

ABSTRACT
Streptococcus pneumoniae (pneumococcus) is a Gram-positive bacterium that causes serious invasive diseases, such as pneumonia, bacteremia, and meningitis, with high morbidity and mortality throughout the world. Before causing invasive disease, S. pneumoniae encounters cellular barriers, which are often composed of endothelial cells, like the alveolar-capillary barrier and the blood-brain barrier. S. pneumoniae adheres to endothelial cells and may invade them, which requires an efficient host response to the intracellular bacteria. The precise intracellular fate of S. pneumoniae during infection still remains a subject of debate. The proteasome-ubiquitin system is largely responsible for the degradation of misfolded, damaged, or no-longer-useful proteins. Recently, the role of the proteasome-ubiquitin system in the clearing of invading bacteria and viruses has been more closely studied. In this study, we show that inhibition of the proteasome-ubiquitin system leads to a marked increase in S. pneumoniae survival inside host cells. Immunofluorescence analysis showed that intracellular pneumococci colocalized with proteasome and ubiquitin in human endothelial cells in vitro. Confocal imaging analysis demonstrated that in the brains of mice intravenously infected with S. pneumoniae, the bacteria were inside endothelial cells, where they colocalized with proteasome and ubiquitin signals. In conclusion, our data indicate that a fully functional proteasome-ubiquitin system in endothelial cells is crucial for efficient killing of intracellular S. pneumoniae.
IMPORTANCE
Bacterial meningitis is a serious invasive disease with high morbidity and mortality. How bacteria traverse the blood-brain barrier in vivo and what mechanisms are employed by the host to prevent invasion are still unclear. Our data show that inhibition of the proteasome-ubiquitin system in vitro leads to a significant increase in S. pneumoniae survival inside brain endothelial cells. Confocal imaging analysis of brain tissue from mice intravenously infected with pneumococci demonstrated that the bacteria are inside brain microvascular endothelial cells, where they associate with the proteasome and ubiquitin. This is, as far as we know, the first report that demonstrates that Streptococcus pneumoniae invades endothelial cells of the blood-brain barrier in vivo. The host requires the proteasome-ubiquitin system for an efficient decimation of intracellular S. pneumoniae.
INTRODUCTION
Streptococcus pneumoniae (pneumococcus) is a Gram-positive human pathogen that causes life-threatening invasive diseases such as pneumonia, bacteremia, and meningitis with high morbidity and mortality throughout the world. Cellular barriers encountered by S. pneumoniae before causing invasive disease are often composed of endothelial cells. For instance, during pneumonia, the bacteria interact with endothelial cells in the alveolar-capillary barrier before translocating from the lungs into the bloodstream and causing bacteremia. S. pneumoniae also interacts with the endothelium of the blood-brain barrier before invading the central nervous system (CNS), leading to meningitis (1). Once attached, in vitro S. pneumoniae organisms can invade endothelial cells, where the majority of them are degraded in the lysosomes (2–4). However, a minor subset of internalized S. pneumoniae cells is most likely recycled out of the cell again, as exemplified by the association with Rab markers (3, 4). Yet another subset of the internalized S. pneumoniae cells is not killed or recycled but translocated from the apical side, through the cell, to the basolateral side through a process called transcytosis (3–6), causing meningitis. A similar degradation by the lysosome has been shown for other bacteria, for instance Staphylococcus aureus and Legionella pneumophila (7, 8).
To shed further light on the process determining S. pneumoniae degradation, we studied the possible involvement of the proteasome-ubiquitin system in the killing of internalized S. pneumoniae. The proteasome-ubiquitin system is largely responsible for the degradation of misfolded, damaged, or no-longer-useful proteins. In order for a protein to be degraded, it needs to be tagged by the small (76-amino-acid) ubiquitin protein, which is covalently bound to the substrate via lysine residues by the action of the E3-ubiquitin ligase. Prior to binding to the substrate, the ubiquitin molecule needs to be activated in an energy-dependent manner by the activating E1 enzyme, followed by transfer of the ubiquitin from the activating E1 enzyme to the conjugating E2 enzyme. The E2-ubiquitin complex is then docked with the E3-ubiquitin ligase. This complex recognizes the protein, which is to be degraded, and transfers the ubiquitin to the substrate (9–12). In general, it seems that mono-ubiquitination (one ubiquitin molecule bound to the substrate) functions mainly in signaling, for example for the regulation of endocytosis, endosomal sorting, and trafficking (13, 14). In contrast, poly-ubiquitination (many ubiquitin molecules bound to a substrate either as monomers or in chains) leads to degradation of the substrate in the proteasome or performs other regulatory functions, including trafficking, kinase activation, and DNA repair (15–17). In recent years, the role of the proteasome-ubiquitin system in the innate immune system has come under close scrutiny (9), especially so with regard to its role in the clearing of invading bacteria (18) and viruses (10). Here we investigated the role of the proteasome-ubiquitin system in the intracellular fate of S. pneumoniae and show that, in vivo, the bacteria invade brain endothelial cells, which need a fully functional proteasome-ubiquitin system for efficient killing of intracellular S. pneumoniae.
RESULTS
A fully functional proteasome-ubiquitin system is required for efficient intracellular killing of S. pneumoniae.
To determine the role of the proteasome-ubiquitin system in the killing of intracellular S. pneumoniae, we used chemical inhibitors to block discrete parts of the proteasome-ubiquitin system in endothelial cells infected with pneumococci. To avoid any effect on the adhesion and uptake mechanisms, all inhibitors were added after S. pneumoniae had invaded and external S. pneumoniae cells had been either washed away or killed with antibiotics, as described in Materials and Methods. This approach, as opposed to adding inhibitors from the start or even pretreating the endothelial cell cultures with the inhibitors, yields less pronounced but more defined, relevant, and reliable effects. Adding the inhibitors after the bacteria have invaded the cells circumvents several problems. The cells are not exposed to the chemicals for a long time and are not impaired in their normal function, and thus decreases in viability are unlikely to be a problem. Also with this approach, we are certain that the effects that we observe are due to differences in intracellular survival and not invasion. It is important to note that even with the short incubation time, we observed the effects described below. We used the chemicals carbobenzoxy-l-leucyl-l-leucyl-l-leucinal (MG132) (19, 20) and tetraethylthiuram disulfide (TET) (21–23) for proteasome inhibition. The MG132 and TET treatments resulted, respectively, in 79% and 253% increases in intracellular S. pneumoniae survival compared to the survival of the organism in the control experiment without inhibitor (Fig. 1A and see Fig. S1 and S2 in the supplemental material). This strongly indicated that a fully functional proteasome is required for efficient killing of intracellular pneumococci. To further dissect the influence of the proteasome-ubiquitin system on S. pneumoniae survival inside the endothelial host cell, we inhibited the first step of the ubiquitination process. The inhibitor UBEI-41 (also known as PYR-41) (20) specifically inhibits the activating E1 enzyme, necessary for the initial activation of the ubiquitin molecule, the first step in a process ultimately resulting in the ubiquitination of target proteins (24). Inhibition of the ubiquitin-activating enzyme with UBEI-41 resulted in a 142% increase in intracellular S. pneumoniae survival (Fig. 1B and S3).
FIG 1 .
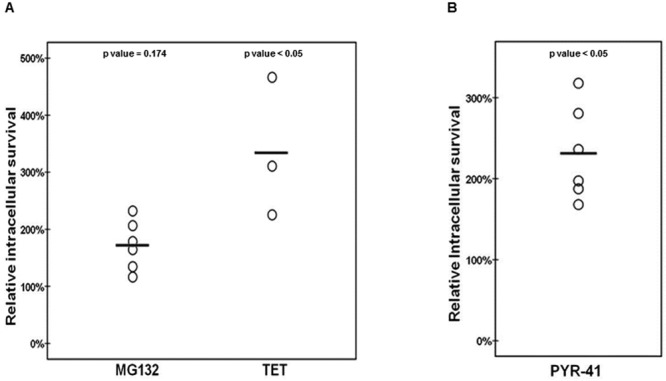
A fully functional proteasome-ubiquitin system in endothelial cells is crucial for efficient clearance of intracellular S. pneumoniae. (A) Relative survival of internalized S. pneumoniae cells during proteasomal inhibition with MG132 or TET. The control is set at 100%, and the experimental conditions are related to this percentage. Each circle represents the relative intracellular survival in an individual experiment, and horizontal lines indicate the averages of results from all experiments (n = 6 and 3, respectively). P values = 0.174 and <0.05, respectively. Relative intracellular survival is defined as intracellular survival divided by the amount of the initial invasion. (B) Relative survival of internalized S. pneumoniae during inhibition of the activating E1 enzyme with the inhibitor UBEI-41. The control is set as 100%, and the experimental conditions are related to this percentage. Each circle is the relative intracellular survival in an individual experiment, and horizontal lines indicate the averages of results from all experiments (n = 6). P value < 0.05. Relative intracellular survival is defined as intracellular survival divided by the amount of the initial invasion.
The capsule of S. pneumoniae does not inhibit the proteasome-mediated killing process.
The capsule is one of S. pneumoniae’s main virulence factors (25). However, it also impedes adhesion and invasion into host cells, and it has been shown that S. pneumoniae in close interaction with host cells loses its capsule in the mouse nasopharynx (26). This is why we, like others in the field (3, 4, 26), used unencapsulated bacteria to study the S. pneumoniae-host cell interactions, especially so since this greatly increases the adhesion and invasion frequency. However, to exclude the possibility that encapsulated strains interact differently with the host cell and are trafficked through different cellular pathways, we investigated the effect of proteasome inhibition with an encapsulated strain. Proteasome inhibition also resulted in a substantial (118%) increase in the intracellular survival of the encapsulated S. pneumoniae cells (Fig. 2). Thus, the involvement of the proteasome system in the intracellular killing of S. pneumoniae is independent of the presence of the capsule, and the bulk of invading encapsulated S. pneumoniae cells also enter a cellular trafficking pathway, which ultimately ends in their degradation.
FIG 2 .
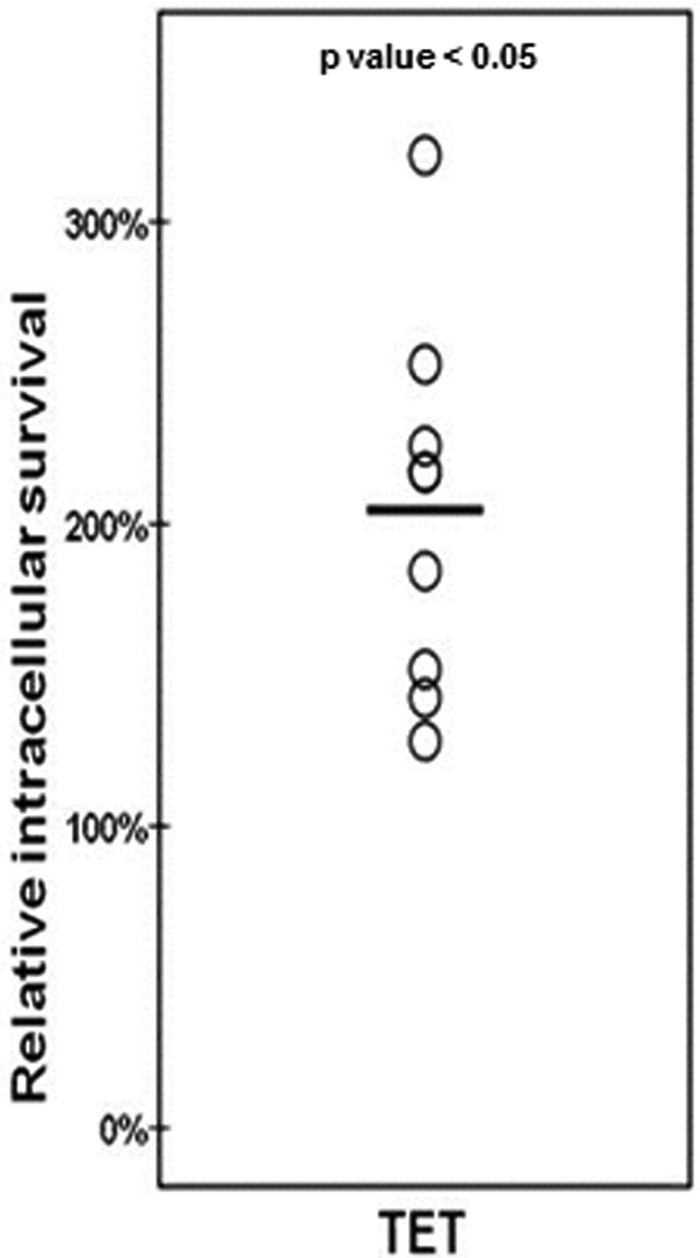
The capsule of S. pneumoniae does not inhibit the proteasome-mediated killing process. Relative rates of survival of internalized encapsulated S. pneumoniae during proteasomal inhibition with TET are shown. The control is set at 100%, and the experimental conditions are related to this percentage. Each circle is the relative intracellular survival in an individual experiment, and the horizontal line indicates the average of results from all experiments (n = 9). P value < 0.05. Relative intracellular survival is defined as intracellular survival divided by the amount of the initial invasion.
Intracellular S. pneumoniae colocalizes with proteasome and ubiquitin in vitro.
Immunofluorescent analysis was used to ascertain whether intracellular pneumococci colocalized with proteasome and or ubiquitin. First we investigated whether a combined treatment of antibiotics and lysozyme would be sufficient for the removal of most extracellular bacteria, which interfere with the specific study of intracellular bacteria. A combination of lysozyme and antibiotics was sufficient to lyse >99% of bacteria grown in broth (Fig. S1). Comparing the amount of extracellular bacteria after treatment with a combination of lysozyme and antibiotic with that after antibiotic treatment alone using the Triton X-100 permeabilization method previously described by Pracht et al. (27) (Fig. S2) clearly showed a drastic reduction in numbers of extracellular bacteria when lysozyme was added. At the same time, there was no apparent change in the amount of intracellular bacteria. Immunofluorescence analysis using this method showed that the intracellular pneumococci colocalized with proteasome (Fig. 3A). Moreover, a ubiquitin signal was detected on the bacteria in both human brain-derived microvascular endothelial cells (HBMEC) and human umbilical vein-derived endothelial cells (HUVEC) (Fig. 3B).
FIG 3 .

Confocal microscopy visualization of intracellular S. pneumoniae cells colocalized with proteasome-ubiquitin in vitro. S. pneumoniae (blue) inside HBMEC and HUVEC (red) colocalizes with proteasome (A) and ubiquitin (B) (both green). The scale of each image is shown by the white bar, which represents 2 µm. For each staining, 10 cells were randomly selected for imaging; each image shows 1 cell.
Intracellular S. pneumoniae cells are detected in mouse brain tissue and also colocalize with ubiquitin and proteasome.
To validate our in vitro findings, confocal microscopy analysis was performed on brain tissue from mice intravenously challenged with pneumococci. Mice were and sacrificed 1 and 14 h after infection, which represent the early and late stages of bacteremia preceding the development of meningitis (1). We focused on the subarachnoid space, as it was previously shown to be the anatomical site of the blood-brain barrier where most S. pneumoniae bacteria were found (1). Remarkably, the confocal imaging analysis, in particular, the XZ and YZ orthogonal views, clearly showed S. pneumoniae inside brain endothelial cells in vivo (Fig. 4A and B and 5A and B). The groups of pneumococci (blue signal, within white circles) detected inside the brain vascular endothelium (red signal) by confocal microscopy were in very close proximity to the proteasome signal (green), and some bacteria of these groups were clearly colocalized with proteasome at 1 h and 14 h postinfection (Fig. 4AB). Furthermore, all the groups of bacteria detected inside the brain endothelium had the ubiquitin signal (green) on their surfaces (Fig. 5AB).
FIG 4 .

S. pneumoniae was associated with the proteasome signal inside the brain vascular endothelium of the subarachnoid space. S. pneumoniae (blue, within white circles) detected inside the brain vascular endothelium (red) by confocal microscopy is associated with the proteasome signal (green) at 1 h (A) and 14 h (B) postinfection. Bacteria within the white circles are also shown in the enlarged images. For each time point of infection, brains from 3 mice were analyzed, and from each mouse, 3 brain sections were used for the immunofluorescence detection. These images are representative of observations for each analyzed mouse brain. The scale of each image is shown by the white bar, which represents 5 µm.
FIG 5 .

Association between the ubiquitin signal and intracellular S. pneumoniae in mouse brains. Confocal microscopy imaging of the ubiquitin signal (green) on the surfaces of intracellular bacteria (blue, within white circles) at 1 h (A) and 14 h (B) postinfection. Bacteria within the white circles are also shown in the enlarged images. For each time point of infection, brains from 3 mice were analyzed, and from each mouse, 3 brain sections were used for the immunofluorescence detection. These images are representative of what was observed in each mouse that was analyzed. The scale of each image is shown by the white bar, which represents 5 µm.
DISCUSSION
In this study, we investigated the involvement of the proteasome-ubiquitin system in the process of killing intracellular S. pneumoniae. Apart from regulating protein turnover, the proteasome system modulates responses to invading and intracellular pathogenic bacteria, and this modulation has previously been shown for many other pathogenic bacteria and viruses (8, 9, 12, 28). In our study, the inhibition of the proteasome-ubiquitin function resulted in a substantial increase in S. pneumoniae survival inside endothelial cells, indicating that the proteasome-ubiquitin system is essential for the host cell’s ability to efficiently kill intracellular S. pneumoniae.
How bacterial pathogens cross the blood-brain barrier is currently a subject of debate. Diverse studies have described (i) the destruction of endothelial cell layers in the cases of, for example, Neisseria meningitidis and the pneumococcal endotoxin pneumolysin (29, 30); (ii) the passage of organisms between brain endothelial cells, causing disruption of cell-cell tight junctions (31); and (iii) trafficking across the blood-brain barrier by transcytosis, an intracellular transport route for the transport of molecules and vesicles through cells from the apical to the basolateral side (4). The visualization of S. pneumoniae after invasion into the host cells was shown only using in vitro cell culture models (2–4, 27). Here we show for the first time to our knowledge that S. pneumoniae invades endothelial cells of the blood-brain barrier in vivo. These findings indicate that S. pneumoniae can invade endothelial cells without causing major disruptions of the endothelium during the translocation across the blood-brain barrier and support the hypothesis of pneumococcal translocation through either a pericellular or a transcytosis route. The bacteria were detected using an antipneumococcal antiserum (see Materials and Methods) raised against whole heat-killed bacteria. In a recent study, a preliminary quantification analysis indicated that the number of pneumococci associated with the brain vascular endothelium detected with anticapsule antibody was not different from the number detected with the antipneumococcal antiserum (1). If pneumococci had lost their capsule once inside brain endothelial cells, the number of bacteria detected with the antipneumococcal antiserum would have been significantly higher than the number of bacteria seen with the anticapsule antibody. When interacting with epithelial cells of the respiratory tract, S. pneumoniae was shown to lose its capsule (26). Our results suggest that, while in the brain, S. pneumoniae might maintain its capsule during translocation over the blood-brain barrier endothelium. Keeping the capsule when translocating over the blood-brain barrier would be an important advantage for the bacterium in evading the host defenses in the cerebrospinal fluid (CSF). However, this would not impede killing by the proteasome-ubiquitin system, as we show that this observation was capsule independent (Fig. 2).
The proteasome-ubiquitin system has also been implicated in the degradation of intracellular Salmonella enterica serovar Typhimurium (28, 32). Perrin et al. reported that proteasomes become associated with the surface of cytosolic Salmonella in macrophages (32) and that poly-ubiquitinated proteins accumulate on the surfaces of these cytosolic bacteria, suggesting that the proteasome-ubiquitin system might be implicated directly in the clearance of intracellular Salmonella cells (32). Intracellular pneumococci associated with both proteasome and ubiquitin signals; however, with confocal microscopy it is not possible to determine whether these bacteria are free in the cytosol or in a vesicle. Autophagy was recently described as a host mechanism of defense against bacteria; in particular, the ubiquitin-mediated recognition of invading bacteria during the selective autophagy process may be crucial for host cell survival (33). Although there are no indications that S. pneumoniae can access the cytosol by escaping the endosome, our study raises the intriguing possibility that this might occur. Once in the cytosol, organisms might be (partially) degraded directly by the proteasome and ubiquitin system, and inhibition of proteasome may interfere with endocytosis, thus resulting in reduced killing of intracellular bacteria.
Alternatively, the role of the proteasome-ubiquitin system in the degradation of bacteria is indirect. Apart from marking proteins for degradation, the ubiquitination system is involved in several essential cellular processes, such as cell cycle progression, DNA repair, and endocytosis (9–11, 32, 34). Given the importance of these biological processes, it is not surprising that microorganisms have developed mechanisms to interfere with various stages of the ubiquitin pathway in order to promote their trafficking inside the host cell. However, as of yet, there are no indications that S. pneumoniae gains any advantage in its association with the proteasome-ubiquitin system.
In conclusion, the data described in this paper clearly show that Streptococcus pneumoniae invades endothelial cells during infection and that a fully functional proteasome-ubiquitin system in endothelial cells is crucial for the efficient killing of intracellular S. pneumoniae. Our study sheds light on S. pneumoniae’s life and death inside endothelial cells, whose balance may ultimately lead to bacterial passage across cellular barriers and the development of invasive disease.
MATERIALS AND METHODS
Bacteremia-derived meningitis model.
All experiments involving animals were performed in strict accordance with Dutch legislation on animal experiments (35 [modified in 1996 with implementation of the European guidelines 86/609/EEG], 36), with the prior approval of and in accordance with guidelines of the Institutional Animal Care and Use Committee of the University of Groningen (DEC number 6152A). The bacteremia-derived meningitis model described by Orihuela et al. (37) was adapted as described before (1). Four groups of 5 female BALB/c mice, 6 to 8 weeks old (Harlan, Horst, The Netherlands), were anesthetized by inhalation of 2.5% isoflurane before the challenge. Intravenous tail vein injection with 200 µl of 107 CFU of the TIGR4 wild type was performed. The mice were sacrificed at 1, 3, 8, and 14 h after bacterial challenge. After sacrifice, to remove unattached bacteria in the bloodstream, perfusion was performed by injecting sterile phosphate-buffered saline (PBS) in the right ventricle via the vena cava until the blood was completely removed. Brains, lungs, and spleens were collected and stored with Shandon Cryomatrix (Thermo Scientific, Runcorn, England) at −80° C.
Bacterial strains and growth conditions.
Encapsulated S. pneumoniae TIGR4, obtained from C. J. Orihuela (37), was used to challenge the mice. Bacteria were grown in Todd-Hewitt broth (Oxoid Thermo Scientific, Basingstoke, United Kingdom), and unencapsulated TIGR4 was grown in M17 medium supplemented with 0.5% glucose (GM17). Bacteria were harvested at an optical density (OD) at 600 nm of 0.25 to 0.30. One milliliter of encapsulated TIGR4 was centrifuged at 10,000 × g for 3 min and resuspended in sterile PBS (Lonza, Verviers, Belgium) to generate a challenge dose of 107 CFU/mouse. For the in vitro experiments, encapsulated S. pneumoniae TIGR4 (38) and its unencapsulated version (39) were used. Bacteria were grown in M17 medium supplemented with 0.5% glucose (GM17). Cultures were incubated in a 5% CO2 incubator at 37°C. S. pneumoniae aliquots were made by growing bacteria in GM17 to an OD at 600 nm of ~0.25, mixed to an 11% glycerol concentration, and then frozen in 1-ml aliquots at −80° C
Inhibitors.
The concentrations indicated below were selected after titrating out nonstressful concentrations. Their respective solvents were used as controls. For ubiquitination and proteasomal inhibition, the following endpoint concentrations were used: tetraethylthiuram disulfide (TET) (Sigma-Aldrich) at 10 µM in H2O, MG132 (Calbiochem) at 10 µM in dimethyl sulfoxide (DMSO), and UBEI-41 (Biogenova) at 10 µM in DMSO.
Intracellular survival of S. pneumoniae in endothelial cells.
HBMEC (obtained from K. S. Kim) and HUVEC (obtained from the Endothelial Cell Facility, UMCG) were cultivated as previously described (40, 41). Prior to infection, confluent HBMEC monolayers in 6- or 12-well plates were washed repeatedly with RPMI 1640 and incubated for 1 h in cell culture medium. One-milliliter aliquots of S. pneumoniae (see above) were centrifuged at 10,000 × g for 3 min, and the bacterial pellet was dissolved in endothelial cell culture medium. Subsequently, ~5 × 106 CFU of S. pneumoniae was added to each well and incubated for 2 h; nonadherent bacteria were removed by repeated washing with RPMI 1640. To assess invasion of the host cell by S. pneumoniae, any remaining extracellular bacteria were eradicated by a 1-h incubation with cell culture medium supplemented with gentamicin (50 µg/ml) and penicillin G (2.5 µg/ml) and then washed repeatedly with RPMI 1640 and lysed. CFU were counted by plating serial dilutions on blood agar plates. To assess intracellular survival, after treatment with gentamicin and penicillin G as described above, cells were washed repeatedly with RPMI 1640 and new culture medium containing gentamicin (13.34 µg/ml), penicillin G (0.67 µg/ml), and inhibitors of the proteasome-ubiquitin system were added at phenotypically nonstressful concentrations, which were previously determined by monitoring the cells microscopically for stress symptoms. All inhibitors and bacteria were used at nonstressful concentrations throughout the assay. Three hours after the addition of inhibitors, the cells were lysed and plated as described above.
Immunofluorescence detection and image processing.
For immunofluorescence detection, endothelial cells were grown on glass coverslips placed inside each well. In addition to antibiotics, 10 mg/ml of lysozyme (Sigma-Aldrich) was added to the cell culture medium in order to lyse extracellular bacteria. After a 2-h incubation with unencapsulated S. pneumoniae (see “Intracellular survival of S. pneumoniae in endothelial cells” above), HBMEC and HUVEC were washed with PBS to remove nonadherent bacteria and fixed with 4% paraformaldehyde (Sigma-Aldrich). The slides were then washed in PBS three times for 5 min each time and preincubated using PBS with 0.3% Triton X-100 (Sigma-Aldrich). For confocal microscopy detection in mice brains, 5-µm thin sections were fixed with acetone for 10 min. After fixation, cells and tissue sections were incubated with primary antibody for 1 h at room temperature (RT). After the washing with PBS, incubation with secondary antibody for 1 h at RT followed. After the washing with PBS, CitiFluor solution (Science Services, Munich, Germany) was added to each tissue section/glass disk. The slides were analyzed with a Leica TC SP8 confocal microscope, and the z-stacks were merged by means of Imaris (Bitplane Scientific Software).
Antibodies and lectin.
All antibodies and lectin were diluted in sterile PBS with 5% fetal calf serum (FCS) (Biochrom, Berlin, Germany). To detect pneumococci, an antipneumococcal antiserum (1) (Eurogentec, Maastricht, The Netherlands), diluted 1:200, was used in combination with an Alexa Fluor 350 goat anti-rabbit antibody (Invitrogen Life Technologies, Carlsbad, CA) at a1:500 dilution. The antipneumococcal antiserum was directly labeled with Alexa Fluor 350 (Zenon rabbit IgG labeling kit; Invitrogen Life Technologies) when used in combination with the rabbit antiubiquitin antibody. For the detection of endothelial cells, DyLight 594-labeled Lycopersicon esculentum lectin (tomato lectin) (Vector Laboratories, Burlingame, CA) at a 1:200 dilution was used (13). To detect proteasome and ubiquitin, respectively, a mouse antiproteasome antibody (Abcam, Cambridge, United Kingdom) and a rabbit antiubiquitin antibody (Acris Antibodies, Herford, Germany) were used, both at a 1:200 dilution, in combination with, respectively, Alexa Fluor 488 goat anti-mouse antibody (Invitrogen Life Technologies) and Alexa Fluor 488 goat anti-rabbit antibody (Invitrogen Life Technologies).
Confocal visualization of S. pneumoniae inside brain endothelial cells in vivo.
After the z-stacks were merged, by means of Imaris, xyz series were generated with the corresponding orthogonal projections in the xz and zy planes (2). The orthogonal views allowed visualization of the endothelial layer of the subarachnoid space from the x, y, and z axes. In order to accurately discriminate the location of the bacteria, the whole-cell/endothelial layer was imaged through the sequential imaging of z-stacks. Only the bacteria observed inside the endothelial layer in all orthogonal planes were considered intracellular.
Statistical analysis.
The SPSS-16 2-tailed independent-sample t test was used to determine the significance, presented as a P value, of the difference between bacterial intracellular survival results of endothelial cells with and without proteasome-ubiquitin inhibitor.
SUPPLEMENTAL MATERIAL
Survival of internalized S. pneumoniae during proteasomal inhibition with MG132. (A) Each circle represents the intracellular survival of S. pneumoniae in one biological replicate for each sample (control, MG132) expressed as numbers of CFU/well. Horizontal lines are the averages of results for all biological replicates for control and MG132 experiments (n = 4 wells). In total, 6 different experiments were performed. (B) S. pneumoniae intracellular survival (as a percentage) in HBMEC with MG132 in comparison with survival in control HBMEC. Instead of showing relative increases in survival (as in Fig. 1 and 2), here the percent increase is defined as relative to 100% (intracellular survival in control cells divided by the intracellular survival in cells treated with MG132). Download
Survival of internalized S. pneumoniae during proteasomal inhibition with TET. (A) Each circle represents the intracellular survival of S. pneumoniae in one biological replicate for each sample (control, TET), expressed as numbers of CFU/well. Horizontal lines are the averages of results for all biological replicates for control and TET experiments (n = 3 wells). Three different experiments were performed. (B) Increases (percentages) in S. pneumoniae intracellular survival in HBMEC with TET in comparison with that in control HBMEC. Instead of showing relative increases in survival (as in Fig. 1 and 2), here the percent increase is defined as relative to 100% (intracellular survival in control cells divided by the intracellular survival in cells treated with TET). Download
Survival of internalized S. pneumoniae during ubiquitin inhibition with UBEI-41. (A) Each circle represents the intracellular survival of S. pneumoniae in one biological replicate for each sample (control, UBEI-41) expressed as numbers of CFU/well. Horizontal lines are the averages of results for all biological replicates for control and UBEI-41 experiments (n = 4 wells). Six different experiments were performed. (B) Increases (percentages) in S. pneumoniae intracellular survival in HBMEC with UBEI-41 in comparison with that in control HBMEC. Instead of showing relative increases in survival (as in Fig. 1 and 2), here the percent increase is defined as relative to 100% (intracellular survival in control cells divided by the intracellular survival in cells treated with UBEI-41). Download
Survival of internalized encapsulated S. pneumoniae during proteasomal inhibition with TET. (A) Each circle represents the intracellular survival of S. pneumoniae in one biological replicate for each sample (control, TET) expressed as numbers of CFU/well. Horizontal lines are the averages of results for all biological replicates for control and TET experiments (n = 2 wells). Nine different experiments were performed. (B) Increases (percentages) in S. pneumoniae intracellular survival in HBMEC with TET in comparison with that in control HBMEC. Instead of showing relative increases in survival (as in Fig. 1 and 2), here the percent increase is defined as relative to 100% (intracellular survival in control cells divided by the intracellular survival in cells treated with TET). Download
Combining antibiotics with lysozyme kills and lyses pneumococci. (A) Immunofluorescence detection of S. pneumoniae (red) grown in GM17 medium to an OD at 600 nm of 0.25 to 0.30. (B) Immunofluorescence detection of S. pneumoniae (red) grown in GM17 medium supplemented with gentamicin (13.34 µg/ml), penicillin G (0.67 µg/ml), and lysozyme (10 mg/ml). Hardly any bacteria were detected because of the lysis induced by the lysozyme with antibiotics. Download
Combining antibiotics and lysozyme degrades extracellular bacteria and allows specific visualization of intracellular pneumococci. (A) In the absence of antibiotics and lysozyme, adherent bacteria were detected with both Alexa Fluor 488 and Alexa Fluor 594 goat anti-rabbit (yellow), while the intracellular bacteria were detected only with Alexa Fluor 594 goat anti-rabbit (red) after permeabilization with Triton X-100. (B) Addition of antibiotics and lysozyme leads to lysis of almost all extracellular bacteria and the specific detection of intracellular bacteria (red). The distinction between intra- and extracellular bacteria was determined using permeabilization with Triton X-100 (19). Download
ACKNOWLEDGMENTS
Part of the work was performed at the UMCG Microscopy and Imaging Center (UMIC). In particular, we thank Klaas Sjollema for all the technical support during the confocal imaging. We thank Melania Minoia for essential support during the setup of the proteasome-ubiquitin experiments. We also thank Henk Moorlag for providing HUVEC and for the cryostat cutting of tissue sections.
Footnotes
Citation Iovino F, Gradstedt H, Bijlsma JJ. 2014. The proteasome-ubiquitin system is required for efficient killing of intracellular Streptococcus pneumoniae by brain endothelial cells. mBio 5(4):e00984-14. doi:10.1128/mBio.00984-14.
REFERENCES
- 1. Iovino F, Orihuela CJ, Moorlag HE, Molema G, Bijlsma JJ. 2013. Interactions between blood-borne Streptococcus pneumoniae and the blood-brain barrier preceding meningitis. PLoS One 8:e68408. 10.1371/journal.pone.0068408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Gradstedt H, Iovino F, Bijlsma JJ. 2013. Streptococcus pneumoniae invades endothelial host cells via multiple pathways and is killed in a lysosome dependent manner. PLoS One 8:e65626. 10.1371/journal.pone.0065626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Radin JN, Orihuela CJ, Murti G, Guglielmo C, Murray PJ, Tuomanen EI. 2005. Beta-arrestin 1 participates in platelet-activating factor receptor-mediated endocytosis of Streptococcus pneumoniae. Infect. Immun. 73:7827–7835. 10.1128/IAI.73.12.7827-7835.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ring A, Weiser JN, Tuomanen EI. 1998. Pneumococcal trafficking across the blood-brain barrier. Molecular analysis of a novel bidirectional pathway. J. Clin. Invest. 102:347–360. 10.1172/JCI2406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Zhang JR, Mostov KE, Lamm ME, Nanno M, Shimida S, Ohwaki M, Tuomanen E. 2000. The polymeric immunoglobulin receptor translocates pneumococci across human nasopharyngeal epithelial cells. Cell 102:827–837. 10.1016/S0092-8674(00)00071-4 [DOI] [PubMed] [Google Scholar]
- 6. Nizet V, Kim KS, Stins M, Jonas M, Chi EY, Nguyen D, Rubens CE. 1997. Invasion of brain microvascular endothelial cells by group B streptococci. Infect. Immun. 65:5074–5081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Schröder A, Kland R, Peschel A, von Eiff C, Aepfelbacher M. 2006. Live cell imaging of phagosome maturation in Staphylococcus aureus infected human endothelial cells: small colony variants are able to survive in lysosomes. Med. Microbiol. Immunol. 195:185–194. 10.1007/s00430-006-0015-0 [DOI] [PubMed] [Google Scholar]
- 8. Chiaraviglio L, Brown DA, Kirby JE. 2008. Infection of cultured human endothelial cells by Legionella pneumophila. PLoS One 3:e2012. 10.1371/journal.pone.0002012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Malynn BA, Ma A. 2010. Ubiquitin makes its mark on immune regulation. Immunity 33:843–852. 10.1016/j.immuni.2010.12.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Blanchette P, Branton PE. 2009. Manipulation of the ubiquitin-proteasome pathway by small DNA tumor viruses. Virology 384:317–323. 10.1016/j.virol.2008.10.005 [DOI] [PubMed] [Google Scholar]
- 11. Haglund K, Dikic I. 2005. Ubiquitylation and cell signaling. EMBO J. 24:3353–3359. 10.1038/sj.emboj.7600808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ciechanover A. 1998. The ubiquitin-proteasome pathway: on protein death and cell life. EMBO J. 17:7151–7160. 10.1093/emboj/17.24.7151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sigismund S, Polo S, Di Fiore PP. 2004. Signaling through monoubiquitination. Curr. Top. Microbiol. Immunol. 286:149–185 [DOI] [PubMed] [Google Scholar]
- 14. Haglund K, Di Fiore PP, Dikic I. 2003. Distinct monoubiquitin signals in receptor endocytosis. Trends Biochem. Sci. 28:598–603. 10.1016/j.tibs.2003.09.005 [DOI] [PubMed] [Google Scholar]
- 15. Dahlmann B. 2007. Role of proteasomes in disease. BMC Biochem. 8(Suppl 1):S3. 10.1186/1471-2091-8-S1-S3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Haglund K, Dikic I. 2005. Ubiquitylation and cell signaling. EMBO J. 24:3353–3359. 10.1038/sj.emboj.7600808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Malynn BA, Ma A. 2010. Ubiquitin makes its mark on immune regulation. Immunity 33:843–852. 10.1016/j.immuni.2010.12.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Rytkönen A, Holden DW. 2007. Bacterial interference of ubiquitination and deubiquitination. Cell Host Microbe 1:13–22. 10.1016/j.chom.2007.02.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Steinhilb ML, Turner RS, Gaut JR. 2001. The protease inhibitor, MG132, blocks maturation of the amyloid precursor protein Swedish mutant preventing cleavage by beta-secretase. J. Biol. Chem. 276:4476–4484. 10.1074/jbc.M008793200 [DOI] [PubMed] [Google Scholar]
- 20. Kubori T, Galán JE. 2003. Temporal regulation of Salmonella virulence effector function by proteasome-dependent protein degradation. Cell 115:333–342. 10.1016/S0092-8674(03)00849-3 [DOI] [PubMed] [Google Scholar]
- 21. Chen D, Cui QC, Yang H, Dou QP. 2006. Disulfiram, a clinically used anti-alcoholism drug and copper-binding agent, induces apoptotic cell death in breast cancer cultures and xenografts via inhibition of the proteasome activity. Cancer Res. 66:10425–10433. 10.1158/0008-5472.CAN-06-2126 [DOI] [PubMed] [Google Scholar]
- 22. Wickström M, Danielsson K, Rickardson L, Gullbo J, Nygren P, Isaksson A, Larsson R, Lövborg H. 2007. Pharmacological profiling of disulfiram using human tumor cell lines and human tumor cells from patients. Biochem. Pharmacol. 73:25–33. 10.1016/j.bcp.2006.08.016 [DOI] [PubMed] [Google Scholar]
- 23. Lövborg H, Oberg F, Rickardson L, Gullbo J, Nygren P, Larsson R. 2006. Inhibition of proteasome activity, nuclear factor-kappaB translocation and cell survival by the antialcoholism drug disulfiram. Int. J. Cancer 118:1577–1580. 10.1002/ijc.21534 [DOI] [PubMed] [Google Scholar]
- 24. Yang Y, Kitagaki J, Dai RM, Tsai YC, Lorick KL, Ludwig RL, Pierre SA, Jensen JP, Davydov IV, Oberoi P, Li CC, Kenten JH, Beutler JA, Vousden KH, Weissman AM. 2007. Inhibitors of ubiquitin-activating enzyme (E1), a new class of potential cancer therapeutics. Cancer Res. 67:9472–9481. 10.1158/0008-5472.CAN-07-0568 [DOI] [PubMed] [Google Scholar]
- 25. Kadioglu A, Weiser JN, Paton JC, Andrew PW. 2008. The role of Streptococcus pneumoniae virulence factors in host respiratory colonization and disease. Nat. Rev. Microbiol. 6:288–301. 10.1038/nrmicro1871 [DOI] [PubMed] [Google Scholar]
- 26. Hammerschmidt S, Wolff S, Hocke A, Rosseau S, Müller E, Rohde M. 2005. Illustration of pneumococcal polysaccharide capsule during adherence and invasion of epithelial cells. Infect. Immun. 73:4653–4667. 10.1128/IAI.73.8.4653-4667.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Pracht D, Elm C, Gerber J, Bergmann S, Rohde M, Seiler M, Kim KS, Jenkinson HF, Nau R, Hammerschmidt S. 2005. PavA of Streptococcus pneumoniae modulates adherence, invasion, and meningeal inflammation. Infect. Immun. 73:2680–2689. 10.1128/IAI.73.5.2680-2689.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Veiga E, Cossart P. 2005. Ubiquitination of intracellular bacteria: a new bacteria-sensing system? Trends Cell Biol. 15:2–5. 10.1016/j.tcb.2004.11.005 [DOI] [PubMed] [Google Scholar]
- 29. Coureuil M, Mikaty G, Miller F, Lécuyer H, Bernard C, Bourdoulous S, Duménil G, Mège RM, Weksler BB, Romero IA, Couraud PO, Nassif X. 2009. Meningococcal type IV pili recruit the polarity complex to cross the brain endothelium. Science 325:83–87. 10.1126/science.1173196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Marriott HM, Mitchell TJ, Dockrell DH. 2008. Pneumolysin: a double-edged sword during the host-pathogen interaction. Curr. Mol. Med. 8:497–509. 10.2174/156652408785747924 [DOI] [PubMed] [Google Scholar]
- 31. Attali C, Durmort C, Vernet T, Di Guilmi AM. 2008. The interaction of Streptococcus pneumoniae with plasmin mediates transmigration across endothelial and epithelial monolayers by intercellular junction cleavage. Infect. Immun. 76:5350–5356. 10.1128/IAI.00184-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Perrin AJ, Jiang X, Birmingham CL, So NS, Brumell JH. 2004. Recognition of bacteria in the cytosol of Mammalian cells by the ubiquitin system. Curr. Biol. 14:806–811. 10.1016/j.cub.2004.09.022 [DOI] [PubMed] [Google Scholar]
- 33. Huang J, Brumell JH. 2014. Bacteria-autophagy interplay: a battle for survival. Nat. Rev. Microbiol. 12:101–114. 10.1038/nrmicro3160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Strous GJ, Govers R. 1999. The ubiquitin-proteasome system and endocytosis. J. Cell Sci. 112:1417–1423 [DOI] [PubMed] [Google Scholar]
- 35. Dutch Government 12 January 1997. Wet op de dierproeven. Dutch Government, Amsterdam, The Netherlands: (In Dutch.) [Google Scholar]
- 36. Dutch Government 31 May 1985. Dierproevenbesluit. Dutch Government, Amsterdam, The Netherlands: (In Dutch.) [Google Scholar]
- 37. Orihuela CJ, Mahdavi J, Thornton J, Mann B, Wooldridge KG, Abouseada N, Oldfield NJ, Self T, Ala’Aldeen DA, Tuomanen EI. 2009. Laminin receptor initiates bacterial contact with the blood brain barrier in experimental meningitis models. J. Clin. Invest. 119:1638–1646. 10.1172/JCI36759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Tettelin H, Nelson KE, Paulsen IT, Eisen JA, Read TD, Peterson S, Heidelberg J, DeBoy RT, Haft DH, Dodson RJ, Durkin AS, Gwinn M, Kolonay JF, Nelson WC, Peterson JD, Umayam LA, White O, Salzberg SL, Lewis MR, Radune D, Holtzapple E, Khouri H, Wolf AM, Utterback TR, Hansen CL, McDonald LA, Feldblyum TV, Angiuoli S, Dickinson T, Hickey EK, Holt IE, Loftus BJ, Yang F, Smith HO, Venter JC, Dougherty BA, Morrison DA, Hollingshead SK, Fraser CM. 2001. Complete genome sequence of a virulent isolate of Streptococcus pneumoniae. Science 293:498–506. 10.1126/science.1061217 [DOI] [PubMed] [Google Scholar]
- 39. Burghout P, Cron LE, Gradstedt H, Quintero B, Simonetti E, Bijlsma JJ, Bootsma HJ, Hermans PW. 2010. Carbonic anhydrase is essential for Streptococcus pneumoniae growth in environmental ambient air. J. Bacteriol. 192:4054–4062. 10.1128/JB.00151-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Stins MF, Badger J, Sik Kim K. 2001. Bacterial invasion and transcytosis in transfected human brain microvascular endothelial cells. Microb. Pathog. 30:19–28. 10.1006/mpat.2000.0406 [DOI] [PubMed] [Google Scholar]
- 41. Asgeirsdóttir SA, Talman EG, de Graaf IA, Kamps JA, Satchell SC, Mathieson PW, Ruiters MH, Molema G. 2010. Targeted transfection increases siRNA uptake and gene silencing of primary endothelial cells in vitro—a quantitative study. J. Control. Release 141:241–251. 10.1016/j.jconrel.2009.09.008 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Survival of internalized S. pneumoniae during proteasomal inhibition with MG132. (A) Each circle represents the intracellular survival of S. pneumoniae in one biological replicate for each sample (control, MG132) expressed as numbers of CFU/well. Horizontal lines are the averages of results for all biological replicates for control and MG132 experiments (n = 4 wells). In total, 6 different experiments were performed. (B) S. pneumoniae intracellular survival (as a percentage) in HBMEC with MG132 in comparison with survival in control HBMEC. Instead of showing relative increases in survival (as in Fig. 1 and 2), here the percent increase is defined as relative to 100% (intracellular survival in control cells divided by the intracellular survival in cells treated with MG132). Download
Survival of internalized S. pneumoniae during proteasomal inhibition with TET. (A) Each circle represents the intracellular survival of S. pneumoniae in one biological replicate for each sample (control, TET), expressed as numbers of CFU/well. Horizontal lines are the averages of results for all biological replicates for control and TET experiments (n = 3 wells). Three different experiments were performed. (B) Increases (percentages) in S. pneumoniae intracellular survival in HBMEC with TET in comparison with that in control HBMEC. Instead of showing relative increases in survival (as in Fig. 1 and 2), here the percent increase is defined as relative to 100% (intracellular survival in control cells divided by the intracellular survival in cells treated with TET). Download
Survival of internalized S. pneumoniae during ubiquitin inhibition with UBEI-41. (A) Each circle represents the intracellular survival of S. pneumoniae in one biological replicate for each sample (control, UBEI-41) expressed as numbers of CFU/well. Horizontal lines are the averages of results for all biological replicates for control and UBEI-41 experiments (n = 4 wells). Six different experiments were performed. (B) Increases (percentages) in S. pneumoniae intracellular survival in HBMEC with UBEI-41 in comparison with that in control HBMEC. Instead of showing relative increases in survival (as in Fig. 1 and 2), here the percent increase is defined as relative to 100% (intracellular survival in control cells divided by the intracellular survival in cells treated with UBEI-41). Download
Survival of internalized encapsulated S. pneumoniae during proteasomal inhibition with TET. (A) Each circle represents the intracellular survival of S. pneumoniae in one biological replicate for each sample (control, TET) expressed as numbers of CFU/well. Horizontal lines are the averages of results for all biological replicates for control and TET experiments (n = 2 wells). Nine different experiments were performed. (B) Increases (percentages) in S. pneumoniae intracellular survival in HBMEC with TET in comparison with that in control HBMEC. Instead of showing relative increases in survival (as in Fig. 1 and 2), here the percent increase is defined as relative to 100% (intracellular survival in control cells divided by the intracellular survival in cells treated with TET). Download
Combining antibiotics with lysozyme kills and lyses pneumococci. (A) Immunofluorescence detection of S. pneumoniae (red) grown in GM17 medium to an OD at 600 nm of 0.25 to 0.30. (B) Immunofluorescence detection of S. pneumoniae (red) grown in GM17 medium supplemented with gentamicin (13.34 µg/ml), penicillin G (0.67 µg/ml), and lysozyme (10 mg/ml). Hardly any bacteria were detected because of the lysis induced by the lysozyme with antibiotics. Download
Combining antibiotics and lysozyme degrades extracellular bacteria and allows specific visualization of intracellular pneumococci. (A) In the absence of antibiotics and lysozyme, adherent bacteria were detected with both Alexa Fluor 488 and Alexa Fluor 594 goat anti-rabbit (yellow), while the intracellular bacteria were detected only with Alexa Fluor 594 goat anti-rabbit (red) after permeabilization with Triton X-100. (B) Addition of antibiotics and lysozyme leads to lysis of almost all extracellular bacteria and the specific detection of intracellular bacteria (red). The distinction between intra- and extracellular bacteria was determined using permeabilization with Triton X-100 (19). Download