

Abstract
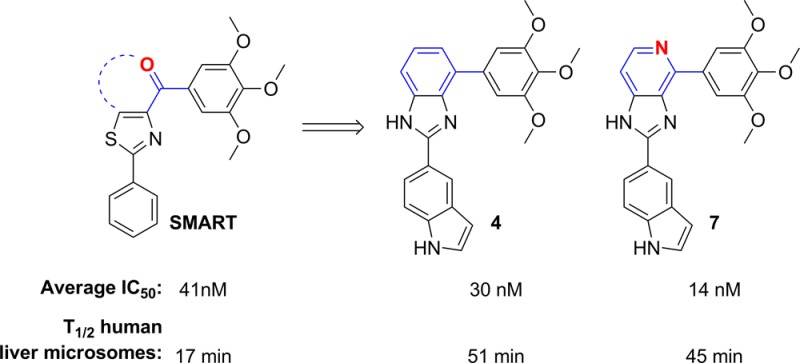
To block the metabolically labile sites of novel tubulin inhibitors targeting the colchicine binding site based on SMART, ABI, and PAT templates, we have designed, synthesized, and biologically tested three focused sets of new derivatives with modifications at the carbonyl linker, the para-position in the C ring of SMART template, and modification of A ring of the PAT template. Structure–activity relationships of these compounds led to the identification of new benzimidazole and imidazo[4,5-c]pyridine-fused ring templates, represented by compounds 4 and 7, respectively, which showed enhanced antitumor activity and substantially improved the metabolic stability in liver microsomes compared to SMART. MOM group replaced TMP C ring and generated a potent analogue 15, which showed comparable potency to the parent SMART compound. Further modification of PAT template yielded another potent analogue 33 with 5-indolyl substituent at A ring.
Introduction
Tubulin/microtubule-interacting drugs are used successfully for treatment of a wide variety of human cancers. They are commonly classified into two major categories: microtubule-stabilizing (e.g., taxanes and epothilones) and microtubule-destabilizing drugs (e.g., vinca alkaloids and colchicine). Three major binding sites on α,β-tubulin subunits have been identified as taxanes-, vinca alkaloid-, and colchicine-binding sites.1 While antimitotic agents interacting with the taxanes- or vinca alkaloid-binding sites in tubulin are tremendously successful in clinical oncology, there are no Food and Drug Administration (FDA)-approved colchicine-binding site drugs currently available for cancer treatment. Most of the colchicine-binding agents have high potency, relatively simple chemical structures for optimization, and selective toxicity toward tumor vasculature and show promising ability to overcome P-glycoprotein (P-gp) efflux pump mediated multidrug resistance.2 Therefore, the colchicine-binding site compounds have attracted great interest from medicinal chemists in recent years.
Several outstanding agents for such an approach are listed in Figure 1. Combretastatin A-4 (CA-4) is the most active member of the combretastatin family, isolated from the African tree Combretum caffrum. CA-4 exhibits strong antitubulin activity by binding to the colchicine-site and underwent phase II and phase III studies in clinical.3 The replacement of the olefinic bridge of CA-4 with a carbonyl group yields phenstatin,4 which has similar potency and mechanism of actions with CA-4. BPR0L0755 and Oxi-61966 are 2-aroylindole and dihydronaphthalene analogues of CA-4, which show strong inhibition on tubulin polymerization. Methylated chalcone SD400, which has an IC50 value of 0.21 nM against K562 human leukemia cells, is a potent tubulin inhibitor.7 Podophyllotoxin is a nonalkaloid toxin lignin, and it also possesses an anticancer property that can be attributed to the inhibition of tubulin polymerization through binding to the colchicine binding site.8 All these agents shared a common 3,4,5-trimethoxyphenyl (TMP) moiety in their chemical structures. This TMP moiety, common to the above-mentioned antitubulin agents, has been shown to be crucial for inhibiting the growth of tumor cells.9
Figure 1.
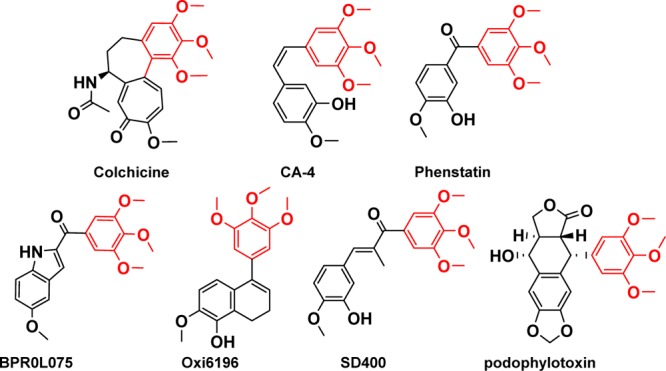
Structures of representative tubulin inhibitors bound to the colchicine-binding site.
Our research group discovered 4-substituted methoxybenzoyl-aryl-thiazole10 (SMART, Figure 2), 2-aryl-4-benzoyl-imidazole (ABI),11 and phenyl amino thiazole (PAT)12 templates as potential anticancer agents targeting tubulin by binding to the colchicine binding site. These agents show low nanomolar inhibition on various cancer cell lines and can effectively overcome a number of clinically relevant multidrug resistant mechanisms that are often associated with the use of existing tubulin inhibitors.13 Considering the structure similarity, the TMP ring plays an important role in these templates to enhance cytotoxic activity, whereas pharmacokinetic (PK) studies showed that these compounds have low bioavailability mainly due to two major metabolic processes in human liver microsomes: the carbonyl reduction and demethylation in the TMP ring.14 As a result, the half-life of the SMART compound 10 is only 17 min.15 These data highlight the need for modifications of carbonyl linker and TMP ring that could reduce metabolic liability at these sites and potentially increase the bioavailability of these agents. Previously we have replaced the carbonyl with a variety of linkers (sulfonyl, sulfinyl, hydrazide, etc.), but those modifications had limited success in overcoming the metabolic stability problem while maintaining the high potency.12 In this article, we presented our latest approach of cyclizing the carbonyl with the B ring (Figure 2), which yielded a new “D” ring on the top of “B” ring and thus blocked the metabolic reduction of the ketone linker to a secondary alcohol. In a separate approach, we focused on the modification of the TMP “C” ring of SMART to specifically block the known sites of demethylation metabolism while maintaining or improving the in vitro antiproliferative activity profile. On the basis of these initial studies, we made a series of modifications focused in TMP C ring at the para-position and produced derivatives with comparable or increased activity. Several strategies including the incorporation of an alkylating group, a hydrogen bond donor, or a hydrophobic group were examined. Finally, we further modified the PAT template and obtained another highly active analogue bearing a 5-indolyl moiety at “A” ring position.
Figure 2.
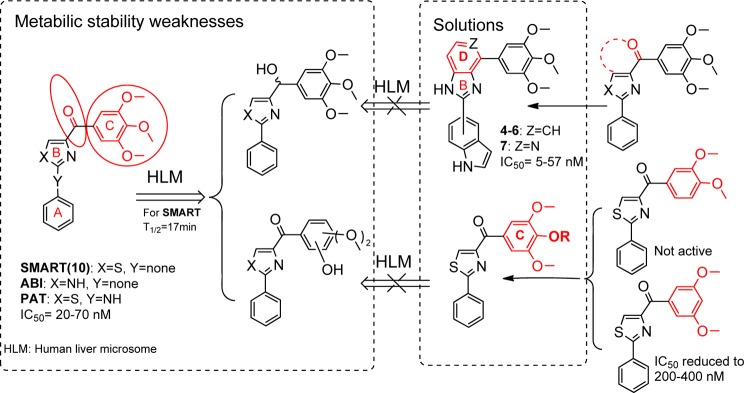
Proposed approaches to solve metabolic liabilities of lead compounds.
Chemistry Modification
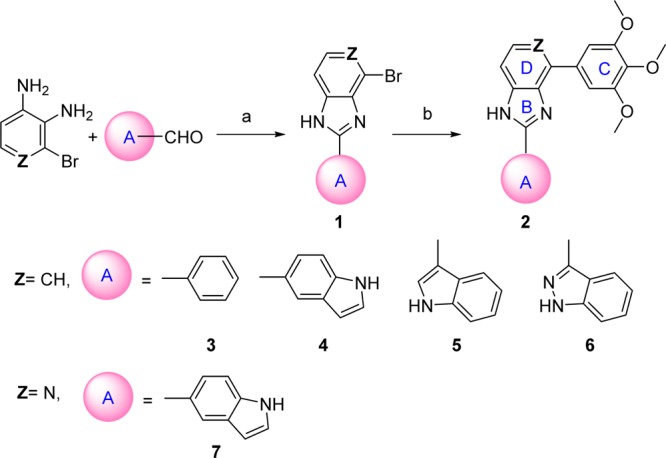
We focused our efforts in preparing three series of modifications. The first two series of compounds were designed based on overcoming two major metabolism-related liabilities: ketone reduction and demethylation in the C ring. As an alternative approach to replacing the carbonyl with other function groups,12 we designed new ring-bioisosteres of the ketone carbonyl (Figure 2). Five analogues in this new series were synthesized as shown in Scheme 1 and their activities were evaluated against both prostate cancer and melanoma cell lines. The synthesis approach included the aldehyde–amine condensation in which the intermediate imidazolidine was oxidized to the imidazoline (1), and this was followed by the Suzuki coupling of (3,4,5-trimethoxyphenyl)boronic acid using Pd(PPh3)4 as a catalyst to give 2.
Scheme 1. Synthesis of the Fused D Ring Antitubulin Compounds.
Reagents and conditions: (a) TsOH, EtOH, reflux; (b) (3,4,5-trimethoxyphenyl)boronic acid, K2CO3, Pd(PPh3)4.
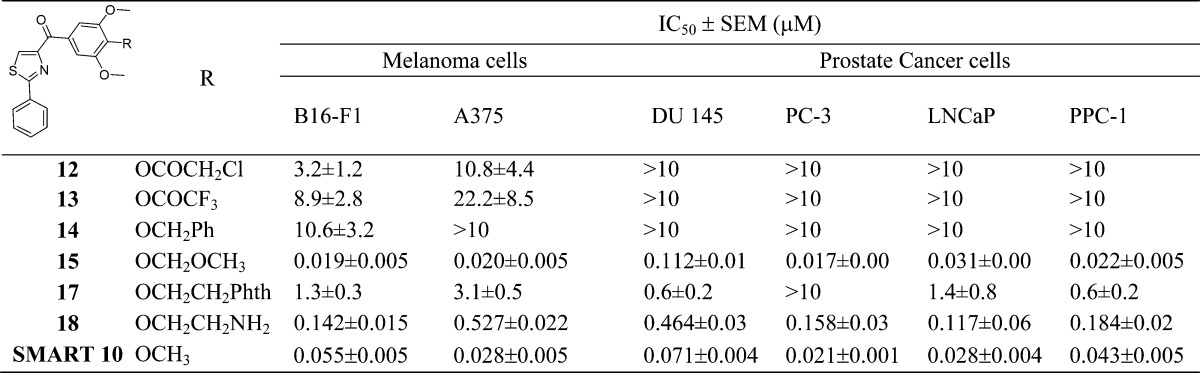
The second aim was focused on modification of the para-position at the C ring. One purpose of this approach is to bypass the potential metabolic problems caused by demethylation. Preliminary modifications of the TMP ring were not successful in our initial studies since the potency was totally lost when single substituted methoxyphenyl (at o-, m-, and p-positions, respectively) or 3,4-dimethoxy substituted phenyl replaced the TMP moiety in the SMART template. Interestingly, 3,5-dimethoxy substituted phenyl maintained a certain level of activity in the 200–400 nM range.10 It indicated that the para-OMe of TMP might be a potential location for further chemical modification. Another important reason for modification at C ring is based on the hypothesis that introducing different functional groups at the para-position of the TMP ring will likely form stronger interactions with Cys-241 in the β-tubulin subunit (Figure 3) and thus increasing the potency of inhibition of tubulin polymerization. Furthermore, the modification of the C ring can help us better understand the potential metabolic demethylation mechanism. We introduced both hydrophobic (OBn, 14, and OMOM, 15) and hydrophilic (OCH2CH2NH2, 18) groups as shown in Scheme 2. Another strategy is coming from the hypothesis that if an alkylating group was introduced at the para-position of the TMP ring, it may form an irreversible covalent bond with the mercapto group of Cys-241 in the colchicine binding domain and induce irreversible mitotic blocks. A well-described mechanism for inhibiting microtubule assembly is a small molecule binding to tubulin via a covalent interaction with a tubulin amino residue. Bai et al.16 reported that 2- and 3-chloroacetyl analogues of dimethylthiocolchicine bound irreversibly to the colchicine binding site primarily with Cys-241 and prevented colchicine binding agents from binding to the same site. The covalent interaction of 2,4-dichlorobenzyl thiocyanate (Figure 3) with tubulin occurs at multiple cysteine residues, especially Cys-241 of β-tubulin.17 Formation of the covalent bond between tubulin and the 2, 4-dichlorobenzyl mercaptan moiety appeared to be reversible. 2-Fluoro-1-methoxy-4-(pentafluorophenyl-sulfonamido)benzene (T138067, Figure 3) irreversibly bound β-tubulin by the thiol group of Cys-241 displacing the para-F atom. It recruits unmodified tubulin dimers into large, amorphous aggregates and thus quickly depletes the pool of tubulin available for microtubule formation.18 On the basis of the above reports, we proposed to modify the template of our tubulin inhibitors by introducing an alkylating functional group to form a covalent bond or enhance the interaction between Cys-241 and TMP ring. Thus, chloroacetic analogue (12) and trifluoroacetate (13) in Scheme 2 were also synthesized and tested.
Figure 3.
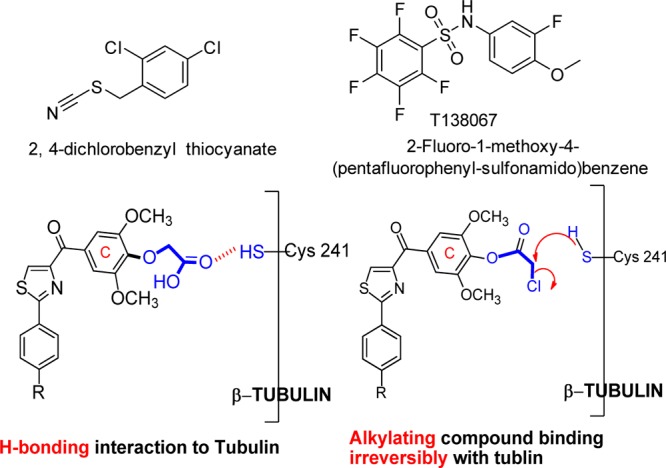
Irreversible tubulin binding agents and hypothesis of interactions between Cys-241 and para-position at the C ring.
Scheme 2. Synthesis of Analogues Focused on Modifications at para-Position of the C Ring.
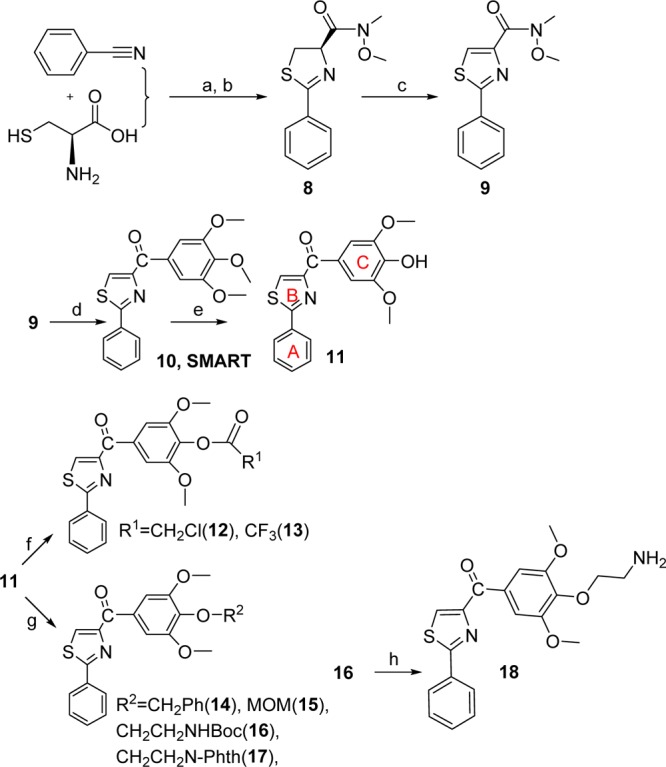
Reagents and conditions: (a) MeOH/pH = 6.4 phosphate buffer, RT; (b) EDCI, HOBt, NMM, CH3OCH3NH·HCl; (c) CBrCl3, DBU; (d) 5-bromo-1,2,3-trimethoxybenzene/BuLi, THF, −78 °C; (e) AlCl3, CH2Cl2; (f) ClCH2COCl, CH2Cl2, NEt3 (12) or (CF3CO)2O, CH2Cl2, DMAP (13); (g) PhCH2Br, K2CO3, DMF (14); MOMCl, Hunig’s base, CH2Cl2 (15); BrCH2CH2NHBoc, DMF, Cs2CO3 (16) or 2-(2-bromoethyl)isoindoline-1,3-dione, K2CO3, DMF 120 °C (17); (h) 4 M HCl in dioxane.
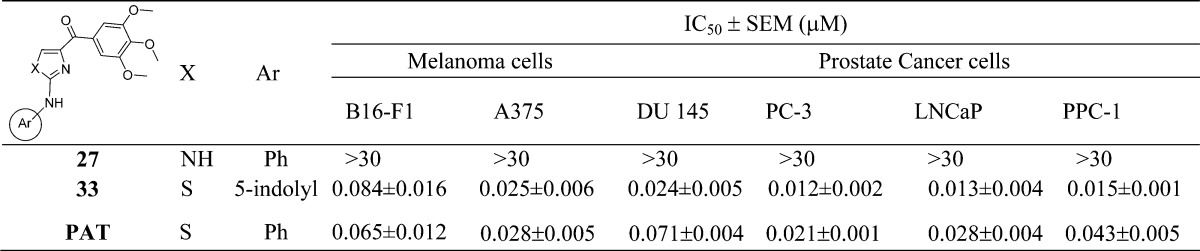
The last aim of modification is based on the “A” ring of PAT template, which we discovered from SMART agents by inserting an amino linker between the “B” and “C” rings. This PAT template increased the oral bioavailability from 3% (SMART series) to 21%.12 Based on our extensive studies on the SMART template, we selected 5-indolyl to be introduced into the bottom A ring and synthesized 33 (Scheme 3). Meanwhile, we replaced the thiazole B ring with imidazole (27) to compare with the parental ABI compound as illustrated in Scheme 3. N-Phenyl-1H-imidazol-2-amine (21) was prepared from amino-acetaldehyde diethyl acetal after three steps. The protections of the imidazole ring with PhSO2 or Boc groups followed by 3,4,5-trimethoxybenzoyl lithium attacking cannot afford the desired target compound while two byproducts 22 and 23 were obtained. Then we chose triphenylmethyl (i.e., trityl or Tri) as a protecting group for the imidazole and prepared two products protected at 4- and N-position (24 and 25) of imidazole. Then the reaction of 24 with 3,4,5-trimethoxybenzoyl lithium followed by deprotection of the trityl generated 27, the imidazole analog of the PAT template.
Scheme 3. Synthesis of Analogues Based on PAT Template.

Reagents and conditions: (a) BrCN, Et2O/hexane; (b) CH3SO3H, EtOH, reflux, 24 h; (c) (1) 6 M HCl, (2) NaOH 25% conc.; (d) PhSO2Cl, Et3N; (e) −78 °C, t-BuLi, 3,4,5-trimethoxybenzoyl chloride; (f) Boc2O, NaOH; (g) (chloromethanetriyl)tribenzene, Et3N, CH2Cl2; (h) HCl; (i) H2, Pd–C, 5%, EtOH, 40 psi; (j) PhCOSCN, Me2CO; (k) MeOH, 1 N NaOH; (l) EtOH, 65 °C; (m) NaOH, MeOH; (n) HBTU, NMM, HNCH3OCH3·HCl, CH2Cl2; (o) 3,4,5-trimethoxyphenylbromide, n-BuLi, THF.
Results and Discussion
Blocking Ketone Reduction by Introducing a New D Ring
In our previous studies,12,15 we made attempts to introduce alternatives to the carbonyl linker in order to avoid potential metabolic problems, but those approaches were unsuccessful. The replacements of the carbonyl linker in the SMART template included double bonds, amides, oximes, hydrazide, acrylonitriles, cyanoimine, sulfonyl amide, sulfur ether, and sulfonyl/sulfinyl compounds but we obtained only limited success. The oxime and hydrazide derivatives demonstrated a 2- to 3-fold improved half-life in human liver microsomes, indicating that metabolic stability of SMART can be extended by blocking ketone reduction. However, these derivatives had less potent antiproliferative activities (micromolar range of IC50). In the current approaches, we designed a new template with the fused D ring (Table 1) on top of the B ring, which maintains the conjugated structure and mimics the carbonyl group but could potentially bypass the ketone reduction. From the proliferative activity data as compared to SMART compound 10, most of the benzo-imidazoles 3–6 showed only moderate activity, except 4, which has a 5-indolyl at the A ring position, showed comparable potency against tested melanoma and prostate cancer cell lines. For further modification, we retained this 5-indolyl at the A ring, utilized pyridine-fused to the imidazole to replace the benzo-imidazole and yielded 7. This compound showed increased potency compared to both parent SMART compound 10 and 4. The IC50 values improved by at least 5-fold against melanoma A375 cells and androgen sensitive prostate cancer LNCaP cells. These novel fused ring templates represented new chemotypes for further optimizing our colchicine binding site inhibitors, which is also expected to remove the potential phase I metabolic reactions caused by ketone reduction.
Table 1. Antiproliferative Activities of Analogues with a Fused D Ring Template.
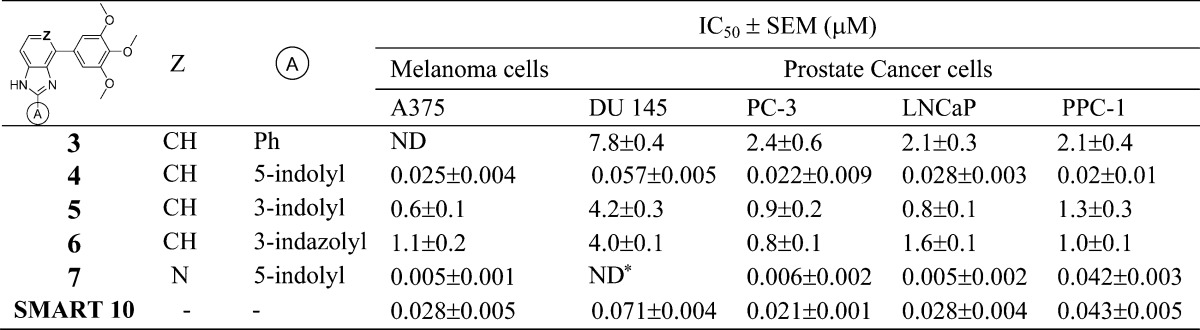
ND: not determined.
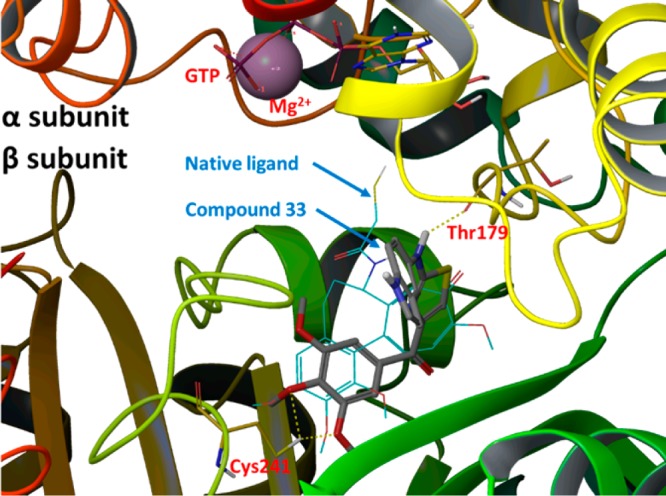
When 4 and 7 were docked into the colchicine binding site in tubulin (Figure 4, PDB code 1SA0), they showed very similar binding poses and overlapped with the native ligand reasonably well. As anticipated, the TMP moiety in 4 or 7 occupied the pocket of the trimethoxy moiety in the native ligand (DAMA–colchicine) but showed some shifting in its position. This slight shift positioned the oxygen atoms in two methoxy groups of the TMP close to Cys-241 of the β-subunit and allowed the formation of two hydrogen bonds (yellow dotted lines). The imidazole NH moiety in 4 or 7 formed another hydrogen bond to Thr-179 in the α-subunit as shown in Figure 4. Interestingly, because of the formation of the new D ring, which forced a planar structure in the middle portion of 4 or 7, the 5-indolyl moiety changed orientation to reach toward the GTP in the α-subunit. The glide docking scores for compounds 4 (−8.58) and 7 (−8.10) were comparable with that of the native ligand, DAMA–colchicine (−9.26), based on this modeling calculation, suggesting they may have comparable effects in tubulin binding.
Figure 4.
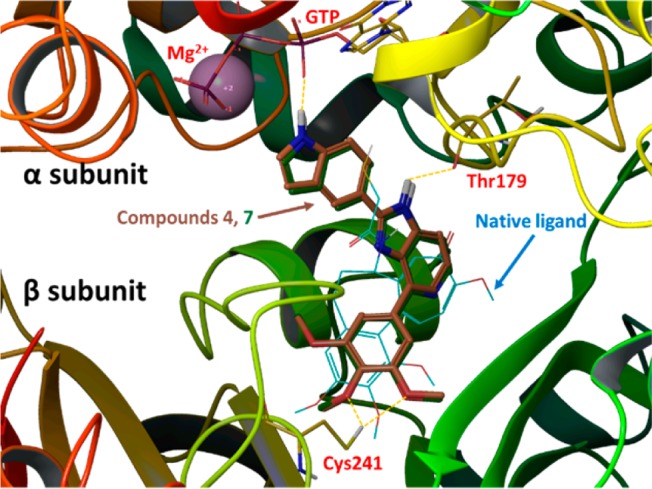
Potential binding poses for 4 (gold tube model; glide docking score −8.58) and 7 (dark green tube model; glide docking score −8.10) in tubulin α,β-dimer (PDB code 1SA0). The native ligand, DAMA–colchicine (glide docking score of −9.26), is shown in blue thin wire model.
C Ring Modification: H-Bonding and Alkylation of the Colchicine Binding Site
The methoxy groups in the TMP ring were known to interact with Cys-241 residue of DAMA–colchicine in the cocrystallized tubulin structure (PDB code 1SA0). We hypothesized that a series of functional groups (R1 and R2) attached to the 4-oxygen atom of the C ring will bind to Cys-241 in β-tubulin. Thus, we designed and synthesized a template that may interact with Cys-241 through either hydrogen bonding or by alkylating at C ring para-position as shown in Scheme 2.
The synthesized new analogues were tested against both melanoma and prostate cancer cells for their antiproliferative activity. Compound 15 showed improved activity compared to the parent SMART compound (i.e., 55 nM (15) vs 19 nM (SMART 10) against B16-F1 cells; Table 2). This discovery encouraged the hypothesis that the para-position of C ring is a tolerant location for further modification. The two atoms extension (i.e., −OCHH2−) of 15 was potent. However, the idea of alkylation at p-position did not work as expected on the inhibition of cancer cell growth. From the result shown in Table 2, the potency of alkylating agent 12 dropped significantly against both melanoma and prostate cancer cells. Compounds 13 and 14 showed similar trends of activity as 12. Compound 17, with a phthalimide protection group, showed micromolar range potency. Introducing an ethyl amine (18) at the p-position remained moderate activity with hundreds of nanomolar IC50s, but it still was less potent than the parent compound SMART.
Table 2. Antiproliferative Activities of Analogues with Modified para-Position of C Ring.
Modifications of the PAT Template
The PAT template was obtained by inserting an amino linker between the A ring and the B ring of the SMART template. This template maintained the potency and improved the oral bioavailability (>30%) compared to SMART (F = 3.3%). The ABI template also showed high potency and improved bioavailability. Thus, we designed to integrate the ABI imidazole ring into the PAT template and obtained 27. However, this new imidazole B ring variant of the PAT compound did not demonstrate activity against any of the tested cell lines. In contrast, the 5-indolyls 4 and 7 showed excellent potency in the first D ring fused analogues and was also introduced into the PAT template to generate 33 (Figure 5). This analogue showed excellent growth inhibition for both prostate cancer and melanoma cells in vitro. The IC50s were increased 2–3-fold on prostate cancer cells compared to the parent PAT compound (Table 3).
Figure 5.
Potential binding poses for 33 (gray tube model; glide docking score of −8.70) and the native ligand DAMA–colchicine (blue thin wire model; glide docking score of −9.26) in tubulin α,β-dimer (PDB Code: 1SA0).
Table 3. Antiproliferative Activities of Modified A Ring on PAT Template.
Molecular modeling studies with 33 (Figure 5) showed three hydrogen bonding interactions between this ligand and the tubulin α,β-dimer, similar to those observed between 4 or 7 and tubulin. However, the 5-indolyl moiety in 33 did not seem to reach the GTP moiety as in 4 or 7, possibly due to the fact that the ketone moiety was not forced into a ring system as seen in 4 or 7. Thus, 33 mainly stays within the β-subunit of tubulin dimer and shows a slightly better glide docking score (−8.70).
In Vitro Metabolic Stability Studies
To determine whether the metabolism of the labile carbonyl linker may be reduced by incorporation into a cyclic structure, we measured the metabolic stability in liver microsomes for two potent compounds (4 and 7). The carbonyl linker in the SMART compound was susceptible to ketone reduction and was replaced by a new D ring in these two newly designed compounds. This modification preserved the potency while improving metabolic stability about 2–3-fold (17 min vs 45 and 51 min in human microsomes, Table 4) compared to the parent SMART compound. Furthermore, the potency of the cyclic D ring compounds 4 and 7 increased. Another active analogue 15 with an extended methoxymethyl (MOM) tail at the para-C ring did not improve its metabolic stability in any of the tested liver microsomes. Another substituent, the aminoethyl of 18, at the same para-O position blocked the metabolic liability of the TMP ring (T1/2 ranged from 110–225 min in the tested liver microsomes species). This result confirmed our hypothesis that the para-position of the C ring could be a modifiable place for improvement of compound stability. However, the selection of functional groups is very important, and it is worth further investigating in future studies.
Table 4. Half-Lives of Tested Compounds in Liver Microsomes of Different Species.
|
T1/2 (min) |
|||
|---|---|---|---|
| compds | human | mouse | rat |
| 4 | 50.7 ± 1.2 | 53.5 ± 2.4 | 72.3 ± 4.6 |
| 7 | 45.3 ± 2.0 | 19.7 ± 0.7 | 30.4 ± 1.9 |
| 15 | 7.8 ± 0.3 | 4.0 ± 0.3 | 9.7 ± 0.4 |
| 18 | 110.0 ± 5.5 | 123.0 ± 7.7 | 225.0 ± 12.6 |
| SMART 10(15) | 17 | ≪5 | 31 |
In Vitro Metabolic Pathways of Compounds 4, 7, 15, and 18
In order to understand why these new analogues demonstrated different metabolic patterns in the liver microsomes, we performed additional experiments using a higher concentration (50 μM) of the tested compounds. We utilized a high resolution mass spectrometer for the identification of the metabolites with a mass error of less than 2 ppm generally. The detailed information regarding the mass spectrum and the chromatogram of each metabolite is presented in the Supporting Information. For compound 15 (Figure 6), the removal of the MOM group to form M1 is the major metabolic pathway (Figure 10A), followed by o-demethylation of the 3′- or 5′-methly group to generate M2. This result is consistent with the short half-life (<10 min) of this compound, as the MOM group seems to be unstable after exposure to liver microsomes. M3 is also the o-demethylation product, however, we were unable to pinpoint the exact site for this demethylation due to limited information available at this stage. M4 is the product resulted from ketone reduction, and it was further hydroxylated to M5 at a position that is currently unidentifiable due to limited information. For compound 18 (Figure 7), M6 (dealkylation) and M8 (ketone reduction) are the major metabolites (Figure 10B) and M7 (deamination) is a minor product. For compound 4 (Figure 8), o-demethylation (M9) and monohydroxylation (M10) are the major products (Figure 10C). M9 and M10 have more than one possible structure as indicated in the chromatograms (Supporting Information). For compound 7 (Figure 9), various metabolites including o-demethylation (M12), monohydroxylation (M14), o-demethylation followed by monohydroxylation (M11), and dihydroxylation (M13) were detected. M14 is the major metabolite (Figure 10D). All of these metabolites have multiple isomeric forms as indicated in the chromatograms (Supporting Information).
Figure 6.

Proposed metabolites and metabolic pathway of 15.
Figure 10.

Kinetics of compounds 15, 18, 4, and 7 and their metabolites in human liver microsomes. (A) Compound 15 and its metabolites; (B) compound 18 and its metabolites; (C) compound 4 and its metabolites; and (D) compound 7 and its metabolites; 50 μM of the test compound was incubated with 1 mg/mL microsomal proteins. Samples at various time points were analyzed by Q-TOF LC–MS.
Figure 7.

Proposed metabolites and metabolic pathway of 18.
Figure 8.

Proposed metabolites and metabolic pathway of 4.
Figure 9.

Proposed metabolites and metabolic pathway of 7.
Compounds Inhibit in Vitro Tubulin Polymerization
We investigated the inhibition of tubulin polymerization of selected potent compounds 4 and 7 with improved metabolic properties and compared them with positive control colchicine and negative control taxol. DMSO was used as a blank control. Bovine brain tubulin (>97% pure) was incubated with the individual compounds (5 or 10 μM) to test their effect on tubulin polymerization (Figure 11). After a 20 min incubation, tubulin polymerization was inhibited to the extent of 30% and 60% by 4 at 5 and 10 μM, respectively (Figure 11A), as compared to vehicle. While about 33% and 81% inhibition was observed for 7 at 5 and 10 μM, respectively (Figure 11B). Both 4 and 7 showed stronger inhibition than colchicine at the two tested concentrations. These data suggest that these compounds exhibit strong antitubulin polymerization activity that corresponds well with their cytotoxicity.
Figure 11.
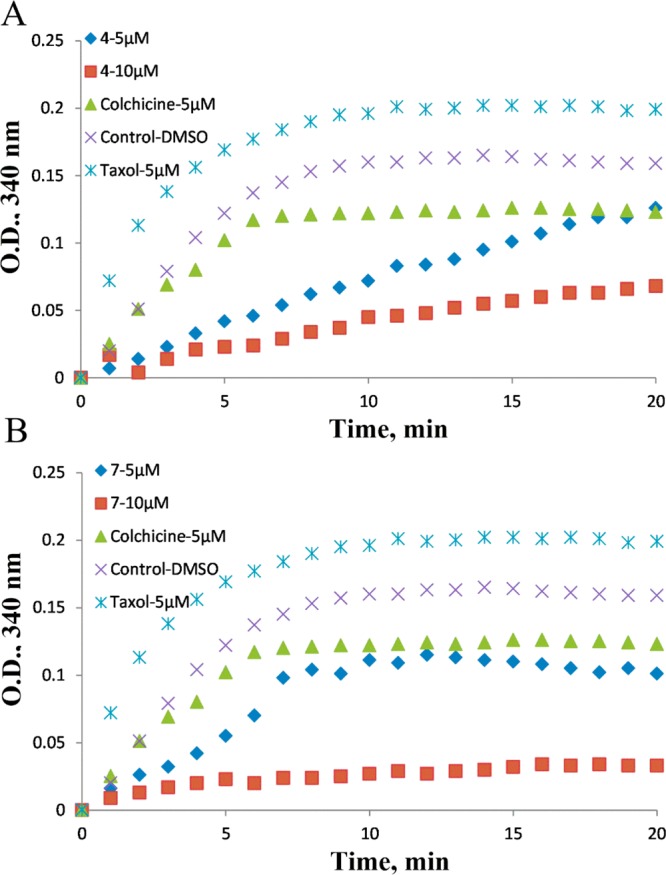
Compounds 4 and 7 inhibit tubulin polymerization in vitro.
Conclusions
In this report, three series of new derivatives targeting modifications of the carbonyl linker, the C ring para-position of the SMART template, and the PAT template were synthesized and screened for their antiproliferative activities. Structure–activity relationships (SAR) of these compounds led to the identification of lead analogues 4 and 7, which showed enhanced anticancer activity in vitro compared to SMART 10 while increasing the metabolic stability on human liver microsomes. Utilizing the MOM group to replace the para-position methoxy on the C ring, which is considered nonreplaceable in many reports, also generated a potent analogue 15, which showed comparable potency to the parent compound 10. Further modification of the PAT template yielded a potent analogue 33 with a 5-indolyl substituent at the A ring.
Experimental Section
General
All reagents were purchased from Sigma-Aldrich Chemical Co., Fisher Scientific (Pittsburgh, PA), AK Scientific (Mountain View, CA), Oakwood Products (West Columbia, SC), etc., and were used without further purification. Moisture-sensitive reactions were carried under an argon atmosphere. Routine thin layer chromatography (TLC) was performed on aluminum backed Uniplates (Analtech, Newark, DE). Melting points were measured with Fisher-Johns melting point apparatus (uncorrected). NMR spectra were obtained on a Bruker ARX 300 (Billerica, MA) spectrometer or Agilent Inova-500 spectrometer. Chemical shifts are reported as parts per million (ppm) relative to TMS in CDCl3. Mass spectral data was collected on a Bruker ESQUIRE electrospray/ion trap instrument in positive and negative ion modes. Elemental analyses were performed by Atlantic Microlab Inc. (Norcross, GA). Unless specified, all the tested compounds described in the article present >95% purity established through combustion analysis.
General Procedure for the Preparation of 3–7
Different aldehydes, 3-bromobenzene-1,2-diamine (3 mmol), p-toluenesulfonic acid (0.3 mmol), and 15 mL of EtOH were refluxed for 24 h under argon atmosphere. The solvent was removed, 25 mL of water was added, and the mixture was extracted with EtOAc (3 × 50 mL). The combined organic layers were dried on MgSO4, filtered, and concentrated in vacuo. The residue was purified by flash chromatography to give the desired 4-bromo-2-substituted-1H-benzo[d]imidazole.
Corresponding bromides obtained from last step (1 equiv), 3,4,5-trimethoxyphenylboronic acid (1 equiv), THF (3 mL)/water (0.3 mL) solution of sodium carbonate (2 equiv), and tetrakistriphenyl phosphinepalladium (0.1 equiv) was refluxed overnight. After adding water to a reaction mixture, it extracted with ethyl acetate. The organic layer was dried on MgSO4, filtered, and concentrated in vacuo. The residue was purified by flash chromatography to give desired fused D ring benzoimidazole compounds 3–6 or imidazo[4,5-c]pyridine (7).
2-Phenyl-4-(3,4,5-trimethoxyphenyl)-1H-benzo[d]imidazole (3)
1H NMR (CDCl3): 3.78, 3.93 (s, s, 6H), 3.91, 3.98 (s, s, 3H), 6.10, 6.82 (s, s, 2H), 7.29–8.08 (m, 8H), 9.70, 9.84 (s, br, 1H). MS (ESI) m/z 359.1 [M – H]−, 361.4 [M + H]+. Anal. (C22H20N2O3) C, H, N.
2-(1H-Indol-3-yl)-4-(3,4,5-trimethoxyphenyl)-1H-benzo[d]imidazole (4)
1H NMR (DMSO-d6): 3.76, 3.97 (s, s, 3H), 3.90, 3.97 (s, s, 6H), 6.96–7.62 (m, H), 7.62, 7.72 (s, s, 2H), 8.16, 8.42 (s, br, 1H), 8.58, 8.66 (d, d, 1H), 11.57, 11.64 (s, s, 1H), 12.16, 12.60 (s, s, 1H). MS (ESI) m/z 398.1 [M – H]−, 400.3 [M + H]+. Anal. (C24H21N3O3) C, H, N.
2-(1H-Indol-5-yl)-4-(3,4,5-trimethoxyphenyl)-1H-benzo[d]imidazole (5)
1H NMR (DMSO-d6): 3.73, 3.87 (s, s, 6H), 3.75, 3.92 (s, s, 3H), 5.70, 5.75 (s, s, 2H), 6.32, 6.49 (s, s, 1H), 6.54, 6.85 (d, d, 1H), 7.22–7.65 (m, 4H), 8.01, 8.42 (br, s, 2H), 11.09, 11.37 (s, s, 1H), 12.36, 12.84 (s, s, 1H). MS (ESI) m/z 398.1 [M – H]−, 400.1 [M + H]+. Anal. (C24H21N3O3) C, H, N.
3-(4-(3,4,5-Trimethoxyphenyl)-1H-benzo[d]imidazol-2-yl)-1H-indazole (6)
1H NMR (DMSO-d6): 3.76 (s, 3H), 3.95 (s, 6H), 6.65 (s, 1H), 6.93 (t, 1H), 7.34–7.34 (m, 2H), 7.45–7.57 (m, 4H), 7.63 (s, 2H), 7.70 (d, 1H), 8.75 (d, 1H), 12.96 (s, 1H), 13.77 (s, 1H). MS (ESI) m/z 399.1 [M – H]−, 401.3 [M + H]+. Anal. (C23H20N4O3) C, H, N.
2-(1H-Indol-5-yl)-4-(3,4,5-trimethoxyphenyl)-1H-imidazo[4,5-c]pyridine (7)
1H NMR (DMSO-d6): 3.77 (s, 3H), 3.96 (s, 6H), 6.61 (s, 1H), 7.46–7.48 (m, 2H), 7.59 (d, 1H), 8.08 (dd, 1H), 8.36 (d, 1H), 8.41 (s, 2H), 8.49 (s, 1H), 11.44(s, 1H), 13.26 (s, 1H). MS (ESI) m/z 399.0 [M – H]−, 401.3 [M + H]+. Anal. (C23H20N4O3) C, H, N.
N-(2,2-Diethoxyethyl)carbodiimide (19)
At 0 °C, to a solution of the aminoacetaldehyde diethyl acetal (5.32 g, 40 mmol) in a mixture of diethyl ether (20 mL) and hexane (20 mL) was added BrCN (4.22 g, 40 mmol). A solid was precipitated from solution. The reaction mixture was magnetically mixed overnight at room temperature. The solid was removed by filtration, and the reaction mixture was concentrated. Flash chromatography of the concentrated residue afforded 2.82 g of the reagent (44.6%). MS (ESI) m/z 156.8 [M – H]−, 180.9 [M + Na]+.
(4-Hydroxy-3,5-dimethoxyphenyl)(2-phenylthiazol-4-yl)methanone (11)
Intermediates 8–9 and SMART 10 were prepared from benzonitrile and cysteine following the same procedure as described in our previous publication.10 Compound 10 (500 mg, 1.4 mmol) was dissolved in CH2Cl2 (50 mL) at RT under argon protection. Anhydrous AlCl3 (374 mg, 2.8 mmol) was added, and the reaction mixture was stirred for 12 h. The reaction was quenched with H2O (30 mL), the organic phase separated, and the aqueous phase extracted with CH2Cl2 (2 × 20 mL). The combined organic phases were washed with brine, dried over Mg2SO4, filtered, and concentrated to dryness under reduced pressure. Compound 11 (410 mg, 85.9% yield) was obtained after flash column purification using hexanes–EtOAc system. 1H NMR (CDCl3): 4.00 (s, 6H), 6.02 (s, 1H), 7.47–7.48 (m, 3H), 7.91 (s, 2H), 8.01–8.03 (m, 2H), 8.27 (s, 1H). MS (ESI) m/z 339.9 [M – H]−, 364.1 [M + Na]+. Anal. (C18H15NO4S) C, H, N.
2,6-Dimethoxy-4-(2-phenylthiazole-4-carbonyl)phenyl 2-Chloroacetate (12)
At 0 °C, 2-chloroacetyl chloride (100 mg, 0.9 mmol) was added to a solution of 11 (100 mg, 0.29 mmol) in CH2Cl2 (30 mL). Then triethylamine (44 mg, 0.44 mmol) was charged in the mixture and stirred until starting material disappeared on TLC. The reaction mixture was quenched with H2O (10 mL), the organic phase separated, and the aqueous phase extracted with CH2Cl2 (2 × 10 mL). The combined organic phases were washed with brine, dried over Mg2SO4, filtered, and concentrated to dryness under reduced pressure. Compound 12 (99 mg, 81.7% yield) was obtained after flash column purification using hexanes–EtOAc system. Mp 147–148 °C. 1H NMR (CDCl3): 3.92 (s, 6H), 4.42 (s, 2H), 7.47–7.49 (m, 3H), 7.82 (s, 2H), 8.00–8.02 (m, 2H), 8.32 (s, 1H). MS (ESI) m/z 418.1 [M – H]−. Anal. (C20H16ClNO5S) C, H, N.
2,6-Dimethoxy-4-(2-phenylthiazole-4-carbonyl)phenyl 2,2,2-Trifluoroacetate (13)
At 0 °C, trifluoroacetyl anhydride (189 mg, 0.9 mmol) was added to a solution of 11 (100 mg, 0.29 mmol) in CH2Cl2 (10 mL). Then DMAP (54 mg, 0.44 mmol) was charged in the mixture and stirred at RT until starting material disappeared on TLC. The reaction mixture was quenched with H2O (10 mL), the organic phase separated, and the aqueous phase extracted with CH2Cl2 (2 × 10 mL). The combined organic phases were washed with brine, dried over Mg2SO4, filtered, and concentrated to dryness under reduced pressure. Compound 13 (89 mg, 70.2% yield) was obtained after flash column purification using hexanes–EtOAc system. Mp 151–153 °C. 1H NMR (CDCl3): 3.94 (s, 6H), 7.48–7.49 (m, 3H), 7.84 (s, 2H), 8.00–8.02 (m, 2H), 8.34 (s, 1H). MS (ESI) m/z 438.1 [M + H]+. Anal. (C20H164F3NO5S) C, H, N.
(4-(Benzyloxy)-3,5-dimethoxyphenyl)(2-phenylthiazol-4-yl)methanone (14)
Under an argon atmosphere, potassium carbonate (49 mg, 0.352 mmol) and benzyl bromide (33 mg, 0.194 mmol) were added to a solution of 11 (60 mg, 0.176 mmol) in 10 mL of dry DMF. The mixture was stirred for 1 h at 100 °C and then transferred into water (10 mL). The compound 14 was extracted with EtOAc, washed with distilled water, dried on magnesium sulfate, and concentrated under vacuum using a rotary evaporator. The crude oily product was purified by flash column, and white solid 14 (51 mg) was obtained. Yield = 67.2%. Mp 119–120 °C. 1H NMR (CDCl3): 3.92 (s, 6H), 5.15 (s, 2H), 7.29–7.37 (m, 3H), 7.48–7.51 (m, 5H), 7.79 (s, 2H), 8.01–8.02 (m, 2H), 8.28 (s, 1H). MS (ESI) m/z 432.1 [M + H]+. Anal. (C25H21NO4S) C, H, N.
(3,5-Dimethoxy-4-(methoxymethoxy)phenyl)(2-phenylthiazol-4-yl)methanone (15)
At 0 °C, MOMCl (27 mg, 0.33 mmol) was added to a solution of 11 (75 mg, 0.22 mmol) in CH2Cl2 (10 mL). Then Hunig’s base (57 mg, 0.44 mmol) was charged in the mixture and stirred at RT until starting material disappeared on TLC. The reaction mixture was quenched with H2O (10 mL), the organic phase separated, and the aqueous phase extracted with CH2Cl2 (2 × 10 mL). The combined organic phases were washed with brine, dried over Mg2SO4, filtered, and concentrated to dryness under reduced pressure. Compound 15 (83 mg, 98.0% yield) was obtained as yellow crystals after flash column purification using hexanes–EtOAc system. Mp 103–104 °C. 1H NMR (CDCl3): 3.62 (s, 3H), 3.95 (s, 6H), 5.26 (s, 2H), 7.47–7.49 (m, 3H), 7.80 (s, 2H), 8.01–8.03 (m, 2H), 8.28 (s, 1H). MS (ESI) m/z 408.1 [M + Na]+. Anal. (C20H19NO5S) C, H, N.
2-(2-(2,6-Dimethoxy-4-(2-phenylthiazole-4-carbonyl)phenoxy)ethyl)isoindoline-1,3-dione (17)
To a solution of 11 (200 mg, 0.59 mmol) and 2-(2-bromoethyl)isoindoline-1,3-dione (223 mg, 0.88 mmol) in DMF (2.5 mL) was added K2CO3 (97 mg, 0.7 mmol), and the reaction mixture was stirred at 120 °C overnight. Then the reaction mixture was quenched in water and extracted with ethyl acetate. The organic layer was concentrated and further purified by column chromatography to get 132 mg of pure desired product 17. Yield = 43.5%. Mp 148–150 °C. 1H NMR (CDCl3) δ 3.71 (s, 6H), 4.14 (t, 2H, J = 5.5 Hz), 4.41 (t, 2H, J = 5.5 Hz), 7.49–7.51 (m, 3H), 7.70 (s, 2H), 7.75 (q, 2H, J = 3.0 Hz), 7.91 (q, 2H, J = 3.0 Hz), 8.01–8.03 (m, 2H), 8.27 (s, 1H). MS (ESI) m/z 537.1 [M + Na]+. Anal. (C28H22N2O6S) C, H, N.
(4-(2-Aminoethyl)-3,5-dimethoxyphenyl)(2-phenylthiazol-4-yl)methanone (18)
To a solution of 11 (23 mg, 0.07 mmol) and tert-butyl (2-bromoethyl)carbamate (23 mg, 0.1 mmol) in DMF (2.5 mL) was added Cs2CO3 (46 mg, 0.2 mmol), and the reaction mixture was stirred for 3 days at RT until TLC showing the reaction had finished. Then the reaction mixture was quenched in ice cold water and extracted with ethyl acetate. The organic layer was concentrated and further purified by column chromatography to get 22 mg of pure desired product tert-butyl (2-(2,6-dimethoxy-4-(2-phenylthiazole-4-carbonyl)phenoxy)ethyl)carbamate 16. Yield = 65.1%. MS (ESI) m/z 483.9 [M – H]−, 485.1 [M + H]+. Boc-protected compound 16 was added to a solution of HCl in dioxane (4 M) and stirred for overnight. The precipitate was collected and washed with diethyl ether to afford HCl salts of compound 18. 1H NMR (acetone-d6): 3.09–3.13 (q, 2H, J = 5.5 Hz), 3.79 (br, 2H), 3.90 (s, 6H), 4.17 (t, 2H, J = 5.5 Hz), 7.55–7.58 (m, 3H), 7.66 (s, 2H), 8.02–8.04 (m, 2H), 8.68 (s, 1H). MS (ESI) m/z 385.1 [M + H]+. Anal. (C20H20N2O3S) C, H, N.
N-Phenyl-1H-imidazol-2-amine (21)
At 0 °C, to a solution of the amino-acetaldehyde diethyl acetal (2.66 g, 20 mmol) in diethyl ether/hexane mixture (20 mL, 1:1) was added BrCN (2.11 g, 20 mmol) in small portions. The reaction mixture was stirred at RT overnight. The solid is removed by filtration and washed with ether. The combined filtrate is concentrated. Purification by flash column chromatography (silica gel, eluting with dichloromethane to 5% methanol in dichloromethane, gradient) affords N-(2,2-diethoxyethyl)carbodiimide 19. 1H NMR 500 MHz (CDCl3): 1.23 (t, 6H, J = 7.0 Hz), 3.16 (t, 2H, J = 6.0 Hz), 3.56 (dt, 2H), 3.64 (br, s, 1H), 3.73 (dt, 2H), 4.58 (t, J = 5.0 Hz, 1H). MS (ESI) m/z 156.8 [M – H]−, 180.9 [M + Na]+. Aniline (1.66 g, 17.8 mmol) was dissolved in ethanol (50 mL), and a solution of 19 (2.82 g, 17.8 mmol) in 5 mL of diethyl ether was added dropwise. Methanesulfonic acid (1.71 g, 17.8 mmol) was then added, and the mixture was refluxed for 24 h. The reaction mixture was poured into NaOH (0.5 M) and extracted with CH2Cl2. Drying with MgSO4 and concentrated in vacuo afforded a product that was subjected to flash chromatography to give the intermediate guanidine 20 (3.3 g, 73.8%). The guanidine (3 g, 12 mmoL) was dissolved in HCl (5 mL, 6 M) at 0 °C and then stirred for 2 h. After the starting material was consumed, NaOH (25%) was added until a precipitate formed (pH 14). This mixture was stirred for 30 min. The reaction was then poured into NaOH (0.5 M), extracted with CH2Cl2, dried, and concentrated. Flash chromatography afforded 21 (1.16 g, 61%). 1H NMR (DMSO-d6): 6.68 (s, 2H), 6.75 (m, 1H), 7.17 (m, 2H), 7.34 (m, 2H), 8.58 (s, 1H). MS (ESI) m/z 157.6 [M – H]−, 160.0 [M + H]+.
3,4,5-Trimethoxy-N-phenyl-N-(1-(phenylsulfonyl)-1H-imidazol-2-yl)benzamide (22)
To a solution of N-phenyl-1H-imidazol-2-amine 21 (40 mg, 0.25 mmol) in CH2Cl2 (10 mL) was added benzenesulfonyl chloride (441 mg, 2.5 mmoL) and triethylamine (252 mg, 2.5 mmol). Reaction mixture was stirred overnight at room temperature. The reaction mixture was quenched by sat. NH4Cl and extracted with CH2Cl2. Drying with MgSO4 and concentrated in vacuo afforded a product that was subjected to flash chromatography to give a benzenesulfonyl protected intermediate (79 mg, 72%). This intermediate was dissolved in THF and cooled down to −78 °C, and then t-BuLi (1.7 M) was charged under Ar2. After stirred for an hour, 3,4,5-trimethoxybenzoyl chloride (47 mg, 0.26 mmoL) was added and stirred overnight. The reaction mixture was poured into NH4Cl (sat.) and extracted with ethyl acetate. Drying with MgSO4 and concentrated in vacuo afforded a crude product that was purified by flash chromatography to give 22 (35%). 1H NMR (CDCl3): 3.78 (s, 6H), 3.87 (s, 3H), 6.91 (s, 2H), 6.97 (s, 1H), 7.18 (m, 2H), 7.20 (d, 1H), 7.25 (m, 2H), 7.38 (m, 2H), 7.40 (d, 1H), 7.54 (br, 1H), 7.59 (t, 2H). MS (ESI) m/z 491.9 [M – H]−, 516.1 [M + Na]+.
N-(1H-Imidazol-2-yl)-3,4,5-trimethoxy-N-phenylbenzamide (23)
To a solution of N-phenyl-1H-imidazol-2-amine 21 (900 mg, 5.66 mmoL) in dioxane and water (30 mL, 3:1) was added Boc2O (2.68 g, 12.3 mmol) and NaOH (0.6 g, 15 mmol) and stirred for 4 h. The mixture was concentrated in vacuo, and the residue was purified by flash chromatography to obtain the Boc protected intermediate. This intermediate (130 mg, 0.502 mmol) was dissolved in THF and cooled down to −78 °C, and then t-BuLi (0.65 mL, 1.7 M, 1.1 mmol) was charged under Ar2. After stirred for an hour, 3,4,5-trimethoxybenzoyl chloride (116 mg, 0.502 mmoL) was added and stirred overnight. The reaction mixture was poured into NH4Cl (Sat.) and extracted with ethyl acetate. Drying with MgSO4 and concentrated in vacuo afforded a crude product that was purified by flash chromatography to give 23 (35%). 1H NMR (CDCl3): 3.65 (s, 6H), 3.79 (s, 3H), 6.56 (s, 2H), 6.90 (m, 2H), 7.27–7.39 (m, 5H), 11.17 (br, 1H). MS (ESI) m/z 351.8 [M – H]−, 376.3 [M + Na]+.
N-Phenyl-4-trityl-1H-imidazol-2-amine (24) and N-Phenyl-1-trityl-1H-imidazol-2-amine (25)
To a solution of N-phenyl-1H-imidazol-2-amine (159 mg, 10 mmoL) in triethylamine and CH2Cl2 stirring under an inert atmosphere at 0 °C was added (chloromethanetriyl)tribenzene (5 equiv). The solution was allowed to warm to RT and stir until complete by TLC. The reaction mixture was then concentrated in vacuo, quenched with saturated aqueous sodium bicarbonate, and extracted with ethyl acetate. Then dried with magnesium sulfate and concentrated in vacuo. The resulting residue is purified by flash chromatography to give two different protected products. Compound 24, 1H NMR (DMSO-d6): 6.0 (s, 1H), 6.75 (m, 1H), 7.29–7.62 (m, 19H), 8.65 (s, 1H), 10.62 (s, 1H). MS (ESI) m/z 399.9 [M – H]−, 403.1 [M + H]+. Compound 25, 1H NMR (DMSO-d6): 6.08 (s, 1H), 6.41 (s, 1H), 6.85 (s, 1H), 7.13–7.52 (m, 20H), 8.65 (s, 1H), 10.62 (s, 1H). MS (ESI) m/z 399.8 [M – H]−, 402.8 [M + H]+.
(2-(Phenylamino)-1-trityl-1H-imidazol-4-yl)(3,4,5-trimethoxyphenyl)methanone (26)
To a solution of N-phenyl-1-trityl-1H-imidazol-2-amine 25 (116 mg, 0.289 mmol) in THF (10 mL) stirring under an inert atmosphere at −78 °C was added t-BuLi (0.34 mL, 1.7 M, 0.58 mmol) and trimethoxybenzoyl chloride (66.5 mg, 0.289 mmol). The reaction mixture was reacted for overnight, then quenched by NH4Cl (sat.) and extracted with ethyl acetate. Drying with MgSO4 and concentrated in vacuo afforded a crude product that was purified by flash chromatography to give 26 (75 mg, 43.7%). 1H NMR (DMSO-d6): 3.71, (s, 3H), 3.78 (s, 6H), 5.87 (s, 1H), 6.94 (s, 2H), 7.18–7.58 (m, 21H). MS (ESI) m/z 594.2 [M – H]−, 596.3 [M + H]+.
(2-(Phenylamino)-1H-imidazol-4-yl)(3,4,5-trimethoxyphenyl)methanone (27)
(2-(Phenylamino)-1-trityl-1H-imidazol-4-yl)(3,4,5-trimethoxyphenyl)methanone was dissolved in a solution of HCl in diethyl ether (2 M) and stirred overnight. Saturated NaHCO3 solution is then added, and the reaction mixture is extracted three times with ether. The combined organic layers are dried (sodium sulfate), filtered, and concentrated in vacuo. The residue is purified by flash chromatography to give pure 27. 1H NMR (DMSO-d6): 3.73 (s, 3H), 3.82 (s, 6H), 6.62 (s, 2H), 7.02 (s, 2H), 7.33 (d, 2H), 7.43–7.51 (m, 3H), 7.54 (br, 1H). MS (ESI) m/z 352.1 [M – H]−, 354.3 [M + H]+. Anal. (C19H19N3O4) C, H, N.
N-((1H-Indol-5-yl)carbamothioyl)benzamide (28)
A mixture of 5-nitro-1H-indole (11 g, 67.9 mmol) and Pd/C (5%; 1 g), dissolved in ethanol (50 mL), was hydrogenated for 3 h at 40 psi. The reaction mixture was filtered, and the excess of ethanol was evaporated under reduced pressure. Solid product was recrystallized from hexane to obtain the pure compound 5-aminoindole. Yield: 92.5%. 1H NMR (500 MHz, CDCl3): δ 3.50 (s, 2 H), 6.37 (s, 1 H), 6.67 (dd, 1 H), 6.95 (s, 1 H), 7.13 (s, 1 H), 7.20 (d, 1 H), 7.96 (br, 1 H). MS (ESI) m/z 133.0 (M + H)+. A solution of 5-aminoindole (8 g, 60.6 mmol) in acetone (150 mL) was reacted with benzoylisothiocyanate (9.88 g, 60. mmol) at RT for about 4 h until TLC showed that the reaction finished to yield compound 28. 1H NMR (CDCl3): δ 6.61 (br, 1 H), 7.26–7.28 (d, 1H), 7.38–7.45 (m, 2H), 7.54–7.59 (m, 2H), 7.65–7.70 (m, 1 H), 7.91–7.94 (m, 2 H), 7.98 (s, 1 H), 8.27 (s, br, 1 H), 9.12 (s, 1 H), 12.51 (s, 1 H). MS (ESI) m/z 318.1 [M + Na]+.
2-(1H-Indol-5-ylamino)-N-methoxy-N-methylthiazole-4-carboxamide (32)
The resulting solid 28 was filtered and treated with 2 N NaOH in THF (120 mL). The mixture was refluxed for about 6 h and allowed to warm to RT. The solvent was evaporated off under vacuum. The residue was diluted with water (20 mL) and neutralized to pH 7 with 1 N HCl. The resulting solid was filtered and dried under vacuum to afford 5-indolylthiourea (29). Compound 29 (0.01 mol) and ethyl bromopyruvate (0.011 mol) were dissolved in 3 mL of ethanol and held at reflux for 2 h. The reaction was cooled, and the crystalline ethyl 2-(1H-indol-5-ylamino)thiazole-4-carboxylate (30) was collected by filtration and washed with ethanol. Refluxing the mixture of ethyl esters with the NaOH–ethanol solution gave 2-(1H-indol-5-ylamino)thiazole-4-carboxylic acid (31), which was used directly in the next steps. To a mixture of the crude acid (2.5 mmol), HBTU (2.6 mmol), and NMM (5.3 mmol) in CH2Cl2 (30 mL) was added HCl salt of HNCH3OCH3 (2.6 mmol), and stirring continued at RT for overnight. The reaction mixture was diluted with CH2Cl2 (20 mL) and sequentially washed with water, satd. NaHCO3, and brine and dried over MgSO4. The solvent was removed under reduced pressure to yield a crude product, which was purified by column chromatography to obtain pure Weinreb amide 2-(1H-indol-5-ylamino)-N-methoxy-N-methylthiazole-4-carboxamide (32) (45.6% yield for overall 5 steps). 1H NMR (CDCl3): 3.42 (s, 3H), 3.77 (s, 3H), 6.54 (m, 1H), 7.26 (m, 1H), 7.29 (m, 2H), 7.40 (d, 2H), 7.61 (m, 1H), 8.30 (br, 1H). MS (ESI) m/z 303.0 [M + H]+.
(2-((1H-Indol-5-yl)amino)thiazol-4-yl)(3,4,5-trimethoxyphenyl)methanone (33)
At −78 °C, a solution of 5-bromo-1,2,3-trimethoxybenzene (1.235 g, 5.0 mmol) in 30 mL of THF was charged with n-BuLi in hexane (2.5 N, 2.4 mL, 6 mmol) under Ar2 protection and stirred for 10 min. Weinreb amide 32 (1 mmol) in 10 mL of THF was added to lithium reagent and allowed to stir at RT for 2 h. The reaction mixture was quenched with satd. NH4Cl, extracted with ethyl ether, and dried with MgSO4. The solvent was removed under reduced pressure to yield a crude product, which was purified by column chromatography to obtain pure compound 33 (51.7% yield). 1H NMR (CDCl3) δ 3.89 (s, 6 H), 3.93 (s, 3 H), 6.55 (m, 1 H), 7.15–7.12 (m, 1 H), 7.28–7.26 (m, 1 H), 7.36 (s, 1 H), 7.39 (s, 1 H), 7.46 (s, 2 H), 7.68 (d, 1 H), 8.29 (br, 1 H). MS (ESI) m/z 432.1 (M + Na)+, 408.0 (M – H)−. Anal. (C23H19N3O4S) C, H, N.
Molecular Modeling
The molecular modeling studies were performed with the published crystal structures of the α,β-tubulin dimer in complex with DAMA–colchicine (Protein Data Bank code 1SA0). Schrodinger Molecular Modeling Suite 2013 (Schrodinger Inc., Portland, OR) was used for the modeling studies with procedures similar to those described before.11,19 Briefly, the structures of the protein–ligand complexes were prepared using the Protein Preparation module, and the active ligand binding sites were defined based on the native ligand. Both native ligand DAMA–colchicine and the designed tubulin inhibitors described in this study were built and prepared for docking using the Ligprep module before they were docked into 1SA0. The Glide docking score obtained from this modeling approach is an estimation of the binding energy (kcal/mol) when a ligand binds to the tubulin dimer. A lower (more negative) number suggests more favorable binding interaction between a ligand and the receptor. Data analyses were performed using the Maestro interface of the software.
Cell Culture and Cytotoxicity Assay of Prostate Cancer and Melanoma
All cell lines were obtained from ATCC (American Type Culture Collection, Manassas, VA, USA), while cell culture supplies were purchased from Cellgro Mediatech (Herndon, VA, USA). We examined the antiproliferative activity of our antitubulin compounds in four human prostate cancer cell lines (LNCaP, DU 145, PC-3, and PPC-1) and three melanoma cell lines (A375, B16-F1, and WM-164). All prostate cancer cell lines were cultured in RPMI 1640, supplemented with 10% fetal bovine serum (FBS). Melanoma cells were cultured in DMEM, supplemented with 5% FBS, 1% antibiotic/antimycotic mixture (Sigma-Aldrich, Inc., St. Louis, MO, USA), and bovine insulin (5 μg/mL; Sigma-Aldrich). The cytotoxic potential of the antitubulin compounds was evaluated using the sulforhodamine B (SRB) assay after 96 h of treatment.
In Vitro Tubulin Polymerization Assay
Bovine brain tubulin (0.4 mg, >97% pure) (Cytoskeleton, Denver, CO) was mixed with 10 μM of the test compounds and incubated in 100 μL of general tubulin buffer (80 mM PIPES, 2.0 mM MgCl2, 0.5 mM EGTA, and 1 mM GTP) at pH 6.9. The absorbance of wavelength at 340 nm was monitored every 1 min for 20 min by the SYNERGY 4 Microplate Reader (Bio-Tek Instruments, Winooski, VT). The spectrophotometer was set at 37 °C for tubulin polymerization.
Microsomal Stability Assay
Metabolic stability studies were performed by incubating the test compounds (0.5 μM) in a total reaction volume of 1.2 mL containing 1 mg/mL microsomal protein in reaction buffer [0.2 M of phosphate buffer solution (pH 7.4), 1.3 mM NADP+, 3.3 mM glucose-6-phosphate, and 0.4 U/mL glucose-6-phosphate dehydrogenase] at 37 °C in a shaking incubator.12 Pooled human liver microsomes were utilized to examine metabolic stability. The NADPH regenerating system (solution A and B) was obtained from Xenotech, LLC (Lenexa, KS). Aliquots (100 μL) from the reaction mixtures to determine metabolic stability were sampled at 5, 10, 20, 30, 60, and 90 min. Acetonitrile (200 μL) containing 200 nM of the internal standard was added to quench the reaction and to precipitate the proteins. Samples were then centrifuged at 10 000 rpm for 15 min at RT, and the supernatant was analyzed directly by LC–MS/MS (AB Sciex API4500). For metabolite identification, the reaction mixture was incubated for 2 h with 50 μM test compound concentration under the previously described conditions.20 The supernatants were analyzed using a Water Xevo G2-S high resolution mass spectrometer.
Acknowledgments
This research was supported by the Van Vleet Endowed Professorship (to D.D.M.) and NIH grants R01CA148706 and 1S10OD010678-01 (to W.L.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. We thank Dr. Christopher Coss for his help with the data collection at GTx, Inc. We thank Dr. Michael Mohler at GTx, Inc., for his proofreading and editorial assistance.
Glossary
Abbreviations Used
- ABI
2-aryl-4-benzoyl-imidazole
- Boc2O
tert-butyl dicarbonate
- CA-4
combretastatin A-4
- DMF
dimethylformamide
- DMSO
dimethyl sulfoxide
- EDCI
1-ethyl-3-(3-(dimethylamino)propyl)carbodiimide
- HOBt
hydroxybenzotriazole
- MOM
methoxymethyl
- NMM
N-methylmorpholine
- NMR
nuclear magnetic resonance
- PAT
phenyl amino thiazole
- P-gp
P-glycoprotein
- PK
pharmacokinetic
- RT
room temperature
- Phth
phthalimide
- SMART
4-substituted methoxybenzoyl-aryl-thiazole
- THF
tetrahydrofuran
- TMP
3,4,5-trimethoxyphenyl
- TsOH
p-toluenesulfonic acid
Supporting Information Available
High resolution mass spectra of the metabolites. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Jordan M. A.; Wilson L. Microtubules as a target for anticancer drugs. Nat. Rev. Cancer 2004, 44253–265. [DOI] [PubMed] [Google Scholar]
- Lu Y.; Chen J.; Xiao M.; Li W.; Miller D. D. An overview of tubulin inhibitors that interact with the colchicine binding site. Pharm. Res. 2012, 29112943–2971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nam N. H. Combretastatin A-4 analogues as antimitotic antitumor agents. Curr. Med. Chem. 2003, 10171697–1722. [DOI] [PubMed] [Google Scholar]
- Pettit G. R.; Toki B.; Herald D. L.; Verdier-Pinard P.; Boyd M. R.; Hamel E.; Pettit R. K. Antineoplastic agents. 379. Synthesis of phenstatin phosphate. J. Med. Chem. 1998, 41101688–1695. [DOI] [PubMed] [Google Scholar]
- Kuo C. C.; Hsieh H. P.; Pan W. Y.; Chen C. P.; Liou J. P.; Lee S. J.; Chang Y. L.; Chen L. T.; Chen C. T.; Chang J. Y. BPR0L075, a novel synthetic indole compound with antimitotic activity in human cancer cells, exerts effective antitumoral activity in vivo. Cancer Res. 2004, 64134621–4628. [DOI] [PubMed] [Google Scholar]
- Sriram M.; Hall J. J.; Grohmann N. C.; Strecker T. E.; Wootton T.; Franken A.; Trawick M. L.; Pinney K. G. Design, synthesis and biological evaluation of dihydronaphthalene and benzosuberene analogs of the combretastatins as inhibitors of tubulin polymerization in cancer chemotherapy. Bioorg. Med. Chem. 2008, 16178161–8171. [DOI] [PubMed] [Google Scholar]
- Liu X.; Go M. L. Antiproliferative properties of piperidinylchalcones. Bioorg. Med. Chem. 2006, 141153–163. [DOI] [PubMed] [Google Scholar]
- Burns R. G. Analysis of the colchicine-binding site of beta-tubulin. FEBS Lett. 1992, 2973205–208. [DOI] [PubMed] [Google Scholar]
- a Alvarez C.; Alvarez R.; Corchete P.; Perez-Melero C.; Pelaez R.; Medarde M. Exploring the effect of 2,3,4-trimethoxy-phenyl moiety as a component of indolephenstatins. Eur. J. Med. Chem. 2010, 452588–97. [DOI] [PubMed] [Google Scholar]; b Brancale A.; Silvestri R. Indole, a core nucleus for potent inhibitors of tubulin polymerization. Med. Res. Rev. 2007, 272209–38. [DOI] [PubMed] [Google Scholar]; c Ray K.; Bhattacharyya B.; Biswas B. B. Role of B-ring of colchicine in its binding to tubulin. J. Biol. Chem. 1981, 256126241–6244. [PubMed] [Google Scholar]
- Lu Y.; Li C. M.; Wang Z.; Ross C. R. II; Chen J.; Dalton J. T.; Li W.; Miller D. D. Discovery of 4-substituted methoxybenzoyl-aryl-thiazole as novel anticancer agents: synthesis, biological evaluation, and structure-activity relationships. J. Med. Chem. 2009, 5261701–1711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J.; Li C. M.; Wang J.; Ahn S.; Wang Z.; Lu Y.; Dalton J. T.; Miller D. D.; Li W. Synthesis and antiproliferative activity of novel 2-aryl-4-benzoyl-imidazole derivatives targeting tubulin polymerization. Bioorg. Med. Chem. 2011, 19164782–4795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C. M.; Chen J.; Lu Y.; Narayanan R.; Parke D. N.; Li W.; Ahn S.; Miller D. D.; Dalton J. T. Pharmacokinetic optimization of 4-substituted methoxybenzoyl-aryl-thiazole and 2-aryl-4-benzoyl-imidazole for improving oral bioavailability. Drug Metab. Dispos. 2011, 39101833–1839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z.; Chen J.; Wang J.; Ahn S.; Li C. M.; Lu Y.; Loveless V. S.; Dalton J. T.; Miller D. D.; Li W. Novel tubulin polymerization inhibitors overcome multidrug resistance and reduce melanoma lung metastasis. Pharm. Res. 2012, 29113040–3052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C. M.; Wang Z.; Lu Y.; Ahn S.; Narayanan R.; Kearbey J. D.; Parke D. N.; Li W.; Miller D. D.; Dalton J. T. Biological activity of 4-substituted methoxybenzoyl-aryl-thiazole: an active microtubule inhibitor. Cancer Res. 2011, 711216–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C. M.; Lu Y.; Narayanan R.; Miller D. D.; Dalton J. T. Drug metabolism and pharmacokinetics of 4-substituted methoxybenzoyl-aryl-thiazoles. Drug Metab. Dispos. 2010, 38112032–2039. [DOI] [PubMed] [Google Scholar]
- Bai R.; Covell D. G.; Pei X. F.; Ewell J. B.; Nguyen N. Y.; Brossi A.; Hamel E. Mapping the binding site of colchicinoids on beta-tubulin. 2-Chloroacetyl-2-demethylthiocolchicine covalently reacts predominantly with cysteine 239 and secondarily with cysteine 354. J. Biol. Chem. 2000, 2755140443–40452. [DOI] [PubMed] [Google Scholar]
- Bai R. L.; Lin C. M.; Nguyen N. Y.; Liu T. Y.; Hamel E. Identification of the cysteine residue of beta-tubulin alkylated by the antimitotic agent 2,4-dichlorobenzyl thiocyanate, facilitated by separation of the protein subunits of tubulin by hydrophobic column chromatography. Biochemistry 1989, 28135606–5612. [DOI] [PubMed] [Google Scholar]
- Shan B.; Medina J. C.; Santha E.; Frankmoelle W. P.; Chou T. C.; Learned R. M.; Narbut M. R.; Stott D.; Wu P.; Jaen J. C.; Rosen T.; Timmermans P. B.; Beckmann H. Selective, covalent modification of beta-tubulin residue Cys-239 by T138067, an antitumor agent with in vivo efficacy against multidrug-resistant tumors. Proc. Natl. Acad. Sci. U.S.A. 1999, 96105686–5691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Slominski A. T.; Kim T. K.; Li W.; Yi A. K.; Postlethwaite A.; Tuckey R. C. The role of CYP11A1 in the production of vitamin D metabolites and their role in the regulation of epidermal functions. J. Steroid Biochem. Mol. Biol. 2013, 10.1016/j.jsbmb.2013.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Xiao M.; Ahn S.; Wang J.; Chen J.; Miller D. D.; Dalton J. T.; Li W. Discovery of 4-aryl-2-benzoyl-imidazoles as tubulin polymerization inhibitor with potent antiproliferative properties. J. Med. Chem. 2013, 5683318–3329. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Chen J.; Ahn S.; Wang J.; Lu Y.; Dalton J. T.; Miller D. D.; Li W. Discovery of novel 2-aryl-4-benzoyl-imidazole (ABI-III) analogues targeting tubulin polymerization as antiproliferative agents. J. Med. Chem. 2012, 55167285–7289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J.; Wang J.; Kim T. K.; Tieu E. W.; Tang E. K.; Lin Z.; Kovacic D.; Miller D. D.; Postlethwaite A.; Tuckey R. C.; Slominski A. T.; Li W. Novel vitamin d analogs as potential therapeutics: metabolism, toxicity profiling, and antiproliferative activity. Anticancer Res. 2014, 3452153–2163. [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.