

ABSTRACT
The global threat to public health posed by emerging multidrug-resistant bacteria in the past few years necessitates the development of novel approaches to combat bacterial infections. Endolysins encoded by bacterial viruses (or phages) represent one promising avenue of investigation. These enzyme-based antibacterials efficiently kill Gram-positive bacteria upon contact by specific cell wall hydrolysis. However, a major hurdle in their exploitation as antibacterials against Gram-negative pathogens is the impermeable lipopolysaccharide layer surrounding their cell wall. Therefore, we developed and optimized an approach to engineer these enzymes as outer membrane-penetrating endolysins (Artilysins), rendering them highly bactericidal against Gram-negative pathogens, including Pseudomonas aeruginosa and Acinetobacter baumannii. Artilysins combining a polycationic nonapeptide and a modular endolysin are able to kill these (multidrug-resistant) strains in vitro with a 4 to 5 log reduction within 30 min. We show that the activity of Artilysins can be further enhanced by the presence of a linker of increasing length between the peptide and endolysin or by a combination of both polycationic and hydrophobic/amphipathic peptides. Time-lapse microscopy confirmed the mode of action of polycationic Artilysins, showing that they pass the outer membrane to degrade the peptidoglycan with subsequent cell lysis. Artilysins are effective in vitro (human keratinocytes) and in vivo (Caenorhabditis elegans).
IMPORTANCE
Bacterial resistance to most commonly used antibiotics is a major challenge of the 21st century. Infections that cannot be treated by first-line antibiotics lead to increasing morbidity and mortality, while millions of dollars are spent each year by health care systems in trying to control antibiotic-resistant bacteria and to prevent cross-transmission of resistance. Endolysins—enzymes derived from bacterial viruses—represent a completely novel, promising class of antibacterials based on cell wall hydrolysis. Specifically, they are active against Gram-positive species, which lack a protective outer membrane and which have a low probability of resistance development. We modified endolysins by protein engineering to create Artilysins that are able to pass the outer membrane and become active against Pseudomonas aeruginosa and Acinetobacter baumannii, two of the most hazardous drug-resistant Gram-negative pathogens.
INTRODUCTION
Endolysins are produced by bacteriophages at the end of their lytic replication cycle to hydrolyze the bacterial cell wall for progeny release. In the last decade, it has been extensively demonstrated that the addition of recombinant purified endolysin to susceptible Gram-positive bacteria, including Staphylococcus, Streptococcus, and Bacillus species, kills the bacteria through cell lysis, both in vitro and in various animal infection models of human disease (1–4). Their rapid action, high specificity, nontoxicity, high efficiency, and low probability of resistance development make them promising, novel enzyme-based antibacterials (5). The expansion of endolysins as antibacterials against important Gram-negative pathogens is hindered by the outer membrane. Although some endolysins show limited membrane-destabilizing properties (6, 7), this outer membrane poses a highly effective permeability barrier for the passage of harmful compounds, including endolysins and many other antibacterial compounds (8). We have previously shown that the use of high hydrostatic pressure (in the range of 150 to 200 MPa) (9) or outer membrane permeabilizers (10) strongly increases the access of endolysins to the Gram-negative peptidoglycan layer. In this study, we developed an efficient methodology of protein engineering of endolysins to generate highly antibacterial, outer membrane-penetrating endolysins termed Artilysins independently of protein transport systems (11). We show that Artilysins cause 4 to 5 log killing of different (clinically relevant) Gram-negative species in vitro and are also effective in vivo.
RESULTS
Artilysins: proof of concept.
The rationale behind Artilysins is based on the modification of endolysins with lipopolysaccharide (LPS)-destabilizing peptides. The LPS layer is stabilized through ionic interactions between divalent cations and phosphate groups and hydrophobic stacking of the lipid A moiety (12). Seven peptides were selected for their (putative) LPS-destabilizing activity based on different physicochemical properties (cationic, hydrophobic or amphipathic) that interfere with these stabilizing forces (see Table S1 in the supplemental material) (13–16). To test this engineering concept, the two most active endolysins (OBPgp279 and PVP-SE1gp146) from our collection of characterized endolysins (7) were used. OBPgp279 originates from Pseudomonas fluorescens phage OBP (phiKZ-like; Myoviridae) and PVP-SE1gp146 from Salmonella enterica serovar Enteritidis phage PVP-SE1 (rV5-like; Myoviridae). Both endolysins feature a modular structure with one (PVP-SE1gp146) or two (OBPgp279) N-terminal peptidoglycan-binding (PBD-like) domains and a C-terminal catalytic domain (glycoside hydrolase 19-like family, lysozyme-like superfamily). This domain configuration is rare among endolysins targeting Gram-negative peptidoglycan and is associated with strongly increased enzymatic activity (7, 17, 18). In addition, OBPgp279 has a moderate intrinsic antibacterial effect on Pseudomonas aeruginosa (1.10 log reduction) (7). Fourteen different Artilysins (LoGT-001 to LoGT-014) (Fig. 1; see also Tables S2 and S3 in the supplemental material) were produced and purified, the effect of the respective fusions on protein yield was determined (see Table S4), and the antibacterial activity of the different Artilysins against P. aeruginosa, Escherichia coli, and Salmonella enterica serovar Typhimurium was evaluated (see Table S5). The fusion of each LPS-destabilizing peptide to endolysin PVP-SE1gp146 resulted in an enhanced bactericidal activity against P. aeruginosa in comparison to wild-type PVP-SE1gp146, but the effect was most prominent for the polycationic nonapeptide (PCNP). The latter is the only peptide that also increases the intrinsic antibacterial effect of OBPgp279, whereas the other peptides appear to inhibit this intrinsic antibacterial effect (Fig. 2A). The highest reduction was obtained with LoGT-001, combining the PCNP tag and OBPgp279, with a 2.61 ± 0.09 log reduction in only 30 min. Thus, the PCNP tag emerged as the most appropriate peptide to confer antibacterial activity to (otherwise nonantibacterial) endolysins.
FIG 1 .

Visual representation of engineered Artilysins. Seven different endolysins (OBPgp279 [YP_004958186.1], PVP-SE1gp146 [YP_004893953.1], phiKZgp144 [NP_803710.1], 201ϕ2-1gp229 [YP_001956952.1], CR8gp3.5 [unpublished], P2gp09 [NP_046765.1], and PsP3gp10 [NP_958065.1) were selected for modification with different peptides. (A) Single N-terminal fusion constructs, (B) double N-terminal fusion constructs, (C) N-terminal extended-linker constructs, (D) C-terminal extended-linker constructs. Abbreviations: PCNP = polycationic peptide, HPP = hydrophobic pentapeptide, Pa1 = Parasin1, Ly1 = lycotoxin1, L = linker unit consisting of GAGA sequence.
FIG 2 .
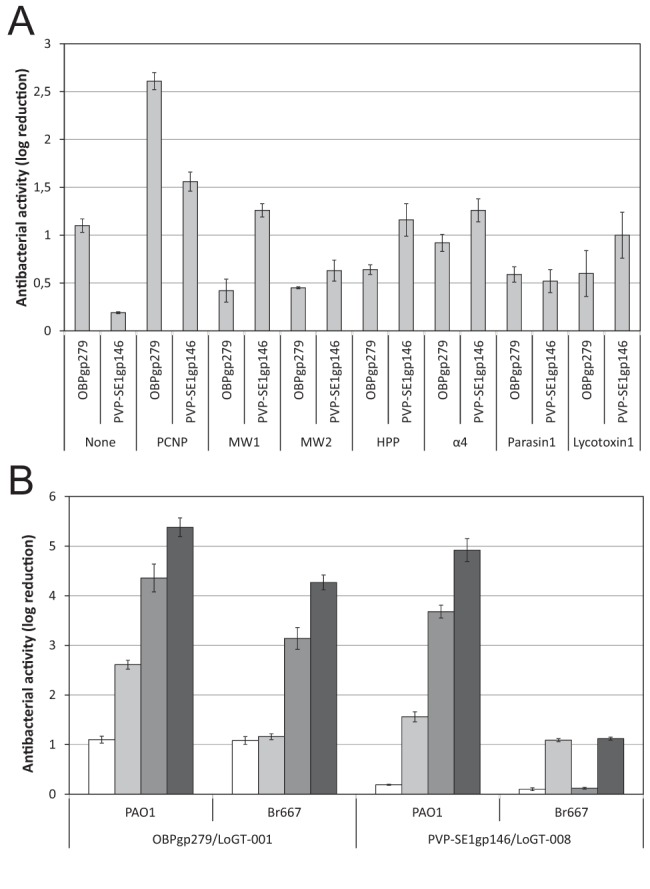
Antibacterial activity of the N-terminal fusion variants of OBPgp279 and PVP-SE1gp146. (A) Fourteen Artilysins with N-terminal fusions of seven different peptides (see Table S1 in the supplemental material) to OBPgp279 and PVP-SE1gp146 are compared to the corresponding unmodified endolysins for their antibacterial activity against P. aeruginosa PAO1. This comparison shows the PCNP to be the most effective peptide among the seven peptides tested here. (B) Comparison of the antibacterial activities of two wild-type endolysins (OBPgp279 and PVP-SE1gp146) and their PCNP-tagged Artilysin counterparts (LoGT-001 and LoGT-008, respectively) with and without 0.5 mM EDTA. Activity was tested on P. aeruginosa strains PAO1 and multidrug-resistant Br667, both in the absence (wt endolysin, white bars; Artilysin, light gray bars) and presence (wt endolysin, dark gray bars; Artilysin, black bars) of 0.5 mM EDTA. These data show that the PCNP fusion for the antibacterial activity of the endolysin added value while not compromising its synergy with EDTA. Averages and standard deviations of results of three replicates are shown.
We also performed the same experiment in the presence of 0.5 mM EDTA, an outer membrane permeabilizer that shows strong synergy with endolysins (10). For both LoGT-001 and LoGT-008, combining the PCNP tag with OBPgp279 and PVP-SE1gp146, respectively, the synergy with EDTA is not compromised by the modification: both the fusion to the PCNP tag and the addition of a low concentration of EDTA confer antibacterial activity against P. aeruginosa PAO1 and Br667 (a multidrug-resistant clinical isolate [19]) to the wild-type endolysins or further improve this activity. A maximal reduction of P. aeruginosa PAO1, at 5.38 ± 0.19 log, was observed with LoGT-001 (Fig. 2B). This indicates that the PCNP tag has a mode of action different from that of EDTA. This synergy constitutes a potential benefit for the topical applicability of Artilysins. Although E. coli and S. Typhimurium were also most strongly affected by PCNP-fused endolysins, the general effect was here significantly smaller, with maximal reductions of 1.70 ± 0.05 (E. coli) and 0.91 ± 0.04 (S. Typhimurium) (see Table S5).
Mode of action of Artilysins.
We used time-lapse microscopy to observe the mode of killing by a PCNP-fused endolysin. LoGT-008 was mixed with P. aeruginosa PAO1 in the presence of 0.5 mM EDTA and immediately incubated on an agar pad for microscopic observation. Within minutes, the first rods lysed abruptly (see movie http://www.biw.kuleuven.be/dp/logt/phagesinteraction/movieS1.avi). Even before observation could start (<1 min), lysis of some cells had likely occurred, as witnessed by the presence of the remaining debris of lysed cells. Other rods lysed one by one at up to 1 h after addition of LoGT-008. The involvement of peptidoglycan degradation in the lysis process was shown by the transient formation of round or lemon-shaped spheroplasts before complete lysis took place. Septal peptidoglycan appeared to be degraded as well, suggested by the fusion of spheroplasts that originate from almost completely constricted cells (Fig. 3). These observations are fully consistent with the Artilysin concept, stating that the LPS layer is disturbed by the polycationic nonapeptide, followed by penetration of the endolysin moiety through the outer membrane and subsequent lysis due to peptidoglycan degradation.
FIG 3 .

Time-lapse series of the action of LoGT-008 on P. aeruginosa PAO1. Cells were mixed with LoGT-008 in the presence of 0.5 mM EDTA and were subsequently recorded using time-lapse microscopy. Time points are indicated (min:s). Scale bar, 2 µm.
Evaluation of different endolysins as fusion partners.
The PCNP tag was also fused to four additional endolysins from our collection, one with a modular structure (201ϕ2-1gp229) and three smaller ones comprising only a single catalytic domain (7, 20) (LoGT-015 to LoGT-018) (Fig. 1; see also Tables S2 and S3 in the supplemental material). We measured the effect of the PCNP fusion on the enzymatic activity of the different endolysins (see Table S6) and analyzed the bactericidal effect of these new Artilysins on P. aeruginosa PAO1 and Br667, Pseudomonas putida, Burkholderia pseudomallei, E. coli, and S. Typhimurium (Fig. 4; see also Table S7). Despite a (52% to 94%) reduction in enzymatic activity in comparison to the corresponding wild-type endolysins, all PCNP-fused endolysins showed improved antibacterial activity. In addition, modularity—and/or its associated high enzymatic activity (7)—is a key factor for the success of the Artilysin approach. Besides the reductions in the cell counts of P. putida (4.89 ± 0.02 log) and B. pseudomallei (4.81 ± 0.05 log), the cell count for the multidrug-resistant P. aeruginosa Br667 strain was also drastically reduced by 4.27 ± 0.15 log within 30 min using a single dose (1.5 µM) of LoGT-001 in the presence of 0.5 mM EDTA (see Table S7).
FIG 4 .
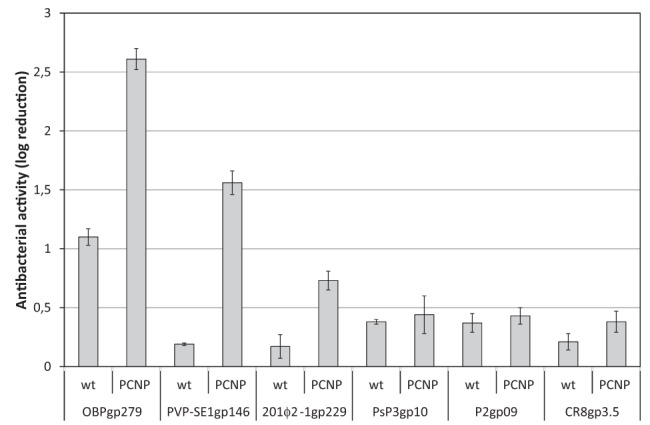
Antibacterial activity of PCNP-Artilysins based on different endolysins. The effect on antibacterial activity of an N-terminal PCNP fusion to three modular (OBPgp279, PVP-SE1gp146, and 201ϕ2-1gp229) and three globular (PsP3gp10, P2gp09, and CR8gp3.5) endolysins is shown for P. aeruginosa PAO1. Averages and standard deviations of results of three replicates are shown. wt, wild type.
Optimization of the Artilysin approach.
To further optimize the potential of the PCNP tag, a set of 31 related variations of the same fusion with both endolysins (OBPgp279 and PVP-SE1gp146) was produced (LoGT-019 to LoGT-049) (Fig. 1; see also Tables S2 and S3 in the supplemental material). These variations encompass various linker lengths between the polycationic tag and the endolysin and the N- or C-terminal fusions of the tag to the endolysin, as well as combinatorial double-tag fusions of the PCNP with additional hydrophobic or amphipathic moieties (see Table S1). With the latter variations, we aimed to respond to differences in LPS structure between P. aeruginosa and E. coli or S. Typhimurium with a corresponding shift from ionic interactions to hydrophobic packing as the major LPS stabilization force (21). The effect on protein yield and enzymatic activity (see Table S8) and on their antibacterial effect against P. aeruginosa, E. coli, S. Typhimurium, and Acinetobacter baumannii, both in the absence and presence of 0.5 mM EDTA (see Table S9), was analyzed. An increasing length of the linker between the N-terminal PCNP tag and the endolysin results in a slightly higher antibacterial effect against P. aeruginosa for Artilysins derived from OBPgp279, with up to a 3.41 ± 0.18 log reduction for LoGT-023 in the absence of EDTA (Fig. 5A). N-terminal fusions generally show higher antibacterial activity than C-terminal fusions (see Table S9). No surviving P. aeruginosa cells could be detected after coincubation with the best Artilysins (LoGT-020, LoGT-021, LoGT-022, and LoGT-023) in the presence of 0.5 mM EDTA, which also almost completely eradicated A. baumannii (up to 5.33 ± 0.20 log). In the case of E. coli, four of eight double-tagged Artilysins (LoGT-037, LoGT-038, LoGT-041, and LoGT-042), combining the PCNP tag with a hydrophobic peptide (HPP; MW1) or amphipathic peptide (MW2), showed an improved bactericidal effect in comparison to the single-tagged Artilysin LoGT-001, with reductions of up to 2.22 ± 0.09 log (Fig. 5B). In the case of S. Typhimurium, no improvement could be obtained using the double-tag approach. Altogether, LoGT-023, comprising a 16-amino-acid linker between the N-terminal PCNP tag and OBPgp279, represents the best optimization of LoGT-001, with strong antibacterial activity against P. aeruginosa (≥5.50 log) and A. baumannii (5.18 ± 0.17 log) and moderate activity against E. coli (2.41 ± 0.08 log) and S. Typhimurium (1.52 ± 0.07 log) (Fig. 5C).
FIG 5 .
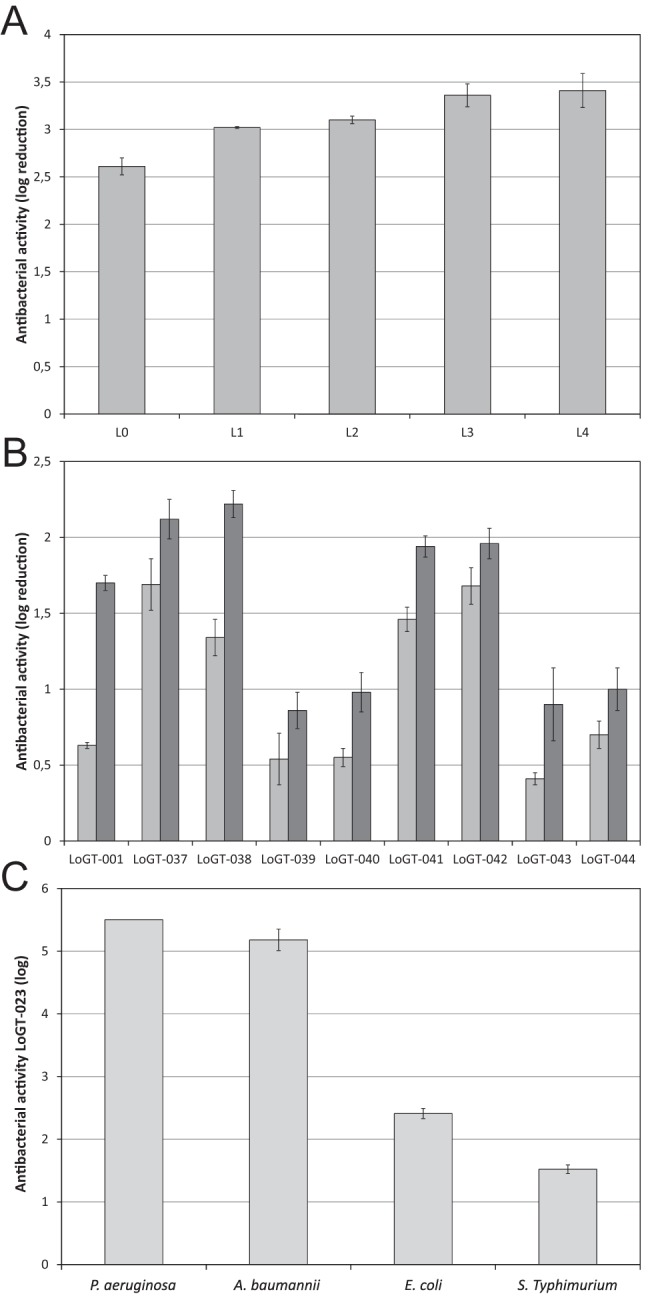
Optimization of PCNP-based Artilysins. (A) The effect of an increasing length of the flexible linker between PCNP and OBPgp279 on the antibacterial activity against P. aeruginosa PAO1 is shown. L0, L1, L2, L3, and L4 correspond to intervening amino acid sequences of AGAS, AGAGAS, AGAGAGAGAS, AGAGAGAGAGAGAS, and AGAGAGAGAGAGAGAGAS. (B) The antibacterial activity of Artilysins comprising a tandem of the PCNP peptide and a second peptide as N-terminal fusions to OBPgp279 is compared to that of PCNP-OBPgp279 (LoGT-001) in the absence (light gray) and presence (dark gray) of 0.5 mM EDTA. (C) LoGT-023, comprising a 16-amino-acid flexible linker between the N-terminal PCNP tag and OBPgp279, generally appeared the most effective Artilysin. The antibacterial activity against P. aeruginosa, A. baumannii, E. coli, and S. Typhimurium in the presence of 0.5 mM EDTA is shown. Averages and standard deviations of results of three replicates are given.
Artilysins are effective on infected human keratinocytes.
Barrier-disrupted skin, such as in burn wounds, is easily infected by different Gram-negative bacteria, most importantly by the opportunistic pathogens P. aeruginosa and A. baumannii (22). Such topical infections in humans and animals are therefore of primary interest for Artilysin applications. LoGT-008 shows MICs of 4 and 8 µg/ml against P. aeruginosa and A. baumannii, respectively. In molar concentrations, these values equate to 0.15 and 0.30 µM, which are in the same range as the value for ciprofloxacin (0.5 µM), a common therapeutically used first-line antibiotic for many Gram-negative infections. To evaluate LoGT-008 against P. aeruginosa in skin infections, we developed a new human neonatal foreskin keratinocyte model (23). This model relies on a confluent keratinocyte monolayer, infected with P. aeruginosa PA14 (105 or 107 CFU/ml). At 1 h after infection, 2 µM PVP-SE1gp146 or LoGT-008 was added. Keratinocyte viability and bacterial reductions were quantified at 4 h after infection. Both PVP-SE1gp146 and LoGT-008 could fully protect (100%) the monolayer from the otherwise cytotoxic effects upon a PA14 infection with 105 CFU/ml (Fig. 6A). This protection is associated with a drastic decrease in bacterial load. A larger bacterial inoculum of 107 CFU/ml was found to cause 97% mortality within 4 h. Under these extreme circumstances, a single dose of LoGT-008 could still rescue 27% of the keratinocytes (significantly more than the 14% rescued by the native endolysin; P < 0.05). Correspondingly, the bacterial load was reduced significantly (P < 0.05) in the presence of LoGT-008 (71% versus 31% for the native endolysin) (Fig. 6B).
FIG 6 .
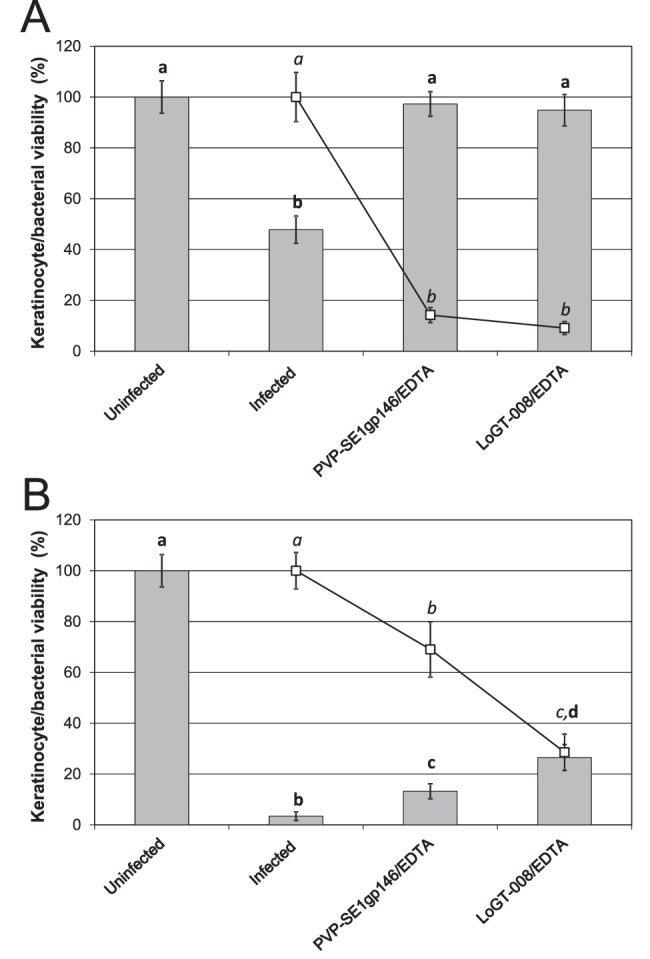
Antibacterial effect of PVP-SE1gp146 and PCNP-PVP-SE1gp146 (LoGT-008) in a P. aeruginosa PA14-infected human keratinocyte monolayer. A mixture of 2 µM of PVP-SE1gp146/LoGT-008 and 0.005 mM of EDTA was applied to a 5-day-old confluent keratinocyte monolayer that was infected with 105 CFU/ml (A) or 107 CFU/ml (B) P. aeruginosa PA14 1 h before treatment. Numbers of surviving bacteria (squares) and surviving keratinocytes (bars) were quantified after 4 h of infection. Proportions of surviving bacteria are shown relative to the infected, untreated control results. Significantly different conditions (P < 0.05) are indicated with bold (keratinocyte survival) or italic (bacterial survival) letters.
Artilysins show in vivo antibacterial efficacy.
Since the PCNP fusions provide endolysins with in vitro antibacterial activity against pseudomonads in both suspension and keratinocyte cultures, we used a C. elegans nematode gut infection model (24) to evaluate the in vivo efficacy of a PCNP-fused Artilysin, LoGT-008, against bacterial infection. When C. elegans grazed on the selected, highly virulent P. aeruginosa PA14 strain, 90% of the nematodes were killed within 5 days after infection (see Fig. S1 in the supplemental material). Nematode survival rates were compared after treatment with the native PVP-SE1gp146 endolysin and the corresponding LoGT-008, using ciprofloxacin as a positive control (Fig. 7). In the presence of 0.5 mM EDTA, PVP-SE1gp146 could rescue 40% ± 7% of the infected worms after 5 days. In the same assay, LoGT-008 showed a significant (P < 0.01) protective effect in the longer term (63% ± 4% survival) versus 45% ± 5% survival after treatment with ciprofloxacin.
FIG 7 .
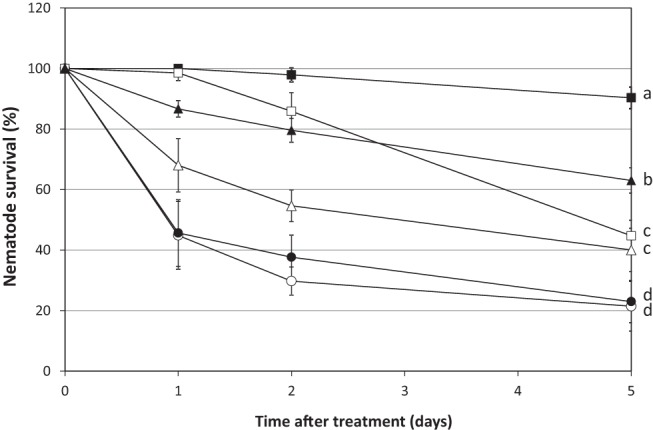
Survival rates of P. aeruginosa PA14-infected C. elegans SS104 nematodes treated with PVP-SE1gp146 or PCNP-PVP-SE1gp146 (LoGT-008) in combination with EDTA. PVP-SE1gp146 (5× MIC; △) or LoGT-008 (5× MIC; ▲) was added to infected nematodes in the presence of 0.5 mM EDTA. Ciprofloxacin (□; 5× MIC) was used as a positive control for antibacterial activity. Other controls included were untreated PA14-infected nematodes (○), untreated nematodes grown on E. coli OP 50 (■), and EDTA-treated infected nematodes (●). A total of 30 infected worms were used for each condition. Nematode viability, in percentages relative to initial values at day 0, was assessed at different time points after compound addition (day 0). Averages and standard deviations of results of three independent experiments are shown. Significantly different conditions (P < 0.05) are marked.
DISCUSSION
The concept of endolysins as antibacterials against Gram-positive pathogens was postulated in 1991 by Gasson (25) and proven in 2001 by Nelson et al., reporting the prevention and elimination of group A streptococcus colonization in the upper respiratory tract of mice (1). Since then, a multitude of endolysins active against diverse Gram-positive pathogens have been described, while Gram-negative targets have been largely neglected (5). We have previously reported the efficacy of a combination of high hydrostatic pressure (9) or outer membrane permeabilizers (10) with endolysins against Gram-negative species. We, and other authors, also described the existence of some endolysins that have moderate intrinsic antibacterial activity against P. aeruginosa, most likely because they can interact with and partially pass across the outer membrane barrier (7, 8). In this study, we showed that tailoring highly active, modular phage endolysins with a polycationic peptide turns them in potent antibacterials that are active against a broad range of important Gram-negative pathogens. Through this engineering, endolysins acquire autonomous antibacterial activity, and in the case of OBPgp279, which itself possesses intrinsic activity, the antibacterial effect is further enhanced to the same extent as for PVP-SE1gp146. Time-lapse microscopy of cells exposed to LoGT-008 confirmed that cells are indeed killed through cell lysis as a consequence of peptidoglycan degradation. Therefore, the endolysin moiety must have been transferred through the outer membrane, most likely because of the competitive displacement of stabilizing divalent cations of the LPS layer by the polycationic nonapeptide. The reduced enzymatic activity after PCNP fusion does not impede the strong antibacterial activity; however, considering that the most active (modular) endolysins generally show the best antibacterial effect, fusions that maintain the high enzymatic activity of such endolysins may represent a further optimization of novel Artilysins.
Extension of the flexible linkers between the penetrating peptide and the endolysins generally improves the antibacterial activity at up to 3.41 ± 0.18 against P. aeruginosa PAO1 in the case of LoGT-023 in the absence of EDTA. N-terminal fusions perform better than C-terminal fusions of OBPgp279 and PVP-SE1gp146; however, it appears that both the optimal fusion side and linker length are best tested empirically. In specific cases, a combinatorial double-tag approach gives some significant improvements as well, as shown for different Artilysins against E. coli. Importantly, in all cases, the strong synergy with EDTA is maintained, giving rise to combinations that reduce cell counts more than 5.50 log (the detection limit) in vitro.
The variation in LPS structure appears to be a key determinant for the sensitivity to Artilysins. We specifically focused on the applicability to P. aeruginosa, an important emerging multidrug-resistant pathogen. P. aeruginosa LPS has a high phosphate content and consequently a higher concentration of divalent cations stabilizing its structure (12). These cations are simultaneously the strength and Achilles’ heel of the P. aeruginosa LPS layer. EDTA and polycationic peptides act on this same site but through two different mechanisms, chelation and competitive displacement, respectively (26). The lower phosphorylation degree in E. coli and S. Typhimurium LPS is compensated by increased hydrophobic packing of the lipid A moiety through a higher degree of acylation with longer acyl chains (12). This variation may explain the higher sensitivity of P. aeruginosa strains compared to that of E. coli and S. Typhimurium and the increased activity against E. coli of some double tags that combine polycationicity and hydrophobicity/amphipathicity.
Given the proteinaceous nature of Artilysins, they may be considered biodegradable compounds that are less likely to accumulate or induce resistant bacteria in the environment. In contrast, many antibiotics persist in the environment, leading to an increased risk of selecting resistant bacteria, especially when environmental bacteria are exposed to subtherapeutic antimicrobial concentrations (27). This detrimental side effect of many nonbiodegradable antimicrobials may be nonexistent for Artilysins.
Recently, Lukacik and coworkers (11) described an alternative approach to ensure transport of an endolysin across the outer membrane by constructing a hybrid of T4 lysozyme and the FyuA binding domain of pesticin. Reduction of the cell count of about 1 log in 24 h depends on the presence of the TonB-dependent outer membrane transporter FyuA. Besides restriction to Yersinia species and uropathogenic E. coli strains that express FyuA, this FyuA dependence may also quickly result in resistance development by shutting down fyuA expression or the accumulation of point mutations in the fyuA genes. Eukaryotic lysozymes have also been subjected to recombinant fusion with diverse peptides or chemical modifications to render them active against Gram-negative bacteria. Various modifications of the cationic hen egg white lysozyme (HEWL) or human lysozyme have been constructed to increase their hydrophobicity, generating amphitropic proteins with both hydrophilic and lipophilic properties (for a review, see reference 28). These modifications expand the activity range of HEWL to Gram-negative E. coli. The highest antibacterial activity was obtained by the covalent attachment of one stearoyl or two palmitoyl acids or by a C-terminal fusion to a hydrophobic pentapeptide (Phe-Phe-Val-Ala-Pro, which has approximately the same length as one palmitoyl acid); however, the maximal bacterial reduction by these HEWL modifications remained below 1 log (29–31).
Multidrug-resistant Gram-negative pathogens are particularly troublesome, because they have intrinsic or acquired resistance to nearly all drugs that can be currently considered for treatment. This worrisome situation also exists, but not to the same extent, for some Gram-positive infections (e.g., Staphylococcus and Enterococcus). The most prevalent Gram-negative pathogens are Enterobacteriaceae, P. aeruginosa, and Acinetobacter, all subjects of this study, and infections are mostly health care associated (32). This report shows that Artilysins can drastically expand the spectrum of endolysins as a novel class of promising anti-infectives (33), making Gram-negative species susceptible to these new enzyme-based antibacterials. The activity of Artilysins in two different settings (C. elegans bacterial infection model and keratinocytes) shows that Artilysins may have a potentially broad field of applications ranging from topical, mucosal, and gastrointestinal medical use to disinfection, veterinary, food, feed, and agricultural applications.
MATERIALS AND METHODS
Bacterial strains.
Pseudomonas aeruginosa PAO1 (ATCC 15692), originally a wound isolate, is now a widely used laboratory strain (34). The clinical P. aeruginosa strains Br667, a multiresistant (10 of 11 antibiotics) burn wound isolate collected in the intensive care unit in the Queen Astrid Military Hospital in Brussels, Belgium, PA14, a highly virulent wound isolate from Boston, MA, and other clinical P. aeruginosa strains were all provided by J.-P. Pirnay (19, 35). Pseudomonas putida G1 was isolated from Russian soil in the neighborhood of Moscow (V. Krylov, Laboratory of Bacteriophage Genetics). Acinetobacter baumannii 25 is an uncharacterized clinical isolate from the Queen Astrid Military hospital, donated by Maya Merabishvilli (Lab MCT, Brussels, Belgium), and the Burkholderia pseudomallei strain used is a clinical strain from the University Hospital of Gasthuisberg (Leuven, Belgium) isolated by J. Verhaegen (Laboratory of Clinical Microbiology and Mycology, UZ Leuven). S. Typhimurium LT2 was provided by the Centre of Food and Microbial Technology (KU Leuven, Belgium). Several different Escherichia coli strains were used in this study: E. coli XL1-Blue MRF′ (Agilent Technologies, Santa Clara, CA) for antibacterial activity testing, E. coli TOP10 (Life Technologies, Carlsbad, CA) for DNA cloning and cell stock storage, and E. coli BL21(DE3) pLysS, BL21-CodonPlus-(DE3)-RIL, and BL21-CodonPlus-(DE3)-RP (Agilent Technologies) as host strains for protein expression. For proper selection, ampicillin (Roche Diagnostics, Mannheim, Germany) (100 µg/ml) or chloramphenicol (Calbiochem, Darmstadt, Germany) (50 µg/ml) was used. All strains were grown at 37°C, except for P. putida G1 (30°C), in lysogeny broth (LB).
Plasmid construction.
Overviews of all LPS-destabilizing peptides and produced Artilysins are given in Tables S1 to S3 in the supplemental material and Fig. 1, respectively. Open reading frames (ORFs) encoding the different endolysins were amplified (Pfu polymerase; Thermo Fisher Scientific, Waltham, MA) with purified genomic DNA isolated from the respective phages as template DNA. Cloning into the pEXP5CT/TOPO expression vector was performed according to the manufacturer’s guidelines (Life Technologies). The polycationic nonapeptide (PCNP) and the hydrophobic pentapeptide (HPP) were fused to endolysins by a standard PCR on purified genomic DNA of the respective phages using forward (N-terminal fusion) or reverse (C-terminal fusion) primers that harbor the peptide-encoding sequence. For N-terminal fusion of the α4, MW1, MW2, Lycotoxin1, and Parasin1 peptides, an adapted version of the ligation-independent cloning (LIC) technique (36) was used. Briefly, peptide-encoding cassettes, created by hybridization of specific primer pairs (20 µM each), were treated with a mixture of dCTP (Promega, Agora, WI) and T4 DNA polymerase (Thermo Fisher Scientific) to introduce LIC-compatible 5′ sticky ends. In parallel, the endolysin-encoding expression vectors were made LIC compatible by introduction of a unique Ecl136II restriction endonuclease site upstream of the endolysin-encoding ORF using a standard PCR (see intermediate constructs in Table S3 in the supplemental material). The procedure was followed by linearization with Ecl136II endonuclease (New England Biolabs, Ipswich, MA) and creation of complementary sticky ends by treatment with dGTP (Promega) and T4 DNA polymerase. A short incubation step of the hybridized peptide-encoding cassettes with the LIC-compatible vectors completed the cloning process. Two different types of double-tag fusions were constructed in this study: type 1, with the PCNP located N-terminally to the second peptide and the endolysin (type 1 conformation: PCNP—second peptide—endolysin), and type 2, with the PCNP between the second peptide and the endolysin (type 2 conformation: second peptide-PCNP-endolysin). For the type 1 configuration, a standard PCR with a PCNP peptide-encoding 5′ primer (Pfu polymerase) was applied on purified pEXP5-CT/α4-endolysin and pEXP5-CT/HPP-endolysin plasmid DNA, respectively. To obtain type 2 double-tag fusions, the same adapted version of the LIC method was used as described for the single-peptide fusion, except that the pEXP5CT/PCNP-endolysin vector was used as the template. All type 1 and type 2 fusion constructs were introduced in the pEXP5CT-TOPO expression vector. To construct plasmids with extended linkers between the PCNP-encoding sequence and the endolysin-encoding ORF, a standard PCR was first used to introduce NheI and NgoMIV restriction sites for directional cloning. Upon double digestion of the obtained pEXP5CT/PCNP-NheI-NgoMIV-endolysin (for N-terminal end) and pEXP5CT/endolysin-NgoMIV-NheI-PCNP (for C-terminal end) constructs (NgoMIV, New England Biolabs; NheI, Thermo Fisher Scientific), cassettes of various lengths with 5′ complementary sticky ends (encoding the following amino acid sequences: larval stage 1 [L1] = GAGA, L2 = GAGAGAGA, L3 = GAGAGAGAGAGA, L4 = GAGAGAGAGAGAGAGA), obtained through hybridization of specific primer pairs (20 µM each), were inserted in the vectors (T4 ligase; Thermo Fisher Scientific).
Recombinant expression and purification.
Recombinant expression of the different constructs was performed in an E. coli-based expression system upon induction of exponentially growing cells (optical density at 600 nm [OD600] = 0.6) with 1 mM isopropyl-beta-d-thiogalactopyranoside (IPTG; Thermo Fisher Scientific), unless otherwise stated. Expression conditions (temperature, time, and expression strain) differed depending on the protein to optimize the soluble-expression levels for each construct (see Table S3 in the supplemental material). Expression was stopped by centrifugation (3,900 × g, 30 min, 4°C), and the obtained cell pellet was resuspended in a 1/25 vol of lysis buffer (20 mM NaH2PO4 [Merck, Darmstadt, Germany]/NaOH, 0.5 M NaCl, pH 7.4). This cell lysate was then subjected to three freeze-thaw cycles (−80°C/22°C) prior to sonication (Vibra-Cell Sonics, Newtown, CT) (8 cycles of a 30-s pulse and 30-s rest; amplitude of 40%). The protein lysate was then filtered through 0.45- and 0.22-µm-pore-size Durapore membrane filters (Millipore, Billerica, MA). Purification of the His6-tagged fusion proteins was performed on an Aktä fast protein liquid chromatography (FPLC) system (GE Healthcare, Buckinghamshire, United Kingdom) controlled by UNICORN 5.1 software with Ni2+-nitrilotriacetic acid (NTA) columns (HisTrap HP; GE Healthcare) (1 ml). The wash buffer used in the purification process contains a low protein-dependent imidazole concentration (50 to 80 mM) for higher purification stringency (see Table S3). After dialysis to an appropriate buffer, the protein concentration was determined spectrophotometrically in silica cuvettes at a wavelength of 280 nm (Ultraspec III spectrophotometer; GE Healthcare).
Muralytic assay.
Outer-membrane-permeabilized cells were used as the substrate (37). Briefly, P. aeruginosa PAO1 cells growing in the mid-exponential phase (OD600 = 0.6) were spun down (3,900 × g, 30 min, 4°C) and subsequently incubated with gentle shaking for exactly 45 min in chloroform-saturated 0.05 M Tris-HCl (pH 7.7). Cells were then washed in a KH2PO4/K2HPO4 buffer (ionic strength = 80 mM, pH 7.2) to remove residual chloroform and concentrated to an OD600 of 1.5. Upon addition of 30 µl of muralytic enzymes (at from 10 to 2,000 nM) to 270 µl of outer-membrane-permeabilized cells, the optical density was measured spectrophotometrically over time at 655 nm using a Microplate Reader 680 system (Bio-Rad). A standardized calculation method to quantify the muralytic activities of lytic enzymes using this assay was described before (38).
Antibacterial assays.
Gram-negative cells growing in the mid-exponential phase (OD600 = 0.6) were diluted 100-fold in 5 mM HEPES/NaOH (pH 7.4) to a final density of ±106 CFU/ml. Each cell culture (100 µl) was incubated for 30 min at room temperature with 50 µl endolysin/Artilysin dialyzed against phosphate-buffered saline (PBS) (pH 7.4) and 50 µl 5 mM HEPES/NaOH (pH 7.4) buffer or 50 µl 2 mM EDTA-Na2 (abbreviated as EDTA; final concentration of 0.5 mM) dissolved in the same buffer. After 30 min of incubation, appropriate dilutions of cell suspensions were plated on LB agar in triplicate. Colonies were counted after overnight incubation at 37°C. The antibacterial activity was quantified as the relative inactivation level in log units [log10(N0/Ni), with N0 = initial number of untreated cells and Ni = number of residual cells counted after treatment). The MIC of LoGT-008/ciprofloxacin against P. aeruginosa PAO1 and A. baumannii 25 in the presence of EDTA in 96-well plates was determined using the broth microdilution method. Cells were diluted according to the McFarland standard in low-salt LB (10 g/liter tryptone, 5 g/liter yeast extract). A dilution series of LoGT-008/ciprofloxacin was added, and the plate was incubated for 18 h at 37°C. All measurements were performed in comparison to a negative control (medium without bacteria) or a positive control (medium without LoGT-008). The MIC was defined as the minimum concentration that completely inhibited bacterial growth.
Time-lapse microscopy.
A temperature-controlled (Okolab Ottaviano, Italy) Ti-Eclipse inverted microscope (Nikon, Champigny-sur-Marne, France) equipped with a TI-CT-E motorized condenser and a CoolSnap HQ2 FireWire charge-coupled-device (CCD) camera was used as described previously (39). P. aeruginosa PAO1 cells were grown to mid-exponential phase (OD600 = 0.6), washed three times with PBS (pH 7.4), and finally concentrated five times by resuspension in PBS–0.5 mM EDTA-Na2. LoGT-008 was added to reach a final concentration of 0.5 mg/ml. For imaging, samples were placed on a PBS–0.5 mM EDTA–Na2–agar pad and a cover glass, essentially as described previously (39), and incubated at 37°C. The time between addition of LoGT-008 to the cells and recording was kept as short as possible (~60 s). Images were acquired using NIS-Elements (Nikon), and the resulting pictures were further handled with ImageJ open source software (downloaded from http://rsbweb.nih.gov/ij/) and CorelDRAW X5.
Human keratinocyte assays.
Keratinocytes were seeded in coated (1 µg/ml collagen type I for a minimum of 2 h at 37°C) 25-cm2 vented culture flasks at 5,000 cells/cm2 and cultured until more than 90% confluence was reached (supplemented EpiLife medium; 6 days, 37°C, 5% CO2, and 95% relative humidity). Confluent grown keratinocytes were infected with clinical P. aeruginosa PA14 strains at an infectivity dose of 105 or 107 CFU/ml. At 1 h after infection, both the unmodified and PCNP-modified endolysins (2 µM final concentration) were added together with EDTA (5 µM final concentration) to the keratinocyte cultures. Uninfected keratinocytes and infected but untreated keratinocytes were included as negative controls. After an additional 3 h of incubation, the infection process was stopped by trypsinization of the residual keratinocytes (TrypLE Select recombinant trypsin [Life Technologies]) for 5 min at 37°C and washing in supplemented EpiLife medium. The cell suspensions were collected and centrifuged at 170 × g for 10 min. After centrifugation, the cells were resuspended in supplemented EpiLife medium. Keratinocyte viability was microscopically evaluated (Bürker chamber) using trypan blue live/dead staining. Living keratinocytes appeared as colorless cells under the microscope, whereas dead ones became blue due to their disintegrated cell membrane. For evaluation of the antibacterial activity of the P. aeruginosa strains, 10-fold dilutions of the keratinocyte culture were plated on LB agar and the residual bacterial CFUs were quantified for each condition after 18 h of incubation at 37°C. The antibacterial activity is expressed in percent reduction relative to the untreated negative control. Multiple pairwise t test comparisons were performed (P < 0.05) to classify the different conditions in significantly different groups for keratinocyte (bold labels) and bacterial (italic labels) viability.
C. elegans infection assay.
Caenorhabditis elegans SS104, genotype glp-4(bn2), was maintained at 16°C on nematode growth medium (NGM) agar covered with a lawn of E. coli OP 50 as a feed source (40). Nematode experiments were performed with synchronized L4-stage nematodes acquired by adult bleaching and hatching of the eggs at 16°C for 4 days. For infection of C. elegans SS104, 50 synchronized L4-stage nematodes were seeded on bacterial lawns of P. aeruginosa PA14. Lawns were prepared by spreading 100 µl of culture at the stationary phase of growth on top of the NGM agar and incubation at 37°C for 18 h before seeding. The infection took place for 24 h at 26°C. Nematode survival after infection was evaluated by the liquid assay as described before (41, 42). Briefly, infected nematodes were washed three times with M9 buffer to remove attached bacteria and transferred to 96-well plates (~10 nematodes per well). Wells contained 100 µl of an NGM/M9 buffer (1:4) mixture, 50 µl (PCNP−) endolysin dialyzed against PBS (pH 7.4), and 50 µl EDTA (0.5 mM final concentration)–PBS (pH 7.4). In addition, PBS buffer (pH 7.4) and EDTA were added as negative controls and ciprofloxacin (Sigma-Aldrich) (5× MIC) as a positive control. Each condition was tested at a 10-fold level. Survival of C. elegans SS104 was determined by monitoring the presence or absence of touch-provoked movement for each nematode during 5 subsequent days. Dead nematodes appear as long, immobile rods; living ones move in a sinusoidal shape (41, 42). Multiple pairwise t test comparisons were performed (P < 0.05) to classify the tested conditions in significantly different groups for C. elegans survival after 5 days.
SUPPLEMENTAL MATERIAL
Survival rate of C. elegans SS104 upon infection. L4-stage synchronized C. elegans SS104 nematodes were infected by seeding them on NGM agar plates overgrown with P. aeruginosa PA14 (blue squares), Us447 (red diamonds), and Br667 (green circles) and subsequent incubation for 24 h at 37°C. Nematode viability was assessed by pinching at different time points for 6 days after infection. Nonpathogenic, food source E. coli OP 50 (purple triangles) was used as a negative control. Since P. aeruginosa PA14 is the most virulent strain infecting C. elegans SS104 (10% survival after 5-day incubation), PA14 was selected for nematode infection assays. Download
Peptides with (putative) LPS-destabilizing activity. A list of known (HPP, α4, Parasin1, Lycotoxin1) or newly designed (PCNP, MW1, MW2) peptides with (putative) LPS-destabilizing activity used in this study is shown. These peptides are either polycationic, hydrophobic, or amphipathic in nature. Abbreviations: PCNP = polycationic nonapeptide; HPP = hydrophobic pentapeptide; α4 = α4-helix of T4 lysozyme.
Overview of engineered Artilysins. Seven different endolysins (OBPgp279 [YP_004958186.1], PVP-SE1gp146 [YP_004893953.1], phiKZgp144 [], 201ϕ2-1gp229 [YP_001956952.1], CR8gp3.5 [unpublished], P2gp09 [], and PsP3gp10 []) were selected for modification with different peptides (see Table S1). L0, L1, L2, L3, and L4 correspond to intervening amino acid sequences of AGAS, AGAGAS, AGAGAGAGAS, AGAGAGAGAGAGAS, and AGAGAGAGAGAGAGAGAS for N-terminal linkers and to ASAG, ASGAGAG, ASGAGAGAGAG, ASGAGAGAGAGAGAG, and ASGAGAGAGAGAGAGAGAG for C-terminal linkers, respectively. All fusions are done to either the N terminus or the C terminus of the endolysin, as indicated.
Cloning methodology and expression and purification conditions. Names of native endolysins and corresponding Artilysins (LoGT-xxx) constructs, primers used for PCR amplification/cassette formation, cloning methodology (see additional details below), expression conditions (temperature/time/expression strain), and washing buffer stringency during purification are summarized. Expression was induced at an OD600 of 0.6 upon addition of 1 mM IPTG, unless indicated otherwise. All proteins were purified with Ni2+-NTA chromatography (HisTrap HP columns; GE Healthcare) (1 ml) using an Aktä-FPLC system. The restriction endonuclease recognition sites used for cloning are boxed in the respective primer sequences. Additional nucleotides necessary for in-frame cloning are indicated in bold. Nucleotides encoding the polycationic nonapeptide are underlined and in bold. Abbreviations: PCNP = polycationic nonapeptide; HPP = hydrophobic pentapeptide; α4 = α4-helix of T4 lysozyme. Intervening sequences of different linkers encode L0 through L4 as follows: L0 = AGAS; L1 = AGAGAS; L2 = AGAGAGAGAS; L3 = AGAGAGAGAGAGAS; L4 = AGAGAGAGAGAGAGAGAS. A superscript character refers to the reference describing the identification and characterization of the corresponding endolysin.
Protein purification yields for N-terminal single-tag fusion variants of endolysins OBPgp279 and PVP-SE1gp146. The total yield for each fusion protein is shown as the amount of purified recombinant protein (in mg) per liter of E. coli expression culture. The protein amount after purification and dialysis was quantified spectrophotometrically at wavelength 280 nm.
Antibacterial activity of the N-terminal fusion variants of OBPgp279 (A and B) and PVP-SE1gp146 (C and D) without (A and C) and with (B and D) outer membrane permeabilizer EDTA (0.5 mM) on Gram-negative bacteria in comparison to unmodified endolysins. Exponentially growing cells (106 CFU/ml) were incubated at room temperature with 1.5 µM OBPgp279/OBPgp279-derived Artilysins or with 5 µM PVP-SE1gp146/PVP-SE1gp146-derived Artilysins for 30 min. Antibacterial activity was quantified as a log10 reduction [log10(N0/Ni), with N0 = number of untreated cells and Ni = number of cells after treatment]. Averages ± standard deviations are given for n = 3 repeats.
Influence of the N-terminal PCNP fusion on enzymatic activity of the selected modular and single-domain endolysins. The specific enzymatic activities (in units/µM) of the unmodified and PCNP-modified endolysins are listed together with the reduction percentage relative to the specific activity of the unmodified endolysin (=100% activity).
Antibacterial activity of PCNP-based Artilysins without (a) and with (b) EDTA (0.5 mM) in comparison to native endolysins against a set of Gram-negative strains. Cell suspensions (±106 CFU/ml) were incubated with endolysins/Artilysins dialyzed against PBS buffer (pH 7.4). The antibacterial activity is shown as a log reduction [log10(N0/Ni), with N0 = initial number of untreated cells and Ni = number of residual, treated cells). Averages and standard deviations of results of three repeats are given. ND = not determined.
Protein yield and residual enzymatic activity of Artilysins having a C-terminal PCNP tag, an N-terminal double tag, or an extended linker between PCNP and endolysin. The total yield for each Artilysin is shown as the amount of purified recombinant protein (in mg) per liter of E. coli expression culture. The yield has been quantified spectrophotometrically. The residual enzymatic activity is expressed relative to the specific activity of the corresponding unmodified endolysin (100%; OBPgp279, 19,979 U/µM; PVP-SE1gp146, 13,614 U/µm). ND = not determined.
Antibacterial activity of modifications of PCNP-fused Artiysins. The antibacterial activity of N-terminal and C-terminal linker variants of OBPgp279 (1.5 µM) and PVP-SE1gp146 (5 µM) in the absence (a) and in the presence (b) of 0.5 mM EDTA was determined. The antibacterial activity of the N-terminal double-tagged fusion variants of OBPgp279 (1.5 µM) and PVP-SE1gp146 (5 µM) in the absence (C) and in the presence (D) of 0.5 mM EDTA was determined. Antibacterial activity is expressed as a log10 reduction [log10(N0/Ni), with N0 = initial number of cells and Ni = number of residual cells after treatment]. Averages and standard deviations are shown here (n = 3). *, the detection level of 10 CFU/ml was reached. ND = not determined.
ACKNOWLEDGMENTS
We thank Jean-Pierre Hernalsteens (GEVI, VUB, Brussels) and Karlien Nerinckx for introducing us to the C. elegans bacterial infection model.
ARTILYSIN is a registered trademark in the European Union, United States, and other countries.
M.W. held a predoctoral fellowship of the “Instituut voor aanmoediging van Innovatie door Wetenschap en Technologie in Vlaanderen” (IWT Flanders). Y.B. and M.W. were supported by IWT Flanders and Y.B. by a postdoctoral fellowship of the “Bijzonder Onderzoeksfonds—KU Leuven.” S.M. is an employee of Lisando GmbH. R.L. acts as scientific adviser to Lisando GmbH.
Footnotes
Citation Briers Y, Walmagh M, Van Puyenbroeck V, Cornelissen A, Cenens W, Aertsen A, Oliveira H, Azeredo J, Verween G, Pirnay J, Miller S, Volckaert G, Lavigne R. 2014. Engineered endolysin-based “Artilysins” to combat multidrug-resistant gram-negative pathogens. mBio 5(4):e01379-14. doi:10.1128/mBio.01379-14.
REFERENCES
- 1. Nelson D, Loomis L, Fischetti VA. 2001. Prevention and elimination of upper respiratory colonization of mice by group A streptococci by using a bacteriophage lytic enzyme. Proc. Natl. Acad. Sci. U. S. A. 98:4107–4112. 10.1073/pnas.061038398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Loeffler JM, Nelson D, Fischetti VA. 2001. Rapid killing of Streptococcus pneumoniae with a bacteriophage cell wall hydrolase. Science 294:2170–2172. 10.1126/science.1066869 [DOI] [PubMed] [Google Scholar]
- 3. Schuch R, Nelson D, Fischetti VA. 2002. A bacteriolytic agent that detects and kills Bacillus anthracis. Nature 418:884–889. 10.1038/nature01026 [DOI] [PubMed] [Google Scholar]
- 4. Schmelcher M, Powell AM, Becker SC, Camp MJ, Donovan DM. 2012. Chimeric phage lysins act synergistically with lysostaphin to kill mastitis-causing Staphylococcus aureus in murine mammary glands. Appl. Environ. Microbiol. 78:2297–2305. 10.1128/AEM.07050-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Nelson DC, Schmelcher M, Rodriguez-Rubio L, Klumpp J, Pritchard DG, Dong S, Donovan DM. 2012. Endolysins as antimicrobials. Adv. Virus Res. 83:299–365. 10.1016/B978-0-12-394438-2.00007-4 [DOI] [PubMed] [Google Scholar]
- 6. Morita M, Tanji Y, Orito Y, Mizoguchi K, Soejima A, Unno H. 2001. Functional analysis of antibacterial activity of Bacillus amyloliquefaciens phage endolysin against gram-negative bacteria. FEBS Lett. 500:56–59. 10.1016/S0014-5793(01)02587-X [DOI] [PubMed] [Google Scholar]
- 7. Walmagh M, Briers Y, dos Santos SB, Azeredo J, Lavigne R. 2012. Characterization of modular bacteriophage endolysins from Myoviridae phages OBP, 201φ2-1 and PVP-SE1. PLoS One 7:e36991. 10.1371/journal.pone.0036991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Delcour AH. 2009. Outer membrane permeability and antibiotic resistance. Biochim. Biophys. Acta 1794:808–816. 10.1016/j.bbapap.2008.11.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Briers Y, Cornelissen A, Aertsen A, Hertveldt K, Michiels CW, Volckaert G, Lavigne R. 2008. Analysis of outer membrane permeability of Pseudomonas aeruginosa and bactericidal activity of endolysins KZ144 and EL188 under high hydrostatic pressure. FEMS Microbiol. Lett. 280:113–119. 10.1111/j.1574-6968.2007.01051.x [DOI] [PubMed] [Google Scholar]
- 10. Briers Y, Walmagh M, Lavigne R. 2011. Use of bacteriophage endolysin EL188 and outer membrane permeabilizers against Pseudomonas aeruginosa. J. Appl. Microbiol. 110:778–785. 10.1111/j.1365-2672.2010.04931.x [DOI] [PubMed] [Google Scholar]
- 11. Lukacik P, Barnard TJ, Keller PW, Chaturvedi KS, Seddiki N, Fairman JW, Noinaj N, Kirby TL, Henderson JP, Steven AC, Hinnebusch BJ, Buchanan SK. 2012. Structural engineering of a phage lysin that targets gram-negative pathogens. Proc. Natl. Acad. Sci. U. S. A. 109:9857–9862. 10.1073/pnas.1203472109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Nikaido H. 2003. Molecular basis of bacterial outer membrane permeability revisited. Microbiol. Mol. Biol. Rev. 67:593–656. 10.1128/MMBR.67.4.593-656.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ibrahim HR, Yamada M, Matsushita K, Kobayashi K, Kato A. 1994. Enhanced bactericidal action of lysozyme to Escherichia coli by inserting a hydrophobic pentapeptide into its C-terminus. J. Biol. Chem. 269:5059–5063 [PubMed] [Google Scholar]
- 14. Düring K, Porsch P, Mahn A, Brinkmann O, Gieffers W. 1999. The non-enzymatic microbicidal activity of lysozymes. FEBS Lett. 449:93–100 [DOI] [PubMed] [Google Scholar]
- 15. Park IY, Park CB, Kim MS, Kim SC. 1998. Parasin I, an antimicrobial peptide derived from histone H2A in the catfish, Parasilurus asotus. FEBS Lett. 437:258–262. 10.1016/S0014-5793(98)01238-1 [DOI] [PubMed] [Google Scholar]
- 16. Yan L, Adams ME. 1998. Lycotoxins, antimicrobial peptides from venom of the wolf spider Lycosa carolinensis. J. Biol. Chem. 273:2059–2066. 10.1074/jbc.273.4.2059 [DOI] [PubMed] [Google Scholar]
- 17. Briers Y, Schmelcher M, Loessner MJ, Hendrix J, Engelborghs Y, Volckaert G, Lavigne R. 2009. The high-affinity peptidoglycan binding domain of Pseudomonas phage endolysin KZ144. Biochem. Biophys. Res. Commun. 383:187–191. 10.1016/j.bbrc.2009.03.161 [DOI] [PubMed] [Google Scholar]
- 18. Briers Y, Volckaert G, Cornelissen A, Lagaert S, Michiels CW, Hertveldt K, Lavigne R. 2007. Muralytic activity and modular structure of the endolysins of Pseudomonas aeruginosa bacteriophages phiKZ and EL. Mol. Microbiol. 65:1334–1344. 10.1111/j.1365-2958.2007.05870.x [DOI] [PubMed] [Google Scholar]
- 19. Pirnay JP, De Vos D, Cochez C, Bilocq F, Pirson J, Struelens M, Duinslaeger L, Cornelis P, Zizi M, Vanderkelen A. 2003. Molecular epidemiology of Pseudomonas aeruginosa colonization in a burn unit: persistence of a multidrug-resistant clone and a silver sulfadiazine-resistant clone. J. Clin. Microbiol. 41:1192–1202. 10.1128/JCM.41.3.1192-1202.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Walmagh M, Boczkowska B, Grymonprez B, Briers Y, Drulis-Kawa Z, Lavigne R. 2013. Characterization of five novel endolysins from gram-negative infecting bacteriophages. Appl. Microbiol. Biotechnol. 97:4369–4375. 10.1007/s00253-012-4294-7 [DOI] [PubMed] [Google Scholar]
- 21. Raetz CR, Whitfield C. 2002. Lipopolysaccharide endotoxins. Annu. Rev. Biochem. 71:635–700. 10.1146/annurev.biochem.71.110601.135414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Church D, Elsayed S, Reid O, Winston B, Lindsay R. 2006. Burn wound infections. Clin. Microbiol. Rev. 19:403–434. 10.1128/CMR.19.2.403-434.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. De Corte P, Verween G, Verbeken G, Rose T, Jennes S, De Coninck A, Roseeuw D, Vanderkelen A, Kets E, Haddow D, Pirnay JP. 2012. Feeder layer- and animal product-free culture of neonatal foreskin keratinocytes: improved performance, usability, quality and safety. Cell Tissue Bank. 13:175–189. 10.1007/s10561-011-9247-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Tan MW, Mahajan-Miklos S, Ausubel FM. 1999. Killing of Caenorhabditis elegans by Pseudomonas aeruginosa used to model mammalian bacterial pathogenesis. Proc. Natl. Acad. Sci. U. S. A. 96:715–720. 10.1073/pnas.96.2.715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Gasson MJ. April 1991. iral products. US patent 6,083,684 A
- 26. Vaara M. 1992. Agents that increase permeability of the outer membrane. Microbiol. Rev. 56:395–411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kümmerer K. 2003. Significance of antibiotics in the environment. J. Antimicrob. Chemother. 52:5–7. 10.1093/jac/dkg293 [DOI] [PubMed] [Google Scholar]
- 28. Masschalck B, Michiels CW. 2003. Antimicrobial properties of lysozyme in relation to foodborne vegetative bacteria. Crit. Rev. Microbiol. 29:191–214. 10.1080/713610448 [DOI] [PubMed] [Google Scholar]
- 29. Ibrahim HR, Kato A, Kobayashi K. 1991. Antimicrobial effects of lysozyme against gram-negative bacteria due to covalent binding of palmitic acid. J. Agric. Food Chem. 39:2077–2082. 10.1021/jf00011a039 [DOI] [Google Scholar]
- 30. Ibrahim HR, Yamada M, Kobayashi K, Kato A. 1992. Bactericidal action of lysozyme against gram-negative bacteria due to insertion of a hydrophobic pentapeptide into its C-terminus. Biosci. Biotechnol. Biochem. 56:1361–1363. 10.1271/bbb.56.1361 [DOI] [PubMed] [Google Scholar]
- 31. Ibrahim HR, Kobayahsi K, Kato A. 1993. Length of hydrocarbon chain and antimicrobial action to gram-negative bacteria of fatty acylated lysozyme. J. Agric. Food Chem. 41:1164–1168. 10.1021/jf00031a029 [DOI] [Google Scholar]
- 32. Centers for Disease Control and Prevention. CDC 2013. Antibiotic resistance threats in the United States. http://www.cdc.gov/drugresistance/threat-report-2013/pdf/ar-threats-2013-508.pdf Accessed: 10 February 2014
- 33. Pastagia M, Schuch R, Fischetti VA, Huang DB. 2013. Lysins: the arrival of pathogen-directed anti-infectives. J. Med. Microbiol. 62:1506–1516 [DOI] [PubMed] [Google Scholar]
- 34. Holloway BW. 1955. Genetic recombination in Pseudomonas aeruginosa. J. Gen. Microbiol. 13:572–581. 10.1099/00221287-13-3-572 [DOI] [PubMed] [Google Scholar]
- 35. Pirnay JP, Bilocq F, Pot B, Cornelis P, Zizi M, Van Eldere J, Deschaght P, Vaneechoutte M, Jennes S, Pitt T, De Vos D. 2009. Pseudomonas aeruginosa population structure revisited. PLoS One 4:e7740. 10.1371/journal.pone.0007740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Haun RS, Serventi IM, Moss J. 1992. Rapid, reliable ligation-independent cloning of PCR products using modified plasmid vectors. BioTechniques 13:515–518 [PubMed] [Google Scholar]
- 37. Lavigne R, Briers Y, Hertveldt K, Robben J, Volckaert G. 2004. Identification and characterization of a highly thermostable bacteriophage lysozyme. Cell. Mol. Life Sci. 61:2753–2759. 10.1007/s00018-004-4301-y [DOI] [PubMed] [Google Scholar]
- 38. Briers Y, Lavigne R, Volckaert G, Hertveldt K. 2007. A standardized approach for accurate quantification of murein hydrolase activity in high-throughput assays. J. Biochem. Biophys. Methods 70:531–533. 10.1016/j.jbbm.2006.10.009 [DOI] [PubMed] [Google Scholar]
- 39. Cenens W, Mebrhatu MT, Makumi A, Ceyssens PJ, Lavigne R, Van Houdt R, Taddei F, Aertsen A. 2013. Expression of a novel P22 ORFan gene reveals the phage carrier state in Salmonella typhimurium. PLoS Genet. 9:e1003269. 10.1371/journal.pgen.1003269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Brenner S. 1974. The genetics of Caenorhabditis elegans. Genetics 77:71–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Stiernagle T. 11 February 2006. Maintenance of C. elegans. In The C. elegans Research Community, WormBook. 10.1895/wormbook.1.101.1 [DOI] [PMC free article] [PubMed]
- 42. Moy TI, Ball AR, Anklesaria Z, Casadei G, Lewis K, Ausubel FM. 2006. Identification of novel antimicrobials using a live-animal infection model. Proc. Natl. Acad. Sci. U. S. A. 103:10414–10419. 10.1073/pnas.0604055103 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Survival rate of C. elegans SS104 upon infection. L4-stage synchronized C. elegans SS104 nematodes were infected by seeding them on NGM agar plates overgrown with P. aeruginosa PA14 (blue squares), Us447 (red diamonds), and Br667 (green circles) and subsequent incubation for 24 h at 37°C. Nematode viability was assessed by pinching at different time points for 6 days after infection. Nonpathogenic, food source E. coli OP 50 (purple triangles) was used as a negative control. Since P. aeruginosa PA14 is the most virulent strain infecting C. elegans SS104 (10% survival after 5-day incubation), PA14 was selected for nematode infection assays. Download
Peptides with (putative) LPS-destabilizing activity. A list of known (HPP, α4, Parasin1, Lycotoxin1) or newly designed (PCNP, MW1, MW2) peptides with (putative) LPS-destabilizing activity used in this study is shown. These peptides are either polycationic, hydrophobic, or amphipathic in nature. Abbreviations: PCNP = polycationic nonapeptide; HPP = hydrophobic pentapeptide; α4 = α4-helix of T4 lysozyme.
Overview of engineered Artilysins. Seven different endolysins (OBPgp279 [YP_004958186.1], PVP-SE1gp146 [YP_004893953.1], phiKZgp144 [], 201ϕ2-1gp229 [YP_001956952.1], CR8gp3.5 [unpublished], P2gp09 [], and PsP3gp10 []) were selected for modification with different peptides (see Table S1). L0, L1, L2, L3, and L4 correspond to intervening amino acid sequences of AGAS, AGAGAS, AGAGAGAGAS, AGAGAGAGAGAGAS, and AGAGAGAGAGAGAGAGAS for N-terminal linkers and to ASAG, ASGAGAG, ASGAGAGAGAG, ASGAGAGAGAGAGAG, and ASGAGAGAGAGAGAGAGAG for C-terminal linkers, respectively. All fusions are done to either the N terminus or the C terminus of the endolysin, as indicated.
Cloning methodology and expression and purification conditions. Names of native endolysins and corresponding Artilysins (LoGT-xxx) constructs, primers used for PCR amplification/cassette formation, cloning methodology (see additional details below), expression conditions (temperature/time/expression strain), and washing buffer stringency during purification are summarized. Expression was induced at an OD600 of 0.6 upon addition of 1 mM IPTG, unless indicated otherwise. All proteins were purified with Ni2+-NTA chromatography (HisTrap HP columns; GE Healthcare) (1 ml) using an Aktä-FPLC system. The restriction endonuclease recognition sites used for cloning are boxed in the respective primer sequences. Additional nucleotides necessary for in-frame cloning are indicated in bold. Nucleotides encoding the polycationic nonapeptide are underlined and in bold. Abbreviations: PCNP = polycationic nonapeptide; HPP = hydrophobic pentapeptide; α4 = α4-helix of T4 lysozyme. Intervening sequences of different linkers encode L0 through L4 as follows: L0 = AGAS; L1 = AGAGAS; L2 = AGAGAGAGAS; L3 = AGAGAGAGAGAGAS; L4 = AGAGAGAGAGAGAGAGAS. A superscript character refers to the reference describing the identification and characterization of the corresponding endolysin.
Protein purification yields for N-terminal single-tag fusion variants of endolysins OBPgp279 and PVP-SE1gp146. The total yield for each fusion protein is shown as the amount of purified recombinant protein (in mg) per liter of E. coli expression culture. The protein amount after purification and dialysis was quantified spectrophotometrically at wavelength 280 nm.
Antibacterial activity of the N-terminal fusion variants of OBPgp279 (A and B) and PVP-SE1gp146 (C and D) without (A and C) and with (B and D) outer membrane permeabilizer EDTA (0.5 mM) on Gram-negative bacteria in comparison to unmodified endolysins. Exponentially growing cells (106 CFU/ml) were incubated at room temperature with 1.5 µM OBPgp279/OBPgp279-derived Artilysins or with 5 µM PVP-SE1gp146/PVP-SE1gp146-derived Artilysins for 30 min. Antibacterial activity was quantified as a log10 reduction [log10(N0/Ni), with N0 = number of untreated cells and Ni = number of cells after treatment]. Averages ± standard deviations are given for n = 3 repeats.
Influence of the N-terminal PCNP fusion on enzymatic activity of the selected modular and single-domain endolysins. The specific enzymatic activities (in units/µM) of the unmodified and PCNP-modified endolysins are listed together with the reduction percentage relative to the specific activity of the unmodified endolysin (=100% activity).
Antibacterial activity of PCNP-based Artilysins without (a) and with (b) EDTA (0.5 mM) in comparison to native endolysins against a set of Gram-negative strains. Cell suspensions (±106 CFU/ml) were incubated with endolysins/Artilysins dialyzed against PBS buffer (pH 7.4). The antibacterial activity is shown as a log reduction [log10(N0/Ni), with N0 = initial number of untreated cells and Ni = number of residual, treated cells). Averages and standard deviations of results of three repeats are given. ND = not determined.
Protein yield and residual enzymatic activity of Artilysins having a C-terminal PCNP tag, an N-terminal double tag, or an extended linker between PCNP and endolysin. The total yield for each Artilysin is shown as the amount of purified recombinant protein (in mg) per liter of E. coli expression culture. The yield has been quantified spectrophotometrically. The residual enzymatic activity is expressed relative to the specific activity of the corresponding unmodified endolysin (100%; OBPgp279, 19,979 U/µM; PVP-SE1gp146, 13,614 U/µm). ND = not determined.
Antibacterial activity of modifications of PCNP-fused Artiysins. The antibacterial activity of N-terminal and C-terminal linker variants of OBPgp279 (1.5 µM) and PVP-SE1gp146 (5 µM) in the absence (a) and in the presence (b) of 0.5 mM EDTA was determined. The antibacterial activity of the N-terminal double-tagged fusion variants of OBPgp279 (1.5 µM) and PVP-SE1gp146 (5 µM) in the absence (C) and in the presence (D) of 0.5 mM EDTA was determined. Antibacterial activity is expressed as a log10 reduction [log10(N0/Ni), with N0 = initial number of cells and Ni = number of residual cells after treatment]. Averages and standard deviations are shown here (n = 3). *, the detection level of 10 CFU/ml was reached. ND = not determined.