

ABSTRACT
Infection with the dimorphic fungus Histoplasma capsulatum results from the inhalation of contaminated soil. Disease outcome is variable and depends on the immune status of the host, number of organisms inhaled, and the H. capsulatum strain. H. capsulatum is divided into seven distinct clades based on phylogenetic analyses, and strains from two separate clades have been identified in North America (denoted as NAm strains). We characterized an H. capsulatum isolate (WU24) from the NAm 1 lineage in relation to two other well-characterized Histoplasma isolates, the Panamanian strain G186A and the NAm 2 strain G217B. We determined that WU24 is a chemotype II strain and requires cell wall α-(1,3)-glucan for successful in vitro infection of macrophages. In a mouse model of histoplasmosis, WU24 exhibited a disease profile that was very similar to that of strain G186A at a high sublethal dose; however, at this dose G217B had markedly different kinetics. Surprisingly, infection with a lower dose mitigated many of the differences during the course of infection. The observed differences in fungal burden, disease kinetics, symptomology, and cytokine responses all indicate that there is a sophisticated relationship between host and fungus that drives the development and progression of histoplasmosis.
IMPORTANCE
Histoplasmosis has a wide range of clinical manifestations, presenting as mild respiratory distress, acute respiratory infection, or a life-threatening disseminated disease most often seen in immunocompromised patients. Additionally, the outcome appears to be dependent on the amount and strain of fungus inhaled. In this study, we characterized a recent clinical H. capsulatum isolate that was collected from an HIV+ individual in North America. In contrast to other isolates from the same lineage, this strain, WU24, infected both macrophages and wild-type mice. We determined that in contrast to many other North American strains, WU24 infection of macrophages is dependent on the presence of cell wall α-(1,3)-glucan. Surprisingly, comparison of WU24 with two previously characterized isolates revealed that many conclusions regarding relative strain virulence and certain hallmarks of histoplasmosis are dependent on the inoculum size.
INTRODUCTION
Histoplasma capsulatum is endemic to soils throughout the world and is one of the most common fungal respiratory pathogens. In areas where the fungus is prevalent, more than 80% of the human population is positive for a Histoplasma antigen skin test, indicating previous exposure or infection (1, 2). Histoplasmosis presents with diverse manifestations, including mild subclinical disease, acute respiratory distress, and systemic dissemination. Disease outcome varies, depending on the immune status of the host and the quantity of organisms inhaled (3–5). H. capsulatum is one of the classic dimorphic fungal pathogens, a group that consists of six primary human pathogens that also includes Blastomyces dermatitidis, Coccidioides immitis, Paracoccidioides brasiliensis, Sporothrix schenckii, and Penicillium marneffei. This group of pathogens is defined by their temperature-dependent transition from a saprophytic mold to a parasitic yeast form (a spherule in the case of C. immitis) upon transition into a mammalian host (6, 7). For H. capsulatum, the ability to convert from the hyphal to unicellular yeast form is required for virulence (3–5, 8–11).
Until recently, H. capsulatum was divided into three distinct subspecies that were determined by geographical distribution and clinical symptoms. This classification system subdivided H. capsulatum into New World human pathogens (H. capsulatum var. capsulatum), African human pathogens (H. capsulatum var. duboisii), and Old World horse pathogens (H. capsulatum var. farciminosum). A more recent comprehensive phylogeographic study of isolates from around the world reclassified H. capsulatum into at least 7 distinct clades that correlate with their region of geographic isolation (12, 13). The composition of the fungal cell wall is also used to further classify H. capsulatum strains into two chemotypes (14, 15). Isolates lacking the polysaccharide α-(1,3)-glucan in their cell wall are designated chemotype I. Chemotype I isolates all belong to the North American 2 (NAm 2) group. All other H. capsulatum clades are classified as chemotype II and have α-(1,3)-glucan in their cell walls, which is required for virulence and immune evasion (16). Spontaneous α-(1,3)-glucan mutants arise during repeated passaging, which enriches for nonclumping yeast. Loss of α-(1,3)-glucan results in virulence attenuation for chemotype II strains (17–20). Chemotype I strains are virulent despite the absence of α-(1,3)-glucan (16, 19, 21, 22).
Most reports on H. capsulatum pathogenesis and virulence focus on two clinical isolates from phylogenetically distinct groups: G186A, a chemotype II strain of Panamanian origin, and G217B, a chemotype I strain of the NAm 2 clade (Table 1). In mouse models of infection, disease progression in the lungs is similar for both strains. Fungal burden in the lungs peaks at 8 days postinoculation (dpi); however, overall burden is higher in animals infected with G217B. When inoculated at high doses, G217B causes greater mortality than G186A (23–25). Although the virulence and pathogenesis of these two strains have been compared, most studies have focused on the acute phase of the disease and have not evaluated disease resolution. Additionally, comparisons have involved a relatively high inoculating dose (1 × 105 to 1 × 107 cells administered intranasally), which is likely much higher than would be found in contaminated soil. Furthermore, virulence of each strain has been determined by counting organ fungal burden or determining the 50% lethal dose and not by comparing other features of disease, including relative lung damage, inflammation, and resolution of infection. A minimal number of Histoplasma particles (either yeast or conidia) are required to establish infection, and the capacity to initiate infection is dramatically reduced when fewer particles are used (4, 26). The effect of inoculum size on disease progression has not been examined previously in a direct strain comparison.
TABLE 1 .
H. capsulatum strains used in this study
In North America, H. capsulatum strains from two different clades, NAm 1 and NAm 2, are routinely isolated from patients. Members of the NAm 1 clade are thought to be associated with HIV-positive or otherwise-immunocompromised patients. NAm 2 infection appears to be more widespread and does not correlate with host immune status (3, 27, 28). The NAm 2 isolate G217B has been extensively studied; however, less is known about NAm 1 strains. The canonical NAm 1 isolate, Downs, was isolated from an 80-year-old woman with disseminated histoplasmosis and is attenuated for virulence in a murine histoplasmosis model (27, 28). This strain has been heavily passaged, which is likely responsible for the loss of virulence. WU24 was collected from an HIV+ patient in St. Louis, MO, which is located within a region where histoplasmosis is endemic. Like the well-characterized Panama isolate, G186A, and the NAm 2 isolate, G217B, the genome of WU24 has been fully sequenced (29), and a sequence comparison between WU24, G186A, and G217B reveals substantial differences. G186A and WU24 have much smaller genomes (30.4 Mb and 30.99 Mb, respectively) than G217B (41-Mb genome), and the G217B genome contains many repetitive elements, including retrotransposons, mobile DNA insertions, and cryptons, throughout the chromosomes (30, 31).
We characterized the NAm 1 isolate, WU24, in comparison to G186A (Panama clade) and G217B (NAm 2 clade) in macrophage virulence assays and a murine model of histoplasmosis. We also evaluated the effect of inoculum size on disease progression and fitness for each strain, as well as their ability to cause inflammation in the lungs. Our observations challenge the current presumptions about the virulence of the NAm 1 clade and illustrate the complexity of assessing relative strain virulence in experimental models of histoplasmosis.
RESULTS
Histoplasma capsulatum clinical isolate WU24 is a chemotype II strain.
WU24 was originally isolated in 2003 from an HIV+ patient in St. Louis, MO, but it has not been fully characterized in the laboratory. In its yeast form at 37°C, this strain grows in large clumps in liquid culture and exhibits a rough colony morphology on solid medium (Fig. 1C), similar to the previously characterized chemotype II isolate G186A (Fig. 1A). We generated a spontaneous α-(1,3)-glucan-negative mutant of WU24 by enriching for nonclumping yeasts, as previously described for G186A (19). This strain, WU24S, exhibited a smooth colony morphology on solid medium (Fig. 1D) that was comparable to that of the chemotype I strain G217B (Fig. 1E) and an α-(1,3)-glucan mutant [ags1(Δ)] of G186A (Fig. 1B). We used a monoclonal antibody against α-(1,3)-glucan to evaluate the presence of this polysaccharide in the cell wall of G217B, G186A, ags1(Δ), WU24, and WU24S by immunofluorescence microscopy (Fig. 1K to O). The cell walls of G186A and WU24 stained brightly with the anti-α-(1,3)-glucan antibody (Fig. 1K and M), while ags1(Δ), WU24S, and G217B had no detectable α-(1,3)-glucan (Fig. 1L, M, and O). The presence of α-(1,3)-glucan leads to classification of WU24 as a chemotype II strain (14, 15).
FIG 1 .
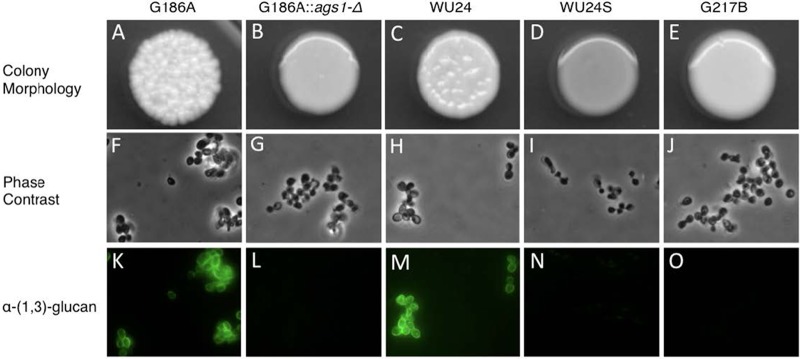
Colony morphology and α-(1,3)-glucan immunostaining. (A to E) Colony morphology of G186A (A) and WU24 (C), both chemotype II strains, which showed rough colony morphology, chemotype I strain G217B (E), which had smooth colony morphology, and also strains G186A ags1(Δ) (B) and spontaneous mutant WU24S (D). (F to J) Phase-contrast microscopy images of yeast broth cultures. (K to O) α-(1,3)-glucan immunostaining. The smooth colony morphology correlates with the absence of α-(1,3)-glucan in the yeast cell wall, as shown by immunofluorescence.
Spontaneous loss of α-(1,3)-glucan impairs the ability of WU24 to kill macrophages in vitro.
We evaluated the requirement of α-(1,3)-glucan for WU24 to kill macrophages in vitro. Bone marrow-derived macrophages (BMDMs) were infected at a multiplicity of infection (MOI) of 0.3 (1 yeast:3 macrophages) with G186A, ags1(Δ), WU24, or WU24S, and macrophage survival was determined after 7 days (Fig. 2). Macrophage survival was calculated as the amount of macrophage DNA remaining after incubation of macrophages with H. capsulatum, normalized to the amount of macrophage DNA from an uninfected macrophage monolayer. At 7 dpi, both WU24 and G816A had destroyed the majority of the monolayer, indicating that WU24 is also able to infect and eliminate macrophages in vitro. As previously reported, ags1(Δ) did not clear the macrophage monolayer, and WU24S was also completely impaired for macrophage killing. The requirement of α-(1,3)-glucan for in vitro macrophage killing of WU24 further supports the classification of this strain as chemotype II.
FIG 2 .
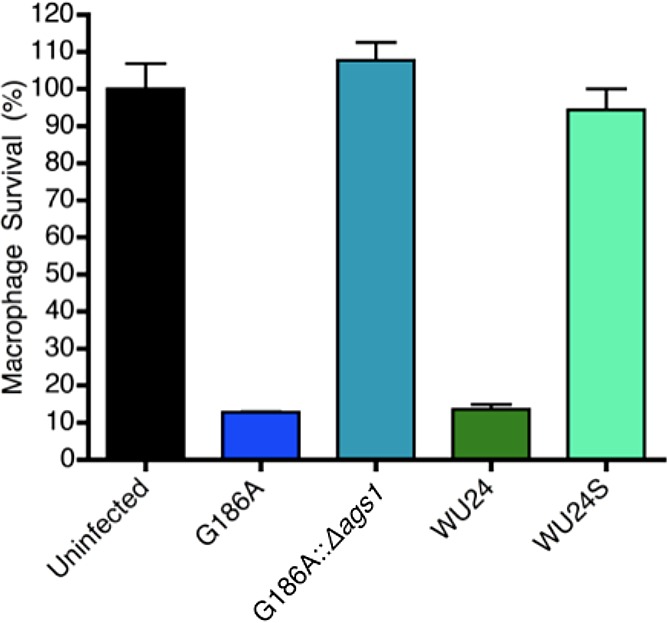
Spontaneous loss of α-(1,3)-glucan impairs the virulence of WU24 yeast cells in macrophages. Macrophage survival was measured as the reduction in macrophage DNA remaining after incubation of macrophages with Histoplasma yeast cells. Macrophage DNA remaining at 7 days postinoculation was normalized to that in uninfected populations of macrophages. Data represent results from three independent assays. Error bars represent the standard errors of the means.
Kinetics of in vitro macrophage killing by H. capsulatum strains.
Previous studies demonstrated that G217B effectively infects and destroys cultured macrophages (18, 32, 33). We compared the kinetics of in vitro macrophage killing among the three H. capsulatum strains (Fig. 3). P388D1 macrophages were infected with G186A, G217B, or WU24 at an MOI of 0.3, and macrophage survival was measured at 3, 6, and 9 dpi. Macrophage DNA at each time point was normalized to the amount in populations of macrophages, as described above. Strain G186A began to clear the macrophage monolayer as early as 3 dpi; however, there was no discernible killing in WU24- or G217B-infected macrophages at this time. At 6 dpi, WU24 and G186A exhibited similar levels of macrophage clearance, and both of these strains had completely destroyed the monolayer by 9 dpi. In G217B-infected macrophages, no detectable killing was observed until 9 dpi. This delay in lysis of host macrophages was not due to any apparent defect in intracellular replication of G217B (see Fig. S1 in the supplemental material). A similar pattern of kinetics was observed in G186A-infected, WU24-infected, and G217B-infected BMDMs (data not shown).
FIG 3 .
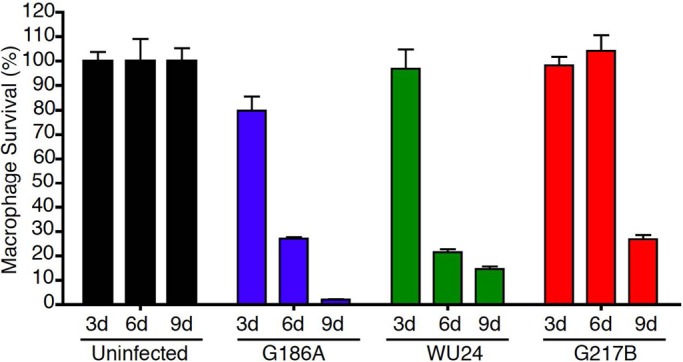
The virulence kinetics of the G217B strain is delayed in vitro. Macrophage survival was measured as the reduction in macrophage DNA remaining after incubation of macrophages with Histoplasma yeast cells. Macrophage DNA remaining at 3, 6, and 9 days postinoculation was normalized to that in uninfected populations of macrophages. Data represent results of two independent assays. Error bars represent the standard errors of the means.
In vivo virulence of H. capsulatum at a high sublethal dose.
Clinical histoplasmosis is often symptomatic but rarely lethal in immunocompetent human hosts. We therefore chose a sublethal dose to characterize the course of disease during experimental infection of mice, which are natural hosts of H. capsulatum. To compare the fungal strain fitness in vivo, C57BL/6 mice were inoculated intranasally with 2.5 × 105 G186A, WU24, or G217B yeast cells, and fungal burdens in the lungs were evaluated throughout the acute phase and resolution phase of disease (Fig. 4A). At days 0 and 4, there was no difference in pulmonary fungal burden between the three strains. Fungal burden peaked in the lungs at 8 dpi for all three strains; however, G217B-infected mice had a higher fungal burden than either WU24-infected or G186A-infected mice. The kinetics of fungal infection with WU24 and G186A were comparable, but the resolution phase of G217B infection lasted longer.
FIG 4 .
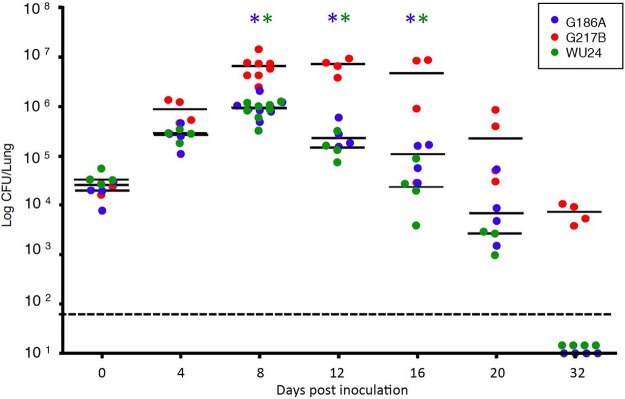
Histoplasma fitness in vivo, evaluated using a high sublethal dose. The chart depicts the number of CFU recovered from the lungs 0 (4 h), 4, 8, 12, 16, 20, and 32 days postinoculation, using 2.5 × 105 yeast cells as the inoculum (high sublethal dose). Data represent results from two independent assays. Bars represent the medians. *, Mann-Whitney test, P < 0.05; blue asterisks, significant differences between G217B and G186A; green asterisks, significant differences between G217B and WU24. The limit of detection is denoted by a dotted line.
Histological examination of infected lungs revealed the presence of monocytic cell infiltrate in the lungs at 8 dpi for all three strains (Fig. 5A, F, and K; see also Fig. S2 in the supplemental material). At 12 dpi, there were no noticeable differences in the pattern or the extent of pulmonary immune cell infiltrate (Fig. 5B, G, and L), even though G217B-infected animals had markedly increased fungal burdens. By 16 dpi, the inflammatory damage in the lungs of WU24-infected mice had started to resolve (Fig. 5M), and it was barely detectable at 20 dpi (Fig. 5N). Despite the differences in fungal burdens, mice infected with G217B or G186A had similar lung pathologies through 16 dpi (Fig. 5C and H). By day 32, only lungs from G217B-infected mice had any residual inflammation (Fig. 5E, J, and O).
FIG 5 .
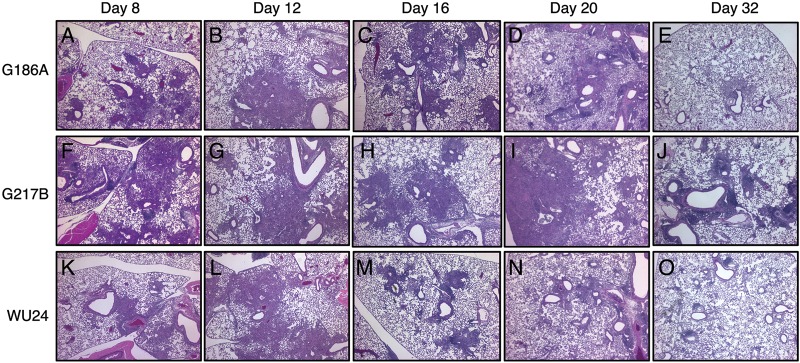
H&E-stained sections of lungs from mice inoculated intranasally with 2.5 × 105 yeast cells. Sections of lungs from mice infected with G186A, G217B, and WU24 were analyzed 8, 12, 16, 20, and 32 days postinoculation. Representative sections of the Histoplasma-infected lungs are shown. The G217B strain caused more noticeable lung damage than did either the G186A or WU24 strains in mice infected using a high sublethal dose.
Other parameters of infection correlated with the kinetics of histopathology caused by these three strains. Mice infected with G217B had noticeable weight loss between days 8 and 16, losing an average of ~20% of their body weight (Fig. 6). G217B-infected mice began to regain weight at 16 dpi. Both G186A-infected and WU24-infected mice lost weight until 14 dpi, but the change in weight was substantially less than that observed for G217B-infected mice. We also compared the induction of several proinflammatory cytokines in the lungs of H. capsulatum-infected animals. The cytokines evaluated included tumor necrosis factor alpha (TNF-α), gamma interferon (IFN-γ), interleukin-1β (IL-1β), and IL-12 (Fig. 7), which have been implicated in controlling Histoplasma infection (34–39). Cytokines were measured from lungs collected at 8 dpi, which correlates to the peak of infection and the infiltration of monocytes into the lung (Fig. 5). Additionally, lung cytokine levels were determined at 20 and 32 dpi, which correspond to the resolution phase of the disease. At 8 dpi, lungs from G217B-infected mice had significantly higher levels of IFN-γ (Fig. 7A) and TNF-α (Fig. 7B) than G186A-infected or WU24-infected mice. The levels of IL-1β were significantly higher in lungs of animals infected with G217B than in those infected with G186A, but these levels were not statistically different from the WU24 group (Fig. 7C). G217B induced higher levels of IL-12 in lungs than did WU24 (Fig. 7D), but not compared to G186A. Substantial differences in proinflammatory cytokine induction were only observed at 8 dpi. This time point correlates with peak fungal burden, which was much higher in animals infected with G217B. There was no difference in cytokine levels between strains during the resolution phase (20 and 32 dpi).
FIG 6 .
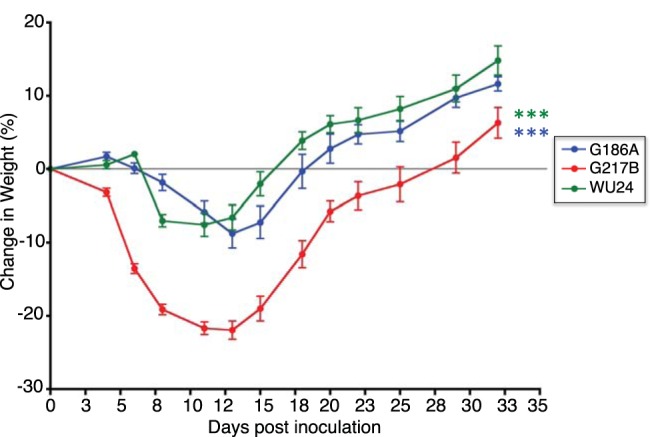
Mice infected with the G217B strain showed a dramatic loss of weight. The percent weight loss/gain of mice infected with G186A, G217B, and WU24 was monitored during the course of infection. ***, P < 0.001, one-way analysis of variance with Bonferroni posttest; blue asterisks, significant differences between G217B and G186A; green asterisks, significant differences between G217B and WU24.
FIG 7 .

Levels of cytokines measured by ELISA in lungs infected with 2.5 × 105 yeasts at 8, 20, and 32 days postinoculation. (A) IFN-γ; (B) TNF-α; (C) IL-1β; (D) IL-12. A two-way repeated-measures analysis of variance was performed. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Collectively, these data indicate that G217B has a fitness advantage at a high sublethal dose, causing more severe disease in mice than do G186A or WU24. Additionally, it is clear that WU24, a member of the NAm1 clade thought to only cause illness in immunocompromised hosts, is able to establish infection in an immunocompetent host. The kinetics and pathology of WU24 infection are very similar to those of another chemotype II strain, G186A.
In vivo virulence of Histoplasma strains at a low sublethal dose.
Given that clinical H. capsulatum infection results from the inhalation of contaminated soil, natural infection in humans probably occurs at doses much lower than what are typically evaluated experimentally. In an attempt to test a more clinically relevant dose of H. capsulatum, we infected male C57BL/6 mice with 2.5 × 103 yeast cells of G186A, WU24, or G217B; this corresponded to an intranasal inoculum that was at least 10 times lower than the doses employed in any previously published study of experimental histoplasmosis. At this low sublethal dose, early infection kinetics reflected the high sublethal dose, with lung fungal burden peaking at 8 dpi. Surprisingly, the amplification of fungal burden was nearly 10 times greater at this dose than with the high sublethal dose (Fig. 8A). Infection with the lower dose resulted in similar pulmonary fungal burdens in G217B-infected, G186A-infected, and WU24-infected animals at each time point. These kinetics, particularly during the resolution phase of the disease, contrasted sharply with those for the infection with a high sublethal dose. G217B was cleared more rapidly at a lower dose, but G186A and WU24 (both chemotype II strains) were cleared more slowly. With the high doses of G186A and WU24, fungal burdens in the lungs dropped to levels that were below the initial inoculum at 20 dpi (Fig. 4); however, at the low dose, fungal burdens at 20 dpi were still greater than the initial inoculum (Fig. 8). By day 32, all three strains were undetectable in the lungs.
FIG 8 .

Histoplasma fitness in vivo, evaluated using a low sublethal dose. The chart depicts the number of CFU recovered from the lungs after 0 (4 h), 4, 8, 12, 16, 20, and 32 days postinoculation, using 2.5 × 103 yeast cells as the inoculum (low sublethal dose). Bars represent medians. *, P < 0.05, Mann-Whitney test. The limit of detection is denoted by a dotted line.
Histological examination of the lungs from these mice revealed small foci made up primarily of monocytic cell infiltrate at 8 dpi (Fig. 9A, F, and K) and 12 dpi (Fig. 9B, G, and L). This monocytic cell infiltration was more evident at 16 dpi for all three strains (Fig. 9C, H and M). At 20 dpi, resolution of inflammation began in the lungs of mice infected with G217B (Fig. 9I) and WU24 (Fig. 9N), but not in G186A-infected mice (Fig. 9D). By day 32 there was no evidence of any residual inflammation in mice infected with any of the three strains (Fig. 9E, J, and O). These results demonstrated that H. capsulatum can establish a pulmonary infection with a relatively small inoculum size. Both the fitness advantage and delayed resolution of inflammation observed in mice infected with a high sublethal dose of G217B were ameliorated when a lower dose was used. Surprisingly, at a lower dose, resolution of inflammation was slowest in the lungs of G186A-infected mice, despite no difference in pulmonary fungal burdens. None of the mice infected at this dose had any evident weight loss (data not shown).
FIG 9 .
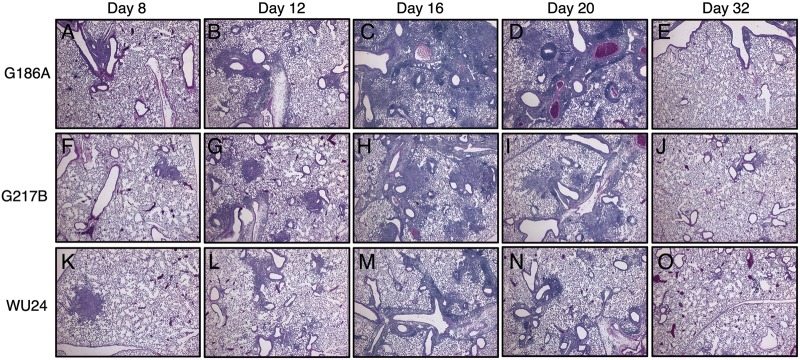
H&E staining of lungs of animals infected with 2.5 × 103 yeast cells. Lung sections from animals infected with G186A (A to E), G217B (F to J), and WU24 (K to O) were analyzed 8, 12, 16, 20, and 32 days postinoculation. Representative lung sections from the Histoplasma-infected mice are shown. The G186A strain caused more noticeable lung damage than did either the G217B or WU24 strain in mice infected with a low sublethal dose.
Because IFN-γ and TNF-α levels were different in the lungs of mice infected with the three different strains at a high sublethal dose, we evaluated the levels of these cytokines in lungs during infection with a lower dose. Comparison of the strain-specific induction of IFN-γ and TNF-α during the complete course of infection (Fig. 10) revealed that there was no difference in cytokine levels at any time during low-dose infection (Fig. 10A and B). These data suggest that the induction of IFN-γ and TNF-α is linked to the number of organisms in the lungs, which was equal for G217B-infected, WU24-infected, and G186A-infected animals at this dose.
FIG 10 .
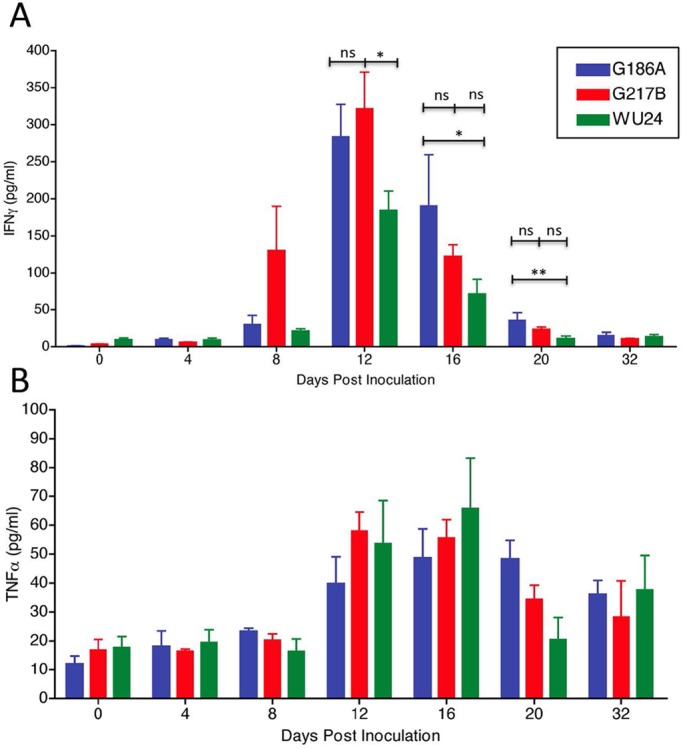
Levels of cytokines measured by ELISA in lungs of animals infected with 2.5 × 103 yeast cells. (A) IFN-γ secretion at 0 (4 h), 4, 8, 12, 16, 20, and 32 days postinoculation. (B) TNF-α secretion at 0 (4 h), 4, 8, 12, 16, 20, and 32 days postinoculation. A two-way repeated-measures analysis of variance was performed. *, P < 0.05; ns, not significant.
DISCUSSION
In this study, we compared virulence in three sequenced H. capsulatum isolates by using in vitro and in vivo models of infection. Based on sequence comparisons, G186A and G217B are predicted to have diverged fairly recently. WU24, a NAm 1 isolate, is the most disparate of the three isolates, but it appears to have many of the same virulence factors as G186A. The NAm 1 clade has historically been associated with immunocompromised individuals, and isolates are typically avirulent in animal models of infection (18, 27, 28, 40). We evaluated the ability of the WU24 strain to cause disease in an immunocompetent host compared to G217B and G186A, which have been well characterized in several in vivo and in vitro histoplasmosis models. Using two different inoculum sizes in a mouse model of sublethal histoplasmosis, we observed isolate-specific and dose-specific differences in disease between the three strains, complicating any conclusions regarding which strain may be more or less virulent.
α-(1,3)-glucan has been linked to strain-specific virulence in H. capsulatum and several other fungal pathogens, including the dimorphic fungi Paracoccidioides brasiliensis (41) and Blastomyces dermatitidis (42); however, the specific function of α-glucan for each pathogen may be different. For example, α-(1,3)-glucan in Cryptococcus neoformans has been shown to anchor the capsule to the cell wall (43), while in H. capsulatum, α-(1,3)-glucan appears to mask yeast cells from recognition by the innate immune β-glucan receptor dectin-1 (16). Interestingly, α-(1,3)-glucan mutants of H. capsulatum chemotype II are not completely killed in vitro by macrophages, yet they are unable to proliferate or lyse macrophages and are markedly attenuated in mouse models of histoplasmosis (18, 19, 21). Despite the requirement of this polysaccharide for virulence in the majority of H. capsulatum strains, isolates from the NAm 2 clade have evolved or maintained virulence in the absence of α-(1,3)-glucan. H. capsulatum exists primarily in the soil, where there is no known advantage to producing α-(1,3)-glucan, yet the majority of isolates produce this cell wall component upon transition into a host. This is not surprising, as collection and identification of H. capsulatum isolates have depended on the organisms’s ability to cause disease in humans. Therefore, strains that do not cause disease would likely not be collected. Comparisons between distinct lineages isolated from the same geographic region should provide insight into how organisms exposed to similar geographical pressures have developed distinct virulence strategies.
We corroborated the presence of α-(1,3)-glucan in the cell wall of WU24 and its requirement for macrophage killing in vitro, experimentally confirming that WU24 is indeed a chemotype II strain. Furthermore, we demonstrated that WU24 is able to cause disease in an immunocompetent host. Previous reports led to the hypothesis that NAm 1 isolates only infect immunocompromised patients; however, this was based on studies of one heavily passaged isolate and three additional isolates that all came from HIV-positive patients (27, 28). Histoplasmosis is more prevalent clinically in patients with a depleted immune system, producing a progressive and disseminated infection that frequently results in death. H. capsulatum also infects and causes primary pulmonary disease in healthy individuals, where it manifests as flu-like symptoms, is usually self-limiting and resolves, and is often not treated clinically. In healthy individuals, the inoculum size has been proposed to play a determining role in disease severity (44), and there is limited anecdotal evidence that individuals with severe disease have been exposed to a large dose of contaminated soil. In our animal models, a sublethal dose resembles subclinical histoplasmosis, producing a symptomatic but nonlethal disease. Our data revealed that inoculum size is an important factor in the degree of virulence, pathogenesis, and fitness of different H. capsulatum strains. This study illustrates the importance of considering multiple parameters in addition to fungal burden to quantify virulence, including the ability to cause pulmonary inflammation, time to fungal clearance from organs, and weight loss. Evaluation of these parameters is especially important when comparing multiple strains.
Comparisons of these three individual strains at different doses in a natural host were informative. At a high dose, G217B replicated to a higher number in the lungs, persisted for a longer duration, showed a delayed resolution of inflammation in the lungs, and led to drastic weight loss in mice, compared to G186A and WU24. These features of disease were not anticipated from in vitro experiments with cultured macrophages, which are actually killed more slowly by G217B. However, the most surprising results came from experimental infections of mice exposed to a lower dose of Histoplasma: G186A proved to be the more virulent strain, persisting for a longer period than when administered at a high dose and appearing to cause more inflammation in the lungs than G217B or WU24. Perhaps at a higher dose, the absence of α-(1,3)-glucan enables easier recognition of G217B by the host immune system, resulting in a heightened immune response that results in a delayed resolution of inflammation, which is echoed in the cytokine profiles. The increased TNF-α in the lungs of G217B-infected mice may explain the increased weight loss in these mice, given that TNF-α is linked to cachexia-associated weight loss (45). This was not seen at the lower dose, where the fungal burden may have been below the threshold to trigger this higher level of inflammation by G217B. In fact, all three strains grew to similar levels in the lung when inoculated at the lower dose, and the corresponding activation of IFN-γ and TNF-α was very similar (and low) for all strains. Interestingly, the kinetics of fungal proliferation, fungal clearance, and inflammation caused by chemotype II strains are substantially shifted at the lower dose, suggesting that the presence of α-(1,3)-glucan may limit immune recognition and cause a longer-lasting disease. For these strains, α-(1,3)-glucan is an essential virulence factor at high or low sublethal doses, as seen in our previous studies with mutants defective in genes that are required for α-(1,3)-glucan production (ags1 and amy1).
Histoplasma is subjected to different evolutionary pressures depending on its morphological state, and the selection and maintenance of yeast-specific virulence factors like α-(1,3)-glucan is only part of the picture. In the mycelial form, the soil environment influences the evolution of specific traits, including the development of conidia, which are considered the infectious particles (along with fragments of mycelia) in this geographically widespread zoonosis. Which is the “right” dose for assessing relative strain virulence in an experimental infection? This is a difficult question to answer with laboratory models of histoplasmosis; even though the mouse is a natural host, intranasal inoculation of yeast cells is typically substituted for aerosol exposure to conidia, which are difficult to generate from most strains. An additional complication is that natural infection is variable and a whole range of doses can be encountered, which may be why multiple clades have evolved within a particular geographic region. For example, the abundance of H. capsulatum in midwestern U.S. soils (46) might lead to more frequent high-dose infections. These conditions may have enriched for the NAm 2 clade, which is a more successful pathogen at a high dose. The low-dose infections could have a more profound impact on fungal persistence, because the immune response is much lower than that after high-dose exposure. This would allow the fungus to establish a long-term infection, which would be an advantage for spreading to a new location via animal host migration.
The development and progression of histoplasmosis have long been known to represent a multifactorial relationship between the immune status of the host and the pathogenicity of the fungus. What is generally unappreciated is the need to consider both the influence of strain background and of the infectious dose when studying disease progression and making conclusions regarding relative virulence. Our results indicate that the Histoplasma strain and dose not only influence the assessment of virulence and the kinetics of infection during the acute phase of the disease but also can alter the resolution phase and the time required for fungal clearance and ultimately host recovery.
MATERIALS AND METHODS
Fungal strains and culture conditions.
The following Histoplasma capsulatum strains were used in this study: NAm 2 isolate G217B (ATCC 26032), Panamanian isolate G186A (ATCC 26029), and NAm 1 isolate WU24 (a clinical strain isolated from an HIV-positive patient). We also used an ags1(Δ) mutant of G186A, which has been previously described (21). We generated a smooth mutant of WU24, WU24S, that was obtained by enrichment of nonclumping yeast strains as described below. All strains were grown in histoplasma macrophage medium (HMM) at 37°C with 95% air–5% CO2 as previously described (47). Solid medium contained 0.6% agarose (SeaKem ME grade) and 25 mM FeSO4. Dispersed Histoplasma yeasts were obtained by growth of liquid cultures on an orbital shaker to late exponential phase, and cells were washed once with warm (37°C) HMM, followed by a low-speed centrifugation (1 min at 600 × g) to remove large yeast clumps and counting with a hemacytometer. To enrich for nonclumping yeast strains, log-phase yeast cells in broth culture were centrifuged for 1 min at 600 × g to remove large yeast clumps. The remaining suspension of cells was removed and subcultured in fresh medium. This process was repeated six times, after which the suspension of cells was plated on HMM plates. The resulting colonies were observed under a dissecting microscope. Colonies with a smooth appearance were selected for further analysis.
Macrophage culture and virulence assay.
P388D1 macrophage-like cells were cultured in F-12 medium (Life Technologies) with 10% fetal bovine serum (HyClone) or in HMM-M when coincubated with Histoplasma yeast cells, as described previously (48). Bone marrow-derived macrophages were collected as previously described (49). Histoplasma virulence in P388D1 cells was determined as previously described (21, 50). Monolayers of P388D1 cells and BMDMs were infected in triplicate with Histoplasma yeasts at a multiplicity of infection of 1:3 (yeasts:macrophages) in 24-well plates and incubated at 37°C in 95% air–5% CO2. Fifty percent of the culture medium was replaced with fresh medium every other day after day 3. After 3, 6, and 9 days of infection, culture medium was removed, and remaining macrophages were lysed with a solution containing 10 mM Tris, 1 mM EDTA, 0.05% SDS. The PicoGreen double-stranded DNA quantification reagent (Molecular Probes) was used to measure the amount of macrophage DNA in each well. Data represent results collected from three independent assays.
Microscopy.
Immunofluorescence and differential interference contrast (DIC) images were obtained using 4×, 40×, and 60× objectives on an Olympus BX-60 microscope and collected using a SPOT-RT slider charge-coupled-device camera and image acquisition software (Diagnostic Instruments). Immunolocalization of cell wall α-(1,3)-glucan was performed by fixing exponentially growing broth cultures of yeasts in 4% formaldehyde in phosphate-buffered saline (PBS) for 60 min at room temperature and staining yeasts with a mouse monoclonal IgM antibody that recognizes α-(1,3)-glucan (number 401925; CalBiochem).
Murine colonization assay.
All animal studies were approved by the University of North Carolina in Chapel Hill Office of Animal Care and Use, protocol 11-195. Male C57BL/6 mice (4 to 6 weeks old; Jackson Laboratories) were sedated with ketamine/xylazine and infected intranasally with sublethal doses of H. capsulatum yeast (2.5 × 105 or 2.5 × 103 dispersed cells in 20 µl of HMM) as described previously (50). Six mice per time point (4 for fungal burden and 2 for histopathology) were inoculated with each strain at each dose. Animals were sacrificed after 4 h and 4, 8, 12, 16, 20, and 32 dpi, and the lungs and spleens were removed and homogenized in 5 ml of cold HMM. Serial dilutions of the homogenates were plated on HMM plates to determine CFU recovered from each organ. The body weight of the animals was monitored every 2 days during the course of the infection.
Cytokine analysis.
Aliquots from the lung homogenates used to determine fungal burden in the lungs of infected mice were centrifuged at 16,000 × g for 2 min and diluted 1/5 (for the high-dose samples) or 1/3 (for the low-dose samples). Levels of TNF-α, IFN-γ, IL-1β, and IL-12, present in mouse lungs infected with the high dose and levels of TNF-α and IFN-γ present in mouse lungs infected with the low-dose were measured. All cytokine analyses were performed using the BD OptEIA enzyme-linked immunosorbent assay (ELISA) set (BD Biosciences). Based on our standard curves, the limit of detection for TNF-α was 8 pg/ml.
Histopathology.
Uninfected mice and mice infected for 8, 12, 16, 20, and 32 days were euthanized with sodium pentobarbital (150 mg/kg of body weight), and their lungs were inflated with 10% formalin via cannulation of the trachea. Lungs were removed and fixed in 10% formalin for at least 96 h and incubated in PBS for 2 h before being embedded in paraffin. Five-micrometer sections of tissue were stained with hematoxylin and eosin (H&E) before being examined.
Statistical analysis.
Data were analyzed using Prism (GraphPad, CA). P values of <0.05 were considered significant.
SUPPLEMENTAL MATERIAL
Destruction of macrophage monolayers by G186A and G217B, as monitored by using phase-contrast microscopy at 72 (A and F), 96 (B and G), 120 (C and H), 144 (D and I), and 168 (E and J) hours postinoculation. Histoplasma yeasts were stained with Uvitex (blue) and visualized via fluorescence microscopy. Download
Lung inflammation of H. capsulatum-infected mice. Mice were inoculated intranasally with 2.5 × 105 yeast cells. At the peak of infection (8 days postinoculation), mononuclear cell infiltration was a consistent pathological feature within the lungs of mice infected with G186A, G217B, or WU24. Download
ACKNOWLEDGMENTS
This work was supported by NIH grants AI025584 and AI099582 to W.E.G.
Footnotes
Citation Sepúlveda VE, Williams CL, Goldman WE. 2014. Comparison of phylogenetically distinct Histoplasma strains reveals evolutionarily divergent virulence strategies. mBio 5(4):e01376-14. doi:10.1128/mBio.01376-14.
REFERENCES
- 1. Edwards LB, Acquaviva FA, Livesay VT, Cross FW, Palmer CE. 1969. An atlas of sensitivity to tuberculin, PPD-B, and histoplasmin in the United States. Am. Rev. Respir. Dis. 99:1–132 [PubMed] [Google Scholar]
- 2. Edwards LB, Acquaviva FA, Livesay VT. 1973. Further observations on histoplasmin sensitivity in the United States. Am. J. Epidemiol. 98:315–325 [DOI] [PubMed] [Google Scholar]
- 3. Eissenberg LG, Goldman WE. 1991. Histoplasma variation and adaptive strategies for parasitism: new perspectives on histoplasmosis. Clin. Microbiol. Rev. 4:411–421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Larsh HW, Cozad GC. 1965. Respiratory infection of mice with Histoplasma capsulatum. Mycopathol. Mycol. Appl. 27:305–310. 10.1007/BF02053788 [DOI] [PubMed] [Google Scholar]
- 5. Durkin M, Kohler S, Schnizlein-Bick C, LeMonte A, Connolly P, Goldberg J, Garringer T, Wheat LJ. 2001. Chronic infection and reactivation in a pulmonary challenge model of histoplasmosis. J. Infect. Dis. 183:1822–1824. 10.1086/320720 [DOI] [PubMed] [Google Scholar]
- 6. Maresca B, Kobayashi GS. 1989. Dimorphism in Histoplasma capsulatum: a model for the study of cell differentiation in pathogenic fungi. Microbiol. Rev. 53:186–209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Rappleye CA, Goldman WE. 2006. Defining virulence genes in the dimorphic fungi. Annu. Rev. Microbiol. 60:281–303. 10.1146/annurev.micro.59.030804.121055 [DOI] [PubMed] [Google Scholar]
- 8. Medoff G, Sacco M, Maresca B, Schlessinger D, Painter A, Kobayashi GS, Carratu L. 1986. Irreversible block of the mycelial-to-yeast phase transition of Histoplasma capsulatum. Science 231:476–479. 10.1126/science.3001938 [DOI] [PubMed] [Google Scholar]
- 9. Klein BS, Tebbets B. 2007. Dimorphism and virulence in fungi. Curr. Opin. Microbiol. 10:314–319. 10.1016/j.mib.2007.04.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Nguyen VQ, Sil A. 2008. Temperature-induced switch to the pathogenic yeast form of Histoplasma capsulatum requires Ryp1, a conserved transcriptional regulator. Proc. Natl. Acad. Sci. U. S. A. 105:4880–4885. 10.1073/pnas.0710448105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Webster RH, Sil A. 2008. Conserved factors Ryp2 and Ryp3 control cell morphology and infectious spore formation in the fungal pathogen Histoplasma capsulatum. Proc. Natl. Acad. Sci. U. S. A. 105:14573–14578. 10.1073/pnas.0806221105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kasuga T, Taylor JW, White TJ. 1999. Phylogenetic relationships of varieties and geographical groups of the human pathogenic fungus Histoplasma capsulatum Darling. J. Clin. Microbiol. 37:653–663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kasuga T, White TJ, Koenig G, Mcewen J, Restrepo A, Castañeda E, Da Silva Lacaz C, Heins-Vaccari EM, De Freitas RS, Zancopé-Oliveira RM, Qin Z, Negroni R, Carter DA, Mikami Y, Tamura M, Taylor ML, Miller GF, Poonwan N, Taylor JW. 2003. Phylogeography of the fungal pathogen Histoplasma capsulatum. Mol. Ecol. 12:3383–3401. 10.1046/j.1365-294X.2003.01995.x [DOI] [PubMed] [Google Scholar]
- 14. Reiss E, Miller SE, Kaplan W, Kaufman L. 1977. Antigenic, chemical, and structural properties of cell walls of Histoplasma capsulatum yeast-form chemotypes 1 and 2 after serial enzymatic hydrolysis. Infect. Immun. 16:690–700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Domer JE. 1971. Monosaccharide and chitin content of cell walls of Histoplasma capsulatum and Blastomyces dermatitidis. J. Bacteriol. 107:870–877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Rappleye CA, Eissenberg LG, Goldman WE. 2007. Histoplasma capsulatum alpha-(1,3)-glucan blocks innate immune recognition by the beta-glucan receptor. Proc. Natl. Acad. Sci. U. S. A. 104:1366–1370. 10.1073/pnas.0609848104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. San-Blas G, Ordaz D, Yegres FJ. 1978. Histoplasma capsulatum: Chemical variability of the yeast cell wall. Sabouraudia 16:279–284. 10.1080/00362177885380381 [DOI] [PubMed] [Google Scholar]
- 18. Eissenberg LG, West JL, Woods JP, Goldman WE. 1991. Infection of P388D1 macrophages and respiratory epithelial cells by Histoplasma capsulatum: selection of avirulent variants and their potential role in persistent histoplasmosis. Infect. Immun. 59:1639–1646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Klimpel KR, Goldman WE. 1987. Isolation and characterization of spontaneous avirulent variants of Histoplasma capsulatum. Infect. Immun. 55:528–533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Klimpel KR, Goldman WE. 1988. Cell walls from avirulent variants of Histoplasma capsulatum lack alpha-(1,3)-glucan. Infect. Immun. 56:2997–3000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Rappleye CA, Engle JT, Goldman WE. 2004. RNA interference in Histoplasma capsulatum demonstrates a role for alpha-(1,3)-glucan in virulence. Mol. Microbiol. 53:153–165. 10.1111/j.1365-2958.2004.04131.x [DOI] [PubMed] [Google Scholar]
- 22. Edwards JA, Alore EA, Rappleye CA. 2011. The yeast-phase virulence requirement for α-glucan synthase differs among Histoplasma capsulatum chemotypes. Eukaryot. Cell 10:87–97. 10.1128/EC.00214-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Tewari RP, Berkhout FJ. 1972. Comparative pathogenicity of albino and brown types of Histoplasma capsulatum for mice. J. Infect. Dis. 125:504–508. 10.1093/infdis/125.5.504 [DOI] [PubMed] [Google Scholar]
- 24. Mayfield JA, Rine J. 2007. The genetic basis of variation in susceptibility to infection with Histoplasma capsulatum in the mouse. Genes Immun. 8:468–474. 10.1038/sj.gene.6364411 [DOI] [PubMed] [Google Scholar]
- 25. Medoff G, Maresca B, Lambowitz AM, Kobayashi G, Painter A, Sacco M, Carratu L. 1986. Correlation between pathogenicity and temperature sensitivity in different strains of Histoplasma capsulatum. J. Clin. Invest. 78:1638–1647. 10.1172/JCI112757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Furcolow ML. 1961. Airborne histoplasmosis. Bacteriol. Rev. 25:301–309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gass M, Kobayashi GS. 1969. Histoplasmosis. An illustrative case with unusual vaginal and joint involvement. Arch. Dermatol. 100:724–727. 10.1001/archderm.100.6.724 [DOI] [PubMed] [Google Scholar]
- 28. Spitzer ED, Keath EJ, Travis SJ, Painter AA, Kobayashi GS, Medoff G. 1990. Temperature-sensitive variants of Histoplasma capsulatum isolated from patients with acquired immunodeficiency syndrome. J. Infect. Dis. 162:258–261. 10.1093/infdis/162.1.258 [DOI] [PubMed] [Google Scholar]
- 29. Broad Institute 2007. Histoplasma capsulatum database. Broad Institute, Cambridge, MA. http://broadinstitute.org/annotation/genome/histoplasma_capsulatum/MultiHome.html
- 30. Goodwin TJD, Butler MI, Poulter RTM. 2003. Cryptons: a group of tyrosine-recombinase-encoding DNA transposons from pathogenic fungi. Microbiology (Reading, Engl.) 149:3099–3109 [DOI] [PubMed] [Google Scholar]
- 31. Edwards JA, Rappleye CA. 2011. Histoplasma mechanisms of pathogenesis—one portfolio doesn’t fit all. FEMS Microbiol. Lett. 324:1–9. 10.1111/j.1574-6968.2011.02363.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Edwards JA, Zemska O, Rappleye CA. 2011. Discovery of a role for Hsp82 in Histoplasma virulence through a quantitative screen for macrophage lethality. Infect. Immun. 79:3348–3357. 10.1128/IAI.05124-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Isaac DT, Coady A, Van Prooyen N, Sil A. 2013. The 3-hydroxy-methylglutaryl coenzyme A lyase HCL1 is required for macrophage colonization by human fungal pathogen Histoplasma capsulatum. Infect. Immun. 81:411–420. 10.1128/IAI.00833-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zhou P, Miller G, Seder RA. 1998. Factors involved in regulating primary and secondary immunity to infection with Histoplasma capsulatum: TNF-alpha plays a critical role in maintaining secondary immunity in the absence of IFN-gamma. J. Immunol. 160:1359–1368 [PubMed] [Google Scholar]
- 35. Zhou P, Sieve MC, Bennett J, Kwon-Chung KJ, Tewari RP, Gazzinelli RT, Sher A, Seder RA. 1995. IL-12 prevents mortality in mice infected with Histoplasma capsulatum through induction of IFN-gamma. J. Immunol. 155:785–795 [PubMed] [Google Scholar]
- 36. Deepe GS, McGuinness M. 2006. Interleukin-1 and host control of pulmonary histoplasmosis. J. Infect. Dis. 194:855–864. 10.1086/506946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Deepe GS. 2005. Modulation of infection with Histoplasma capsulatum by inhibition of tumor necrosis factor-alpha activity. Clin. Infect. Dis. 41:S204–S207. 10.1086/429999 [DOI] [PubMed] [Google Scholar]
- 38. Allendoerfer R, Deepe GS. 1997. Intrapulmonary response to Histoplasma capsulatum in gamma interferon knockout mice. Infect. Immun. 65:2564–2569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Deepe GS. 2007. Tumor necrosis factor-alpha and host resistance to the pathogenic fungus, Histoplasma capsulatum. J. Investig. Dermatol. Symp. Proc. 12:34–37. 10.1038/sj.jidsymp.5650026 [DOI] [PubMed] [Google Scholar]
- 40. Keath EJ, Painter AA, Kobayashi GS, Medoff G. 1989. Variable expression of a yeast-phase-specific gene in Histoplasma capsulatum strains differing in thermotolerance and virulence. Infect. Immun. 57:1384–1390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. San-Blas G, San-Blas F, Serrano LE. 1977. Host-parasite relationships in the yeastlike form of Paracoccidioides brasiliensis strain IVIC Pb9. Infect. Immun. 15:343–346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Hogan LH, Klein BS. 1994. Altered expression of surface alpha-1,3-glucan in genetically related strains of Blastomyces dermatitidis that differ in virulence. Infect. Immun. 62:3543–3546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Reese AJ, Doering TL. 2003. Cell wall alpha-1,3-glucan is required to anchor the Cryptococcus neoformans capsule. Mol. Microbiol. 50:1401–1409. 10.1046/j.1365-2958.2003.03780.x [DOI] [PubMed] [Google Scholar]
- 44. Larsh HW. 1960. Natural and experimental epidemiology of histoplasmosis. Ann. N. Y. Acad. Sci. 89:78–90. 10.1111/j.1749-6632.1960.tb20132.x [DOI] [Google Scholar]
- 45. Cerami A, Beutler B. 1988. The role of cachectin/TNF in endotoxic shock and cachexia. Immunol. Today 9:28–31. 10.1016/0167-5699(88)91353-9 [DOI] [PubMed] [Google Scholar]
- 46. Campbell CC. 1965. The epidemiology of histoplasmosis. Ann. Intern. Med. 62:1333–1336. 10.7326/0003-4819-62-6-1333 [DOI] [PubMed] [Google Scholar]
- 47. Worsham PL, Goldman WE. 1988. Selection and characterization of ura5 mutants of Histoplasma capsulatum. Mol. Gen. Genet. 214:348–352 [DOI] [PubMed] [Google Scholar]
- 48. Eissenberg LG, Schlesinger PH, Goldman WE. 1988. Phagosome-lysosome fusion in P388D1 macrophages infected with Histoplasma capsulatum. J. Leukoc. Biol. 43:483–491 [DOI] [PubMed] [Google Scholar]
- 49. Sullivan JT, Young EF, McCann JR, Braunstein M. 2012. The mycobacterium tuberculosis, SecA2 system subverts phagosome maturation to promote growth in macrophages. Infect. Immun. 80:996–1006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Sebghati TS, Engle JT, Goldman WE. 2000. Intracellular parasitism by Histoplasma capsulatum: fungal virulence and calcium dependence. Science 290:1368–1372. 10.1126/science.290.5495.1368 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Destruction of macrophage monolayers by G186A and G217B, as monitored by using phase-contrast microscopy at 72 (A and F), 96 (B and G), 120 (C and H), 144 (D and I), and 168 (E and J) hours postinoculation. Histoplasma yeasts were stained with Uvitex (blue) and visualized via fluorescence microscopy. Download
Lung inflammation of H. capsulatum-infected mice. Mice were inoculated intranasally with 2.5 × 105 yeast cells. At the peak of infection (8 days postinoculation), mononuclear cell infiltration was a consistent pathological feature within the lungs of mice infected with G186A, G217B, or WU24. Download