

ABSTRACT
The last step of peptidoglycan polymerization involves two families of unrelated transpeptidases that are the essential targets of β-lactam antibiotics. d,d-transpeptidases of the penicillin-binding protein (PBP) family are active-site serine enzymes that use pentapeptide precursors and are the main or exclusive cross-linking enzymes in nearly all bacteria. However, peptidoglycan cross-linking is performed mainly by active-site cysteine l,d-transpeptidases that use tetrapeptides in Mycobacterium tuberculosis, Clostridium difficile, and β-lactam-resistant mutants of Enterococcus faecium. We have investigated reprogramming of the E. faecium peptidoglycan assembly pathway by a switch from pentapeptide to tetrapeptide precursors and bypass of PBPs by l,d-transpeptidase Ldtfm. Mutational alterations of two signal transduction systems were necessary and sufficient for activation of the l,d-transpeptidation pathway, which is essentially cryptic in wild-type strains. The first one is a classical two-component regulatory system, DdcRS, that controls the activity of Ldtfm at the substrate level. As previously described, loss of DdcS phosphatase activity leads to production of the d,d-carboxypeptidase DdcY and conversion of the pentapeptide into the tetrapeptide substrate of Ldtfm. Here we show that full bypass of PBPs by Ldtfm also requires increased Ser/Thr protein phosphorylation resulting from impaired activity of phosphoprotein phosphatase StpA. This enzyme negatively controlled the level of protein phosphorylation both by direct dephosphorylation of target proteins and by dephosphorylation of its cognate kinase Stk. In combination with production of DdcY, increased protein phosphorylation by this eukaryotic-enzyme-like Ser/Thr protein kinase was sufficient for activation of the l,d-transpeptidation pathway in the absence of mutational alteration of peptidoglycan synthesis enzymes.
IMPORTANCE
The mechanism of acquisition of high-level ampicillin resistance involving bypass of the penicillin-binding proteins (PBPs) by l,d-transpeptidase Ldtfm was incompletely understood, as production of tetrapeptide precursors following transcriptional activation of the ddc locus by the DdcRS two-component regulatory system was necessary but not sufficient for full activation of the l,d-transpeptidation pathway. Here, we identified the release of a negative control of Ser/Thr protein phosphorylation mediated by phosphatase StpA as the additional factor essential for ampicillin resistance. Thus, bypass of PBPs by Ldtfm requires the modification of signal transduction regulatory systems without any gain of function by mutational alteration of peptidoglycan biosynthetic enzymes. In contrast, previously characterized mechanisms of antibiotic resistance involve horizontal gene transfer and mutational alteration of drug targets. Activation of the l,d-transpeptidation pathway reported in this study is an unprecedented mechanism of emergence of a new metabolic pathway since it involved the recruitment of preexisting functions following modifications of regulatory circuits.
INTRODUCTION
Enterococcus faecium and Enterococcus faecalis are commensal bacteria but also important nosocomial pathogens (1). These bacteria have an unusually high capacity for resistance to antibiotics since they combine several mechanisms of intrinsic resistance with the acquisition of resistance determinants by horizontal gene transfer (2–4). Intrinsic β-lactam resistance is mediated by species-specific low-affinity penicillin-binding proteins (PBPs), such as PBP5, that are thought to be sufficient for peptidoglycan cross-linking under conditions in which the d,d-transpeptidase catalytic domain of all other PBPs is inactivated by β-lactams (5–7). Although β-lactam resistance is conveyed by a single PBP in the enterococci, expression of resistance depends upon additional host factors (8, 9). In E. faecalis, factors essential for PBP5-mediated resistance to β-lactams of the cephalosporin class have been detected by genetic analyses but their functions remain largely unknown. These factors include the CroRS two-component regulatory system, which is thought to regulate genes essential for PBP5 activity, although none of the targets of CroRS-mediated transcriptional regulation identified so far has a demonstrated role in peptidoglycan synthesis or cephalosporin resistance (10–13). A serine/threonine protein kinase (IreK) was identified as essential for ceftriaxone resistance in E. faecalis (14). Subsequent characterization of the associated protein phosphatase, IreP, showed that the phosphorylation of one or several IreK substrates is essential for cephalosporin resistance (15). Recently, a substrate of IreK and IreP, designated IreB, has been identified and shown to negatively control cephalosporin resistance (16). Additional proteins essential for ceftriaxone resistance include bifunctional transglycosylase-transpeptidase (class A) PBPs for elongation of glycan chains, either PonA or PBPF (17, 18), one of the two E. faecalis MurA synthetases (UDP-N-acetylglucosamine 1-carboxyvinyl transferase) performing the first committed stem of peptidoglycan synthesis (19), and alanine transferase of the Fem family for synthesis of the l-Ala–l-Ala side chain of peptidoglycan precursors (20). Genome-wide identification of ampicillin resistance determinants in E. faecium identified a different set of proteins contributing to intrinsic β-lactam resistance, including the l,d-transpeptidase Ldtfm, the d,d-carboxypeptidase DdcP, and the glycosyltransferase Pgt (21).
In previous studies, we have identified a PBP5-independent β-lactam resistance mechanism in E. faecium based on in vitro selection of mutants resistant to ampicillin (a β-lactam of the penam class) (22). We started from E. faecium D344S, a derivative of E. faecium D344R that had spontaneously lost the pbp5 gene (22). Parental strain D344S is hypersusceptible to ampicillin because of the absence of low-affinity PBP5 (22). In five consecutive selection steps with increasing concentrations of ampicillin, we obtained five mutants, M1, M2, M3, M4, and M512, that gradually acquired high-level ampicillin resistance (22, 23). In mutant M512, 4→3 cross-links formed by PBPs were replaced by 3→3 cross-links generated by an ampicillin-insensitive l,d-transpeptidase, Ldtfm (22, 24). This enzyme cleaves the l-Lys3–d-Ala4 peptide bond of the acyl donor and links the carbonyl group of l-Lys3 to the acyl acceptor (24). Ldtfm and PBPs were found to be structurally unrelated and to harbor different active-site residues for nucleophilic attack of the carbonyl group of the acyl donor (Cys versus Ser, respectively) (24–26). Ldts and PBPs also differ in their spectra of inactivation by β-lactams (27). PBPs are potentially inactivated by all β-lactams, including members of the penam (ampicillin), cephalosporin (ceftriaxone), carbapenem (imipenem), and monobactam (aztreonam) classes, depending upon the specific β-lactam and PBP under consideration (28). In contrast, l,d-transpeptidases are efficiently inactivated by a single class of β-lactams, the carbapenems (27, 29).
The proportion of 3→3 cross-links generated by Ldtfm is very low in wild-type strains of E. faecium (22). Ldtfm is constitutively produced by parental strain D344S (23, 30), but the participation of the l,d-transpeptidase to peptidoglycan cross-linking is marginal since this enzyme requires a tetrapeptide stem in the acyl donor substrate (24) instead of the pentapeptide stem assembled in the cytoplasm (31). Thus, activation of the l,d-transpeptidation pathway requires pentapeptide-to-tetrapeptide conversion (23). The corresponding enzyme, DdcY, was identified as a metallo-d,d-carboxypeptidase that cleaves the C-terminal residue (d-Ala5) of a UDP-MurNAc pentapeptide to generate the precursor of the l,d-transpeptidation pathway (30). DdcY is encoded by a cryptic locus, ddc, that is sporadically disseminated in E. faecium strains and encodes a two-component regulatory system (DdcRS) in addition to DdcY (30). Activation of the ddc locus in M512 results from a mutation in the ddcS sensor kinase gene that impairs the phosphatase activity of the enzyme and leads to constitutive expression of the locus (30). In this investigation, we identified a second locus involved in the activation of the l,d-transpeptidation pathway, which encodes a eukaryotic-enzyme-like serine/threonine protein kinase (Stk) and its cognate phosphatase (StpA). We show that decreased protein dephosphorylation by StpA and the previously characterized activation of the ddc locus are both sufficient and necessary for high-level β-lactam resistance mediated by l,d-transpeptidation in E. faecium.
RESULTS
Identification of mutations necessary and sufficient for high-level ampicillin resistance.
We have previously shown that acquisition of high-level resistance to ampicillin by mutant M512 requires activation of the ddc locus, which encodes the d,d-carboxypeptidase DdcY, for formation of the tetrapeptide-containing substrate of the l,d-transpeptidase (30). In mutant M512, activation of the locus results from a T161A substitution in the sensor kinase DdcS that impairs the phosphatase activity of the protein (30). Here, we show that acquisition of ampicillin resistance involves the mutational alteration of a second locus encoding a putative serine/threonine kinase (Stk) and a phosphatase (StpA) (Fig. 1A). The mutation was detected in mutant M1 (first selection step) and leads to a Thr-to-Arg substitution at position 101 of StpA, which is invariant in related phosphatases (Fig. 1B).
FIG 1 .

Roles of ddcY and stpA in ampicillin resistance. (A) Map of the E. faecium locus encoding the protein phosphatase StpA and the serine/threonine kinase Stk. (B) Sequence alignment of protein phosphatases. Identical and conserved amino acids are highlighted in black and gray, respectively. The positions of amino acid substitutions detected in the mutants are indicated below the alignment. In mutant Ma, a frameshift was detected in codon 236 of stpA. (C) MICs of ampicillin for E. faecium D344S and resistant mutants.
Acquisition of the stpA mutation by strain D344S was associated with a moderate increase in the MIC of ampicillin (from 0.06 to 0.5 µg/ml) (Fig. 1C). In order to evaluate the contributions of DdcY and StpA to resistance, the ddcY gene was cloned under the control of constitutive promoter P2 of expression vector pJEH11 and the resulting plasmid (pJEH11ΩddcY) was introduced into D344S. Production of the d,d-carboxypeptidase in this host resulted in a moderate level of resistance to ampicillin (MIC, 0.5 µg/ml). In contrast, plasmid pJEH11ΩddcY conferred high-level resistance on mutant M1 (MIC, >2,000 µg/ml). Thus, the T101R substitution in StpA and production of DdcY were both necessary and sufficient for high-level ampicillin resistance.
Selection of high-level ampicillin resistance in a d,d-carboxypeptidase-producing background.
Spontaneous mutants of D344S/pJEH11ΩddcY were obtained on agar plates containing ampicillin (4 or 8 µg/ml) at a frequency of ca. 5 × 10−7. The characterization of a total of five independent mutants in each case revealed the expression of high-level ampicillin resistance (MIC, >2,000 µg/ml) and the presence of a mutation in stpA (mutants Ma to Me, Fig. 1B and C). The mutations led to amino acid substitutions in four mutants and to a frameshift mutation in the remaining mutant. Three of the four substitutions affected invariant residues, and the remaining substitution (S192P) was located within a highly conserved motif (Fig. 1B). The frameshift mutation resulted from a base deletion in codon 236 located close to the 3′ end of the stpA gene (246 codons), which encodes a conserved region of the protein. These results indicate that mutations in stpA are reproducibly selected by ampicillin in a d,d-carboxypeptidase-producing background.
Modulation of the ampicillin resistance level by the balance between kinase and phosphatase activities.
Overproduction of Stk in D344S following the introduction of plasmid pJEH11Ωstk increased the MIC of ampicillin from 0.06 to 0.5 µg/ml (Fig. 1C). Thus, the resistance phenotypes resulting from overproduction of Stk (D344S/pJEH11Ωstk) and impaired StpA phosphatase activity (mutant M1) were similar. This observation implies that the level of ampicillin resistance increases with the level of serine and threonine phosphorylation. In agreement, expression of a functional copy of stpA impaired the expression of ampicillin resistance in mutant M512 (data not shown). Thus, the level of ampicillin resistance is modulated by a balance between kinase and phosphatase activities.
In vitro activities of purified Stk and StpA.
A soluble fragment of the kinase Stk (residues 1 to 338) was produced in Escherichia coli, purified, and analyzed by Western blotting. Recombinant Stk was purified as a phosphoprotein, as shown by the detection of phosphorylated proteins (Fig. 2A) or phosphothreonine residues (Fig. 2B). Mass spectrometry analyses revealed several forms of the proteins containing one to five phosphate groups (see Fig. S1A and B in the supplemental material). StpA fully dephosphorylated Stk, as shown by the complete disappearance of the phosphorylated forms of the kinase by Western blotting (Fig. 2C and D) and mass spectrometry (see Fig. S1 in the supplemental material). Upon incubation with ATP, purified Stk phosphorylated myelin basic protein (MBP) (Fig. 2A and B). The phosphoprotein bands corresponding to phospho-MBP disappeared upon the addition of the phosphatase StpA. These results indicate that StpA and Stk display the protein phosphatase and kinase activities inferred from sequence similarity. These results also indicate that kinase Stk is a substrate of StpA.
FIG 2 .
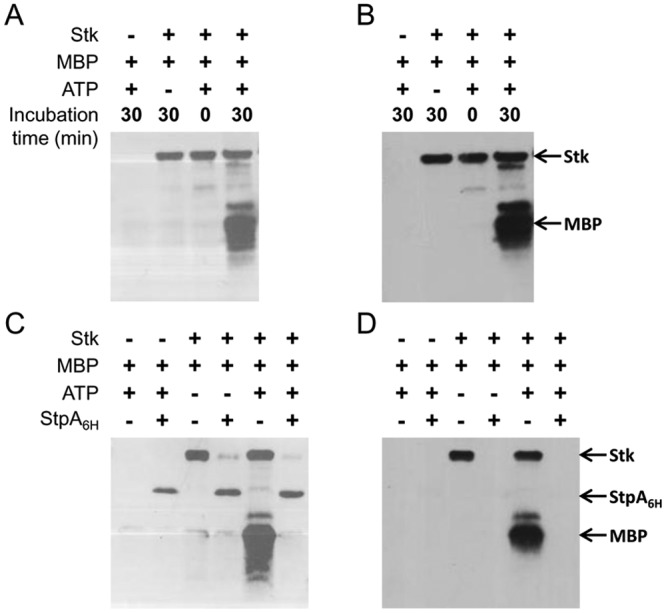
In vitro
activities of Stk (A and B) and StpA (C and D). Phosphoproteins were revealed with a commercial kit (Pro-Q; Invitrogen) (A and C) or with phosphothreonine-specific antibodies (B and D).
Autophosphorylation of purified Stk in vitro.
Since recombinant Stk was purified as a phosphoprotein from E. coli extracts, Stk was dephosphorylated in vitro by the addition of StpA in a 1:10 phosphatase-to-kinase ratio (see Fig. S2 in the supplemental material). The phosphatase StpA was removed by size exclusion chromatography, and the absence of phosphate groups from purified Stk was confirmed by Western blotting and mass spectrometry analyses. Incubation of dephosphorylated Stk with ATP and Mn2+ resulted in the phosphorylation of Stk. The reaction led to a monophosphorylation of Stk, whereas the protein recovered from E. coli extracts contained multiple phosphate groups (compare Fig. S1 and S2 in the supplemental material). These results indicate that Stk catalyzes the phosphorylation of at least one of its threonine residues. The difference between the phosphorylation patterns observed in vitro and in vivo indicates that E. coli might produce an unknown kinase responsible for Stk phosphorylation, although this host does not produce any Stk homologue (32).
Impact of stpA mutations on the in vitro activity of the phosphatase.
Recombinant forms of StpA detected in ampicillin-resistant mutants M1, Ma, Mb, Mc, Md, and Me were produced in E. coli. The truncated form of StpA (mutant Ma) and StpA containing the S192P substitution (mutant Mc) were not soluble and could not be functionally characterized. Purification of the four remaining proteins afforded soluble enzymes with reduced phosphatase activity (M1, Mb, and Me) or no detectable activity (Md) (Table 1). The amino acid substitutions detected in the former mutants mainly led to lower kcat values with marginal impacts on the Km of para-nitrophenylphosphate (pNPP). The values of these catalytic constants were determined in the presence of a high concentration of Mn2+ (2 mM). Further analysis performed with the wild-type enzyme showed that Mn2+ is essential for activity and that this cation cannot be replaced by Mg2+, Ca2+, or Zn2+ (data not shown). The phosphatase activity increased with the Mn2+ concentration, and a relatively high concentration of the cation (≥2 mM) was required for maximal activity (see Fig. S3 in the supplemental material). Reduction of the Mn2+ concentration from 2 mM to 50 µM led to a 100-fold decrease in the catalytic efficiency of wild-type StpA (Table 1). Reduction of the Mn2+ concentration also reduced the activity of StpA from mutants Mb and Me. Together, these results indicated that the mutations detected in the stpA gene from ampicillin-resistant mutants impaired the phosphatase activity of the protein, as suggested by the positions of the mutations. Of note, sufficient Mn2+ is expected to be present in vivo for metalation of StpA since brain heart infusion (BHI) broth contains 20 µM Mn2+ (33) and E. faecium accumulates Mn2+ because of active uptake (34, 35).
TABLE 1 .
Catalytic constants for pNPP hydrolysis by StpA from parental strain D344S and ampicillin-resistant mutants
| Substitution in StpA (strain) | Catalytic constanta in presence of Mn2+ at: |
|||||
|---|---|---|---|---|---|---|
| 2 mM |
50 µM |
|||||
|
Km
(mM) |
kcat
(min−1) |
kcat/Km
(min−1 mM−1) |
Km
(mM) |
kcat
(min−1) |
kcat/Km
(min−1 mM−1) |
|
| None (D344S) | 0.66 ± 0.09 | 1,200 ± 100 | 1,800 ± 290 | 0.69 ± 0.17 | 12 ± 1 | 17 ± 4.5 |
| T101R (M1) | 1.2 ± 0.2 | 0.83 ± 0.04 | 0.69 ± 0.12 | NDb | ND | ND |
| D136Y (Mb) | 1.8 ± 0.5 | 140 ± 15 | 80 ± 23 | 1.5 ± 0.4 | 0.09 ± 0.01 | 0.06 ± 0.02 |
| D18Y (Md) | ND | ND | <0.003 | ND | ND | ND |
| S120R (Me) | 1.3 ± 0.1 | 28 ± 0.1 | 22 ± 1.6 | 2.6 ± 0.7 | 0.23 ± 0.02 | 0.09 ± 03 |
Regression values ± standard errors were obtained by fitting experimental data to the Michaelis-Menten equation V = kcat ES/(Km + S), where V is the initial velocity and E and S are the initial enzyme and substrate concentrations, respectively.
ND, not determined.
StpA and Stk do not control expression of the ddc locus or ldtfm l,d-transpeptidase gene.
We have previously shown that the ddc locus encoding DdcY (d,d-carboxypeptidase) and DdcRS (two-component regulatory system) is cryptic in parental strain D344S (30). Activation of the locus in mutant M512 is due to the T161A substitution in DdcS, which impairs the phosphatase activity of the sensor kinase. Here we show that expression of the ddc locus is not under StpA control since DdcY and DdcS were not detected in extracts of D344S and M1, whereas the two proteins were produced at similar high levels by M512 and M512/pJEH11ΩstpA (Fig. 3). Likewise, overproduction of Stk in D344S did not lead to any increase in the levels of DdcY and DdcS production (data not shown). Production of the l,d-transpeptidase Ldtfm was not affected by impaired StpA and DdcS phosphatase activities (Fig. 3) or by Stk overproduction (data not shown). These results show that the contribution of impaired StpA phosphatase activity or increased Stk activity to ampicillin resistance did not depend upon increased production of the l,d-transpeptidase Ldtfm or activation of the ddc locus for production of the tetrapeptide substrate of this cross-linking enzyme.
FIG 3 .
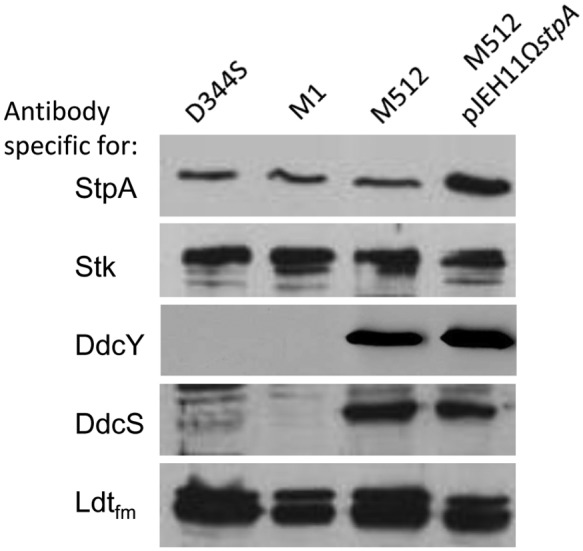
Level of production of proteins encoded by the stpA-stk, ddc, and ldtfm loci in E. faecium strains. Crude E. faecium extracts were analyzed by Western blotting with polyclonal antibodies raised against the protein phosphatase StpA, the serine/threonine kinase Stk, the d,d-carboxypeptidase DdcY, the sensor kinase DdcS, and the l,d-transpeptidase Ldtfm.
In vivo activity of StpA.
Western blot analysis of E. faecium protein extracts was performed to determine whether the in vivo production of StpA affects the level of threonine phosphorylation. The intensity of three protein bands detected by antiphosphothreonine antibodies (100, 40, and 35 kDa) was increased in mutant M1 (StpA T101R) in comparison to that in parental strain D344S (StpA) (Fig. 4). As expected, the phosphoprotein patterns were similar in M1 and M512, whereas production of StpA by M512/pJEH11ΩstpA led to a decrease in the intensity of the same set of phosphoprotein bands. These results indicate that StpA negatively controls the level of threonine phosphorylation in vivo.
FIG 4 .
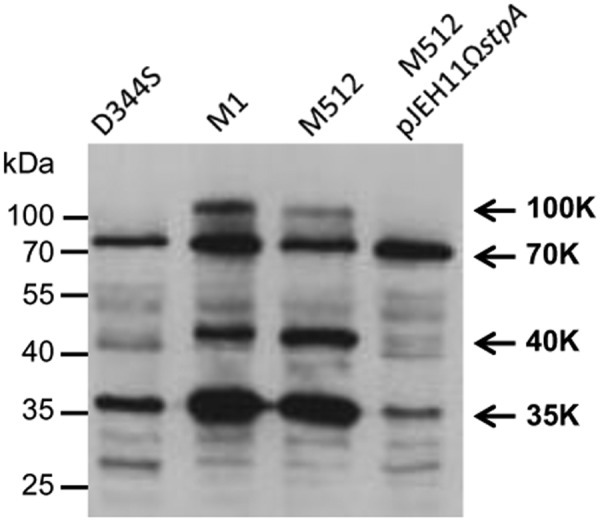
Western blot analysis of phosphoproteins in crude E. faecium extracts. Phosphoproteins were detected with antiphosphothreonine polyclonal antibodies. Arrows indicate the positions of the main phosphoproteins (100, 70, 40, and 35 kDa), which were named according to their relative electrophoretic mobility.
In vitro dephosphorylation of phosphoproteins by purified StpA.
Comparison of the level of protein phosphorylation in crude E. faecium extracts (Fig. 4) does not allow determination of whether the negative control mediated by StpA relies on dephosphorylation of phosphoproteins or an indirect effect involving negative control of the kinase activity of Stk. In order to assay directly for the phosphoprotein phosphatase activity of StpA, E. faecium extracts were incubated with purified StpA and analyzed by Western blot assay (Fig. 5A). The assay was performed with extracts from D344S, M1, M512, and D344S/pJEH11Ωstk. The latter strain was included to maximize the phosphorylation of putative StpA substrates because of high-level production of the Ser/Thr kinase. Moderate overproduction of Stk by D344S/pJEH11Ωstk was experimentally established by the increased intensity of a 100-kDa protein band detected with anti-Stk antibodies (Fig. 5B) and detection of recombinant Stk with antipolyhistidine antibodies (Fig. 5C). The 100-kDa phosphoprotein band (Fig. 5A) had the same electrophoretic mobility as Stk (Fig. 5B) and recombinant Stk containing a 6×His tag (Fig. 5C). Purification of Stk from E. faecium D344S/pJEH11Ωstk (see Fig. S4 in the supplemental material) indicated that the 100-kDa phosphoprotein band corresponds to Stk.
FIG 5 .
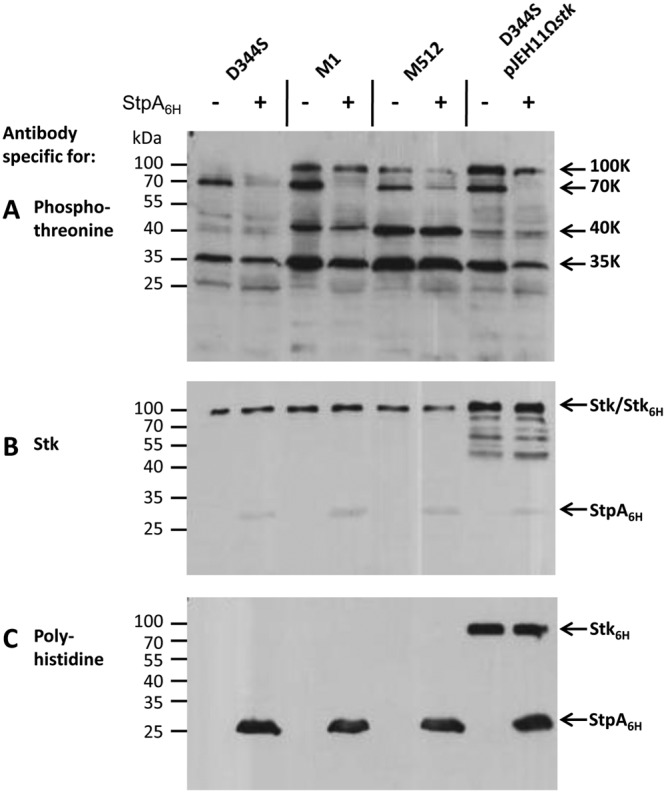
Dephosphorylation of E. faecium phosphoproteins by purified StpA. Proteins from crude cell extracts (10 µg) were incubated with (+) or without (−) purified phosphatase StpA (3 µg) for 30 min at room temperature. Proteins were separated by SDS-PAGE and transferred onto PVDF membranes. Phosphoproteins were detected with antiphosphothreonine (A), anti-Stk (B), and antipolyhistidine (C) antibodies. Arrows indicate the positions of the main phosphoproteins (100, 70, 40, and 35 kDa).
Incubation of the extracts with purified StpA resulted in a decrease in the intensity of the Stk 100-kDa phosphoprotein band (Fig. 5A). This decrease resulted from dephosphorylation of phospho-Stk by StpA, as previously shown for purified StpA and Stk (Fig. 2). Dephosphorylation of the phosphoprotein bands at 40 and 35 kDa by StpA was not detected (Fig. 5A). Thus, modulation of the phosphorylation levels of these proteins by StpA is indirect and may involve a negative control of the kinase activity of Stk. Upon incubation with StpA, the intensity of the 70-kDa phosphoprotein band decreased in all four extracts. Thus, the phosphoprotein band at 70 kDa contains a substrate of StpA. These results suggest that a subset of the proteins phosphorylated by Stk can be dephosphorylated by StpA.
DISCUSSION
The last step of peptidoglycan polymerization involves two families of unrelated transpeptidases of dd and ld specificities that catalyze the formation of 4→3 and 3→3 cross-links (36). The relative contributions of the two enzyme types greatly vary among bacterial species and may also vary along the cell cycle. In E. coli, two l,d-transpeptidases catalyze the formation of a minority (ca. 5%) of the cross-links during the exponential growth phase but the contribution of l,d-transpeptidases to peptidoglycan cross-linking increases in the stationary phase (13%) (37, 38). In Mycobacterium tuberculosis (39, 40) and M. abscessus (41), l,d-transpeptidation is the predominant mode of peptidoglycan cross-linking in both growth phases (70 to 80%), although l,d-transpeptidase paralogs appear to be differently regulated during the cell cycle (42, 43). In wild-type E. faecium, the l,d-transpeptidase Ldtfm is constitutively produced but the enzyme makes a marginal contribution to peptidoglycan cross-linking (3%) (23, 30). The activity of Ldtfm is controlled at the substrate level by the production of the d,d-carboxypeptidase DdcY, which cleaves the C-terminal d-Ala of the stem pentapeptide to form the essential tetrapeptide donor of the l,d-transpeptidation reaction (23, 30). In contrast to the ldtfm gene, which is present in all of the E. faecium isolates that have been examined (24), the ddcY and upstream ddcRS genes are present in only a minority of the isolates belonging to this species (30). The ddc locus is cryptic, and its activation results from the loss of the phosphatase activity of the DdcS sensor kinase in response to selection for ampicillin resistance (30). Production of DdcY is not sufficient for the complete bypass of PBPs by Ldtfm since the production of DdcY was associated with an only moderate (8-fold) increase in the ampicillin MIC (Fig. 1C). In this study, we show that the full bypass of PBPs also requires an increase in Ser/Thr protein phosphorylation resulting from impaired StpA phosphatase activity. Association of a mutation in stpA with expression of ddcY was necessary and sufficient for high-level ampicillin resistance (Fig. 1C). In support of this conclusion, we constructed strains that sequentially acquired a mutation in stpA and a plasmid encoding DdcY in both orders (Fig. 1C). The stpA mutations led to impaired phosphatase activity, as inferred from their nature and positions (Fig. 1B) and a direct assay of StpA phosphatase activity (Table 1). In agreement, homologous residues of the phosphatase Stp from Streptococcus agalactiae were located in the enzyme-active site (44) (see Fig. S5 in the supplemental material). Amino acid substitutions in StpA from E. faecium mutants led to >200-fold reductions in kcat for hydrolysis of pNPP in the presence of 50 µM MnCl2. The impact of the D136Y and S120R substitutions was less (43-fold and 7-fold reductions, respectively) at the nonphysiological Mn2+ concentration of 2 mM. This observation suggests that substitutions D136Y and S120R impaired Mn2+ binding. Together, these results indicate that amino acid substitutions in StpA affected the chemical step of the dephosphorylation reaction rather than substrate recognition. Thus, decreased protein dephosphorylation was essential for acquisition of ampicillin resistance.
Several assays were used to study the protein kinase and phosphatase activities of purified Stk and StpA. First, we showed that Stk catalyzes its own phosphorylation on Thr residues and subsequently transfers the phosphate group to a model protein (MBP) (Fig. 2; see Fig. S2 in the supplemental material). Phospho-Stk and phospho-MBP were dephosphorylated upon the addition of StpA (Fig. 2). This first assay established that Stk and StpA display protein kinase and phosphatase activities, respectively, as expected from sequence similarity to previously characterized enzymes (45). In a second assay, we compared the level of protein phosphorylation in crude cell extracts from E. faecium strains harboring different alleles of stpA (Fig. 4). This analysis revealed that impaired StpA activity increased the phosphorylation of several phosphoprotein bands (Fig. 4), including Stk (Fig. 5), indicating that StpA negatively modulates the level of protein phosphorylation in vivo. A third assay was used to determine whether StpA directly dephosphorylates proteins. In this assay, crude cell extracts were prepared from an E. faecium strain harboring various alleles of stpA and from an additional Stk-overproducing strain to maximize protein phosphorylation (Fig. 5). Incubation of the extracts resulted in the decreased phosphorylation of three phosphoprotein bands (100, 70, and 35 kDa). The extent of phosphorylation of a 40-kDa protein was greater in mutants with impaired phosphatase activity, but the intensity of the phosphoprotein band did not decrease upon incubation of the extract with purified StpA in vitro. For this protein band, modulation of the level of phosphorylation appears to depend upon the negative control of Stk kinase activity by StpA. Other phosphoproteins were directly dephosphorylated by StpA. For these phosphoproteins, negative control of the level of phosphorylation mediated by StpA may involve both protein dephosphorylation and negative control of the kinase Stk.
Together, our results show that the mutational alteration of two signal transduction systems is necessary and sufficient for reprogramming of the peptidoglycan assembly pathway by the production of tetrapeptide-containing precursors. The first signal transduction system is a classical two-component regulatory system, DdcRS, that controls the production of DdcY for conversion of pentapeptide into tetrapeptide precursors as previously described (30). The second system, StpA-Stk, controls the level of phosphorylation of several proteins. Several lines of evidence indicate that increased protein phosphorylation resulting from the mutation of stpA was required for resistance although this did not affect the level of DdcY production. First, expression of ddcY under the control of a heterologous promoter in plasmid pJEH11 was sufficient for high-level ampicillin resistance (MIC, >2,000 g/ml) in mutant M1, which produced a derivative of StpA with reduced phosphatase activity, but not in the parental strain D344S, which harbors a wild-type copy of stpA (Fig. 1C). Thus, production of DdcY and impaired phosphatase activity were both required for ampicillin resistance. Second, Western blot analysis indicated that DdcY was produced at the same level by derivatives of D344S and M1 harboring pJEH11ΩddcY, indicating, as might have been expected, that the heterologous promoter of vector pJEH11 is functional in both hosts independently of the stpA allele (see Fig. S6 in the supplemental material). Conversely, DdcY was not produced by D344S and M1, indicating that the stpA mutation present in M1 did not lead to transcriptional activation of the chromosomal copy of ddcY. Thus, the essential role of the stpA mutation did not depend upon DdcY production. In agreement, derivatives of D344S/pJEH11ΩddcY selected on ampicillin (mutants Ma to Me) harbored mutations in stpA (Fig. 1C) that impaired the phosphatase activity of StpA (Fig. 1B and Table 1) and did not affect the level of DdcY production (see Fig. S6 in the supplemental material). Third, acquisition of a mutation in stpA by M1 and mutants Ma to Me did not result in the production of DdcS (Fig. 3; see Fig. S6 in the supplemental material). Thus, the chromosomal locus ddc was not regulated in response to modulation of protein phosphorylation by StpA. Together, these results establish that alterations of DdcRS and StpA-Stk are both required for resistance and that these signal transduction systems control the activity of distinct sets of proteins.
Production of DdcY resulting from the introduction of plasmid pJEH11ΩddcY into M1 or loss of DdcS phosphatase activity by M512 resulted in similar resistance phenotypes. Thus, DdcY is the only target of DdcRS-mediated regulation that is essential for ampicillin resistance. In contrast, StpA may affect the level of phosphorylation of multiple proteins potentially involved in resistance, as shown by modulation of the level of phosphorylation of the 100-, 70-, 40-, and 35-kDa protein bands (Fig. 4 and 5). Furthermore, additional fainter phosphoprotein bands were detected by anti-phospho-Thr antibodies upon prolonged exposure of Western blots (data not shown) and Ser phosphorylation was not investigated. In agreement, phosphoproteome analyses of Firmicutes suggest that the number of Ser/Thr phosphoproteins may be on the order of 100 to 200 in E. faecium (46, 47). The role of protein phosphorylation in peptidoglycan synthesis and ampicillin resistance remains to be determined.
It is striking that activation of the l,d-transpeptidation pathway in E. faecium involves modifications of representatives of the two main signal transduction pathways of prokaryotes, the two-component regulatory system relying on His-to-Asp phosphotransfer (48) and the one-component eukaryotic-enzyme-like Ser/Thr phosphorylation system (45), in the absence of modification of cell wall biosynthesis proteins. In contrast, previously characterized mechanisms of acquisition of antibiotic resistance involve horizontal gene transfer, for example, van clusters for vancomycin resistance (49), or mutational alteration of an antibiotic target, for example, PBP5, in E. faecium (8, 9). Acquisition of high-level ampicillin resistance resulting from bypass of the PBPs by Ldtfm was found here to depend upon the alteration of regulatory circuits without any gain of function through gene transfer or modification biosynthetic enzymes. This is an unprecedented example of the emergence of a new metabolic pathway through the recruitment of functions via the modification of regulatory circuits.
MATERIALS AND METHODS
Bacterial strains, selection of mutants, and antibiotic susceptibility assay.
All cultures were performed at 37°C in BHI agar or broth (Difco Laboratories). Mutants M1 and M512 were derived from E. faecium D344S by serial selection on medium containing increasing concentrations of ampicillin (22, 23). Derivatives of pJEH11 were introduced into strains of E. faecium by conjugation (17) and electroporation (18) as previously described, except that a field of 1 kV (2-mm gap) and a shunt resistance of 1,000 Ω were used for the latter technique. Transformants and transconjugants were selected on gentamicin (128 µg/ml). Mutants Ma to Me were obtained by plating D344S/pJEH11ΩddcY on agar containing ampicillin at 4 or 8 µg/ml. Colonies appeared after 5 days of incubation at a frequency of about 5 × 10−7. MICs of ampicillin (Bristol-Myers, Paris, France) were determined by the agar dilution method after 48 h of incubation (23).
Plasmid construction.
For a description of the construction of recombinant plasmids encoding the E. faecium d,d-carboxypeptidase DdcY, serine/threonine kinase Stk, serine/threonine phosphatase StpA, see Text S1 in the supplemental material.
Production and purification of kinase Stk and phosphatase StpA.
Recombinant Stk and StpA containing a C-terminal 6×His tag were produced in E. coli BL21(DE3). Bacteria were grown in BHI broth containing ampicillin (100 µg/ml) at 37°C to an optical density at 600 nm of 0.9. Protein production was induced with 0.5 mM IPTG (isopropyl-β-d-thiogalactopyranoside) for 19 h at 16°C. Cells were disrupted by sonication in 40 ml of 50 mM Tris-HCl (pH 8.0) containing 300 mM NaCl, and cells debris was removed by centrifugation. Proteins were purified by affinity chromatography on Ni2+-nitrilotriacetate (Sigma). Elution was performed with 500 mM imidazole in 50 mM Tris buffer (pH 8.0) containing 300 mM NaCl. Proteins were further purified by size exclusion chromatography on a Superdex 75 HL26/60 column (GE Healthcare) equilibrated with 50 mM Tris-HCl (pH 8.0) containing 150 mM NaCl. Stk and StpA eluted as single peaks corresponding to monomers. Proteins were stored at −20°C in the same buffer supplemented with 50% glycerol.
Western blot analysis.
Polyclonal anti-DdcY and anti-DdcS rabbit antisera were previously described (30). Anti-Ldtfm, anti-Stk, and anti-StpA antibodies were obtained by three subcutaneous injections at 2-week intervals of 500 µg of purified protein (24) into rats for Ldtfm or into rabbits for Stk and Stp. Antiphosphothreonine rabbit antibodies were purchased from Invitrogen. Bacteria were lysed in 100 mM Tris-HCl (pH 7.0) with 0.18-µm glass beads (6 × 30 s; FastPrep; QBIOgene, Illkirch, France). Proteins were separated by SDS-PAGE, electrotransferred to a nitrocellulose membrane (Hybond, Amersham Biosciences, Little Chalfont, United Kingdom), and incubated with antisera at dilutions of 1/1,000 (Anti-DdcY), 1/200 (anti-DdcS and anti-Ldtfm), 1/2,000 (anti-Stk), 1/1,000 (anti-StpA), and 1/2,500 (antipolyhistidine from Sigma). Antiphosphothreonine antibodies were used at dilutions of 1/250 and 1/1,000 for analyses of crude cell extracts and purified proteins, respectively. Western blot assays were incubated in Tris-buffered saline (TBS)-Tween (10 mM Tris [pH 7.5], 150 mM NaCl, 0,025% Tween 20, 2.5% nonfat dry milk) for all antibodies except for antiphosphothreonine antibodies (20 mM Tris [pH 7.5], 137 mM NaCl, 0.1% Tween 20, 3% bovine serum albumin). Goat anti-rabbit or anti-rat IgGs coupled to peroxidase (SouthernBiotech, Birmingham, AL) were used as secondary antibodies, and proteins were detected by chemiluminescence (ECL kit; Pierce, Amersham Biosciences).
Kinase and phosphatase assays.
Autophosphorylation of Stk and phosphorylation of MBP (Sigma) were tested in kinase buffer (50 mM Tris-HCl [pH 7.5], 25 mM NaCl, 1 mM MnCl2, 1 mM MgCl2, 1 mM dithiothreitol, 0.1 mM EDTA) in the presence or absence of ATP (2 mM) at room temperature. Dephosphorylation of Stk and MBP (Sigma) by StpA was tested by adding purified StpA (4 µM) to the kinase assay mixture. For dephosphorylation of phosphoproteins from E. faecium crude cell extract, purified StpA (3 µg) was incubated with E. faecium crude cell extract (10 µg) in 20 µl of 100 mM phosphatase buffer (pH 6.4) containing 2 mM MnCl2 for 30 min at room temperature. Reactions were quenched with Laemmli SDS sample buffer and boiled for 5 min. Proteins were separated by 12% SDS-PAGE and transferred to polyvinylidene difluoride (PVDF) transfer membrane (Immobilon-FL; Millipore). Phosphorylated proteins were detected with a Pro-Q Diamond phosphoprotein blot stain kit (Invitrogen) according to the manufacturer’s instructions. Blots were washed in TBS-Tween for serial detection of phosphoproteins with the Pro-Q kit and antiphosphothreonine antibodies.
Hydrolysis of pNPP (Sigma) by StpA was determined at 37°C in 50 mM Tris-HCl (pH 8.0) containing various concentrations of MnCl2. Reactions were initiated by the addition of MnCl2, and absorbance at 405 nm (ε = 12,500 M−1 cm−1) was monitored with a Cary 100 Bio spectrophotometer (Varian).
SUPPLEMENTAL MATERIAL
Mass spectrometry analysis of Stk phosphorylation. (A) The catalytic domain of Stk was produced in E. coli, purified, and analyzed by electrospray ionization mass spectrometry in the positive-ion mode. Shown is a representative portion of the spectrum for (M + 42H)42+ (peaks B, C, D, E, and F) and (M + 41H)41+ (peaks B′, C′, D′, E′, and F′) molecular ions. (B) Deduced average mass of phosphorylated forms of the protein. (C) Incubation of the catalytic domain of Stk with StpA and Mn2+ resulted in complete dephosphorylation of Stk. Minor peaks at m/z = 924.91 and 947.71 correspond to an adduct of Stk with a buffer molecule (Tris). No change in the phosphorylation pattern was observed for incubation of StpA and Stk in the absence of Mn2+ (D) or for incubation of Stk with Mn2+ in the absence of StpA (E). Download
Purification of dephosphorylated Stk and autophosphorylation assay. Stk (69 µM) was dephosphorylated by StpA (6.9 µM) in 50 mM Tris-HCl (pH 8.0) containing 50 µM MnCl2 for 1 h at 37°C. Stk and StpA were separated by size exclusion chromatography on a Superdex 75 HL26/60 column (GE Healthcare) as described for the kinase domain of Stk (A). Dephosphorylated Stk (fraction 4; 34 µM) was incubated with or without ATP (1 mM) and MnCl2 (1 mM), and phosphoproteins were detected by Western blotting with antiphosphothreonine antibodies (B) or by mass spectrometry (C). Download
Turnover number for hydrolysis of pNPP (4 mM) by purified StpA at various Mn2+ concentrations (30 µM to 5 mM). Values are means ± standard deviations of three determinations. Download
Purification of Stk from E. faecium D344S/pJEH11Ωstk. Lane A, crude cell extract from E. faecium D344S/pJEH11Ωstk producing full-length Stk with a C-terminal 6×His tag. The protein was purified by Ni2+ affinity (lane B) and size exclusion (lane C) chromatography as described in Text S1. Proteins were detected with antiphosphothreonine (left), anti-Stk (middle), and antipolyhistidine (right) antibodies. Download
Structure of S. agalactiae protein phosphatase Stp highlighting conserved residues discussed in the text. (a) ClustalW alignment of the sequence of E. faecium StpA and S. agalactiae Stp. The positions of amino acid substitutions (mutants M1, Mb, Mc, Md, and Me) and the frameshift mutation (mutant Ma) detected in StpA are highlighted in gray. (b) Structure of S. agalactiae Stp (44). Amino acid substitutions detected in StpA from E. faecium mutants Mb to Me affect conserved residues that are highlighted in pink sticks in the S. agalactiae Stp structure. The frameshift mutation in mutant Ma is located in codon 240 specifying Ala in S. agalactiae Stp. The downstream portion of the β-sheet, which is not present in StpA from mutant Ma, is highlighted in pink. Orange spheres represent Mn2+ ions. Download
Levels of d,d-carboxypeptidase DdcY and sensor kinase DdcS production by E. faecium strains. Crude cell extracts were analyzed by Western blotting with anti-DdcY and anti-DdcS antibodies. Mutants Ma to Me are ampicillin-resistant derivatives of D344S/pJEH11ΩddcY. Download
Supplemental methods including descriptions of plasmid construction and the methods used for mass spectrometry analysis of phosphorylated proteins and purification of full-length Stk from E. faecium. Download
ACKNOWLEDGMENT
This work was supported by National Institute of Allergy and Infectious Diseases grant RO1 AI046626.
Footnotes
Citation Sacco E, Cortes M, Josseaume N, Rice LB, Mainardi J-L, Arthur M. 2014. Serine/threonine protein phosphatase-mediated control of the peptidoglycan cross-linking l,d-transpeptidase pathway in Enterococcus faecium. mBio 5(4):e01446-14. doi:10.1128/mBio.01446-14.
REFERENCES
- 1. Arias CA, Murray BE. 2012. The rise of the Enterococcus: beyond vancomycin resistance. Nat. Rev. Microbiol. 10:266–278. 10.1038/nrmicro2761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hollenbeck BL, Rice LB. 2012. Intrinsic and acquired resistance mechanisms in Enterococcus. Virulence 3:421–433. 10.4161/viru.21282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Paulsen IT, Banerjei L, Myers GS, Nelson KE, Seshadri R, Read TD, Fouts DE, Eisen JA, Gill SR, Heidelberg JF, Tettelin H, Dodson RJ, Umayam L, Brinkac L, Beanan M, Daugherty S, DeBoy RT, Durkin S, Kolonay J, Madupu R, Nelson W, Vamathevan J, Tran B, Upton J, Hansen T, Shetty J, Khouri H, Utterback T, Radune D, Ketchum KA, Dougherty BA, Fraser CM. 2003. Role of mobile DNA in the evolution of vancomycin-resistant Enterococcus faecalis. Science 299:2071–2074. 10.1126/science.1080613 [DOI] [PubMed] [Google Scholar]
- 4. Willems RJ, Hanage WP, Bessen DE, Feil EJ. 2011. Population biology of Gram-positive pathogens: high-risk clones for dissemination of antibiotic resistance. FEMS Microbiol. Rev. 35:872–900. 10.1111/j.1574-6976.2011.00284.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Fontana R, Grossato A, Rossi L, Cheng YR, Satta G. 1985. Transition from resistance to hypersusceptibility to beta-lactam antibiotics associated with loss of a low-affinity penicillin-binding protein in a Streptococcus faecium mutant highly resistant to penicillin. Antimicrob. Agents Chemother. 28:678–683. 10.1128/AAC.28.5.678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Leimanis S, Hoyez N, Hubert S, Laschet M, Sauvage E, Brasseur R, Coyette J. 2006. PBP5 complementation of a PBP3 deficiency in Enterococcus hirae. J. Bacteriol. 188:6298–6307. 10.1128/JB.00334-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Canepari P, Lleo MM, Cornaglia G, Fontana R, Satta G. 1986. In Streptococcus faecium penicillin-binding protein 5 alone is sufficient for growth at sub-maximal but not at maximal rate. J. Gen. Microbiol. 132:625–631 [DOI] [PubMed] [Google Scholar]
- 8. Rice LB, Bellais S, Carias LL, Hutton-Thomas R, Bonomo RA, Caspers P, Page MG, Gutmann L. 2004. Impact of specific pbp5 mutations on expression of beta-lactam resistance in Enterococcus faecium. Antimicrob. Agents Chemother. 48:3028–3032. 10.1128/AAC.48.8.3028-3032.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sifaoui F, Arthur M, Rice L, Gutmann L. 2001. Role of penicillin-binding protein 5 in expression of ampicillin resistance and peptidoglycan structure in Enterococcus faecium. Antimicrob. Agents Chemother. 45:2594–2597. 10.1128/AAC.45.9.2594-2597.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Comenge Y, Quintiliani R, Jr, Li L, Dubost L, Brouard JP, Hugonnet JE, Arthur M. 2003. The CroRS two-component regulatory system is required for intrinsic beta-lactam resistance in Enterococcus faecalis. J. Bacteriol. 185:7184–7192. 10.1128/JB.185.24.7184-7192.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Le Breton Y, Muller C, Auffray Y, Rincé A. 2007. New insights into the Enterococcus faecalis CroRS two-component system obtained using a differential-display random arbitrarily primed PCR approach. Appl. Environ. Microbiol. 73:3738–3741. 10.1128/AEM.00390-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Muller C, Le Breton Y, Morin T, Benachour A, Auffray Y, Rincé A. 2006. The response regulator CroR modulates expression of the secreted stress-induced SalB protein in Enterococcus faecalis. J. Bacteriol. 188:2636–2645. 10.1128/JB.188.7.2636-2645.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Snyder H, Kellogg SL, Skarda LM, Little JL, Kristich CJ. 2014. Nutritional control of antibiotic resistance via an interface between the phosphotransferase system and a two-component signaling system. Antimicrob. Agents Chemother. 58:957–965. 10.1128/AAC.01919-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kristich CJ, Wells CL, Dunny GM. 2007. A eukaryotic-type Ser/Thr kinase in Enterococcus faecalis mediates antimicrobial resistance and intestinal persistence. Proc. Natl. Acad. Sci. U. S. A. 104:3508–3513. 10.1073/pnas.0608742104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kristich CJ, Little JL, Hall CL, Hoff JS. 2011. Reciprocal regulation of cephalosporin resistance in Enterococcus faecalis. mBio 2(6):e00199–00111. 10.1128/mBio.00199-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hall CL, Tschannen M, Worthey EA, Kristich CJ. 2013. IreB, a Ser/Thr kinase substrate, influences antimicrobial resistance in Enterococcus faecalis. Antimicrob. Agents Chemother. 57:6179–6186. 10.1128/AAC.01472-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Arbeloa A, Segal H, Hugonnet JE, Josseaume N, Dubost L, Brouard JP, Gutmann L, Mengin-Lecreulx D, Arthur M. 2004. Role of class A penicillin-binding proteins in PBP5-mediated beta-lactam resistance in Enterococcus faecalis. J. Bacteriol. 186:1221–1228. 10.1128/JB.186.5.1221-1228.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Rice LB, Carias LL, Rudin S, Hutton R, Marshall S, Hassan M, Josseaume N, Dubost L, Marie A, Arthur M. 2009. Role of class A penicillin-binding proteins in the expression of beta-lactam resistance in Enterococcus faecium. J. Bacteriol. 191:3649–3656. 10.1128/JB.01834-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Vesić D, Kristich CJ. 2012. MurAA is required for intrinsic cephalosporin resistance of Enterococcus faecalis. Antimicrob. Agents Chemother. 56:2443–2451. 10.1128/AAC.05984-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bouhss A, Josseaume N, Severin A, Tabei K, Hugonnet JE, Shlaes D, Mengin-Lecreulx D, van Heijenoort J, Arthur M. 2002. Synthesis of the l-alanyl-l-alanine cross-bridge of Enterococcus faecalis peptidoglycan. J. Biol. Chem. 277:45935–45941. 10.1074/jbc.M207449200 [DOI] [PubMed] [Google Scholar]
- 21. Zhang X, Paganelli FL, Bierschenk D, Kuipers A, Bonten MJ, Willems RJ, van Schaik W. 2012. Genome-wide identification of ampicillin resistance determinants in Enterococcus faecium. PLoS Genet. 8(6):e1002804. 10.1371/journal.pgen.1002804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mainardi JL, Legrand R, Arthur M, Schoot B, van Heijenoort J, Gutmann L. 2000. Novel mechanism of beta-lactam resistance due to bypass of dd-transpeptidation in Enterococcus faecium. J. Biol. Chem. 275:16490–16496. 10.1074/jbc.M909877199 [DOI] [PubMed] [Google Scholar]
- 23. Mainardi JL, Morel V, Fourgeaud M, Cremniter J, Blanot D, Legrand R, Frehel C, Arthur M, Van Heijenoort J, Gutmann L. 2002. Balance between two transpeptidation mechanisms determines the expression of beta-lactam resistance in Enterococcus faecium. J. Biol. Chem. 277:35801–35807. 10.1074/jbc.M204319200 [DOI] [PubMed] [Google Scholar]
- 24. Mainardi JL, Fourgeaud M, Hugonnet JE, Dubost L, Brouard JP, Ouazzani J, Rice LB, Gutmann L, Arthur M. 2005. A novel peptidoglycan cross-linking enzyme for a beta-lactam-resistant transpeptidation pathway. J. Biol. Chem. 280:38146–38152. 10.1074/jbc.M507384200 [DOI] [PubMed] [Google Scholar]
- 25. Biarrotte-Sorin S, Hugonnet JE, Delfosse V, Mainardi JL, Gutmann L, Arthur M, Mayer C. 2006. Crystal structure of a novel beta-lactam-insensitive peptidoglycan transpeptidase. J. Mol. Biol. 359:533–538. 10.1016/j.jmb.2006.03.014 [DOI] [PubMed] [Google Scholar]
- 26. Lecoq L, Dubée V, Triboulet S, Bougault C, Hugonnet JE, Arthur M, Simorre JP. 2013. Structure of Enterococcus faecium l,d-transpeptidase acylated by ertapenem provides insight into the inactivation mechanism. ACS Chem. Biol. 8:1140–1146. 10.1021/cb4001603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Mainardi JL, Hugonnet JE, Rusconi F, Fourgeaud M, Dubost L, Moumi AN, Delfosse V, Mayer C, Gutmann L, Rice LB, Arthur M. 2007. Unexpected inhibition of peptidoglycan ld-transpeptidase from Enterococcus faecium by the beta-lactam imipenem. J. Biol. Chem. 282:30414–30422. 10.1074/jbc.M704286200 [DOI] [PubMed] [Google Scholar]
- 28. Sauvage E, Kerff F, Terrak M, Ayala JA, Charlier P. 2008. The penicillin-binding proteins: structure and role in peptidoglycan biosynthesis. FEMS Microbiol. Rev. 32:234–258. 10.1111/j.1574-6976.2008.00105.x [DOI] [PubMed] [Google Scholar]
- 29. Triboulet S, Dubée V, Lecoq L, Bougault C, Mainardi JL, Rice LB, Ethève-Quelquejeu M, Gutmann L, Marie A, Dubost L, Hugonnet JE, Simorre JP, Arthur M. 2013. Kinetic features of l,d-transpeptidase inactivation critical for β-lactam antibacterial activity. PLoS One 8(7):e67831. 10.1371/journal.pone.0067831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Sacco E, Hugonnet JE, Josseaume N, Cremniter J, Dubost L, Marie A, Patin D, Blanot D, Rice LB, Mainardi JL, Arthur M. 2010. Activation of the l,d-transpeptidation peptidoglycan cross-linking pathway by a metallo-d,d-carboxypeptidase in Enterococcus faecium. Mol. Microbiol. 75:874–885. 10.1111/j.1365-2958.2009.07014.x [DOI] [PubMed] [Google Scholar]
- 31. Barreteau H, Kovac A, Boniface A, Sova M, Gobec S, Blanot D. 2008. Cytoplasmic steps of peptidoglycan biosynthesis. FEMS Microbiol. Rev. 32:168–207. 10.1111/j.1574-6976.2008.00104.x [DOI] [PubMed] [Google Scholar]
- 32. Mieczkowski C, Iavarone AT, Alber T. 2008. Auto-activation mechanism of the Mycobacterium tuberculosis PknB receptor Ser/Thr kinase. EMBO J. 27:3186–3197. 10.1038/emboj.2008.236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Jacobsen FE, Kazmierczak KM, Lisher JP, Winkler ME, Giedroc DP. 2011. Interplay between manganese and zinc homeostasis in the human pathogen Streptococcus pneumoniae. Metallomics 3:38–41. 10.1039/c0mt00050g [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Daly MJ, Gaidamakova EK, Matrosova VY, Vasilenko A, Zhai M, Venkateswaran A, Hess M, Omelchenko MV, Kostandarithes HM, Makarova KS, Wackett LP, Fredrickson JK, Ghosal D. 2004. Accumulation of Mn(II) in Deinococcus radiodurans facilitates gamma-radiation resistance. Science 306:1025–1028. 10.1126/science.1103185 [DOI] [PubMed] [Google Scholar]
- 35. Lisher JP, Giedroc DP. 2013. Manganese acquisition and homeostasis at the host-pathogen interface. Front. Cell. Infect. Microbiol. 3:91. 10.3389/fcimb.2013.00091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Mainardi JL, Villet R, Bugg TD, Mayer C, Arthur M. 2008. Evolution of peptidoglycan biosynthesis under the selective pressure of antibiotics in Gram-positive bacteria. FEMS Microbiol. Rev. 32:386–408. 10.1111/j.1574-6976.2007.00097.x [DOI] [PubMed] [Google Scholar]
- 37. Pisabarro AG, de Pedro MA, Vázquez D. 1985. Structural modifications in the peptidoglycan of Escherichia coli associated with changes in the state of growth of the culture. J. Bacteriol. 161:238–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Magnet S, Dubost L, Marie A, Arthur M, Gutmann L. 2008. Identification of the l,d-transpeptidases for peptidoglycan cross-linking in Escherichia coli. J. Bacteriol. 190:4782–4785. 10.1128/JB.00025-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lavollay M, Arthur M, Fourgeaud M, Dubost L, Marie A, Veziris N, Blanot D, Gutmann L, Mainardi JL. 2008. The peptidoglycan of stationary-phase Mycobacterium tuberculosis predominantly contains cross-links generated by l,d-transpeptidation. J. Bacteriol. 190:4360–4366. 10.1128/JB.00239-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kumar P, Arora K, Lloyd JR, Lee IY, Nair V, Fischer E, Boshoff HI, Barry CE., III 2012. Meropenem inhibits d,d-carboxypeptidase activity in Mycobacterium tuberculosis. Mol. Microbiol. 86:367–381. 10.1111/j.1365-2958.2012.08199.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lavollay M, Fourgeaud M, Herrmann JL, Dubost L, Marie A, Gutmann L, Arthur M, Mainardi JL. 2011. The peptidoglycan of Mycobacterium abscessus is predominantly cross-linked by l,d-transpeptidases. J. Bacteriol. 193:778–782. 10.1128/JB.00606-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Betts JC, Lukey PT, Robb LC, McAdam RA, Duncan K. 2002. Evaluation of a nutrient starvation model of Mycobacterium tuberculosis persistence by gene and protein expression profiling. Mol. Microbiol. 43:717–731. 10.1046/j.1365-2958.2002.02779.x [DOI] [PubMed] [Google Scholar]
- 43. Cordillot M, Dubée V, Triboulet S, Dubost L, Marie A, Hugonnet JE, Arthur M, Mainardi JL. 2013. In vitro cross-linking of Mycobacterium tuberculosis peptidoglycan by l,d-transpeptidases and inactivation of these enzymes by carbapenems. Antimicrob. Agents Chemother. 57:5940–5945. 10.1128/AAC.01663-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Rantanen MK, Lehtiö L, Rajagopal L, Rubens CE, Goldman A. 2007. Structure of Streptococcus agalactiae serine/threonine phosphatase. The subdomain conformation is coupled to the binding of a third metal ion. FEBS J 274:3128–3137. 10.1111/j.1742-4658.2007.05845.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Pereira SF, Goss L, Dworkin J. 2011. Eukaryote-like serine/threonine kinases and phosphatases in bacteria. Microbiol. Mol. Biol. Rev. 75:192–212. 10.1128/MMBR.00042-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Misra SK, Milohanic E, Aké F, Mijakovic I, Deutscher J, Monnet V, Henry C. 2011. Analysis of the serine/threonine/tyrosine phosphoproteome of the pathogenic bacterium Listeria monocytogenes reveals phosphorylated proteins related to virulence. Proteomics 11:4155-4165. 10.1002/pmic.201100259 [DOI] [PubMed] [Google Scholar]
- 47. Sun X, Ge F, Xiao CL, Yin XF, Ge R, Zhang LH, He QY. 2010. Phosphoproteomic analysis reveals the multiple roles of phosphorylation in pathogenic bacterium Streptococcus pneumoniae. J. Proteome Res. 9:275-282. 10.1021/pr900612v [DOI] [PubMed] [Google Scholar]
- 48. Jung K, Fried L, Behr S, Heermann R. 2012. Histidine kinases and response regulators in networks. Curr. Opin. Microbiol. 15:118-124. 10.1016/j.mib.2011.11.009 [DOI] [PubMed] [Google Scholar]
- 49. Arthur M, Molinas C, Depardieu F, Courvalin P. 1993. Characterization of Tn1546, a Tn3-related transposon conferring glycopeptide resistance by synthesis of depsipeptide peptidoglycan precursors in Enterococcus faecium BM4147. J. Bacteriol. 175:117-127 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Mass spectrometry analysis of Stk phosphorylation. (A) The catalytic domain of Stk was produced in E. coli, purified, and analyzed by electrospray ionization mass spectrometry in the positive-ion mode. Shown is a representative portion of the spectrum for (M + 42H)42+ (peaks B, C, D, E, and F) and (M + 41H)41+ (peaks B′, C′, D′, E′, and F′) molecular ions. (B) Deduced average mass of phosphorylated forms of the protein. (C) Incubation of the catalytic domain of Stk with StpA and Mn2+ resulted in complete dephosphorylation of Stk. Minor peaks at m/z = 924.91 and 947.71 correspond to an adduct of Stk with a buffer molecule (Tris). No change in the phosphorylation pattern was observed for incubation of StpA and Stk in the absence of Mn2+ (D) or for incubation of Stk with Mn2+ in the absence of StpA (E). Download
Purification of dephosphorylated Stk and autophosphorylation assay. Stk (69 µM) was dephosphorylated by StpA (6.9 µM) in 50 mM Tris-HCl (pH 8.0) containing 50 µM MnCl2 for 1 h at 37°C. Stk and StpA were separated by size exclusion chromatography on a Superdex 75 HL26/60 column (GE Healthcare) as described for the kinase domain of Stk (A). Dephosphorylated Stk (fraction 4; 34 µM) was incubated with or without ATP (1 mM) and MnCl2 (1 mM), and phosphoproteins were detected by Western blotting with antiphosphothreonine antibodies (B) or by mass spectrometry (C). Download
Turnover number for hydrolysis of pNPP (4 mM) by purified StpA at various Mn2+ concentrations (30 µM to 5 mM). Values are means ± standard deviations of three determinations. Download
Purification of Stk from E. faecium D344S/pJEH11Ωstk. Lane A, crude cell extract from E. faecium D344S/pJEH11Ωstk producing full-length Stk with a C-terminal 6×His tag. The protein was purified by Ni2+ affinity (lane B) and size exclusion (lane C) chromatography as described in Text S1. Proteins were detected with antiphosphothreonine (left), anti-Stk (middle), and antipolyhistidine (right) antibodies. Download
Structure of S. agalactiae protein phosphatase Stp highlighting conserved residues discussed in the text. (a) ClustalW alignment of the sequence of E. faecium StpA and S. agalactiae Stp. The positions of amino acid substitutions (mutants M1, Mb, Mc, Md, and Me) and the frameshift mutation (mutant Ma) detected in StpA are highlighted in gray. (b) Structure of S. agalactiae Stp (44). Amino acid substitutions detected in StpA from E. faecium mutants Mb to Me affect conserved residues that are highlighted in pink sticks in the S. agalactiae Stp structure. The frameshift mutation in mutant Ma is located in codon 240 specifying Ala in S. agalactiae Stp. The downstream portion of the β-sheet, which is not present in StpA from mutant Ma, is highlighted in pink. Orange spheres represent Mn2+ ions. Download
Levels of d,d-carboxypeptidase DdcY and sensor kinase DdcS production by E. faecium strains. Crude cell extracts were analyzed by Western blotting with anti-DdcY and anti-DdcS antibodies. Mutants Ma to Me are ampicillin-resistant derivatives of D344S/pJEH11ΩddcY. Download
Supplemental methods including descriptions of plasmid construction and the methods used for mass spectrometry analysis of phosphorylated proteins and purification of full-length Stk from E. faecium. Download