhibition leads to concurrent effects on many oncogenic proteins and pathways, counteracting the numerous pathological traits displayed by cancer cells [2]. Numerous preclinical studies attest to the potential of HSP90 inhibition to result in tumor growth inhibition, reduction in metastatic potential and in sensitization of tumors to the effect of other therapies. There are several reviews that speak to these effects and we direct the readers to them for more information (Figure 1) [1-4].
Figure 1. The chaperone HSP90 has received significant attention in cancer because the many client proteins it regulates are involved in numerous processes that are dysregulated in cancer.

Inhibition of HSP90 function by small molecules results in client protein inactivation leading to tumor growth inhibition, apoptosis and reduction of its metastatic potential. HSP90: Heat shock protein 90.
The path to HSP90 inhibition was initially paved by two natural products, geldanamycin (GM) (Figure 2) [5] and radicicol (RD) (Figure 3) [6]. Both GM and RD were found to inhibit HSP90 by competing with ATP for binding to its N-terminal regulatory pocket. Unfortunately, these two compounds were precluded from reaching the clinic given their poor in vivo stability and toxicities stemming from their reactive chemical structures. Nonetheless, these pathfinder molecules served as tools for better understanding the biology of HSP90 in tumors, and ultimately sustained the process of bridging the gap between the HSP90 biology and subsequent call for HSP90 drugs. They also provided valuable pharmacophores for next-generation inhibitors, and as we shall see, several HSP90 clinical agents have incorporated in their structures the benzoquinone found in GM (Figure 2) or the resorcinol found in RD (Figure 3).
Figure 2. Chemical structures of ansamycin-based HSP90 inhibitors: GM and its derivatives 17-AAG, 17-DMAG and IPI-504 (benzoquinone moiety shown in blue and the methoxy group at C17 is shown in red).
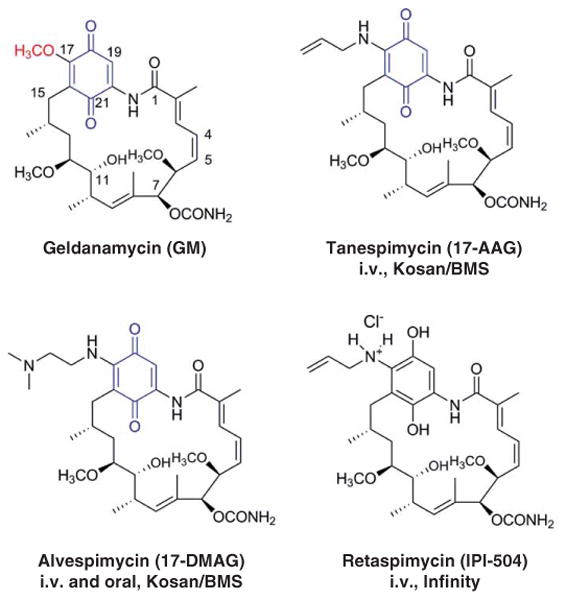
17-DMAG: 17-desmethoxy-17-N,N-dimethylaminoethylaminogeldanamycin; GM: Geldanamycin; i.v.: Intravenous.
Figure 3. Chemical structures of resorcinol-based HSP90 inhibitors: RD and the resorcinol incorporating NVP-AUY922, AT13387, Ganetespib and KW2478 (in blue is shown the resorcinol moiety).
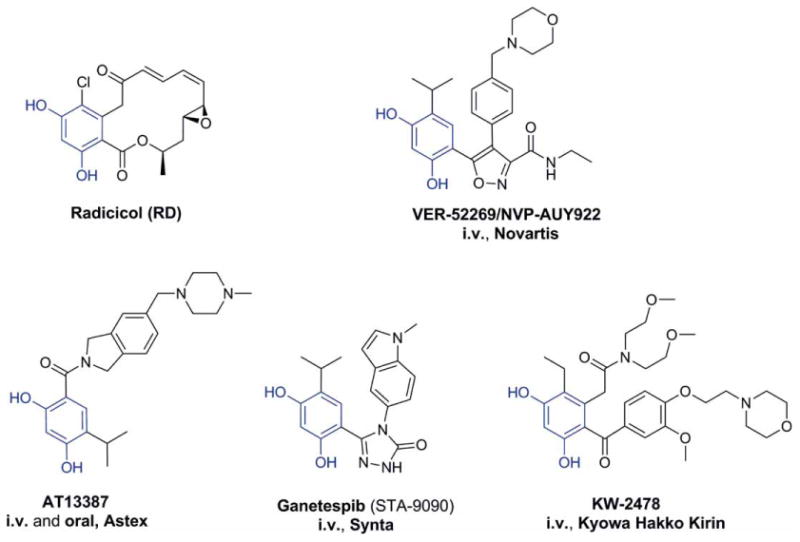
HSP90: Heat shock protein 90; i.v.: Intravenous; RD: Radicicol.
2. First-generation HSP90 inhibitors in clinic – lessons learned
Medicinal chemistry provided the path to first-generation HSP90 inhibitors with clinical potential. Replacement of the non-essential C-17 methoxy group of GM via substitution with various amines provided many semisynthetic derivatives, and among these was 17-AAG (Figure 2; 17-allyl-17-desmethoxygeldanamycin – tanespimycin), the first HSP90 inhibitor to have entered clinical trials (Table 1). 17-AAG retained the important biological features of GM but had an improved toxicity profile [7]. It has been explored in various dosing schedules and formulations as a single agent therapy [8-11]. The most promising clinical activity observed in these diverse Phase I trials was stable disease (SD). In Phase II trials, there were no objective responses noted with single agent tanespimycin in prostate cancer, melanoma or renal cell cancer, despite the presence of known target client proteins in these malignancies [12-15]. The lack of efficacy was attributed to multiple factors, including suboptimal doses and scheduling of tanespimycin in an attempt to avoid treatment-related toxicities, formulation issues and poor patient enrichment for those who might best benefit from therapy based on preclinical experience (e.g., HER2-amplified breast cancer, because HER2 is a sensitive HSP90 onco-client, Figure 1) [1-4].
Table 1. Summary of clinical data – first-generation HSP90 inhibitors.
| HSP90 inhibitor | Clinical stage | Combination | Disease | Response | Ref. |
|---|---|---|---|---|---|
| Tanespimycin | Phase I | No | Advanced solid tumors | SD in metastatic melanoma | [8] |
| Phase II | No | Prostate, papillary and clear cell RCC, melanoma V600E harboring BRAF mutation | SD in melanoma | [13,14] | |
| Phase I/II | Bortezomib | Naïve, pretreated, refractory MM | Objective responses | [17] | |
| Phase II | Trastuzumab | HER2 BC progressing on trastuzumab | Objective responses | [16] | |
| Alvespimycin | Phase I | No | Solid tumors, AML | Objective responses in CRPC, AML and metastatic melanoma and SD in peritoneal mesothelioma, head and neck cancer, renal cancer, chondrosarcoma | [23,24] |
| Phase I | Trastuzumab | HER2 BC progressing on trastuzumab | Objective responses and SD | [25] | |
| Retaspimycin | Phase I | No | MM | SD | [27] |
| Phase II | No | CRPC | No response | [28] | |
| Phase II | Trastuzumab | HER2 BC progressing on trastuzumab | SD | [34] | |
| Phase I/II | No | NSCLC progressing on EGFR inhibitor | Objective responses in patients with ALK rearrangement, EGFR and KRAS wild-type and SD in patients ALK rearrangement | [29,32] | |
| Phase Ib | Docetaxel | NSCLC progressing on chemotherapy | Objective responses in patients with squamous histology, KRAS wild type and smoking history | [34] | |
| Phase III | No | GIST progressing after TKI | No response | [33] |
ALK: Anaplastic lymphoma kinase; AML: Acute myeloid leukemia; BC: Breast cancer; CRPC: Castrate-resistant prostate cancer; GIST: Gastrointestinal stromal tumor; KRAS: Kirsten rat sarcoma viral oncogene homolog; MM: Multiple myeloma; NSCLC: Non-small cell lung cancer; RCC: Renal cell carcinoma; SD: Stable disease; TKI: Tyrosine kinase inhibitor.
On the contrary, activity has been reported when tanespimycin was combined with other cytotoxic or biologic agents (Table 1). Most prominently, a Phase II trial of tanespimycin in combination with trastuzumab in patients with HER2-positive metastatic breast cancer (MBC) progressing on prior trastuzumab, was the first to report Response Evaluation Criteria in Solid Tumors (RECIST)-defined efficacy for tanespimycin in solid tumors [16]. The reported overall response rate (ORR) and clinical benefit rate (CBR, defined as complete response [CR] + partial response [PR] + SD) were 22 and 59%, respectively [16], and median progression free survival (PFS) and overall survival (OS) were 6 and 17 months, respectively. Tanespimycin has also been evaluated in combination with bortezomib in patients with multiple myeloma (MM). In a Phase I/II trial, response rates (RRs) of 41, 20 and 14% were reported in bortezomib-naïve, -pre-treated and -refractory patients, respectively [17]. This led to a Phase III trial of this combination which was later suspended due to nonclinical reasons [18].
Nevertheless, 17-AAG failed to advance further given its poor pharmaceutical and toxicity profile. Besides the limiting solubility and formulation challenges, 17-AAG contains a benzoquinone moiety that undergoes reductive metabolism and detoxification by nicotinamide adenine dinucleotide phosphate (NADPH): quinone oxidoreductase (NQ01) (also called DT-diaphorase) before it acts against HSP90 [1-4,11]. It is this benzoquinone that causes increased hepatotoxicity and perhaps also constitutes a mechanism of drug resistance in patients with a mutation in NQ01 [19]. Naturally, all this information combined with the delayed hepatotoxicity of 17-AAG in the clinic with the twice-a-week continuous dosing schedule provided a further impetus for the development of non-ansamycin HSP90 inhibitors with an improved toxicity profile.
The solubility issue observed for the front-runner 17-AAG was surmounted when Kosan Biosciences discovered 17-desmethoxy-17-N,N-dimethylaminoethylaminogeldanamycin (17-DMAG) (Figure 2; 17-desmethoxy-17-N,N-dimethylaminoethylaminogeldanamycin-alvespimycin) [20]. This compound contains the ionizable N,N-dimethylethylamine group instead of the methoxy group at C-17. Importantly, the introduction of the ionizable amino group provided the much-needed improvement in water solubility, oral bio-availability and equal, if not greater, antitumor activity than 17-AAG [21]. These combined benefits allowed 17-DMAG to advance to clinical trials in 2004 where it was investigated both as an oral and as an intravenous (i.v.) agent (Table 1) [22-24]. Objective responses, including some CRs, have been reported in patients with castrate-resistant prostate cancer (CRPC), acute myeloid leukemia and metastatic melanoma [23,24]. SD > 6 months has also been reported in three patients with chondrosarcoma, CRPC and clear cell renal cancer, respectively [24]. Toxicities reported in these trials included liver, lung, ocular and cardiac toxicities in addition to common side effects such as diarrhea, fatigue and nausea. Similar to tanespimycin, alvespimycin may be most beneficial in combination with trastuzumab. A Phase I trial of this combination reported one PR in a patient with HER2-positive MBC and 6 of the 28 other patients had SD lasting > 6 months [25]. Despite these objective responses, the clinical development of alvespimycin was halted in 2008 by Kosan due to strategic reasons [11]. However, this drug is still currently under evaluation by the National Cancer Institute for the treatment of patients with relapsed chronic lymphocytic leukemia (CLL), small lymphocytic lymphoma, or B-cell prolymphocytic leukemia (NCT01126502).
An improved GM derivative, IPI-504/retaspimycin (Figure 2), was introduced by Infinity. This reduced quinone form of tanespimycin is more water soluble than tanespimycin or alvespimycin and also, by lacking the benzoquinone, less prone to hepatotoxicity [26]. Phase I clinical trials have been conducted in patients with non-small cell lung cancer (NSCLC), MM and metastatic gastrointestinal stromal tumor (GIST) (Table 1) [11,27]. Modest antitumor activity, including metabolic responses with FDG-PET, was reported in 2 out of 4 patients with NSCLC and 4 out of 18 patients with GIST. Single agent Phase II trials were also conducted in patients with CRPC, NSCLC and GIST [28-30]. Results were disappointing in CRPC with no radiological or prostate-specific antigen responses and two deaths related to liver failure and ketoacidosis [28].
Based on preclinical studies that the echinoderm microtubule-associated protein-like 4-anaplastic lymphoma kinase (EML4-ALK) oncogenic fusion protein was highly sensitive to HSP90 inhibition by IPI-504 [31], a Phase II study of IPI-504 in refractory NSCLC stratified by ALK rearrangement status was initiated. The ORR was 67% in patients with ALK rearrangement compared to 8.3% in those without the ALK rearrangement [32]. In another Phase I/II trial of IPI-504 in patients with NSCLC who had progressed on prior EGFR tyrosine kinase inhibitors (TKIs), two out of three patients with ALK rearrangement had a PR and a third patient had SD > 7.2 months with 24% reduction in tumor size [29]. The RR in EGFR-mutated patients in this trial was poor (RR 4%). Most common side effects were diarrhea, nausea and fatigue, and grade 3 liver abnormalities were reported in nine patients [29]. Promising activity was also noted in patients with GIST which led to the Phase III retaspimycin in GIST trial. While the dose and schedule of IPI-504 was the same as the NSCLC trial, hepatotoxicity was very prominent in this trial with four treatment-related deaths leading to early study closure after 47 of the 195 planned patients were enrolled [33].
When combined with trastuzumab in patients with HER2-positive MBC, results from a Phase II trial revealed positive data, with 16 patients (62%) achieving SD with median on-study duration of 2.4 months (range 1.1 – 8.2 months). Contrary to expectations, there were no objective responses which was attributed to the lower dose of IPI-504 in this study (300 mg/m2 weekly compared to 400 mg/m2 twice weekly in the NSCLC trial) [34]. A Phase Ib combination trial of IPI-504 with docetaxel was conducted in 23 patients with NSCLC who had progressed on one or two prior lines of chemotherapy [34]. The ORR was 26% (6 PR, 7 SD) with higher responses in patients with squamous histology (43%) and those with a smoking history (33%) [34]. This has led to the ongoing randomized Phase II trial of this combination compared to docetaxel plus placebo in current and former smokers with pretreated NSCLC (NCT0 1262400). Another Phase Ib/II trial is evaluating the safety and combination of IPI-504 plus everolimus in patients with KRAS mutant NSCLC (NCT01427946).
3. HSP90 clinical agents: second generation
Taken as a whole, the GM derivatives have certainly fueled research efforts by both industry and academia to develop newer small-molecule synthetic HSP90 inhibitors with better bioavailability and toxicity profiles to permit administration of sufficiently high levels of inhibitor required to derive a therapeutic benefit. The quest for these much-improved small synthetic molecules resulted in the development of better HSP90 inhibitors coined as second generation. Studies with the first-generation HSP90 inhibitors also provided knowledge on tumor subtypes more likely to benefit from HSP90 inhibitors facilitating the clinical path of upcoming agents. From a pharmacophore perspective, most second-generation HSP90 inhibitors in clinical development either incorporate the resorcinol moiety of RD (Section 3.1, Figure 3) or mimic the purine-scaffold implemented in the design of the first synthetic HSP90 inhibitor, purine-scaffold (PU3) (Section 3.2, Figure 4), while just a few have a distinct scaffold (Section 3.3, Figure 5). We will next glimpse over the clinical development status of such inhibitors currently in clinical evaluation.
Figure 4. Purine and purine-like inhibitors of HSP90: PU3, PU-H71, MPC-3100 and BIIB021 (in blue is shown the purine and purine-like core) are presented.
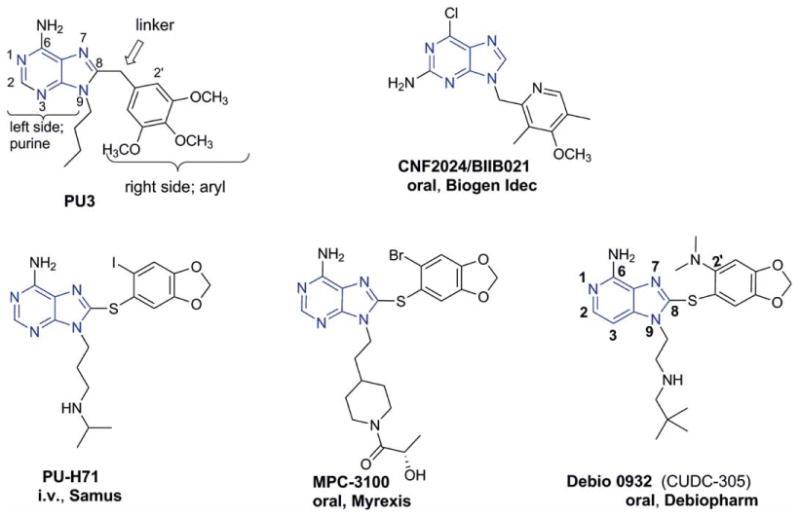
HSP90: Heat shock protein 90; i.v.: Intravenous.
Figure 5. Other HSP90 inhibitors: SNX-5422, HSP990 and XL888 are presented.
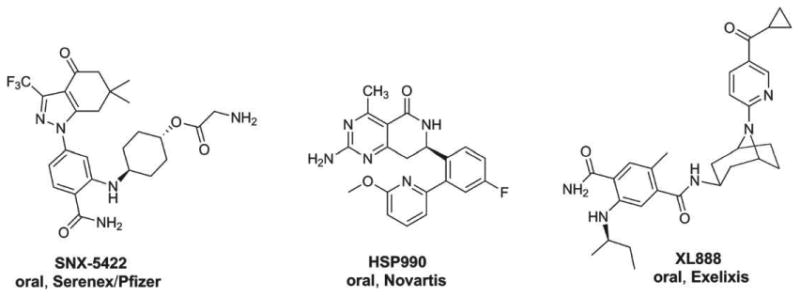
HSP90: Heat shock protein 90.
3.1 Resorcinol derivatives
The resorcinol core of RD, a critical element for binding to HSP90, is found in a number of clinical agents, namely NVP-AUY922, AT-13387, STA-9090 and KW-2478 (Figure 3, the resorcinol moiety is shown in blue), which are described below.
3.1.1 NVP-AUY922
NVP-AUY922 traces its origin to a high-throughput screen that employed inhibition of yeast HSP90 ATPase activity using the malachite green detection of inorganic phosphate [35]. The most potent hit emerging from this screen was the resorcinol-containing pyrazole CCT018159 – a molecule shown later by X-ray crystallography to bind to the ATP pocket in the N-terminal domain of yeast HSP90, similar to RD [36]. Scientists at Vernalis and the Cancer Research UK Center for Cancer Therapeutics used a structure-based approach to optimize this initial hit, and from the fruits of their labor emerged the isoxazole NVP-AUY922 (Figure 3), a potent HSP90 inhibitor [37].
Subsequently, this lead candidate developed by Novartis made it to clinical trials where it is being evaluated as a single agent as well as in combination therapy for various malignancies in multiple Phase I and Phase II trials (Table 2). The Phase I trial in advanced solid tumors established the maximum tolerated dose (MTD) of weekly i.v. infusions at 70 mg/m2 [38]. Dose-limiting toxicities (DLTs) included atrial flutter, darkening of vision, diarrhea and fatigue. Interestingly, 20% of the patients developed night blindness and 7% had grade 3 or higher eye disorders at the MTD. Although there were no objective responses, 16 patients developed SD and 9 also had a partial metabolic response on flurodeoxyglucose positron emission tomography (FDG-PET) scans [38]. A Phase II expansion trial in HER2-positive and estrogen receptor-positive breast cancer also reported two partial metabolic responses on FDG-PET and one of these was also a confirmed PR by RECIST [39]. A Phase II trial of monotherapy NVP-AUY922, conducted in patients with NSCLC progressing on two or more chemotherapy regimens (including patients with EGFR mutations who had progressed on EGFR TKI therapy), reported responses in patients with EGFR mutations (20% RR) and those with ALK rearrangements (32% RR) [40]. This led to the Phase II study of NVP-AUY922 plus erlotinib in patients with EGFR mutant lung cancer with acquired resistance to EGFR TKIs. Twenty-five patients were treated on this Phase II study. The study met its primary end point (defined as CR + PR at 8 weeks) with 5 of 22 patients (23%) demonstrating PR. Notably, three of these five patients had the T790M acquired mutation after EGFR TKI therapy, thus suggesting that the activity of this combination was not limited to patients with EGFRT790M mutation alone [41]. In a study of the combination of trastuzumab plus NVP-AUY922 in patients with HER2-positive MBC previously progressing on two prior anti-HER2 regimens, the ORR was 23% (5/22 patients) [42]. NVP-AUY922 is also being studied in Phase I/II studies in combination with other agents such as bortezomib in MM (NCT00708292), a phosphatidylinositol 3-kinase (PI3K) inhibitor BYL719 in advanced or metastatic gastric cancer (NCT01613950), an ALK inhibitor LDK378 in ALK-rearranged NSCLC (NCT01772797), with pemetrexed or docetaxel in NSCLC with EGFR mutations (NCT01646125) and cetuximab in KRAS-mutated colorectal cancer (NCT01294826).
Table 2. Summary of clinical data – second-generation HSP90 inhibitors.
| HSP90 inhibitor | Clinical stage | Combination | Disease | Response | Ref. |
|---|---|---|---|---|---|
| NVP-AUY922 | Phase I | No | Advanced solid tumors | SD | [38] |
| Phase II | No | NSCLC progressing on chemotherapy | Objective responses in patients with ALK rearrangement, EGFR/KRAS and ALK wild-type and mEGFR | [40] | |
| Phase II | Erlotinib | mEGFR NSCLC progressing on EGFR inhibitors | Objective responses in EGFRT790M and SD | [41] | |
| Phase Ib/II | Trastuzumab | HER2+ MBC refractory to trastuzumab | Objective responses | [42] | |
| Phase II | No | HER2+ or ER+ BC | Objective responses | [39] | |
| Phase I/II | Bortezomib | MM | Ongoing | NCT00708292 | |
| Phase I/II | PI3K inhibitor | Advanced or metastatic gastric cancer | Ongoing | NCT01613950 | |
| Phase I/II | ALK inhibitor | ALK-rearranged NSCLC | Ongoing | NCT01772797 | |
| Phase I/II | Docetaxel | NSCLC with EGFR mutations | Ongoing | NCT01646125 | |
| Phase I/II | Cetuximab | KRAS-mutated colorectal cancer | Ongoing | NCT01294826 | |
| AT-13387 Ganetespib | Phase I | No | Refractory solid tumors | Objective response in GIST and SD in follicular cell thyroid cancer, metastatic uveal melanoma and GIST | [45] |
| Phase I/II | No | CRPC progressing on abiraterone | Ongoing | NCT01685268 | |
| Abiraterone | CRPC progressing on abiraterone | Ongoing | NCT01685268 | ||
| Phase I/II | No | NSCLC | Ongoing | NCT01712217 | |
| Crizotinib | NSCLC | Ongoing | NCT01712217 | ||
| Phase I/II | No | Unresectable and/or metastatic GIST progressing on TKI | Ongoing | NCT01294202 | |
| Imatinib | Unresectable and/or metastatic GIST progressing on TKI | Ongoing | NCT01294202 | ||
| Phase I | No | Unselected solid tumors | SD and 1 PR in colorectal cancer, PR in metastatic melanoma and SD in NSCLC | [49,50] | |
| Phase I/II | No | Hematological malignancies | No formal responses | [51,52] | |
| Phase II | No | NSCLC genotypic characterization of tumors for the presence of EGFR or KRAS mutation or the absence of these mutations | Responses in crizotinib-naïve patients with the EML4-ALK rearrangement | [53] | |
| Phase II | No | NSCLC crizotinib-naïve patients with the EML4-ALK rearrangement | Ongoing | NCT01562015 | |
| Phase II | Docetaxel | NSCLC progressing on chemotherapy; AML; RCC | Objective responses | [58] | |
| Phase III | Docetaxel | NSCLC progressing on chemotherapy | Ongoing | NCT01798485 | |
| Phase II | No | Unselected MBC | Objective not met but activity noted in trastuzumab refractory HER2+ and TNBC | [54] | |
| Phase II | No | GIST progressing on TKI | SD | [55] | |
| Phase I | Paclitaxel and trastuzumab | HER2+ trastuzumab refractory MBC | Ongoing | NCT02060253 | |
| KW-2478 | Phase I | No | Lymphomas and leukemias | Completed | NCT00457782 |
| Phase I/II | Bortezomib | Relapsed and refractory MM | Objective responses | [61] | |
| BIIB021 | Phase I | No | CLL and advanced solid tumors | SD | [11] |
| Phase II | No | GIST progressing on TKI | Metabolic responses as noted on FDG-PET | [66] | |
| Phase I | Trastuzumab | HER2+ BC | Objective responses with metabolic responses on FDG-PET as well | [67] | |
| PU-H71 | Phase I | No | Advanced solid tumors and lymphoma | Ongoing | NCT01393509 |
| MPC-3100 | Phase I | No | Recurrent or refractory cancer | SD | [73] |
| CUDC-305 | Phase I | No | Advanced solid tumors | SD and PR in 2 patients (mKRAS NSCLC and BC) | [76] |
| Phase I/II | Chemotherapy | NSCLC progressing on chemotherapy | ongoing | NCT01714037 | |
| SNX-5422 | Phase I | No | Advanced solid tumors | Terminated due to ocular toxicity | [78] |
| Phase I/II | No | Several HER2+ tumor types (NSLC, esophagogastric, breast, urothelial) | Ongoing | NCT01848756 | |
| NVP-HSP990 | Phase I | No | Advanced solid tumors | No responses (drug development halted) | [81,82] |
| XL888 | Phase I | No | Advanced solid tumors | Terminated | [11] |
| Phase I | Vemurafenib | Unresectable mBRAF melanoma | Ongoing | NCT01657591 |
ALK: Anaplastic lymphoma kinase; AML: Acute myeloid leukemia; BC: Breast cancer; CLL: Chronic lymphocytic leukemia; CRPC: Castrate-resistant prostate cancer; EML4-ALK: Echinoderm microtubule-associated protein-like 4-anaplastic lymphoma kinase; FDG: Fluorodeoxyglucose; GIST: Gastrointestinal stromal tumors; NSCLC: non-small cell lung cancer; MBC: Metastatic breast cancer; MM: Multiple myeloma; PET: Positron emission tomography; PI3K: Phosphatidylinositol 3-kinase; PR: Partial response; SD: Stable disease; TKI: Tyrosine kinase inhibitor; TNBC: Triple-negative breast cancer.
3.1.2 AT-13387
Astex Pharmaceuticals applied a fragment-based drug discovery approach to find fragments with HSP90 binding affinity. This fragment screening consisted of a combination of nuclear magnetic resonance and high-throughput X-ray crystallography. One of the fragments identified was a phenolic chemotype [43] which was picked up and optimized via structure-guided design to ultimately led to the discovery of AT-13387 (Figure 3) [44].
This compound is being evaluated in Phase I trials in patients with advanced solid tumors (Table 2). The various dosing schedules being studied include weekly or twice-weekly infusions [45], every 3 weeks of a 28-day cycle and a twice a week (2 days in a row) for the first 3 weeks of a 28-day cycle [46]. A dose of 260 mg/m2 has been identified as the once-weekly MTD [45]. One durable RECIST PR (8 months) was reported in an imatinib-relapsed metastatic GIST patient with c-kit mutations in exons 11 and 17. Three SD ≥ 6 months (follicular cell thyroid carcinoma, metastatic uveal melanoma, GIST) were also observed. Phase II studies are currently ongoing. HSP70 induction, a surrogate readout of HSP90 inhibition, was noted in peripheral blood mononuclear cells (PBMCs) at all doses and exhibited dose dependence. In addition to diarrhea and fatigue that has been reported with this class of agents, this compound also showed interesting toxicity of reversible visual changes of blurred vision, diplopia, flashes and light–dark accommodation [45]. It is currently under evaluation in three Phase I/II trials either alone or in combination with: i) abiraterone in the treatment of CRPC which is no longer responding to abiraterone (NCT01685268); ii) crizotinib in the treatment of NSCLC (NCT01712217); and iii) imatinib in patients with unresectable and/or metastatic GIST whose tumor has progressed following treatment with a maximum of three TKIs (NCT01294202).
3.1.3 STA-9090 (ganetespib)
STA-9090 (Figure 3) is a novel resorcinolic triazolone inhibitor which was discovered and developed by Synta Pharmaceuticals [47]. It has been or is currently being tested in over 25 clinical trials in both hematologic and solid tumors, including a Phase III trial in NSCLC as summarized below [11,48].
Two Phase I clinical trials in advanced solid malignancies evaluated ganetespib in different dosing schedules: intravenously weekly for 3 weeks in a 28-day cycle or twice-weekly dosing for 3 weeks in a 28-day cycle, respectively. The recommended Phase II dose for the weekly dosing was established at 200 mg/m2 [49]. Ganetespib was well tolerated with predominantly grade 1 or 2 toxicities that were easily manageable. DLT included grade 3 amylase elevation, grade 3 diarrhea and grade 3 and 4 asthenia. One patient with metastatic colorectal cancer achieved a PR and 23 patients achieved SD. Disease control rate defined as PR + SD > 16 weeks was noted in 24.5% of the patients (Table 2) [49]. DLT of elevated liver enzymes was reported in the twice-weekly dosing trial. In this trial, one patient with metastatic melanoma achieved PR and two others with NSCLC achieved SD [50]. Two additional Phase I studies evaluated single agent ganetespib in hematologic malignancies, concluding that the doses for further study in this population are 200 mg/m2 once weekly [51] and 90 mg/m2 twice weekly [52]. The most common adverse events in both these trials were mild-to-moderate diarrhea and fatigue that were reversible and easily manageable. DLT in the once-weekly dosing trial was documented as elevated liver enzymes in one patient. Although there were no formal responses, three patients had a hematologic response [51]. In the twice-weekly dosing trial, DLTs included hyperbilirubinemia/hyponatremia, QTc prolongation and transaminitis [52].
Single agent ganetespib has also been evaluated in Phase II trials in patients with NSCLC [53], MBC [54] and GIST [55]. The drug was well tolerated, with the most frequent toxicities being grade 1/2 diarrhea, nausea and fatigue. In advanced NSCLC, ganetespib activity was evaluated in three cohorts based on prospective genotypic characterization of tumors for the presence of EGFR or KRAS mutation or the absence of these mutations. Ganetespib showed encouraging single agent activity in the non-mutant EGFR or KRAS cohort, specifically in the crizotinib-naïve patients whose tumors harbored the EML4-ALK rearrangement. Among eight such patients, there were four PRs and a median PFS of 8.1 months. This formed the basis for a Phase II study of ganetespib monotherapy in patients with crizotinib-naïve ALK-positive disease which has been initiated (NCT015 62015).
There is a strong preclinical rationale for combining HSP90 inhibitors with taxanes, due to synergistic antitumor activity [56,57]. A randomized Phase II GALAXY-I trial recently reported extended OS with ganetespib and docetaxel compared to docetaxel alone in the second-line setting in patients with advanced NSCLC who are at least 6 months from initial diagnosis of advanced disease [58]. Encouraged by these results the GALAXY-2 trial, a Phase III trial of this combination has been initiated and is currently accruing patients (NCT01798485).
It is well established that HER2 is a very sensitive client protein of HSP90 inhibition [1-4] and proof of concept for HSP90 inhibition was provided by tanespimycin in combination with trastuzumab in HER2-positive trastuzumab refractory MBC [16]. A Phase II trial of single agent ganetespib was conducted in an unselected cohort of MBC patients [54]. Although the study did not meet its prespecified criteria for ORR in the first-stage of the Simon's 2-stage model in this heavily pretreated unselected population, activity was noted in trastuzumab refractory HER2-positive and triple-negative breast cancer (TNBC) [54]. Clinically, anecdotal evidence of single agent activity has been reported in TNBC patients treated on Phase I and Phase II trials including an ongoing Phase II monotherapy trial (ENCHANT, NCT01677455) [59]. Ganetespib also showed enhanced cytotoxic effects when combined with chemotherapeutic agents such as doxorubicin and taxanes [59] and the ENCHANT-1 trial has been expanded to evaluate the combination of ganetespib and weekly paclitaxel, and Synta Pharmaceuticals is planning another trial of ganetespib and docetaxel in TNBC. A Phase I trial of ganetespib plus paclitaxel plus trastuzumab is ongoing in HER2-positive trastuzumab-refractory MBC (NCT020 60253).
Patients with GIST who had failed prior therapy with imatinib and sunitinib were treated with single agent ganetespib weekly for 3 weeks in a 28-day cycle [55]. Correlatives included positron emission tomography (PET) imaging, and HSP90 client proteins were evaluated using pre- and post-treatment biopsies. The primary end point was CBR (CR + PR + SD > 16 weeks). In this study, 12 of 23 patients in this Simons 2-stage model achieved SD, and 7 of the 123 patients reported a > 20% decrease in the standardized uptake value (SUV) as measured by PET imaging. However, paired tumor biopsies in four patients did not show prolonged inhibition of activated KIT or its downstream pathways. These data, therefore, suggested that once-weekly treatment might not be optimal in patients with GIST and therefore accrual is limited to patients with platelet-derived growth factor receptor alpha (PDGFRA) mutations to allow evaluation of other schedules and combinations.
3.1.4 KW-2478
In their pursuit for small-molecule HSP90 leads, Kyowa Hakko Kirin Pharma used a binding assay where HSP90 was fixed onto plates and the compounds to be screened were then added to the wells to identify competitive inhibitors of a labeled RD. Their endeavors resulted in the identification of several resorcinol-based leads which were subsequently optimized via X-ray crystallography, cell-based assays and in vivo models to eventually provide the clinical candidate KW-2478 (Figure 3) [60].
A Phase I study of this compound was reported in patients with relapsed/refractory MM, CLL or B-cell non-Hodgkin's lymphoma [60]. KW-2478 (14 – 99 mg/m2) was administered intravenously over 60 min once-daily on days 1 – 5 of a 14-day cycle. No DLTs were reported up to 99 mg/m2. Drug-related toxicities included grade 1/2 hypertension in one patient and grade 3 QTc prolongation in another. Another Phase I/II trial of KW-2478 in combination with bortezomib, when administered on days 1, 4, 8 and 11 of a 21-day cycle in patients with relapsed and refractory MM, was recently reported at the Annual Society of Hematology meeting in 2013 (Table 2) [61]. A total of 95 patients were treated: 15 on the Phase I trial and 80 on the Phase II trial. The most common adverse events included diarrhea, nausea, vomiting, fatigue and peripheral neuropathy. Five patients had grade 4 thrombocytopenia and three patients had grade 4 neutropenia. In the Phase I trial, HSP70 induction was observed in the PBMC of all patients. Of the 80 patients on the Phase II trial, 79 patients were evaluable for response using the International Myeloma Working Group response criteria. ORR was 39% and PFS was 26.4 weeks. In bortezomib-naïve patients (n = 50), the ORR was 48% [61].
3.2 Purine derivatives
The development of HSP90 inhibitors based on the purine scaffold was achieved via structure-based design, using the X-ray crystal structures of GM, RD and ADP bound to the N-terminal nucleotide-binding domain of HSP90 [62].
These molecules illustrate the ATP-binding pocket as having a distinguishing Bergerat fold, which is seen only in the GHKL ATPase protein family [63]. This knowledge played an important role and provided the blueprint for the design of synthetic HSP90 inhibitors. The first purine-based inhibitor, which was also the first reported synthetic HSP90 inhibitor, was PU3 (Figure 4). It was rationally designed by Chiosis et al. using the co-crystal structures of HSP90 with its ligands (i.e., GM, RD and adenine nucleotides) [64]. Importantly, PU3 was the first synthetic molecule to demonstrate phenotypic properties similar to GM [64]. The essential motif found in PU3, purine-linker-aryl (Figure 4), has been incorporated by numerous research groups in the design and development of potent drug candidates with favorable pharmaceutical properties. These efforts have culminated in the advancement to clinical trials of CNF2024/BIIB021, PU-H71, MPC-3100 and Debio 0932 (CUDC-305) (Figure 4).
3.2.1 CNF2024/BIIB021
Conforma Therapeutics discovered BIIB021 (Figure 4), a compound which distinguishes from the other purine-based compounds by that the aryl moiety that is attached to the N9 position of the purine and not the typical C8 position [65]. This was possible without a loss in activity because the NH2 group found on the 6-position of the purine scaffold was moved to the 2-position of the purine, and hence this change maintained the optimal six bond distance between the NH2 and the aryl group needed for potent binding of the pharmacophore (purine-linker-aryl, Figure 4). This HSP90 inhibitor was initially developed by Conforma Therapeutics and later by Biogen Idec, and it was the first synthetic inhibitor to enter clinical evaluation (Table 2).
BIIB021 has been evaluated in Phase I trials in various dosing regimens in patients with CLL and advanced solid tumors [11]. The monotherapy MTD was 800 mg twice weekly. DLTs included syncope and dizziness, and grade 3 or 4 toxicities included fatigue, hyponatremia and hypoglycemia. In patients with CLL, there was one incidence of grade 3 liver toxicity [11]. Response evaluation revealed SD in 11 of 16 patients with solid tumors and 1 patient with CLL had a 39% reduction in the lymph node size.
A Phase II trial was conducted in patients with GIST who had progressed on prior imatinib and sunitinib [66]. Two dosing schedules: 600 mg twice weekly and 400 mg three times a week were evaluated in 12 and 11 patients, respectively. Although the treatment was well tolerated with no evidence of liver toxicity, there were no radiological responses noted by RECIST criteria, and the duration of response was very brief (25 – 138 days). Interestingly, metabolic responses were noted by FDG-PET in 3 of 12 patients on the 600 mg twice weekly cohort and 2 of 9 patients on the 400 mg three times a week cohort [66]. The combination of BIIB021 with trastuzumab was tested in a Phase I trial in patients with HER2-positive MBC [67]. The MTD was established at 600 mg twice weekly. DLTs included diarrhea and partial seizure that presented as aphasia, and other adverse events included fatigue, nausea, dizziness, headache and rash. Of the 30 patients enrolled, 2 patients had confirmed PR by RECIST and an additional 10 patients had SD. Three patients had metabolic PR (> 25% decline in the SUVmax) with FDG-PET and an additional 16 patients had SD as well. Although other Phase II combination studies were planned, the clinical development of this drug has been halted by Biogen Idec due to the company's exit from the oncology field [68].
3.2.2 PU-H71
PU-H71 (Figure 4) is a purine-based compound discovered at Memorial Sloan-Kettering Cancer Center and developed by Samus Therapeutics and NCI [62,69]. A unique feature of this molecule is that it has an endogenous iodine atom (127I), which has been conveniently replaced with the PET radionuclide 124I to result in the imaging agent 124I-PU-H71 [70]. In fact, the PET agent is identical to PU-H71, and its physical half-life of 4.02 days allows serial imaging for monitoring tumor PU-H71 concentrations for multiple days. 124I-PU-H71 has been tested clinically in a Phase 0, first-in-human trial in patients with advanced solid tumors to determine the microdose biodistribution of PU-H71 in patients, including tumor uptake and retention, as well as gather tracer radiation dosimetry data important for clinical development of this promising tumor-imaging agent. Preliminary results have confirmed the feasibility of this approach in detecting PU-H71 in tumors using HSP90-targeted PET [70]. Tracer uptake at the metastatic tumor sites in this study correlated well with baseline CT and/or FDG-PET scans. Accrual to this trial is ongoing (Table 2).
PU-H71 is also being evaluated in twice weekly, 2 weeks on, 1 week off in a 21-day cycle in a Phase I trial of patients with advanced solid tumors and lymphoma (NCT01 393509). This trial also incorporates the 124I-PU-H71 PET as a noninvasive means to determine tumor pharmacokinetics (PK) and intratumoral drug concentration. A mix of 124I-PU-H71 and unlabeled PU-H71 is given during cycle 2 followed by serial PET imaging. Thus far, data from this ongoing study show that 124I-PU-H71 is useful in visualizing PU-H71 uptake, and tumor concentrations as measured by biopsies correlates well with tumor concentrations calculated by 124I-PU-H71 PET imaging [71]. This agent is also being studied at a different dosing schedule at the NCI in patients with advanced solid tumors and low-grade non-Hodgkin's lymphoma patients who have not responded to standard treatment (NCT01581541).
3.2.3 MPC-3100
Myrexis Inc. discovered MPC-3100 (Figure 4) after initiating a drug discovery program aimed at finding HSP90 inhibitors using the purine-scaffold [72]. Despite the large number of analogs synthesized, they retained the 1,3-benzodioxole moiety found in PU-H71 because no other functionalities had more favorable PK profiles. In the end, the extensive SAR carried out on the pendant N-9 piperidine moiety and of the 1,3-benzodioxole ring, complemented by in vitro and PK data, provided MPC-3100 as their best drug candidate [72]. Nonetheless, the presence of bromine instead of iodine rendered MPC-3100 of reduced activity when compared to PU-H71.
MPC-3100 was evaluated in a Phase I trial in 26 patients with recurrent or refractory cancer (Table 2) [73]. Patients received MPC-3100 either daily for 21 days with a week off in a 28-day cycle (doses: 50,100, 165, 245 or 340 mg/m2) or daily continuously for 28 days (at 240 or 320 mg every 12 h). The most common adverse events were grade 1 or 2 diarrhea, nausea, vomiting and fatigue. The DLT was supraventricular tachycardia at a dose of 245 mg/m2. Grade 1 – 3 gastrointestinal and grade 1/2 visual adverse events were observed at total daily doses > 600mg/day; however, these were reversible and resolved following discontinuation of study drug [73].
3.2.4 Debio 0932 (CUDC-305)
Curis developed CUDC-305 (Figure 4) after studying the structure requirements and compound design of the purine scaffold established by others [62,69,74]. This compound is similar to PU-H71; however, the nitrogen at 3-position is replaced by a carbon and the iodine by dimethylamine, and it also appears that replacement of N with C diminishes activity [75].
Debio-0932 is an oral HSP90 inhibitor that has been evaluated in a Phase I trial in patients with advanced solid tumors and lymphoma (Table 2) [76]. It was evaluated either as daily dosing or every other day dosing. The drug was well tolerated at doses up to 1600 mg every 2 days and 1000 mg/day. The most common adverse events were diarrhea, asthenia and decreased appetite with no ocular or cardiotoxicity being reported. One DLT of febrile neutropenia was observed with the every-other-day dosing and two DLTs of diarrhea and asthenia were reported with the daily dosing. Overall, this inhibitor showed promising activity with PR in two patients, one with KRAS-mutant lung cancer and the other with breast cancer. Specifically, of the eight patients with lung cancer, one had PR and four others had SD [76]. The daily dosing cohort is being expanded to enroll an additional 30 patients and a Phase I/II trial of Debio-0932 in combination with standard-of-care in the first- and second-line treatment of NSCLC has been initiated (NCT01714037).
3.3 Other chemotypes
3.3.1 SNX-5422/PF-04929113
Researchers at Serenex discovered SNX-5422 (Figure 5), which is a compound based on a novel dihydroindazolone scaffold. To identify their initial hits, a chemoproteomics-based strategy was used which employed the screening of their chemical library versus a diverse array of possible targets [77]. This screen found compound–protein interactions which involved HSP90. The subsequent synthetic design was complemented by structural information and ultimately resulted in the lead candidate SNX-2112. To improve the oral bioavailability of SNX-2112, a glycine ester moiety was introduced resulting in the prodrug SNX-5422 (Figure 5). SNX-5422 was advanced by Serenex in 2007 to clinical trials and was further developed with Pfizer. The development was discontinued initially due to concerns of ocular toxicity, including irreversible retinal damage seen in animal models and reported in a Phase I study (Table 2) [78]. However, more recently, this drug has now been acquired by Esanex Inc., a privately owned Lilly-Ventures biotech company and the program for SNX-5422 has been restarted [79]. It is currently being evaluated as a Phase I/II trial in patients with advanced HER2-positive cancers, including metastatic NSCLC, urothelial cancers, esophagogastric and breast cancers (NCT01848756). Patients will receive 100 mg/m2 every other day orally for 3 weeks in a 28-day cycle. Ophthalmological examinations, including visual acuity, visual field, ophthalmoscopy, dark adaptation are planned at screening, at end of cycle 1, at end of cycle 3 and then every 3 months thereafter. Another Phase I trial is evaluating the combination of SNX-5422 with erlotinib in patients with NSCLC that are resistant to EGFR TKIs (NCT01851096).
3.3.2 HSP990 (NVP-HSP990)
Novartis discovered HSP990 (Figure 5) via high-throughput screening (HTS) and subsequent follow-up structure-based lead optimization [80]. It was consequently advanced to clinical trials as an oral agent. HSP990 was evaluated in a Phase I trial in advanced solid tumors in weekly (n = 53; 2.5 – 60 mg) and twice weekly (n = 11; 25 mg) dosing schedules (Table 2). The most common side effects reported were diarrhea, increased liver enzymes, anemia and cholestasis. DLTs included grade 3 diarrhea, grade 3 QTc prolongation, grade 4 alanine aminotransferase/aspartate aminotransferase, grade 3 tremors and grade 2 neurologic toxicity, including ataxia, confusion and visual hallucination. Dose escalation was limited by neurologic toxicity and the MTD was established at 50 mg weekly [81]. Nonetheless, the further advancement of NVP-HSP990 has been stopped due to its failure to achieve clinically meaningful responses at the MTD [82].
3.3.3 XL888
Exelixis discovered XL888 (Figure 5), a novel tropane-derived inhibitor of HSP90. First, the initial hits were found via an HTS campaign of 4.1 million compounds using an in-house chemical library [83]. The challenges encountered during the structure-based approach used for their optimization efforts pertained to reducing the high molecular weights (> 650 amu) and polar surface areas (> 135 Å2), which were outside the ideal range for orally bioavailable drugs [83].
The first Phase I safety study of XL888 was terminated by the sponsor Exelixis [11] with no publicly disclosed specific reason (Table 2). However, a Phase I study of XL888 in combination with vemurafenib in patients with unresectable BRAF-mutated stage III/IV melanoma is underway (NCT01 657591). This study is supported by preclinical data which demonstrated that signaling proteins involved in intrinsic and acquired resistance to BRAF inhibitors are clients of HSP90 and inhibiting HSP90 restores sensitivity to vemurafenib [84].
4. Biomarkers and diagnostics – unmet need in HSP90 therapy
As with the development of any novel targeted therapy, identification of biomarkers and companion diagnostic assays are crucial not only to ascertain target inhibition but also to better identify patients who are most likely to respond or be resistant to therapy.
With HSP90 inhibitors, target inhibition in clinic has been evaluated by either measuring the levels of client oncoproteins in surrogate tissues pre- and post-therapy (RAF-1, AKT, C-KIT, CDK4) [11] or by evaluating the upregulation of other co-chaperones such as HSP70 or HSP27 or other factors involved in the heat shock response using PBMCs [85]. Effective target inhibition certainly helped optimize drug dosing and scheduling in many of these trials. However, although HSP70 upregulation in the serum or PBMCs has served as a pharmacodynamic (PD) readout of HSP90 inhibition, it has not predicted tumor response [11,86-88]. This comes perhaps as no surprise since HSP90 inhibitors are known to be preferentially retained in tumor cells and not normal cells [89-91], and also there seems to be a fundamental difference between the HSP90 species in normal and tumor cells [90,91]. Perhaps for the same reasons, the clinical utility of other serum biomarkers such as soluble insulin growth factor binding protein and HER2 extracellular domain also remains to be validated [92].
Few trials have undertaken pre- and post-treatment tumor biopsies from patients on HSP90 inhibitor therapy for PD analysis. Hence data are limited and inconsistent due to small sample sizes, low sensitivity of traditional immunohistochemical staining methods employed in these trials and tumor heterogeneity [11]. One potential way to overcome these barriers is to utilize noninvasive molecular imaging that not only allows visualization of the target but also permits assessment of whole body tumor burden and heterogeneity, evaluates tumor response and characterizes the PK and PD changes in the tumors. 18F-FDG PET has been utilized in highly glycolytic tumors such as GIST as a PD correlate of antitumor activity [11,93]. Metabolic PR and SD have also been reported using FDG-PET imaging [38,66]. PET with either radiolabeled antibodies against other specific targets such as HER2, VEGF receptor, androgen receptor, and the like [94-96] or with radiolabeling the therapeutic drug itself have also been explored [70,71].
Last, although HSP90 tumor expression might seem to be an appropriate strategy to best select patients who might respond to therapy, available data do not suggest a predictive role for this marker. To date, the only reliable way to predict response to HSP90 inhibition has been to identify those patients whose cancer is driven by a particular oncogene that is a very sensitive HSP90 client (e.g., HER2 in breast cancer or chimeric ALK in NSCLC) [97]. Nonetheless, clinical success in other tumor types that harbor HSP90 onco-clients has been limited so far and the failure of HSP90 inhibitors in these tumors remains poorly understood.
5. Conclusion
The past few years have shown intense activity in the discovery and optimization of synthetic HSP90 inhibitors. Most of these second-generation HSP90 inhibitors that have progressed to clinical development either incorporate the resorcinol moiety of the natural product RD or mimic the purine-scaffold implemented in the design of the first synthetic HSP90 inhibitor, PU3. Studies with these new agents have brought evidence that the liver toxicity associated with the first-generation GM derivatives was pharmacophore-(i.e., benzoquinone) and not target (i.e., HSP90)-mediated. In fact a number of the new agents show manageable and reversible toxicity profiles (predominantly diarrhea and fatigue) and hold promise for further advancement in the clinic. Diarrhea seems to be an on-target effect [54] and lasts for 24 – 48 h and can be easily controlled with anti-diarrheal medications prophylactically. Ocular toxicity has also emerged as a concerning side effect with a few but not all second-generation inhibitors [45,78], but whether it is a class effect/on or off target effect, remains to be further elucidated.
The clinical development of the second-generation inhibitors continued in tumor subtypes where first-generation inhibitors demonstrated response. As such, the clinical activity observed for all HSP90 chemotypes in HER2-positive breast tumors and NSCLC with ALK translocations provides convincing data for the addiction of these tumors to HSP90. Also validated is the ability of these HSP90 agents to provide efficacy enhancement in well-chosen combination strategies. Noteworthy is resensitization of tumors to the effect of other targeted therapies, such as the case for trastuzumab in HER2-positive MBC progressing on trastuzumab, bortezomib in MM refractory to bortezomib and erlotinib in mEGFR NSCLC progressing on EGFR inhibitors. Of value is also their ability to provide enhanced cytotoxic effects with chemotherapy such as taxanes in refractory NSCLC patients. In fact, combination strategies might enhance antitumor activity even in those tumors where despite strong preclinical rationale, single agent HSP90 inhibitor has failed to demonstrate efficacy, perhaps due to the lack of a sensitive client protein or lack of sustained inhibition due to ineffective dose and scheduling or due to tumor evolution and drug resistance [57].
6. Expert opinion
But more importantly, the clinical studies so far also highlight the limitations of the current HSP90 field. They cement the notion that in unselected patient populations, HSP90 studies have limited probability of success, underscoring the need to augment efforts toward a better understanding of HSP90-addicted tumor types. However, finding out the particularities of a tumor that renders it addicted to HSP90 remains elusive. So far, identification of sensitive tumors in clinic appears to be trial-and-error-based. The activity seen in HER2-positive breast cancer and chimeric ALK NSCLC has a strong foundation provided by preclinical studies. Nonetheless, these preclinical studies predicted many other tumor types to harbor HSP90-addicted onco-proteins, and their clinical benefit has been limited thus far.
Further, not all tumors that express the ‘highly sensitive client’ are responsive to HSP90 therapy (e.g., not all HER2-positive tumors respond equally well to an HSP90 inhibitor). This is likely a consequence of the complex molecular networks regulated by HSP90 in a tumor-by-tumor manner; these may interconnect distinctly and as determined by the tumor's specific genetic background. As such, inhibition of HSP90 in tumors expressing a ‘sensitive HSP90 client’ may result in distinct outcomes.
Studies also indicate that the sensitivity of a tumor to pharmacologic inhibition may not be driven by the expression of total HSP90 in the tumor cell, but rather by the presence of actively chaperoning HSP90 species [91,98,99]. These species, whose presence are regulated by co-chaperone recruitment and likely also by post-translational modifications, maintain multiple oncogenic pathways dependent on HSP90 activity. In fact, recent proteomic and biochemical studies support the notion that tumor selection in the HSP90 field should be based on more sophisticated functional proteomic insights that measure not only the network of tumor-driving HSP90 clientele but also the abundance and the biochemical nature of the HSP90 fraction available for inhibitor binding [91,98,99].
Ideally, clinical trials of targeted cancer therapy also require knowledge of whether effective tumor concentrations are achieved and whether the target is appropriately modulated at the site of action, the tumor cell. Knowledge on both such topics is, however, sorely lacking in the HSP90 field. Although preclinical studies indicate the antitumor activity of HSP90 inhibitors to be largely mediated by their ability to engage the target over the time of treatment, as described above, no validated assays are available to answer this question for HSP90 inhibitors in clinic. Therefore it is difficult to conclude that HSP90 inhibition fails to provide benefit in a particular disease setting, when we cannot answer the obvious: whether the HSP90 inhibitor in question given at a dose and on a schedule actually optimally engaged the target in that setting.
Selection of a proper dose and schedule that are needed to achieve antitumor efficacy is also poorly understood in HSP90 therapy. Plasma PK generally provides data relevant to the design of therapeutic dosing, with the plasma area under the curve often used as a metric of systemic drug exposure. However, for HSP90 inhibitors, the concentration and duration of retention of drug in tumor tissues, and not in blood, determine their antitumor effect. Animal and clinical data have shown that blood concentrations do not predict nor mimic tumor concentrations of HSP90 inhibitors [11,69,70]. Nevertheless, only one agent, PU-H71, currently incorporates in its clinical development an assay that provides real-time measurement of the inhibitor concentrations in individual tumors [70].
To conclude, the HSP90 multifaceted chaperone, although not fully understood, remains an enticing target for cancer therapy. In retrospect, the rush for better HSP90 drugs has left unanswered much on patient selection and proper target engagement and this is perhaps slowing down the advancement of these inhibitors in the clinic. Thus, the field should focus more attention on these topics if the fulfillment of the target's promise is to be achieved soon. As such, we believe the successful transition of the current HSP90 inhibitors to approval will be limited to those that not only have a favorable therapeutic index but also incorporate companion diagnostics to inform on a more judicious implementation and clinical use.
Article highlights.
Several second-generation heat shock protein 90 (HSP90) inhibitors have entered the clinical oncology arena. These are based on a restricted number of favorable pharmacophores.
The dose-dependent hepatotoxicity noted with the first-generation geldanamycin is not observed in the second-generation HSP90 inhibitors, indicating it to be chemical- rather than target-related in nature. The second-generation HSP90 inhibitors, except a select few, are in general well tolerated with mostly grade 1/2 toxicities.
The clinical development path of second-generation inhibitors follows in the footsteps of path-finding first-generation agents and has focused on a small select subset of tumors.
Combining HSP90 inhibitors with other potent anticancer therapies holds promise not only due to synergistic antitumor activity but also due to the potential of prolonging or preventing the development of drug resistance.
A clear understanding on HSP90 target engagement in the several ongoing studies is still sorely lacking. Dose and schedule appears to be mostly toxicity- rather than target saturation-driven.
Understanding of tumors that are more likely to draw a benefit from HSP90 therapy remains limited. The field should move from the ‘HSP90 client protein selection criteria’ toward a more sophisticated biomarker based on a functional proteomic approach.
This box summarizes key points contained in the article.
Acknowledgments
Declaration of interest: This work was supported in part by the Jane H. Gordon Breast Cancer Research Fund, Leukemia and Lymphoma Society, UL1RR024996 of the Clinical and Translational Science Center of Weill Cornell Medical College, R01 CA172546, R01 CA155226, R21 CA158609, Conquer Cancer Foundation (ASCO) Young Investigator Award and Terri Brodeur Breast Cancer Foundation. R Peter is supported by the Operational Program of Human Resources Development 2007 – 2013 of the Romanian Ministry of Labor, Family and Social Protection through the Financial Agreement POSDRU/89/1.5/S/62557. YY Janjigian is supported by The Society of Memorial Sloan-Kettering Cancer Center. The Memorial Sloan-Kettering Cancer Center holds the intellectual rights to PU-H71 and [124I]-PU-H71. Samus Therapeutics, of which G Chiosis has partial ownership, has licensed PU-H71 and [124I]-PU-H71.
Bibliography
Papers of special note have been highlighted as either of interest (•) or of considerable interest (••) to readers.
- 1.Zuehlke A, Johnson JL. Hsp90 and co-chaperones twist the functions of diverse client proteins. Biopolymers. 2010;93(3):211–17. doi: 10.1002/bip.21292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2•.Workman P, Burrows F, Neckers L, et al. Drugging the cancer chaperone HSP90: combinatorial therapeutic exploitation of oncogene addiction and tumor stress. Ann N Y Acad Sci. 2007;1113:202–16. doi: 10.1196/annals.1391.012. This is a good review on the therapeutic potential of heat shock protein 90 (HSP90) in cancer. [DOI] [PubMed] [Google Scholar]
- 3•.Patel HJ, Modi S, Chiosis G, et al. Advances in the discovery and development of heat-shock protein 90 inhibitors for cancer treatment. Expert Opin Drug Discov. 2011;6(5):559–87. doi: 10.1517/17460441.2011.563296. This is an overview on the discovery of HSP90 inhibitors of distinct chemotypes. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4•.Whitesell L, Lindquist SL. HSP90 and the chaperoning of cancer. Nat Rev Cancer. 2005;5(10):761–72. doi: 10.1038/nrc1716. Good overview of the biology of HSP90 in cancer. [DOI] [PubMed] [Google Scholar]
- 5•.Neckers L, Schulte TW, Mimnaugh E. Geldanamycin as a potential anti-cancer agent: its molecular target and biochemical activity. Invest New Drugs. 1999;17(4):361–73. doi: 10.1023/a:1006382320697. A pioneering paper on the discovery of the natural products geldanamycin (GM) and radicicol (RD) as HSP90 inhibitors. [DOI] [PubMed] [Google Scholar]
- 6•.Sharma SV, Agatsuma T, Nakano H. Targeting of the protein chaperone, HSP90, by the transformation suppressing agent, radicicol. Oncogene. 1998;16(20):2639–45. doi: 10.1038/sj.onc.1201790. A pioneering paper on the discovery of the natural products GM and RD as HSP90 inhibitors. [DOI] [PubMed] [Google Scholar]
- 7.Schulte TW, Neckers LM. The benzoquinone ansamycin 17-allylamino-17-demethoxy geldanamycin binds to HSP90 and shares important biologic activities with geldanamycin. Cancer Chemother Pharmacol. 1998;42(4):273–9. doi: 10.1007/s002800050817. [DOI] [PubMed] [Google Scholar]
- 8•.Banerji U, O'Donnell A, Scurr M, et al. Phase I pharmacokinetic and pharmacodynamic study of 17-allylamino, 17-demethoxygeldanamycin in patients with advanced malignancies. J Clin Oncol. 2005;23(18):4152–61. doi: 10.1200/JCO.2005.00.612. This was the first trial that also evaluated pre- and 24 h post-treatment tumor biopsies and demonstrated target inhibition. [DOI] [PubMed] [Google Scholar]
- 9.Goetz MP, Toft D, Reid J, et al. Phase I trial of 17-allylamino-17-demethoxygeldanamycin in patients with advanced cancer. J Clin Oncol. 2005;23(6):1078–87. doi: 10.1200/JCO.2005.09.119. [DOI] [PubMed] [Google Scholar]
- 10.Grem JL, Morrison G, Guo XD, et al. Phase I and pharmacologic study of 17-(allylamino)-17-demethoxygeldanamycin in adult patients with solid tumors. J Clin Oncol. 2005;23(9):1885–93. doi: 10.1200/JCO.2005.12.085. [DOI] [PubMed] [Google Scholar]
- 11•.Jhaveri K, Taldone T, Modi S, et al. Advances in the clinical development of heat shock protein 90 (Hsp90) inhibitors in cancers. Biochim Biophys Acta. 2012;1823(3):742–55. doi: 10.1016/j.bbamcr.2011.10.008. A recent review of the current state of the HSP90 inhibitors in the clinic. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Heath EI, Hillman DW, Vaishampayan U, et al. A phase II trial of 17-allylamino-17-demethoxy geldanamycin in patients with hormone-refractory metastatic prostate cancer. Clin Cancer Res. 2008;14(23):7940–6. doi: 10.1158/1078-0432.CCR-08-0221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13•.Solit DB, Osman I, Polsky D, et al. Phase II trial of 17-allylamino-17-demethoxygeldanamycin in patients with metastatic melanoma. Clin Cancer Res. 2008;14(24):8302–7. doi: 10.1158/1078-0432.CCR-08-1002. This trial was performed in melanoma patients harboring V600E BRAF mutations. Although the post 17-AAG treatment tumor biopsies showed HSP70 induction and decrease in cyclin D1 expression, the effects on RAF kinase were very short lived. It suggested that a better suppression of the MAPK pathway, either with an HSP90 inhibitor of an extended tumor pharmacokinetics or through combining the HSP90 inhibitor with an inhibitor of the MAPK pathway, could result in a more prolonged pathway inhibition, as needed for efficacy. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ronnen EA, Kondagunta GV, Ishill N, et al. A phase II trial of 17-(Allylamino)-17-demethoxy geldanamycin in patients with papillary and clear cell renal cell carcinoma. Invest New Drugs. 2006;24(6):543–6. doi: 10.1007/s10637-006-9208-z. [DOI] [PubMed] [Google Scholar]
- 15.Pacey S, Gore M, Chao D, et al. A Phase II trial of 17-allylamino, 17-demethoxygeldanamycin (17-AAG, tanespimycin) in patients with metastatic melanoma. Invest New Drugs. 2012;30(1):341–9. doi: 10.1007/s10637-010-9493-4. [DOI] [PubMed] [Google Scholar]
- 16••.Modi S, Stopeck A, Linden H, et al. HSP90 inhibition is effective in breast cancer: a phase II trial of tanespimycin (17-AAG) plus trastuzumab in patients with HER2-positive metastatic breast cancer progressing on trastuzumab. Clin Cancer Res. 2011;17(15):5132–9. doi: 10.1158/1078-0432.CCR-11-0072. This was the pivotal trial in HER2-positive metastatic breast cancer (MBC) that demonstrated antitumor efficacy for 17-AAG in combination with trastuzumab. [DOI] [PubMed] [Google Scholar]
- 17•.Richardson PG, Chanan-Khan AA, Lonial S, et al. Tanespimycin and bortezomib combination treatment in patients with relapsed or relapsed and refractory multiple myeloma: results of a phase 1/2 study. Br J Haematol. 2011;153(6):729–40. doi: 10.1111/j.1365-2141.2011.08664.x. This trial demonstrated efficacy for 17-AAG in combination with bortezomib that justified the Phase III trial of this combination. [DOI] [PubMed] [Google Scholar]
- 18.PressRelease. Bristol-Myers Squibb Halts Development of Tanespimycin. [Last Accessed 8 January 2014];2008 Available from: http://www.myelomabeacon.com/news/2010/07/22/tanespimycin-development-halted/
- 19.Neckers L, Workman P. Hsp90 molecular chaperone inhibitors: are we there yet? Clin Cancer Res. 2012;18(1):64–76. doi: 10.1158/1078-0432.CCR-11-1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20•.Tian ZQ, Liu Y, Zhang D, et al. Synthesis and biological activities of novel 17-aminogeldanamycin derivatives. Bioorg Med Chem. 2004;12(20):5317–29. doi: 10.1016/j.bmc.2004.07.053. Describes the identification of 17-DMAG. [DOI] [PubMed] [Google Scholar]
- 21.Hollingshead M, Alley M, Burger AM, et al. In vivo antitumor efficacy of 17-DMAG (17-dimethylaminoethylamino-17-demethoxygeldanamycin hydrochloride), a water-soluble geldanamycin derivative. Cancer Chemother Pharmacol. 2005;56(2):115–25. doi: 10.1007/s00280-004-0939-2. [DOI] [PubMed] [Google Scholar]
- 22.Kummar S, Gutierrez ME, Gardner ER, et al. Phase I trial of 17-dimethylaminoethylamino-17-demethoxygeldanamycin (17-DMAG), a heat shock protein inhibitor, administered twice weekly in patients with advanced malignancies. Eur J Cancer. 2010;46(2):340–7. doi: 10.1016/j.ejca.2009.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lancet JE, Gojo I, Burton M, et al. Phase I study of the heat shock protein 90 inhibitor alvespimycin (KOS-1022, 17-DMAG) administered intravenously twice weekly to patients with acute myeloid leukemia. Leukemia. 2010;24(4):699–705. doi: 10.1038/leu.2009.292. [DOI] [PubMed] [Google Scholar]
- 24.Pacey S, Wilson RH, Walton M, et al. A phase I study of the heat shock protein 90 inhibitor alvespimycin (17-DMAG) given intravenously to patients with advanced solid tumors. Clin Cancer Res. 2011;17(6):1561–70. doi: 10.1158/1078-0432.CCR-10-1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25•.Jhaveri K, Miller K, Rosen L, et al. A phase I dose-escalation trial of trastuzumab and alvespimycin hydrochloride (KOS-1022; 17 DMAG) in the treatment of advanced solid tumors. Clin Cancer Res. 2012;18(18):5090–8. doi: 10.1158/1078-0432.CCR-11-3200. This trial confirms the activity of another GM HSP90 inhibitor in combination with trastuzumab in HER2-positive MBC. [DOI] [PubMed] [Google Scholar]
- 26.Sydor JR, Normant E, Pien CS, et al. Development of 17-allylamino-17-demethoxygeldanamycin hydroquinone hydrochloride (IPI-504), an anti-cancer agent directed against Hsp90. Proc Natl Acad Sci USA. 2006;103(46):17408–13. doi: 10.1073/pnas.0608372103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Siegel D, Jagannath S, Vesole DH, et al. A phase 1 study of IPI-504 (retaspimycin hydrochloride) in patients with relapsed or relapsed and refractory multiple myeloma. Leuk Lymphoma. 2011;52(12):2308–15. doi: 10.3109/10428194.2011.600481. [DOI] [PubMed] [Google Scholar]
- 28.Oh WK, Galsky MD, Stadler WM, et al. Multicenter phase II trial of the heat shock protein 90 inhibitor, retaspimycin hydrochloride (IPI-504), in patients with castration-resistant prostate cancer. Urology. 2011;78(3):626–30. doi: 10.1016/j.urology.2011.04.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29••.Sequist LV, Gettinger S, Senzer NN, et al. Activity of IPI-504, a novel heat-shock protein 90 inhibitor, in patients with molecularly defined non-small-cell lung cancer. J Clin Oncol. 2010;28(33):4953–60. doi: 10.1200/JCO.2010.30.8338. This was the first trial that suggested a role for HSP90 inhibitors in NSCLC especially those with anaplastic lymphoma kinase rearrangement. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wagner AJ, Morgan JA, Chugh R, et al. Inhibition of heat shock protein 90 (Hsp90) with the novel agent IPI-504 in metastatic GIST following failure of tyrosine kinase inhibitors (TKIs) or other sarcomas: clinical results from phase I trial. J Clin Oncol. 2008;26 abstract 10503. [Google Scholar]
- 31.Normant E, Paez G, West KA, et al. The Hsp90 inhibitor IPI-504 rapidly lowers EML4-ALK levels and induces tumor regression in ALK-driven NSCLC models. Oncogene. 2011;30(22):2581–6. doi: 10.1038/onc.2010.625. [DOI] [PubMed] [Google Scholar]
- 32.Sequist LV, Natale RB, Senzer NN, et al. Association between anaplastic lymphoma kinase rearrangements (rALK) and the clinical activity of IPI-504 (retaspimycin hydrochloride), a novel Hsp90 inhibitor, in patients with non-small cell lung cancer (NSCLC) J Clin Oncol. 2010;28 doi: 10.1200/JCO.2010.30.8338. abstract 7517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Demetri G, Le Cesne A, Von Mehren M, et al. Final results from a phase III study of IPI-504 (retaspimycin hydrochloride) versus placebo in patients (pts) with gastrointestinal stromal tumors (GIST) following failure of kinase inhibitor therapies. Gastrointestinal Cancers Symposium. 2010;28 abstract-64. [Google Scholar]
- 34.Modi S, Saura C, Henderson C, et al. A multicenter trial evaluating retaspimycin HCL (IPI-504) plus trastuzumab in patients with advanced or metastatic HER2-positive breast cancer. Breast Cancer Res Treat. 2013;139(1):107–13. doi: 10.1007/s10549-013-2510-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35•.Rowlands MG, Newbatt YM, Prodromou C, et al. High-throughput screening assay for inhibitors of heat-shock protein 90 ATPase activity. Anal Biochem. 2004;327(2):176–83. doi: 10.1016/j.ab.2003.10.038. Describes the strategy that led to the identification of a lead compound that ultimately translated into NVP-AUY922. [DOI] [PubMed] [Google Scholar]
- 36.Cheung KM, Matthews TP, James K, et al. The identification, synthesis, protein crystal structure and in vitro biochemical evaluation of a new 3,4-diarylpyrazole class of Hsp90 inhibitors. Bioorg Med Chem Lett. 2005;15(14):3338–43. doi: 10.1016/j.bmcl.2005.05.046. [DOI] [PubMed] [Google Scholar]
- 37.Jensen MR, Schoepfer J, Radimerski T, et al. NVP-AUY922: a small molecule HSP90 inhibitor with potent antitumor activity in preclinical breast cancer models. Breast Cancer Res. 2008;10(2):R33. doi: 10.1186/bcr1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Samuel TA, Sessa C, Britten C, et al. AUY922, a novel HSP90 inhibitor: Final results of a first-in-human study in patients with advanced solid malignancies. J Clin Oncol. 2010;28(Suppl) abstract 2528. [Google Scholar]
- 39.Schroder C, Pederson JV, Chua S, et al. Use of biomarkers and imaging to evaluate the treatment effect of AUY922, an Hsp90 inhibitor, in patients with HER2+ or ER+ metastatic breast cancer. J Clin Oncol. 2011;29(Suppl) abstract e11024. [Google Scholar]
- 40.Garon EB, Moran T, Barlesi F, et al. Phase II study of the HSP90 inhibitor AUY922 in patients with previously treated, advanced non-small cell lung cancer (NSCLC) J Clin Oncol. 2012;30(Suppl) abstract 7543. [Google Scholar]
- 41.Johnson ML, Hart EM, Rademaker A, et al. A phase II study of HSP90 inhibitor AUY922 and erlotinib (E) for patients (pts) with EGFR-mutant lung cancer and acquired resistance (AR) to EGFR tyrosine kinase inhibitors (EGFR TKIs) J Clin Oncol. 2013;31 doi: 10.1200/JCO.2014.59.7328. abstract 8036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kong A, Rea D, Ahmed S, et al. Phase IB/II study of the HSP90 inhibitor AUY922, in combination with trastuzumab, in patients with HER2 positive advanced breast cancer. J Clin Oncol. 2012;30(Suppl) abstract 530. [Google Scholar]
- 43.Murray CW, Carr MG, Callaghan O, et al. Fragment-based drug discovery applied to Hsp90. Discovery of two lead series with high ligand efficiency. J Med Chem. 2010;53(16):5942–55. doi: 10.1021/jm100059d. [DOI] [PubMed] [Google Scholar]
- 44•.Woodhead AJ, Angove H, Carr MG, et al. Discovery of (2,4-dihydroxy-5-isopropylphenyl)-[5-(4-methylpiperazin-1-ylmethyl)-1,3-dihydroisoindol-2-yl] methanone (AT13387), a novel inhibitor of the molecular chaperone Hsp90 by fragment based drug design. J Med Chem. 2010;53(16):5956–69. doi: 10.1021/jm100060b. References 43 and 44 describe Astex's approach using fragment screening against HSP90 and then subsequent structure-guided design to discover AT13387. [DOI] [PubMed] [Google Scholar]
- 45.Mahadevan D, Rensvold DM, Kurtin SE, et al. First-in-human phase I study: Results of a second-generation non-ansamycin heat shock protein 90 (HSP90) inhibitor AT13387 in refractory solid tumors. J Clin Oncol. 2012;30(Suppl) abstract 3028. [Google Scholar]
- 46.Do KT, Speranza G, Chen AP, et al. Phase l study assessing a two-consecutive-day (QD × 2) dosing schedule of the HSP90 inhibitor, AT13387, in patients with advanced solid tumors. J Clin Oncol. 2012;30 abstract 3087. [Google Scholar]
- 47•.Ying W, Du Z, Sun L, et al. Ganetespib, a unique triazolone-containing Hsp90 inhibitor, exhibits potent antitumor activity and a superior safety profile for cancer therapy. Mol Cancer Ther. 2012;11(2):475–84. doi: 10.1158/1535-7163.MCT-11-0755. Discloses the chemical structure and some preclinical data on ganetespib. [DOI] [PubMed] [Google Scholar]
- 48.Choi HK, Lee K. Recent updates on the development of ganetespib as a Hsp90 inhibitor. Arch Pharm Res. 2012;35(11):1855–9. doi: 10.1007/s12272-012-1101-z. [DOI] [PubMed] [Google Scholar]
- 49.Goldman JW, Raju RN, Gordon GA, et al. A first in human, safety, pharmacokinetics, and clinical activity phase I study of once weekly administration of the Hsp90 inhibitor ganetespib (STA-9090) in patients with solid malignancies. BMC Cancer. 2013;13:152. doi: 10.1186/1471-2407-13-152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cho D, Heath EI, Cleary JM, et al. A phase I dose escalation study of the Hsp90 inhibitor ganetespib (STA-9090) administered twice weekly in patients with solid tumors: updated report. J Clin Oncol. 2011;29 abstract 3051. [Google Scholar]
- 51.Lancet JE, Smith BD, Bradley R, et al. A phase 1/2 study of the potent Hsp90 inhibitor STA-9090 administered once weekly in subjects with hematologic malignancies. Blood. 2010 abstract 3294. [Google Scholar]
- 52.Padmanabhan S, Kelly K, Heaney M, et al. A phase I study of the potent Hsp90 Inhibitor STA-9090 administered twice weekly in subjects with hematologic malignancies. Blood. 2010;116 abstract 2898. [Google Scholar]
- 53•.Socinski MA, Goldman J, El-Hariry I, et al. A multicenter phase II study of ganetespib monotherapy in patients with genotypically defined advanced non–small cell lung cancer. Clin Cancer Res. 2013;19(11):3068–77. doi: 10.1158/1078-0432.CCR-12-3381. This trial shows promising activity for single agent second-generation HSP90 inhibitor in NSCLC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jhaveri K, Chandarlapaty S, Lake D, et al. A phase II open-label study of ganetespib, a novel heat shock protein 90 inhibitor for patients with metastatic breast cancer. Clin Breast Cancer. 2013 doi: 10.1016/j.clbc.2013.12.012. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 55.Demetri G, Heinrich MC, Chmielowski B, et al. An open-label phase II study of the Hsp90 inhibitor ganetespib (STA-9090) in patients (pts) with metastatic and/or unresectable GIST. J Clin Oncol. 2011;29(Suppl) abstract 10011. [Google Scholar]
- 56.Solit DB, Basso AD, Olshen AB, et al. Inhibition of heat shock protein 90 function down-regulates Akt kinase and sensitizes tumors to Taxol. Cancer Res. 2003;63(9):2139–44. [PubMed] [Google Scholar]
- 57•.Jhaveri K, Modi S. Hsp90 inhibitors for cancer therapy and overcoming drug resistance. Adv Pharmacol. 2012;65:471–517. doi: 10.1016/B978-0-12-397927-8.00015-4. This review highlights the importance of combinatorial approaches with HSP90 inhibitors in an attempt to overcome or prevent resistance. [DOI] [PubMed] [Google Scholar]
- 58.Ramalingam S, Goss GD, Andric ZG, et al. A randomized study of ganetespib, a heat shock protein 90 inhibitor, in combination with docetaxel versus docetaxel alone for second-line therapy of lung adenocarcinoma (GALAXY-1) J Clin Oncol. 2013;31(Suppl) abstract CRA8007. [Google Scholar]
- 59.Proia DA, Zhang C, Sequeira M, et al. Preclinical activity profile and therapeutic efficacy of the Hsp90 inhibitor ganetespib in triple-negative breast cancer. Clin Cancer Res. 2013;20(2):413–24. doi: 10.1158/1078-0432.CCR-13-2166. [DOI] [PubMed] [Google Scholar]
- 60.Cavenagh JD, Yong K, Byrne J, et al. The safety, pharmacokinetics and pharmacodynamics of KW-2478, a novel Hsp90 antagonist, in patients with B-cell malignancies: a first-in-man, phase I, multicentre, open-label, dose escalation study. Myeloma-Therapy Blood. 2008;112 abstract 2777. [Google Scholar]
- 61.Cavenagh J, Baylon HG, Caguioa PB, et al. A phase 1/2 study of KW-2478, an Hsp 90 inhibitor, in combination with bortezomib (BTZ) in patients (Pts) with relapsed/refractory (R/R) multiple myeloma (MM) Myeloma: Therapy, excluding Transplantation: Poster I Blood. 2013;122 abstract 1967. [Google Scholar]
- 62•.Taldone T, Chiosis G. Purine-scaffold Hsp90 inhibitors. Curr Top Med Chem. 2009;9(15):1436–46. doi: 10.2174/156802609789895737. This review describes the discovery and development of purine-based HSP90 inhibitors. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chene P. ATPases as drug targets: learning from their structure. Nat Rev Drug Discov. 2002;1(9):665–73. doi: 10.1038/nrd894. [DOI] [PubMed] [Google Scholar]
- 64•.Chiosis G, Timaul MN, Lucas B, et al. A small molecule designed to bind to the adenine nucleotide pocket of Hsp90 causes Her2 degradation and the growth arrest and differentiation of breast cancer cells. Chem Biol. 2001;8(3):289–99. doi: 10.1016/s1074-5521(01)00015-1. Reports the design and target validation of the first small-molecule synthetic HSP90 inhibitor, PU3. [DOI] [PubMed] [Google Scholar]
- 65•.Kasibhatla SR, Hong K, Biamonte MA, et al. Rationally designed high-affinity 2-amino-6-halopurine heat shock protein 90 inhibitors that exhibit potent antitumor activity. J Med Chem. 2007;50(12):2767–78. doi: 10.1021/jm050752+. Design and synthesis of BIIB021 and the SAR pertaining to this purine-based compound. [DOI] [PubMed] [Google Scholar]
- 66.Dickson MA, Okuno SH, Keohan ML, et al. Phase II study of the HSP90-inhibitor BIIB021 in gastrointestinal stromal tumors. Ann Oncol. 2013;24(1):252–7. doi: 10.1093/annonc/mds275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Modi S, Ismail-Khan R, Munster P, et al. Phase 1 dose-escalation study of the heat shock protein 90 inhibitor BIIB021 with trastuzumab in HER2+ metastatic breast cancer. Cancer Res. 2010;70(24 Suppl 2) abstract P3-14-02. [Google Scholar]
- 68.Mitchell P. Biogen Idec restructures, sharpens neurology focus. Nat Biotechnol. 2011;29(1):7–8. doi: 10.1038/nbt0111-7. [DOI] [PubMed] [Google Scholar]
- 69•.He H, Zatorska D, Kim J, et al. Identification of potent water soluble purine-scaffold inhibitors of the heat shock protein 90. J Med Chem. 2006;49(1):381–90. doi: 10.1021/jm0508078. This paper describes the synthesis and the identification of PU-H71. [DOI] [PubMed] [Google Scholar]
- 70.Dunphy M, Chiosis G, Beattie B, et al. Progress in first-in-human trial of Hsp90-targeted PET imaging in cancer patients. J Nucl Med. 2013;54(Suppl 2) abstract 279. [Google Scholar]
- 71.Gerecitano JF, Modi S, Gajria D, et al. Using 124I-PU-H71 PET imaging to predict intratumoral concentration in patients on a phase I trial of PU-H71. J Clin Oncol. 2013;31(Suppl) abstract 11076. [Google Scholar]
- 72•.Kim SH, Bajji A, Tangallapally R, et al. Discovery of (2S)-1-[4-(2-{6-amino-8-[(6-bromo-1,3-benzodioxol-5-yl)sulfanyl]-9H-purin-9-yl}et hyl)piperidin -1-yl]-2-hydroxypropan-1-one (MPC-3100), a purine-based Hsp90 inhibitor. J Med Chem. 2012;55(17):7480–501. doi: 10.1021/jm3004619. Design and synthesis of MPC-3100 and the SAR pertaining to this purine-based compound. [DOI] [PubMed] [Google Scholar]
- 73.Samlowski WE, Papadopoulos K, Olszanski AJ, et al. Phase 1 study of HSP90 inhibitor MPC-3100 in subjects with refractory or recurrent cancer. Molecular Targets and Cancer Therapeutics. 2011 abstract A96. [Google Scholar]
- 74•.Bao R, Lai CJ, Qu H, et al. CUDC-305, a novel synthetic HSP90 inhibitor with unique pharmacologic properties for cancer therapy. Clin Cancer Res. 2009;15(12):4046–57. doi: 10.1158/1078-0432.CCR-09-0152. The pharmacological profile of the HSP90 inhibitor CUDC-305 is described. [DOI] [PubMed] [Google Scholar]
- 75•.Taldone T, Patel PD, Patel M, et al. Experimental and structural testing module to analyze paralogue-specificity and affinity in the Hsp90 inhibitors series. J Med Chem. 2013;56(17):6803–18. doi: 10.1021/jm400619b. Describes the contribution of the four human HSP90 paralogues to the activity of several HSP90 inhibitors. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Isambert N, Hollebecque A, Berge Y, et al. A phase I study of Debio 0932, an oral HSP90 inhibitor, in patients with solid tumors. J Clin Oncol. 2012;30 abstract 3026. [Google Scholar]
- 77.Fadden P, Huang KH, Veal JM, et al. Application of chemoproteomics to drug discovery: identification of a clinical candidate targeting Hsp90. Chem Biol. 2010;17(7):686–94. doi: 10.1016/j.chembiol.2010.04.015. [DOI] [PubMed] [Google Scholar]
- 78.Rajan A, Kelly RJ, Trepel JB, et al. A phase I study of PF-04929113 (SNX-5422), an orally bioavailable heat shock protein 90 inhibitor, in patients with refractory solid tumor malignancies and lymphomas. Clin Cancer Res. 2011;17(21):6831–9. doi: 10.1158/1078-0432.CCR-11-0821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Esanex: out of stealth mode (barely). HSP90 Inhibitors Central. [Last Accessed 8 January 2014];2013 Available from: http://www.hsp90central.com/1/post/2013/01/esanex-out-of-stealth-mode-barely.html.
- 80.Menezes DL, Taverna P, Jensen MR, et al. The novel oral Hsp90 inhibitor NVP-HSP990 exhibits potent and broad-spectrum antitumor activities in vitro and in vivo. Mol Cancer Ther. 2012;11(3):730–9. doi: 10.1158/1535-7163.MCT-11-0667. [DOI] [PubMed] [Google Scholar]
- 81.Mattos-Arruda LD, Siu LL, Cortes J, et al. Phase I dose-escalation, open-label study of HSP990 administered orally in adult patients with advanced solid malignancies. J Clin Oncol. 2013;31(Suppl) doi: 10.1038/bjc.2014.653. abstract 2561∧. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Vernalis. Interim results for the six months ended 30 June 2012. [Last Accessed 9 January 2014];2012 Available from: http://www.vernalis.com/media-centre/latest-releases/2012-releases/642-interim-results-for-the-six-months-ended-30-june-2012.
- 83•.Bussenius J, Blazey CM, Aay N, et al. Discovery of XL888: a novel tropane-derived small molecule inhibitor of HSP90. Bioorg Med Chem Lett. 2012;22(17):5396–404. doi: 10.1016/j.bmcl.2012.07.052. The discovery of the HSP90 inhibitor XL888 after following up on tropane-derived high-throughput screening hits via an iterative SAR development process. [DOI] [PubMed] [Google Scholar]
- 84.Paraiso KH, Haarberg HE, Wood E, et al. The HSP90 inhibitor XL888 overcomes BRAF inhibitor resistance mediated through diverse mechanisms. Clin Cancer Res. 2012;18(9):2502–14. doi: 10.1158/1078-0432.CCR-11-2612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85•.Whitesell L, Lin NU. HSP90 as a platform for the assembly of more effective cancer chemotherapy. Biochim Biophys Acta. 2012;1823(3):756–66. doi: 10.1016/j.bbamcr.2011.12.006. Good review on the introduction of HSP90 inhibitors to combination regimens in oncology. [DOI] [PubMed] [Google Scholar]
- 86.Ramalingam SS, Egorin MJ, Ramanathan RK, et al. A phase I study of 17-allylamino-17-demethoxygeldanamycin combined with paclitaxel in patients with advanced solid malignancies. Clin Cancer Res. 2008;14(11):3456–61. doi: 10.1158/1078-0432.CCR-07-5088. [DOI] [PubMed] [Google Scholar]
- 87.Goetz MP, Toft DO, Ames MM, et al. The Hsp90 chaperone complex as a novel target for cancer therapy. Ann Oncol. 2003;14(8):1169–76. doi: 10.1093/annonc/mdg316. [DOI] [PubMed] [Google Scholar]
- 88.Eiseman JL, Lan J, Lagattuta TF, et al. Pharmacokinetics and pharmacodynamics of 17-demethoxy 17-[[(2-dimethylamino) ethyl] amino] geldanamycin (17DMAG, NSC 707545) in C.B-17 SCID mice bearing MDA-MB-231 human breast cancer xenografts. Cancer Chemother Pharmacol. 2005;55(1):21–32. doi: 10.1007/s00280-004-0865-3. [DOI] [PubMed] [Google Scholar]
- 89•.Vilenchik M, Solit D, Basso A, et al. Targeting wide-range oncogenic transformation via PU24FCl, a specific inhibitor of tumor Hsp90. Chem Biol. 2004;11(6):787–97. doi: 10.1016/j.chembiol.2004.04.008. Describes for the first time the prolonged tumor retention profile observed for certain HSP90 inhibitors and comments on how this translates into tumor pharmacodynamics. [DOI] [PubMed] [Google Scholar]
- 90••.Kamal A, Thao L, Sensintaffar J, et al. A high-affinity conformation of Hsp90 confers tumour selectivity on Hsp90 inhibitors. Nature. 2003;425(6956):407–10. doi: 10.1038/nature01913. This paper demonstrates that although HSP90 is expressed in all cells, HSP90 in cancer cells, but not in normal cells, is predominantly found in multichaperone complexes that have high ATPase activity and high affinity for certain HSP90 inhibitors. It, thus, provided a biochemical rationale for the observed therapeutic index of HSP90 inhibitors. [DOI] [PubMed] [Google Scholar]
- 91••.Moulick K, Ahn JH, Zong H, et al. Affinity-based proteomics reveal cancer-specific networks coordinated by Hsp90. Nat Chem Biol. 2011;7(11):818–26. doi: 10.1038/nchembio.670. It demonstrates that the presence of a tumor-enriched HSP90 species and not of total HSP90 indicates the addiction of a tumor to HSP90 and its sensitivity to HSP90 inhibitors. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Eiseman JL, Guo J, Ramanathan RK, et al. Evaluation of plasma insulin-like growth factor binding protein 2 and Her-2 extracellular domain as biomarkers for 17-allylamino-17-demethoxygeldanamycin treatment of adult patients with advanced solid tumors. Clin Cancer Res. 2007;13(7):2121–7. doi: 10.1158/1078-0432.CCR-06-2286. [DOI] [PubMed] [Google Scholar]
- 93.Demetri GD, George S, Morgan JA, et al. Inhibition of the heat shock protein 90 (Hsp90) chaperone with the novel agent IPI-504 to overcome resistance to tyrosine kinase inhibitors (TKIs) in metastatic GIST: updated results of a phase I trial. J Clin Oncol. 2007;25 abstract 10024. [Google Scholar]
- 94.Holland JP, Caldas-Lopes E, Divilov V, et al. Measuring the pharmacodynamic effects of a novel Hsp90 inhibitor on HER2/neu expression in mice using Zr-DFO-trastuzumab. PLoS ONE. 2010;5(1):e8859. doi: 10.1371/journal.pone.0008859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Nagengast WB, de Korte MA, Oude Munnink TH, et al. 89Zr-bevacizumab PET of early antiangiogenic tumor response to treatment with HSP90 inhibitor NVP-AUY922. J Nucl Med. 2010;51(5):761–7. doi: 10.2967/jnumed.109.071043. [DOI] [PubMed] [Google Scholar]
- 96.Oude Munnink TH, Korte MA, Nagengast WB, et al. (89)Zr-trastuzumab PET visualises HER2 downregulation by the HSP90 inhibitor NVP-AUY922 in a human tumour xenograft. Eur J Cancer. 2010;46(3):678–84. doi: 10.1016/j.ejca.2009.12.009. [DOI] [PubMed] [Google Scholar]
- 97.Jhaveri KL. Biomarkers that predict sensitivity to heat shock protein 90 inhibitors (HSP90i) J Clin Oncol. 2012;30(Suppl) doi: 10.1016/j.clbc.2015.11.004. abstract 10618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98•.Darby JF, Workman P. Chemical biology: many faces of a cancer-supporting protein. Nature. 2011;478(7369):334–5. doi: 10.1038/478334b. Good overview of the challenges associated with understanding the sensitivity of tumors to HSP90 inhibition. [DOI] [PubMed] [Google Scholar]
- 99••.Beebe K, Mollapour M, Scroggins B, et al. Posttranslational modification and conformational state of heat shock protein 90 differentially affect binding of chemically diverse small molecule inhibitors. Oncotarget. 2013;4(7):1065–74. doi: 10.18632/oncotarget.1099. Demonstrates that chemically distinct HSP90 inhibitors select for overlapping but not identical subpopulations of total cellular HSP90 despite the fact that they bind to the same pocket. It shows the importance of HSP90's post-translational modifications on the anticancer activity of small-molecule inhibitors. [DOI] [PMC free article] [PubMed] [Google Scholar]