

Abstract
We present an enhanced version of the FLAMEnGO (Fuzzy Logic Assignment of Methyl Group) software, a structure-based method to assign methyl group resonances in large proteins. FLAMEnGO utilizes a fuzzy logic algorithm coupled with Monte Carlo sampling to obtain a probability-based assignment of the methyl group resonances. As an input, FLAMEnGO requires the protein X-ray structure or an NMR structural ensemble with data such as methyl-methyl NOESY, paramagnetic relaxation enhancement (PRE), methine-methyl TOCSY data. Version 2.0 of this software (FLAMEnGO 2.0) has a user-friendly graphic interface and presents improved modules that enable the input of partial assignments and additional NMR restraints. We tested the performance of FLAMEnGO 2.0 on maltose binding protein (MBP) and the C-subunit of the cAMP-dependent protein kinase A (PKA-C). FLAMEnGO 2.0 can be used as a standalone method or to assist in the completion of partial resonance assignments and can be downloaded at www.chem.umn.edu/groups/veglia/forms/flamengo2-form.html.
Keywords: FLAMEnGO 2.0, Automatic assignment of methyl groups, Methyl-TROSY, Sparse and ambiguous data
1. Introduction
Recent development of methyl-TROSY based methodology on selectively protonated methyl groups in a highly deuterated background has significantly expanded the molecular weight range in which high-resolution NMR studies can be performed [1-3]. However, such studies require methyl group resonance assignments to provide site-specific probes to analyze structure, conformational dynamics, and molecular interactions. For small and medium size proteins, through-bond assignment strategies have been developed to correlate backbone carbon chemical shifts with methyl chemical shifts, either via TOCSY- or COSY-type transfer schemes [4-6]. For large proteins, however, these experiments are often insensitive. To assign the methyl groups in these cases, one can utilize extensive site-specific mutagenesis [7,8]. Alternatively, it is possible to apply a “divide and conquer” approach, where a protein's domains are expressed individually, making it more amenable to current assignment procedures [9]. The above methods, however, are cumbersome and time consuming.
To address this problem, we developed a new methodology based on a previous algorithm proposed by the Matthews laboratory, MAP-XS [10,11]. MAP-XS utilizes a pre-existing protein X-ray structure and exploits methyl-methyl NOESY networks to assign the methyl group resonances. Building on this approach, we previously developed a new and more efficient algorithm based on fuzzy logic and Monte Carlo sampling (FLAMEnGO or Fuzzy Logic Assignment of Methyl Group) that dramatically improved the accuracy (correctness) of the resonance assignment [12].
Here, we report version 2.0 of FLAMEnGO (version 2.0) with new and improved modules that incorporate methyl-methyl NOESY, paramagnetic relaxation enhancement (PRE), and methine-methyl TOCSY data, as well as partial assignments obtained from mutagenesis or through-bond experiments. FLAMEnGO 2.0 was tested on maltose binding protein (MBP) and then applied to the more challenging C subunit of the cAMP-dependent protein kinase A (PKA-C). For the kinase, the application of FLAMEnGO 2.0 resulted in 100% assignment of Ile, 95% of Leu, and 100% of Val residue methyl groups. Overall, 98% of total methyl groups were assigned; ∼80% of these were found to be correct when compared to the assignment obtained using through-bond correlation experiments. The assignments obtained with FLAMEnGO 2.0 and sparse data are validated using methyl resonance assignments through-bond correlation experiments.
2. Material and methods
2.1. Protein expression and sample preparation
Expression and purification of PKA-C and Rllα (R213 K) subunit were carried out as described previously [13,14]. Briefly, cell pellets of catalytic subunit and Rllα(R213 K) were combined and lysed in buffer A (30 mM Mops, 200 μM ATP, 15 mM MgCl2, 5 mM 2-mercaptoethanol, pH 8). After removing cell debris by centrifuga-tion, the supernatant was incubated overnight with HIS-Select Ni2+ affinity gel (SIGMA, 1 ml resin/1L culture) at 4 °C. After incubation, the flow-through was collected and buffer B (buffer A containing 25 mM KCI) was used to wash the Ni2+ resin. Finally, PKA-C was eluted using elution buffer A (buffer B containing 1 mM cAMP), and the Rllα subunit was eluted using buffer B (buffer A containing 250 mM imidazole). The three isoforms of PKA-C were further separated by HiTrap SP column (GE) chromatography, with a gradient from buffer A (20 mM KH2PO4, pH 6.5) to 30% buffer B (20 mM KH2PO4, 1.0 M KCl, pH 6.5) and a flow rate of 2.0 mL/ min. The ternary complex PKA-C/AMPPNP/PKI5-4 was prepared using 12 mM of AMP-PNP in NMR buffer (20 mM KH2PO4, 90 mM KCl, 60 mM MgCl2, 10 mM DTT, pH 6.5). The final concentration of PKA-C was about 0.5 mM.
2.2. NMR spectroscopy
All NMR experiments were carried out on Bruker Avance III spectrometers operating at 850 or 900 MHz 1H frequencies. The temperature was held constant at 27 °C. To map the methyl group fingerprint, we used constant-time (CT) [1H, 13C]-HMQC with wild-type PKA-C selectively labeled with [15N,13C]-Leu or [15N,13C]-Val., To obtain the PRE restraints, PKA-C mutants (K16C and I244C) were expressed in 15N labeled M9 medium supplemented with 70 mg/L of 13Cme α-ketobutyrate and 90 mg/L of 13Cme α-ketois-ovalerate. Approximately 200 μM sample solutions were reacted with (1-Oxyl-2,2,5,5-tetramethylpyrroline-3-methyl) methane-thiosulfonate, MTSSL, (10 times the protein concentration) and incubated for 3 h at 4 °C. The MTSSL excess was removed by filtration. To quantify the PREs, two separate experiments were performed with both oxidized and reduced kinase as described previously [12].
To obtain the NOESY restraints, wild-type PKA-C was expressed in 15N labeled deuterated M9 medium (80% 2H2O and 100% 2H-glu-cose) supplemented with 70 mg/L of 2-ketobutyric acid-4-13C,3,3-d2 and 90 mg/L of 2-keto-3-(methyl-d3)-butyric acid-4-13C. The final concentration of the enzyme was ∼0.5 mM and experiments were performed at 27 °C. The optimal mixing time (0.25 s) was chosen after analyzing the 2D NOESY build-up at different mixing times ranging from 0.1 to 0.5 s. The 3D 13C-13C-1H HMQ.C-NOESY-HMQC was collected with 32 scans using 2048 (proton), 80 (carbon), and 120 (carbon) complex points. For the methine-methyl TOCSY restraints, we used the same sample and carried out a 3D TOCSY-HMQC experiment to correlates intra-residue methine proton resonances with the methyl groups. The 3D TOCSY-HMQC spectrum was carried out using a 50 ms mixing time and 16 scans, with 2048 (proton), 90 (carbon), and 136 (proton) complex points.
For the backbone triple resonance and HMCM(CG)CBCA experiments, the kinase was expressed in 15N labeled deuterated M9 medium (80% 2H2O and 100% 2H7,13C6-glucose) supplemented with 70 mg/L of 2-ketobutyric acid-13C4,3,3-d2 and 90 mg/L of 2-keto-3-methyl-d3-3-d1-13C-butyric acid. The final sample concentration was ∼0.8 mM. TROSY-based HNCA and HN(CO)CA experiments were collected with 24 scans, using 2048 (proton), 70 (nitrogen), and 80 (carbon) complex points; while TROSY-based HN(CA)CB and HN(CO)CACB spectra were collected with 40 scans, using 2048 (proton), 70 (nitrogen), and 100 (carbon) complex points. The 3D methyl “out-and-back” HMCM(CG)CBCA experiment was performed with 32 scans, and 2048 (proton), 80 (methyl carbon) and 100 (Cp and Ca) complex points.
2.3. Theoretical basis of the fuzzy logic algorithm
In the first version of FLAMEnGO, we started from a random assignment for the methyl-TROSY spectra, using a 3D NOESY peak list generated from the methyl-methyl distances obtained from the crystal structure and fixing the NOE cutoff distance (12). The peak list was then compared to the corresponding list obtained from experimental 3D NOESY. The congruence between simulated and experimental lists was established using a global scoring function:
where the global score, G(x), is the maximum of the total matching function with an assignment, a, among all possible assignments, A, at a fixed NOE cutoff distance, x. This maximum of G(x) was calculated using a Monte Carlo algorithm sampling over all of the possible assignments. The total matching function includes the individual matching functions for the different restraints:
Note that all of the restraints (NOE, chemical shift, PRE, and TOCSY) are scored for each NOE cutoff distance and assignment. All of the original matching functions are reported in our previous work [12]. The new implementations for FLAMEnGO 2.0 are summarized in the following synopsis.
-
Higher tolerance for sparse and ambiguous data. For large proteins undergoing intermediate conformational exchange, some methyl group resonances may be missing in the methyl-TROSY spectrum. In addition, severe peak overlap may prevent the unambiguous determination of the PRE restraints. In the old version of FLAMEnGO, those resonances were treated as unambiguous information. In contrast, here the missing information is assigned arbitrarily and introduced after the calculation. This logic is implemented in the new total matching function:
where m indicates each single detectable methyl resonance, M indicates all of methyl resonances, r indicates the restraint associated to m, and B represents the total possible restraints. These states that contribute to the matching function will only arise from observable resonances with experimental restraints. Therefore, unobserved resonances are not taken into account and the algorithm does not try to find an assignment for these resonances.
-
Improved assignment of Val and Leu resonances using methine-methyl TOCSY correlations. In this new version, methine-methyl TOCSY correlations are used to assign methyl groups that belong to the same Val or Leu residues. For the methine-methyl TOCSY experiments on PKA-C (Supplementary Fig. 1), we utilized a sample containing a protonated methine group for all Val and Leu residues in a highly deuterated background. Therefore, a pair of methyl resonances originating from the same residue will share the same methine resonance. These restraints are quite powerful because they are independent of the structural models, reducing the sampling space and speeding up the assignment protocol. The matching function for the TOCSY restraints is formulated as it follows:
where C represents all pairs of methyl resonances, m1 and m2, assigned to the same residues, f(m) is a function that associates an m methyl group with the corresponding methine proton, and LW is the linewidth of the methine resonance in the 1H dimension. The matching function calculates the agreement between the frequencies of methine resonances associated to an assigned pair of methyl groups.
-
Improved version of the fuzzy logic step for NOE matching function. Due to errors in phasing the 3D spectra or lack of resolution in the 3D spectra, NOE cross peaks between two methyl groups can have proton and carbon chemical shifts that differ slightly from those of the methyl groups. In the previous version of the algorithm [12], these differences in chemical shifts affected the score, even if these peaks were unambiguously assigned and located in well-resolved regions of the spectrum. To avoid this problem, we assigned scores for cross peaks located in a well-resolved region higher than those for cross peaks located in crowded regions. To take this into account, we modified the matching function:
where p1 is the peak position of the experimental NOE, E, and p2 is the peak position of the synthetic NOE, S.
where pi = (xi, yi, zi) is the coordinate of the peak and LWj is the line-width in the j dimension. In the NOE matching function, the first conditional statement indicates a perfect match is considered. The second statement, gives a % confidence on the assignment.
3. Results
For ease of use, a GUI FLAMEnGO interface was built to allow users to incorporate several different types of NMR restraints and adjust parameters during the calculation (Fig. 1A). In the initial window, the user enters the input files consisting of the structural coordinates, the assigned chemical shifts, predicted chemical shifts, and the assignment swap file. The optional NMR restraints include NOE data (methyl-methyl or amide-methyl NOE data), PRE data (qualitative or quantitative data), TOCSY data (spin systems), and assigned amide shifts (only required for incorporation of amide-methyl NOE data). Moreover, a partial assignment can also be included in the list of arbitrarily assigned resonances. In the pop-out tab, one may set the number of steps in the Monte Carlo search, the range and interval for NOE distance cutoffs, as well as the amino acid types. At the end of the first run, the program plots the global score as a function of the NOE cutoff distance (Fig. 1B). In this auto-assignment algorithm, several calculations need to be performed to maximize the global score curve and provide a probability-based assignment. Since the global score function is non-decreasing, a negative slope may result from insufficient sampling steps are carried out. The latter is a more likely scenario when the calculations are carried out with large proteins, where the conformational space to be sampled is larger. In this case, multiple calculations are needed to prevent the search algorithm from being trapped in local minima and to maximize the global score function. Using the mouse, the user can pick the maximum of the global score function, setting the optimal NOE distance cutoff. At this point, a small window appears to set the number of calculations to be performed toward reaching the final probability-based assignment, which is then saved in a separate file (Fig. 1B).
Fig. 1.
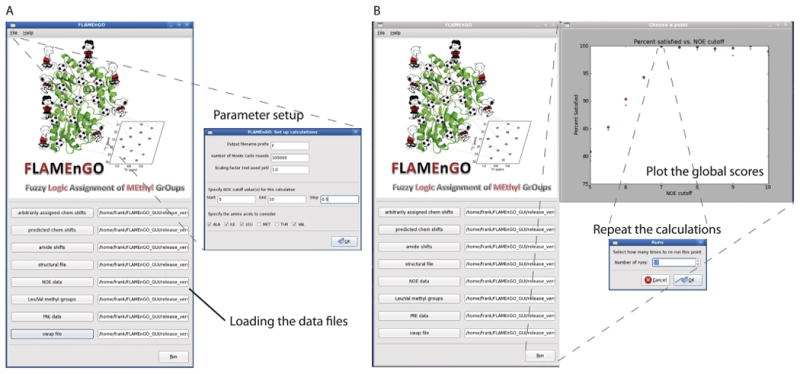
GUI interface of FLAMEnGO 2.0. (A) Main window of the GUI interface used to uplload various NMR restraints and the parameters for the calculations. (B) Output window, where the program plots the global score for each NOE distance cutoff. Note that multiple runs of calculations are necessary to optimize the assignments (see text). The optimal NOE distance cutoff can be chosen for multiple calculations, generating a probability-based assignment.
The new version of the algorithm was first tested with synthetic data obtained from MBP. The purpose of this test was to: (a) assess the accuracy of the new algorithm with sparse and ambiguous NOE data, (b) establish how the search algorithm handles multiple experimental restraints, and (c) test the tolerance for deviations of the X-ray structure from the solution structure. To generate the list of synthetic restraints (NOESY peak list), we used the crystal structure of MBP(1DMB) as an input. Chemical shift assignments and 3D methine-methyl TOCSY data were obtained from BMRB (access #7114). The NOE distance cutoff was chosen to be 7 Å. Sparse NOE data sets were generated by removing 70% of the cross peaks randomly. The effects of spin labels at positions S145C and S306C were back-calculated assuming that methyl groups within 15 Å of the spin label were completely quenched, those between 15 and 35 Å were partially quenched, and that the rest were not affected. Different combinations of data sets were input into versions 1.0 and 2.0 of FLAMEnGO and the results were compared. During the Monte Carlo sampling, we found that the new modules allow the algorithm to converge faster than the old version (Supp Fig. 2A). To test the tolerance of FLAMEnGO 2.0 for structural deviations, different conformers from the solution NMR structural ensemble of MBP (2H25) were used as inputs. Indeed, we found that structural deviations lower the accuracy of the assignment when only NOE restraints are used. However, this problem can be solved by supplementing additional restraints such as PRE and TOCSY data (Suppl. Fig. 2B).
Once validated with MBP, we applied FLAMEnGO 2.0 to the assignments of the methyl groups of PKA-C. This enzyme is quite challenging for through-bond NMR techniques, since in addition to its relatively large size (∼41 kDa), it shows prominent conformational dynamics in the intermediate time scale [15-17]. As an input structure, we utilized the coordinates of the structure 1ATP, rebuilding the hydrogen atoms using CHARMM19 [18]. To predict the 1H and 13C chemical shifts of the methyl groups, we used CH3Shift [19]. The deviations from the experimental and the calculated methyl chemical shifts were minimized using the chemical shift matching function described previously (Fig. 2A) [12]. Residue-type assignments were performed using selective amino acid labeling and mapped with CT-HMQC experiments.
Fig. 2.
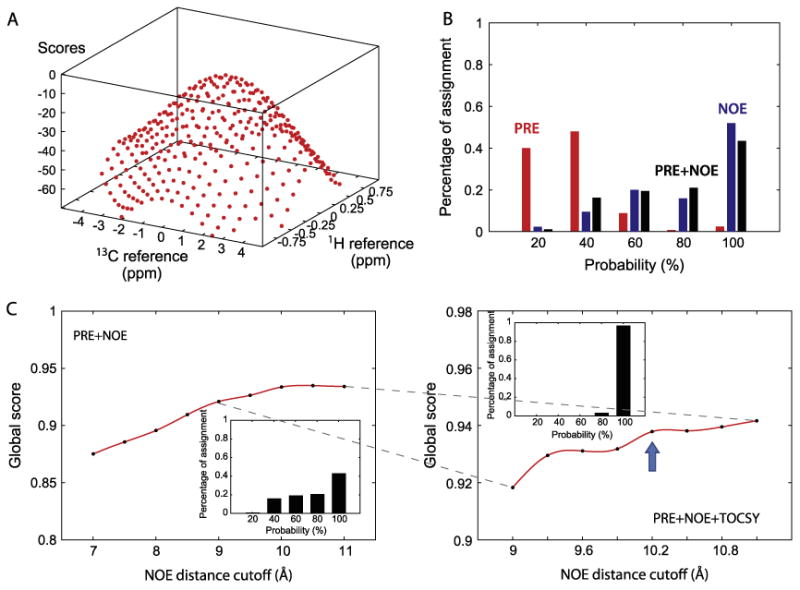
Application of FLAMEnGO 2.0 to PKA-C. (A) The surface of scores obtained by matching experimental chemical shifts with predicted chemical shifts at different references. (B) Percentage assignment versus probability using different combinations of experimental restraints (i.e., NOE, PRE or NOE + PRE). Each data set is used as an input for five independent calculations. In the histogram, the percent assignment from the PRE data is indicated in red, NOE data in blue, and PRE + NOE data in black. (C) Determination of the optimal NOE distance cutoff obtained by arraying different NOE distance cutoffs. Calculations of the global score using PRE and NOE data at coarse intervals of the NOE cutoffs (left). In this case, the optimal cutoff was found to be 10 Å with probability-based assignments reported in the inset. Global score calculations performed with PRE, NOE, and TOCSY data using finer intervals of NOE cutoffs (right). The optimal cutoff distance was found to be 10.2 Å, with assignment statistics shown in the insert. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Three sets of experimental restraints (PRE, methyl-methyl NOESY, and methine-methyl TOCSY) were utilized to determine the most probable assignment. Long-range distance PRE restraints were obtained by engineering MTSSL spin labels at two PKA-C sites, K16C and I244C. The intensity ratios of resonances from oxidized samples to those from reduced samples provide distance restraints for the methyl groups with respect to the positions of two spin labels (Suppl. Fig. 3). The average positions of these two spin labels in the crystal structure were calculated using XPLOR-NIH [20]. Short-range distance restraints were obtained from the 3D HMQC-NOESY-HMQC data on methyl groups. The 3D TOCSY-HMQC intra-residue correlations between methine and two methyl groups were used to assign pairs of methyl resonances belonging to the same spin system. Finally, two site-specific assignments were obtained using mutagenesis (I244 and I315) and enforced during the calculations.
In the first iteration, the probabilistic assignment of methyl group resonances was generated using each group of experimental restraints separately to evaluate their individual performance. In the second iteration, all of the restraints were combined into a single run. Once the optimum NOE cutoff distance was found, the calculation was repeated to provide a probabilistic confidence value for each residue. We found that the PRE data alone are not sufficient to generate an assignment with high confidence (Fig. 2B). In contrast, when only NOE restraints are included, the confidence is significantly higher, although the assignment thus generated does not independently satisfy the experimental PRE restraints (92% percent of the assignment). Once both PRE and NOE data are combined, the overall confidence slightly decreases (Fig. 2B), but most of the experimental PRE restraints are well satisfied (99% of the assignment). The latter does not mean that the assignment is less accurate; rather the increased number of restraints may converge to the correct assignment. The incorporation of TOC-SY restraints with PRE and NOE data boosts the confidence value dramatically (Fig. 2C). To calculate the final statistics and assignment, 100 randomized calculations using all the experimental data were performed at the optimized NOE cutoff value (10.2 Å). The ten sets of assignments with the highest scores were used to determine the final assignment. The preliminary result shows that 92% of methyl groups are assigned and 80% of the assignments have above 90% confidence.
Site-directed mutagenesis on V191 and L82 was also performed to assess the overall accuracy of the assignment. Both V191 methyl groups are assigned with 100% confidence, but both methyl groups of L82 were unassigned. Although site-specific labeling (V191C) is consistent with the assignment (Supp Fig. 4A), the identification of L82 methyl groups was ambiguous (Supp Fig. 4B) and these methyl groups were erroneously assigned to L27 with a 70% confidence level. Upon closer inspection, we found that the mis-assignment was due to the lack of experimental restraints for L27. We then repeated the calculations, fixing the assignments for both V191 and L82. Imposing these restraints did not change the general outcome, and we obtained a global score with a confidence level greater than 90%, confirming that the L82 assignment via mutagenesis is consistent with the entire experimental data set. The same scenario was observed for the other mutagenesis data. Their inclusion in the calculations resulted in 100% assignments for Ile, 100% for Val, and 95% for Leu methyl groups (Fig. 3). Overall, FLAMEnGO 2.0 calculations resulted in ∼98% of total methyl group assignments.
Fig. 3.
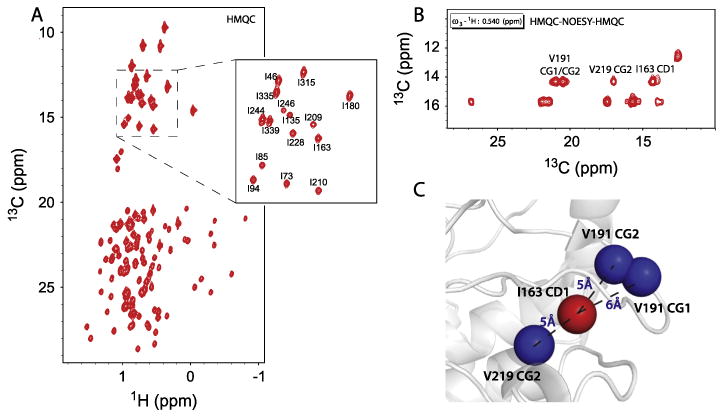
(A) The methyl-TROSY spectrum of PKA-C is shown with selected resonances labeled with highest global scores obtained from 100 independent calculations. (B) A 3D slice of the HMQC-NOESY-HMQC spectrum of PKA-C showing assigned cross peaks for the I163 methyl group (δ1). (C) Example of NOE distance restraints obtained from the NOE cross peaks of the I163 methyl group (δ1) with the corresponding distances utilized in FLAMEnGO 2.0. The restraints are shown in the crystal structure (1ATP).
Finally, to validate the accuracy of the assignment provided by FLAMEnGO 2.0 for PKA-C, we carried out several through-bond experiments to correlate the α and β carbon resonances (assigned with HNCA, HN(CO)CA, HN(CO)CACB, and HNCACB) with the methyl groups (HMCM(CG)CBCA) (Fig. 4) (4,18,19). These experiments resulted in a total of 109 out of a possible 121 correlations established between the methyl groups and the corresponding α and β carbon resonances from the backbone based experiments: 37/40 for Val, 51/60 for Leu, 21/21 for Ile. The through-bond correlation experiments confirmed the assignment of ∼79% of the available residues from FLAMEnGO 2.0 and identified 16% as incorrect. (Table 1). Most of these mis-assigned resonances (∼73%) are due to spatially close methyl groups (Fig. 5), where the correct assignments were obtained by simply swapping their assignments. Other mis-assigned resonances are related to methyl groups positioned at the surface of the protein, which do not have inter-methyl NOE restraints.
Fig. 4.
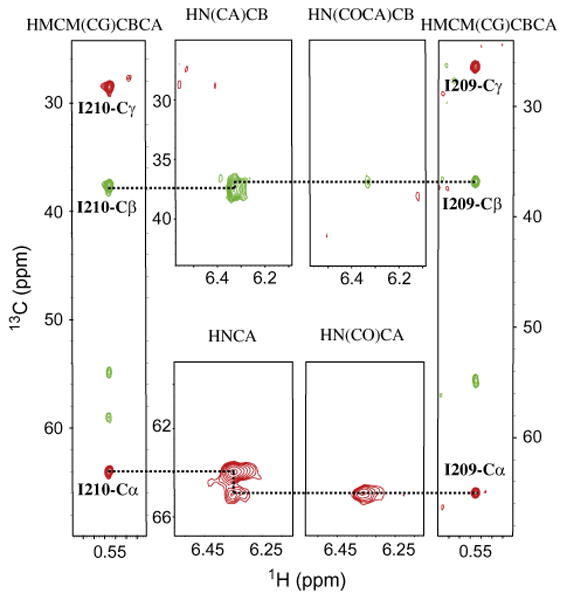
Methyl assignments of I210 and I209 using FLAMEnGO 2.0 are confirmed by sequential walks using triple-resonance and methyl out-and-back experiments.
Table 1. Validation of the statistics-based assignment of FLAMEnGO 2.0.a.
| FLAMEnGO (%) | >90% confidence | <90% confidence | |||||
|---|---|---|---|---|---|---|---|
|
|
|
||||||
| Matchedb (%) | Unassigned in the through-bond correlation spectrac (%) | Erroneous assignmentin FLAMEnGOd (%) | Matched (%) | Unassigned in the through-bond correlation spectra (%) | Erroneous assignment in FLAMEnGO (%) | ||
| ILE (21) | 100 | 90 | 0 | 10 | 0 | 0 | 0 |
| LEU (64) | 95 | 44 | 2 | 6 | 23 | 12 | 8 |
| VAL (40) | 100 | 68 | 6 | 9 | 16 | 1 | 0 |
Validation compared with though-bond experiments.
Matched indicate assignments that were verified using through-bond correlations.
Unassigned indicates that peaks are present in the spectra but the through-bond correlations are absent.
Erroneous indicates misassigned peaks in the FLAMEnGO 2.0 calculations.
Fig. 5.
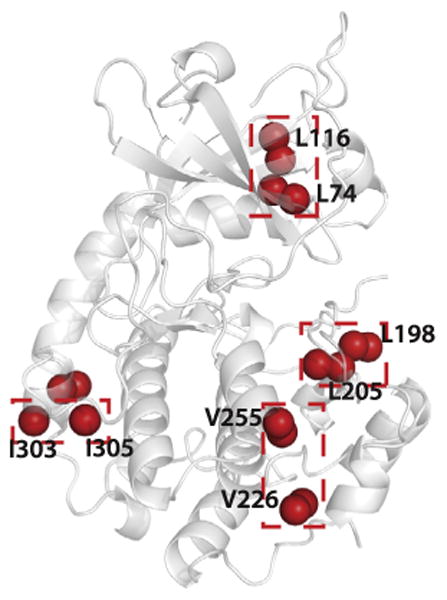
Distribution of mis-assigned methyl groups (red) resonances mapped on the crystal structure of PKA-C ternary complex with ATP and protein kinase inhibitor peptide (1ATP). The swapped assignments are indicated by red boxes.
To compare the performance of FLAMEnGO 2.0 with the MAP-XS auto-assignment software, we used identical input data and constraints. Since MAP_XS does not utilize qualitative PRE restraints, the PRE data was not used in the calculations. Twenty-five iterations of the auto-assignment were carried out and the assignments with the top ten scores were used. When compared to the through-bond experiments, only 59% of the available resides were assigned, with 1.6% of the residues assigned incorrectly. In contrast, FLAMEnGO 2.0 gives ∼79% of the methyl group assignments with an additional 15% of residues whose assignments are swapped (Supp Fig. 5). The latter is due to residues that are spatially close in the crystal structures and impossible to discriminate based on our approach.
4. Discussion
FLAMEnGO 2.0 presents several improvements over the original version, providing better handling of missing resonances, including TOCSY-based restraints, and with an improved fuzzy logic algorithm. Incorporation of PRE restraints [21] was implemented so that the intensity reduction pattern was considered for the calculations. In addition, the new implementation takes into account that many resonances are not detectable in the methyl-TROSY spectra due to exchange broadening. In this case, missing resonances are excluded from the calculations, randomly assigned to methyl groups with zero probability and excluded from the unambiguous list. The latter will not interfere with the assignment of the other resonances. Another significant aspect of the new algorithm is the implementation of the methine-methyl TOCSY data, which improve the assignment procedure for Leu and Val residues by linking them to the shared methine proton. Finally, the new version of the fuzzy logic algorithm takes into account the resolution of the methyl-resonances in the TROSY spectrum, with higher confidence attributed to well-resolved regions.
For the assignment of PKA-C, a combination of 3D NOESY, PRE, and methine-methyl TOCSY restraints were utilized, each with their own experimental challenges. In the case of PKA-C, the NOESY experiment was the least sensitive, which may be due to conformational dynamics of the enzyme and incomplete deutera-tion of the sample. In fact, only 26% of the predicted NOE cross peaks were detectable in a NOESY at 250 ms mixing time. The optimized NOE cutoff distance for the global score function was determined to be ∼10Å, which is unlikely to reflect the experimental distance. A possible explanation is the existence of a deviation between the crystal structure and the solution-state structure. However, our tests show that FLAMEnGO 2.0 has high tolerance for these structural deviations. For long-distance restraints, PRE data are the most sensitive experiments. The total PRE restraints contain 78% of the simulated data, assuming no peak overlapping ambiguity. With the assignment of PKA-C, we have selected a structural form that has been previously shown to be relatively rigid to reduce ambiguity as to the methyl group position in the structure. Finally, it should be noted that the methine-methyl TOCSY experiment is a quite sensitive experiment, less ambiguous than the NOESY cross peaks for crowded regions. For PKA-C, 97% of the expected TOCSY cross peaks were detected with only a 4 day experiment.
The assignment from FLAMEnGO 2.0 was validated using the through-bond approach. Mis-assignments were mapped onto the crystal structure (Fig. 5), and it was found that these incongruences originate from spatially close methyl groups whose assignments are swapped. In the case of the kinase this was expected, as only 26% of the theoretical NOE contacts were observed. Other mis-assignments were found on the surface of the protein, due to the lack of NOE cross peaks. This result also highlights the quality of the NOE cross peaks, which, however sparse, are essential for an accurate assignment. FLAMEnGO was also compared with MAP-XS (Supp Fig. 5). We found that FLAMEnGO provides a higher percentage of assignment than does MAP-XS. Note that FLAMEnGO also provides ∼15% of incorrect assignments due to the proximity of residues in the crystal structure. In this case, site-specific labeling can be utilized to further reduced the ambiguity and increase the number of methyl groups assigned. Overall, the logic used to assign the NOE connectivity pattern is different in the two algorithms. While MAP-XS utilizes a strict binary logic, FLAMEnGO's fuzzy logic is more flexible and enables a more exhaustive search. Due to the varied nature of the logic used in both programs, there were a significant number of correct assignments that were not degenerate between the two programs, and one would benefit from using both during the assignment process.
Overall, the computational approach with sparse and ambiguous data proved to be as accurate as the through-bond approach, and in our case, with a significant reduction of experimental time. In fact, for the kinase, the total experimental time necessary to generate meaningful inputs for FLAMEnGO 2.0 was approximately 12 days. In contrast, the through-bond approach required a total of 26 days for the acquisition of TROSY-based HNCA, HN(CO)CA, HNCACB, HN(CO)CACB experiments for the Cα and Cβ chemical shift assignments as well as the HMCM(CG)CBCA experiments (4) for methyl to Cα/Cβ correlations.
5. Conclusions
In conclusion, the enhanced version of the FLAMEnGO algorithm has higher tolerance for sparse and ambiguous data and can incorporate multiple data sets from different spectroscopic techniques, making it more flexible and efficient. FLAMEnGO 2.0 can be used to assist the resonance assignment when incomplete through-bond correlations are available, or it can be used as a standalone method for larger macromolecular systems such as proteosome, where through-bond correlations experiments fail [22,23].
Supplementary Material
Acknowledgments
This work was supported by the National Institute of Health (GM 72701 and GM 100310). We would like to thank Unhyun Lee and Kaylee Steen for making and purifying mutant constructs of PKA-C. Moreover, we also appreciate the discussion with Dr. Kaustubh Mote, Dr. Alessandro Cembran, and Dr. Vitaly Vostrikov.
Footnotes
Appendix A. Supplementary materials: Supplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/jjmr.2014.04.012.
References
- 1.Religa TL, Sprangers R, Kay LE. Dynamic regulation of archaeal proteasome gate opening as studied by TROSY NMR. Science. 2010;328(5974):98–102. doi: 10.1126/science.1184991. [DOI] [PubMed] [Google Scholar]
- 2.Sprangers R, Kay LE. Quantitative dynamics and binding studies of the 20Sproteasome by NMR. Nature. 2007;445(7128):618–622. doi: 10.1038/nature05512. [DOI] [PubMed] [Google Scholar]
- 3.Tugarinov V, Kay LE. Methyl groups as probes of structure and dynamics in NMR studies of high-molecular-weight proteins. ChemBioChem. 2005;6(9):1567–1577. doi: 10.1002/cbic.200500110. [DOI] [PubMed] [Google Scholar]
- 4.Tugarinov V, Kay LE. Ile, leu, and val methyl assignments of the 723-residue malate synthase G using a new labeling strategy and novel NMR methods. J Am Chem Soc. 2003;125(45):13868–13878. doi: 10.1021/ja030345s. [DOI] [PubMed] [Google Scholar]
- 5.Yang D, Zheng Y, Liu D, Wyss DF. Sequence-specific assignments of methyl groups in high-molecular weight proteins. J Am Chem Soc. 2004;126(12):3710–3711. doi: 10.1021/ja039102q. [DOI] [PubMed] [Google Scholar]
- 6.Tugarinov V, Venditti V, Marius Clore G. A NMR experiment for simultaneous correlations of valine and leucine/isoleucine methyls with carbonyl chemical shifts in proteins. J Biomol NMR. 2014;58(1):1–8. doi: 10.1007/s10858-013-9803-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Amero C, Asuncion Dura M, Noirclerc-Savoye M, Perollier A, Gallet B, Plevin MJ, et al. A systematic mutagenesis-driven strategy for site-resolved NMR studies of supramolecular assemblies. J Biomol NMR. 2011;50(3):229–236. doi: 10.1007/s10858-011-9513-5. [DOI] [PubMed] [Google Scholar]
- 8.Kato H, van Ingen H, Zhou BR, Feng H, Bustin M, Kay LE, et al. Architecture of the high mobility group nucleosomal protein 2-nucleosome complex as revealed by methyl-based NMR. Proc Natl Acad Sci U S A. 2011;108(30):12283–12288. doi: 10.1073/pnas.1105848108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Velyvis A, Schachman HK, Kay LE. Assignment of ile, leu and val methyl correlations in supra-molecular systems: an application to aspartate transcarbamoylase. J Am Chem Soc. 2009;131(45):16534–16543. doi: 10.1021/ja906978r. [DOI] [PubMed] [Google Scholar]
- 10.Xu Y, Liu M, Simpson PJ, Isaacson R, Cota E, Marchant J, et al. Automated assignment in selectively methyl-labeled proteins. J Am Chem Soc. 2009;131(27):9480–9481. doi: 10.1021/ja9020233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xu Y, Matthews S. MAP-XSII: an improved program for the automatic assignment of methyl resonances in large proteins. J Biomol NMR. 2013;55(2):179–187. doi: 10.1007/s10858-012-9700-z. [DOI] [PubMed] [Google Scholar]
- 12.Chao F, Shi L, Masterson LR, Veglia G. FLAMEnGO: a fuzzy logic approach for methyl group assignment using NOESY and paramagnetic relaxation enhancement data. J Magn Reson. 2012;214(0):103–110. 1. doi: 10.1016/j.jmr.2011.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bastidas AC, Deal MS, Steichen JM, Keshwani MM, Guo Y, Taylor SS. Role of N-terminal myristylation in the structure and regulation of cAMP- dependent protein kinase. J Mol Biol. 2012;42(2):215–229. doi: 10.1016/j.jmb.2012.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hemmer W, McGlone M, Taylor SS. Recombinant strategies for rapid purification of catalytic subunits of cAMP-dependent protein kinase. Anal Biochem. 1997;245(2):115–122. doi: 10.1006/abio.1996.9952. [DOI] [PubMed] [Google Scholar]
- 15.Masterson LR, Mascioni A, Traaseth NJ, Taylor SS, Veglia G. Allosteric cooperativity in protein kinase A. Proc Natl Acad Sci U S A. 2008;105(2):506–511. doi: 10.1073/pnas.0709214104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Masterson LR, Cheng C, Yu T, Tonelli M, Kornev A, Taylor SS, et al. Dynamics connect substrate recognition to catalysis in protein kinase A. Nat Chem Biol. 2010;6(11):821–828. doi: 10.1038/nchembio.452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Masterson LR, Yu T, Shi L, Wang Y, Gustavsson M, Mueller MM, et al. CAMP-dependent protein kinase A selects the excited state of the membrane substrate phospholamban. J Mol Biol. 2011;412(2):155–164. doi: 10.1016/j.jmb.2011.06.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Brooks BR, Bruccoleri RE, Olafson BD, States DJ, Swaminathan S, Karplus M. CHARMM: a program for macromolecular energy, minimization, and dynamics calculations. J Comput Chem. 1983;4(2):187–217. [Google Scholar]
- 19.Sahakyan AB, Vranken WF, Cavalli A, Vendruscolo M. Structure-based prediction of methyl chemical shifts in proteins. J Biomol NMR. 2011;50(4):331–346. doi: 10.1007/s10858-011-9524-2. [DOI] [PubMed] [Google Scholar]
- 20.Schwieters CD, Kuszewski JJ, Tjandra N, Clore GM. The xplor-NIH NMR molecular structure determination package. J Magn Reson. 2003;160(1):65–73. doi: 10.1016/s1090-7807(02)00014-9. [DOI] [PubMed] [Google Scholar]
- 21.Venditti V, Fawzi NL, Clore GM. Automated sequence and stereo-specific assignment of methyl-labeled proteins by paramagnetic relaxation and methyl-methyl nuclear Overhauser enhancement spectroscopy. J Biomol NMR. 2011;51(3):319–328. doi: 10.1007/s10858-011-9559-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Religa TL, Ruschak AM, Rosenzweig R, Kay LE. Site-directed methyl group labeling as an NMR probe of structure and dynamics in supramolecular protein systems: applications to the proteasome and to the ClpP protease. J Am Chem Soc. 2011;133(23):9063–9068. doi: 10.1021/ja202259a. [DOI] [PubMed] [Google Scholar]
- 23.Amero C, Asunción Durá M, Noirclerc-Savoye M, Perollier A, Gallet B, Plevin MJ, Vernet T, Franzetti B, Boisbouvier J. A systematic mutagenesis-driven strategy for site-resolved NMR studies of supramolecular assemblies. J Biomol NMR. 2011;50(3):229–236. doi: 10.1007/s10858-011-9513-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.