

ABSTRACT
Soil microbial diversity represents the largest global reservoir of novel microorganisms and enzymes. In this study, we coupled functional metagenomics and DNA stable-isotope probing (DNA-SIP) using multiple plant-derived carbon substrates and diverse soils to characterize active soil bacterial communities and their glycoside hydrolase genes, which have value for industrial applications. We incubated samples from three disparate Canadian soils (tundra, temperate rainforest, and agricultural) with five native carbon (12C) or stable-isotope-labeled (13C) carbohydrates (glucose, cellobiose, xylose, arabinose, and cellulose). Indicator species analysis revealed high specificity and fidelity for many uncultured and unclassified bacterial taxa in the heavy DNA for all soils and substrates. Among characterized taxa, Actinomycetales (Salinibacterium), Rhizobiales (Devosia), Rhodospirillales (Telmatospirillum), and Caulobacterales (Phenylobacterium and Asticcacaulis) were bacterial indicator species for the heavy substrates and soils tested. Both Actinomycetales and Caulobacterales (Phenylobacterium) were associated with metabolism of cellulose, and Alphaproteobacteria were associated with the metabolism of arabinose; members of the order Rhizobiales were strongly associated with the metabolism of xylose. Annotated metagenomic data suggested diverse glycoside hydrolase gene representation within the pooled heavy DNA. By screening 2,876 cloned fragments derived from the 13C-labeled DNA isolated from soils incubated with cellulose, we demonstrate the power of combining DNA-SIP, multiple-displacement amplification (MDA), and functional metagenomics by efficiently isolating multiple clones with activity on carboxymethyl cellulose and fluorogenic proxy substrates for carbohydrate-active enzymes.
IMPORTANCE
The ability to identify genes based on function, instead of sequence homology, allows the discovery of genes that would not be identified through sequence alone. This is arguably the most powerful application of metagenomics for the recovery of novel genes and a natural partner of the stable-isotope-probing approach for targeting active-yet-uncultured microorganisms. We expanded on previous efforts to combine stable-isotope probing and metagenomics, enriching microorganisms from multiple soils that were active in degrading plant-derived carbohydrates, followed by construction of a cellulose-based metagenomic library and recovery of glycoside hydrolases through functional metagenomics. The major advance of our study was the discovery of active-yet-uncultivated soil microorganisms and enrichment of their glycoside hydrolases. We recovered positive cosmid clones in a higher frequency than would be expected with direct metagenomic analysis of soil DNA. This study has generated an invaluable metagenomic resource that future research will exploit for genetic and enzymatic potential.
INTRODUCTION
Soil microorganisms catalyze Earth’s biogeochemical reactions, including the degradation of organic matter and recycling of nutrients. Soils host diverse microhabitats with varied physicochemical gradients and environmental conditions. In this context, soil microorganisms live in consortia, interacting physically and biochemically with other members of the soil biota (1). Attesting to the heterogeneity, interactivity, and connectivity of the soil niche, traditional culture-based techniques grossly underestimate microbial diversity. Readily cultured microorganisms typically represent a very small proportion of soil microbial communities (2); the “uncultured majority” harbor an enormous reservoir of uncharacterized organisms, genes, and enzymatic processes (3). An outstanding methodological question remains: how best to access the biotechnological potential contained within the DNA of soil’s uncultured microorganisms?
Degradation of plant organic matter by the combined action of glycoside hydrolase (GH) enzymes is an important soil function. The GH group of enzymes is distributed across a wide variety of organisms. They catalyze the hydrolysis of glycosidic bonds in complex carbohydrates (e.g., cellulose and hemicellulose) to release simple sugars (e.g., pentoses and hexoses), and as a result, GHs include important enzymes for biotechnological applications. Because glycosidic bonds are considered among the most stable linkages that occur naturally, GHs are credited as some of the most proficient catalysts (4). Recent research suggests a broad diversity of bacteria contribute to plant polymer degradation (5–8), supporting the use of cultivation-independent methods, such as metagenomics, as most strategic for the recovery of genes and enzymes from these microorganisms.
Metagenomics captures the genomes of environmental community microbes, circumventing the need for cultivation and enabling the exploration of microbial genetic diversity and biotechnological potential (9). Metagenomic analyses have exposed new microbial pathways and reactions, yielding novel enzymes and products of economic importance. Given that metagenomic studies demonstrate that the majority of total genetic diversity space remains unexplored, “it will be far more efficient and productive to seek new enzymes from metagenome libraries than to tweak the activities of existing ones” (10). Indeed, there are several recent examples of GHs (e.g., cellulases) recovered by functional screening of metagenomic libraries from terrestrial environments (e.g., see references 11, 12, 13, and 14). These studies reflect a laborious limitation of bulk DNA metagenomic library construction: in the absence of suitable selections for phenotype, many clones (e.g., tens of thousands) must be screened prior to recovering targets of interest. In addition, recovered clones are theoretically the most abundant target genes in the microbial community of interest. Targeted metagenomic approaches, such as those involving an enrichment culture step (15), thus offer the potential to filter for sequences specific to an activity of environmental or industrial relevance.
Stable-isotope probing (SIP) is a culture-independent method for targeting microorganisms that assimilate a particular growth substrate (16–18). For the analysis of genomic DNA of active organisms, a SIP substrate (e.g., 13C labeled or 15N labeled) is incorporated into the DNA (DNA-SIP) or RNA (RNA-SIP) of active organisms, and isopycnic ultracentrifugation can differentiate labeled nucleic acids from an abundant background of unlabeled community genomes. Combining SIP with metagenomics provides access to the genomes of less-abundant community members and offers insight into complex environmental processes, such as biodegradation (as reviewed in references 19, 20, and 21).
Several studies have combined DNA-SIP and metagenomic sequencing to identify high proportions of genes from active microorganisms, such as those using glycerol (22), C1 compounds (23–26), and biphenyl (27, 28). Previous SIP studies reported that in an agricultural soil (clay loam soil, pH 6.6), cellulose was metabolized by Bacteroidetes, Chloroflexi, and Planctomycetes; cellobiose and glucose were degraded predominantly by Actinobacteria (8). The results also suggested that cellulolytic bacteria are different from saccharolytic bacteria and that oxygen availability defined the different taxonomic groups involved. Under anoxic conditions, cellulose was metabolized by Actinobacteria, Bacteroidetes, and Firmicutes; carbon from cellobiose and glucose were assimilated by Firmicutes. Others found that members of the Burkholderiales, Caulobacteriales, Rhizobiales, Sphingobacteriales, Xanthomonadales, and Group 1 Acidobacteria were associated with three different soils amended with cellulose (29). A recent survey of active bacteria in an Arctic tundra sample found Clostridium and Sporolactobacillus involved in 13C-glucose assimilation and Betaproteobacteria, Bacteroidetes, and Gammaproteobacteria involved in the assimilation of carbon derived from 13C-cellulose (30). Others have used SIP and labeled cellulose to identify Dyella, Mesorhizobium sp., Sphingomonas sp., and an uncultured deltaproteobacterium (affiliated with Myxobacteria) linked to cellulose degradation (6).
The ability to identify genes based on function, instead of sequence homology, is arguably the most powerful application of metagenomics for the recovery of novel genes (31) and a natural partner of the SIP approach for targeting active-yet-uncultured microorganisms (21). Previous studies were focused on the analysis of single substrates or individual samples. In addition, only one previous study combined SIP and functional metagenomic screens, expressing labeled DNA within a surrogate Escherichia coli host for identification of enzyme activity (22). In this study, we expand on previous efforts to combine SIP and metagenomics (as reviewed in reference 21), enriching soil microorganisms active in degrading plant-derived carbohydrates and screening GHs through activity-based functional metagenomics. We combined SIP, high-throughput sequencing of labeled 16S rRNA genes and metagenomic DNA, multiple-displacement amplification (MDA), and functional metagenomics to identify active microorganisms and associated GH enzymes. We also isolated GH-positive clones from a cosmid library in a much higher frequency than would be expected with traditional efforts using conventional metagenomics.
RESULTS AND DISCUSSION
Characterization of active soil bacteria.
We used DNA-SIP as a targeted approach for enriching active soil microorganisms involved in the metabolism of five plant-derived carbohydrates (glucose, cellobiose, xylose, arabinose, and cellulose). Three disparate soil samples were obtained from the CM2BL soil collection based on maximal physicochemical diversity (Table 1) (http://www.cm2bl.org/). In particular, soil pH was low for the Arctic tundra and temperate rainforest soil samples, suggesting that the microbial composition and diversity of these two samples would be fundamentally different from those in agricultural soil (32, 33). The water-filled pore space (WFPS) was maintained between 50% and 60% to avoid decreased aerobic microbial activity at WFPS values of >60% (34, 35).
TABLE 1 .
Location and physicochemical characteristics of the soil samples selected for DNA stable-isotope probing incubationsa
| Sample | Location | Latitude and longitude |
Bulk density (g/cm3) |
Amt of carbon (% dry wt) |
pH | Moisture (% dry wt) |
Amt of nitrogen (% dry wt) |
Soil type | ||
|---|---|---|---|---|---|---|---|---|---|---|
| Total | Inorganic | Organic | ||||||||
| Arctic tundra (1AT) | Daring Lake, North-West Territories, Canada |
64°52′N, 111°35′W |
0.2 | 46.9 | BDLb | 46.9 | 3.9 | 417.7 | 1.42 | Organic |
| Temperate rainforest (7TR) |
Pacific coastal rainforest, Vancouver Island, Canada |
48°36′N, 124°13′W |
0.6 | 10.8 | BDL | 10.8 | 4.9 | 69.8 | 0.35 | Coarse sandy loam |
| Agricultural soil-wheat (11AW) |
Elora Research Station, Ontario, Canada |
43°38′N, 80°24′W |
1.1 | 1.85 | 0.12 | 1.7 | 7.4 | 17.9 | 0.19 | Silt loam |
For more details, see http://www.cm2bl.org/.
BDL, below detection limit.
Because 13C-labeled cellulose was commercially unavailable at the time of this research, both native cellulose and 13C-labeled cellulose were produced as the substrates for SIP incubations by Gluconacetobacter xylinus, generating predominantly amorphous cellulose (36), which is more readily degraded than crystalline cellulose (37). To ensure detectable labeling, similar to a previous experimental approach (8), glucose, cellobiose, arabinose, and xylose were added weekly (1.5 mmol of C) for 3 weeks, reaching levels approximately 5 to 500 times higher than those normally detected in soils (38, 39). Although substrate concentrations were higher than typical bulk soil concentrations, higher polysaccharide substrate concentrations would be expected in the root rhizosphere and in areas of active plant matter decomposition (as reviewed in reference 39), suggesting that our incubation conditions would not be unrealistic for some naturally occurring soils. These concentrations were chosen to ensure that labeled isotope was more abundant than endogenous soil carbon sources for the success of DNA-SIP, enabling the separation and purification of labeled DNA for subsequent molecular analyses (16, 40). Similar substrate concentrations and incubation times with glucose and cellulose were used previously (30), demonstrating minimal-yet-detectable labeling of DNA in an Arctic tundra soil sample.
Metabolism of labeled substrates in DNA-SIP incubations was confirmed by higher headspace CO2 production in all substrate-amended serum vials compared to uninoculated controls for each of the three soils (Fig. 1). In all cases, cellulose-amended vials demonstrated reduced CO2 production compared to the other substrates, further justifying an extended incubation time for this comparably recalcitrant substrate. The average amount of CO2 released after 6 days was 13% of the headspace, which, after subtraction of the average CO2 produced in uninoculated vials, was approximately equivalent to 1.4 mmol of carbon. This represents 93% of the total weekly carbon added (~1.5 mmol of carbon).
FIG 1 .
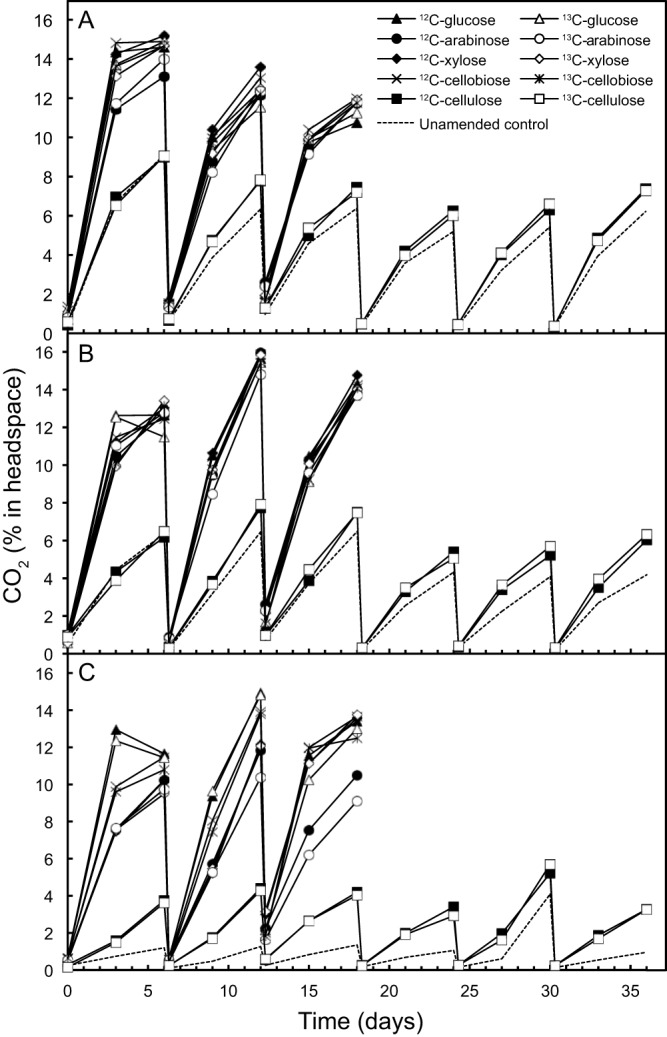
Carbon dioxide production for Arctic tundra (1AT) (A), temperate rainforest (7TR) (B), and agricultural (11AW) (C) soils. Soil samples were amended with labeled (13C) or unlabeled (12C) substrates, and serum bottles were aerated weekly to replenish oxygen and deplete carbon dioxide. The “control” represents a soil sample incubated without substrate.
In addition to monitoring CO2 production in all vials, separate soil incubations were prepared with a defined helium-oxygen headspace and glucose amendment in order to monitor O2 consumption. As expected, the addition of glucose stimulated O2 consumption, but the headspace remained oxic for each of the weekly incubation periods over the first 3 weeks (see Fig. S1 in the supplemental material), indicating that weekly aeration of experimental vials was sufficient to deplete CO2 and replenish O2. Maintaining oxic conditions was important to ensure that the DNA-SIP incubation recovered DNA from microorganisms involved in aerobic degradation of complex carbohydrates in addition to capturing DNA from microorganisms involved in anaerobic metabolism (41). Indeed, recent oxic incubations demonstrated activity of anaerobic clostridia (8, 30, 42), presumably because anoxic microenvironments exist even within oxic experimental microcosms.
Confirmation of isotope labeling.
At the two time points of all incubations (1 and 3 weeks for all substrates, except for cellulose, which was sampled at 3 and 6 weeks), DNA was retrieved for the analysis of bacterial community composition by agarose gel electrophoresis and denaturing gradient gel electrophoresis (DGGE) (43). All DNA extracts from microcosm soils were subjected to density gradient ultracentrifugation and recovered in 12 fractions, which were analyzed in agarose gels. The results demonstrated that all soils possessed more DNA in 13C-incubated heavy fractions (i.e., 1 to 7) than in 12C-control fractions (i.e., 8 to 12) from glucose, cellobiose, arabinose, and xylose SIP incubations (see Fig. S2 to S6 in the supplemental material). For cellulose, only temperate rainforest and agricultural soil incubations resulted in heavier DNA visible in agarose gels corresponding to 13C-labeled sample heavy DNA fractions (see Fig. S6) for the 6-week time point. Similar results were observed for all earlier time points but with less DNA associated with heavy fractions for 13C-incubated samples compared to the later time points (data not shown). Although extended incubation times were important, one caveat of extended incubation times for SIP incubations (e.g., for cellulose) is that labeled carbon might have been distributed more broadly within the microbial community, which may result in less-specific enrichment of substrate-degrading microbial genomes in the resulting data and libraries.
The presence of distinct fingerprint profiles in heavy fractions for 13C-incubated samples, but not for the corresponding 12C-control fractions, demonstrates isotopic enrichment of nucleic acids (16). Bacterial DGGE fingerprints corresponding to all late-time-point fractions demonstrated unique patterns associated with the heavy fractions (e.g., fractions 1 to 7) for all 13C-incubated SIP microcosms (see Fig. S2 to S6 in the supplemental material). Although some cross-gradient fingerprint variations were associated with 12C-control DNA, these differences were likely GC content shifts because they were pronounced only in the lightest fractions (e.g., fractions 10 to 12) and were distinct from shifts associated with fractionated 13C-DNA. Substrate- and soil-specific heavy fraction patterns were consistent for early- and late-time-point samples (data not shown), which indicated that detected active bacteria were stable over time rather than changing due to food web dynamics (40).
Heavy DNA fingerprints were used to identify fractions containing 13C-labeled DNA for subsequent 16S rRNA gene sequencing, bulk DNA sequencing, and functional metagenomics. Based on DGGE patterns, we identified fraction 5 and/or 6 as being representative of heavy DNA and fraction 10 as representing light DNA for all soils, substrates, and incubation times (see Fig. S2 to S6 in the supplemental material). Although fractions 1 to 5 also may have captured DNA from labeled microorganisms, these fractions were not analyzed further because the vanishingly small proportions of DNA recovered from these gradient fractions would have made PCR and subsequent metagenomic library preparation problematic.
Taxonomic characterization of heavy DNA.
We selected representative gradient fractions from all soils, substrates, and incubation times for profiling of the bacterial V3 region of 16S rRNA genes. Based on DGGE data, we selected fractions 6 (heavy) and 10 (light) for Arctic tundra and fractions 5 (heavy) and 10 (light) for temperate rainforest and the agricultural soil. In addition, we sequenced V3 regions of 16S rRNA genes from DNA extracted from the initial soil samples used to establish SIP incubations to determine whether light fractions resembled the original soil community as would be expected. Following paired-end-read assembly, we analyzed 630,000 assembled sequences (10,000 sequences per sample) using an AXIOME management of the QIIME pipeline and additional custom analyses (e.g., multiresponse permutation procedure [MRPP] and indicator species analysis). Good’s coverage (44) for the heavy fraction samples ranged from 84 to 92%, and light fraction samples ranged from 68 to 85%, which indicates that this level of sequencing captured the majority of bacterial taxa in these samples. β diversity was assessed by weighted UniFrac distances visualized within principal coordinate analysis (PCoA) plots. The results indicated that all samples from within each of the three soil treatments were grouped distinctly according to soil type (Fig. 2A), which was highly significant based on MRPP analysis (A = 0.18 [chance-corrected within-group agreement], T = −20.4 [test statistic], P < 0.001). Both the Arctic tundra and temperate rainforest soil profiles grouped more closely with one another, which is likely a result of both soils sharing low pH (Table 1), a major determinant of soil bacterial diversity and taxonomic composition (45, 46). In addition, all heavy and light fraction profiles for the three soils were clustered distinctly (Fig. 2A), which was also highly significant (A = 0.40, T = −28.3, P < 0.001). Native soil phylogenetic profiles clustered with their respective light fractions, indicating that the “background” bacterial community remained relatively constant throughout the SIP incubation. Although the two time points for some 13C-labeled substrates grouped together (Fig. 2B), the differences between heavy and light fractions were much greater than those observed between the five substrates used in this study.
FIG 2 .
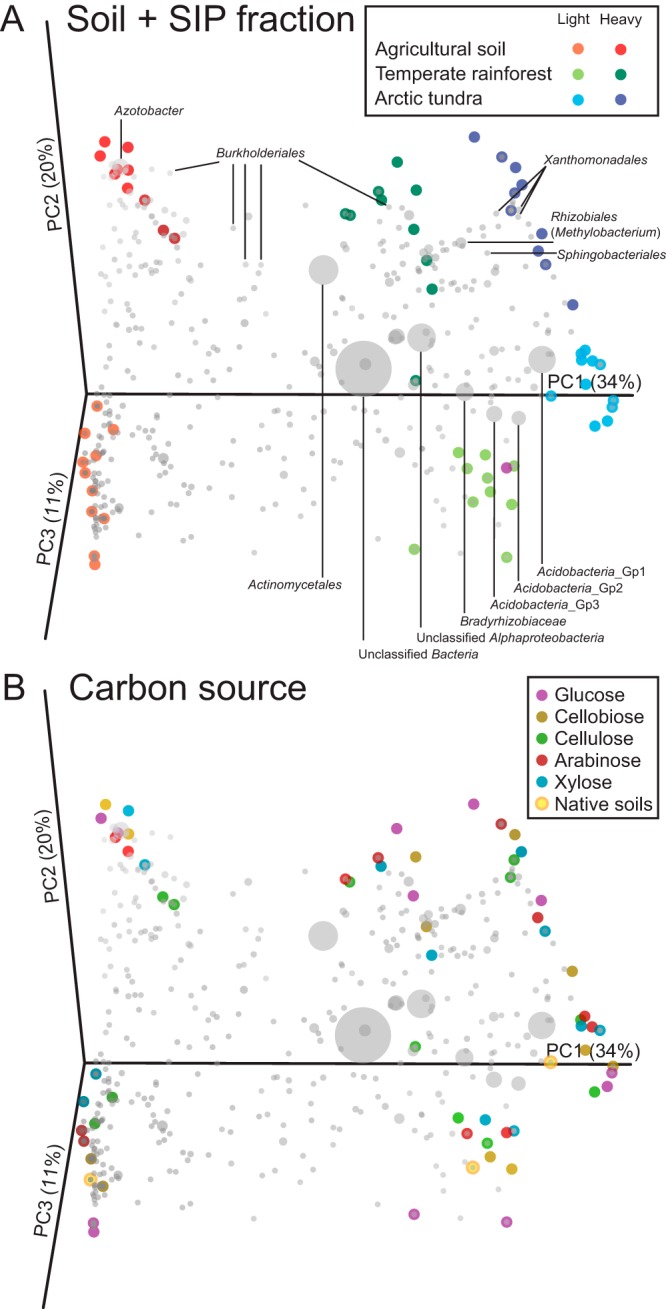
Principal coordinate analysis (PCoA) biplots of weighted UniFrac distances for 16S rRNA gene sequences generated by assembled paired-end Illumina reads. Samples separated by soil type and fraction (A) as well as by carbon source (B). Native soils were associated with their respective light fractions. Gray spheres represent taxonomic affiliations of OTUs that correlated most strongly within the ordination space.
Many operational taxonomic units (OTUs) were affiliated with SIP-derived heavy DNA, but multiple permutations of the analysis were required to summarize indicator OTUs for different sample subsets. We used indicator species analysis (47), with an indicator value (IV) threshold of 70% and a >250 minimum sequence sum threshold to identify the strongest significant OTUs (P < 0.01) associated with (i) all heavy DNA samples (versus all light DNA samples) (Fig. 3; see Table S1 in the supplemental material), (ii) all heavy DNA samples within each soil type (versus all light DNA samples for the same soil type) (see Table S2 in the supplemental material), (iii) each substrate across all heavy DNA samples from all soil types (versus the heavy DNA for the other substrates from all soil types) (see Table S3 in the supplemental material), and (iii) each substrate from heavy DNA within each soil type (versus the other substrates for the same soil type heavy DNA) (see Tables S4 to S6 in the supplemental material).
FIG 3 .

Cleveland plot of operational taxonomic unit (OTU) abundance for OTUs possessing the highest indicator values (i.e., >70%) for an association with DNA-SIP heavy DNA (black squares [average abundance]) for all substrates and soils combined, in comparison to light DNA (gray squares [average abundance]). Taxonomic affiliations are included for phyla, with additional classifications for order (o_), family (f_), and genus (g_). For additional details, see Table S1 in the supplemental material.
When we compared OTUs associated with all heavy DNA samples versus all light DNA samples from all soils, indicator species analysis revealed multiple poorly classified indicators, in addition to genus-classified OTUs associated with the Salinibacterium (Actinobacteria), Devosia (Alphaproteobacteria), Telmatospirillum (Alphaproteobacteria), Phenylobacterium (Alphaproteobacteria), and Asticcacaulis (Alphaproteobacteria) genera (Fig. 3; see Table S1 in the supplemental material). The indicator species analysis from all heavy DNA samples versus all light DNA samples within each soil type showed that the predominant genus-classified OTUs identified in heavy fractions from tundra soil (1AT) were Salinibacterium (Actinobacteria), Rhodanobacter (Gammaproteobacteria), Conexibacter (Actinobacteria), Telmatospirillum (Alphaproteobacteria), Asticcacaulis (Alphaproteobacteria), and Burkholderia (Betaproteobacteria), in addition to OTUs within orders such as Sphingomonadales and Acidobacteriales (see Table S2 in the supplemental material). The temperate rainforest soil (7TR) heavy DNA was dominated by OTUs classified to the genera Paucibacter (Betaproteobacteria), Burkholderia (Betaproteobacteria), Spirochaeta (Spirochaetes), Salinibacterium (Actinobacteria), Telmatospirillum (Alphaproteobacteria), Labrys (Alphaproteobacteria), Mesorhizobium (Alphaproteobacteria), and Phenylobacterium (Alphaproteobacteria), in addition to uncharacterized genera from other phyla, such as Verrucomicrobia (see Table S2). The agricultural soil wheat (11AW) heavy DNA OTUs were represented by the genera Pseudomonas (Gammaproteobacteria), Devosia (Alphaproteobacteria), Pseudoxanthomonas (Gammaproteobacteria), Salinibacterium (Actinobacteria), Ramlibacter (Betaproteobacteria), Ochrobactrum (Alphaproteobacteria), Paenibacillus (Firmicutes), and Aeromicrobium (Actinobacteria) and further unclassified members of the orders Pseudomonadales, Rhizobiales, Xanthomonadales, Actinomycetales, Burkholderiales, and Bacillales (see Table S2), among others.
Orders associated with the metabolism of cellulose were dominated by Actinomycetales and Caulobacterales (genus Phenylobacterium) (see Table S3 in the supplemental material). Members of the Alphaproteobacteria were associated with the metabolism of arabinose, and members of the order Rhizobiales were strongly associated with the metabolism of xylose. There were no specific indicator species associated with glucose or cellobiose across all soils (see Table S3), which might also suggest that abundant soil OTUs were also active in assimilating these substrates.
The predominant indicator species for the agricultural soil fed with [13C]glucose were associated with Paenibacillus (Bacillales) (see Table S4 in the supplemental material). The use of cellulose was associated with Mesorhizobium (Alphaproteobacteria), Devosia (Alphaproteobacteria), and Cellvibrio (Gammaproteobacteria), in addition to other poorly classified OTUs from the Sphingomonadales and Actinomycetales. The use of cellulose in temperate rainforest soil was associated with the Myxococcales (Deltaproteobacteria) (see Table S5 in the supplemental material). An OTU affiliated with Caulobacterales was associated with the metabolism of glucose in Arctic tundra. Nevskia (Gammaproteobacteria), and two OTUs affiliated with the Acidobacteria were associated with tundra cellulose assimilation (see Table S6 in the supplemental material). No other OTUs were significant indicators for the remaining substrates (i.e., cellobiose, arabinose, and xylose) for the three soils, which might indicate that active taxa were also abundant soil bacteria.
Although our DNA-SIP incubation revealed many poorly classified indicator taxa (see Tables S1 to S6 in the supplemental material), many of the indicator species associated with heavy DNA were expected based on previous studies. For example, Salinibacterium was associated with frozen soils from glaciers (48) and Antarctic permafrost (49). This genus has been associated with the metabolism of a variety of carbon sources, including sucrose, glucose, cellobiose, mannose, melibiose, maltose, galactose, arabinose, and fructose (48, 50). In addition, Devosia species were isolated from greenhouse soil and beach sediments, testing positive for the hydrolysis of esculin, β-galactosidase, β-glucosidase, and N-acetyl-β-glucosaminidase, although unable to degrade carboxymethyl cellulose (CMC) (51, 52). Phenylobacterium and Burkholderia are abundant in forest soils (53) and the genus Asticcaulis was identified among aerobic chemoorganoheterotrophs in tundra wetlands, able to use glucose, sucrose, xylose, maltose, galactose, arabinose, lactose, fructose, rhamnose, and trehalose, among other carbon sources (54). The genus Spirochaeta has some species that are free-living saccharolytic and obligate or facultative anaerobes and were isolated from diverse environments, mainly from extreme aquatic environments (55, 56). Spirochaeta americana was reported to be a consumer of d-glucose, fructose, maltose, sucrose, starch, and d-mannitol (56), and Spirochaeta thermophila was reported to be a cellulolytic organism; the study of its genome revealed a high proportion of genes encoding more than 30 GHs (55).
MG-RAST analysis and functional annotation.
We used next-generation sequence analysis of bulk 13C-labeled DNA to survey the prevalence of annotated GHs within three pooled samples that were targeted for subsequent functional metagenomic screens. Guided by the UniFrac-based PCoA plot (Fig. 2), we pooled heavy DNA samples representing all substrates (except cellulose) associated with low pH (i.e., temperate rainforest, Arctic tundra), heavy DNA for all substrates (except cellulose) from the agricultural soil, and the cellulose-enriched DNA from the three soils. Analysis of paired-end reads was performed by MG-RAST using annotations derived from the Swiss-Prot/Uniprot database. Only 19.4% (low-pH library), 19.6% (cellulose library), and 22.0% (agricultural library) of sequences were annotated by Swiss-Prot in MG-RAST using an E value threshold of 0.01, which is an important consideration for any subsequent analysis of annotation data based on this minority of sequences. Nonetheless, using a custom Perl script to convert Swiss-Prot annotations to CAZy GH identifiers, we detected 81 distinct GH families for the pooled-cellulose library and 80 GH families for each of the low-pH and agricultural soil composite libraries. The distribution of annotated GHs varied between samples, and the most abundant families in the three pooled samples were GH1, -2, -3, -5, -9, -13, -23, -28, and -35 (see Table S7 in the supplemental material). In addition, the three next-generation sequence data sets were very similar in their distributions (i.e., r > 0.99) for the three libraries (Fig. 4), and all had representation among GH families commonly associated with known cellulases (GH1, -3, -5 to -9, -12, -45, -48, and -61), hemicellulases (GH8, -10 to -12, -26, -28, -53, and -74), and debranching enzymes (GH51, -54, -62, -67, -78, and -74) as reviewed elsewhere (57, 58). The GH families involved in the hydrolysis of cellulose that were most abundant in our data were GH families 3, 5, and 9 (Fig. 4; see Table S7). However, given that most GH family annotations were not represented by known CAZy identifiers and that only ~20% of our paired-end reads were annotated by Swiss-Prot, the abundance and distribution of functional GH families in our pooled DNA is underrepresented. As a result, we used functional screens of large-insert metagenomic libraries for the recovery of GHs to help circumvent the limitations of sequence-based analysis of our heavy DNA samples.
FIG 4 .
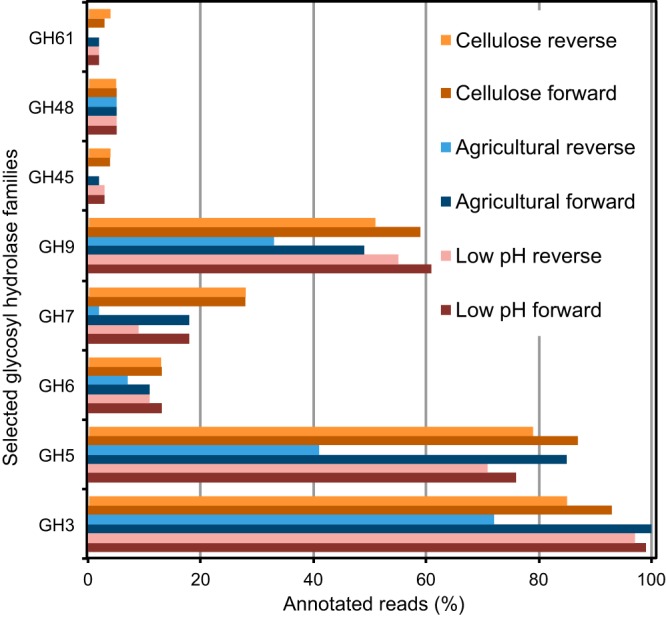
Glycoside hydrolase (GH) families associated with pooled heavy DNA. Functional annotation of the metagenomic data revealed diverse GH gene representation within the pooled heavy DNA.
Enriched metagenomic library.
Pooled high-molecular-weight DNA from the 13C-cellulose-enriched SIP incubations for the three soils were captured in cosmid libraries and screened for GHs involved in the degradation of cellulose and other plant-derived polymers based on activity in E. coli. Multiple-displacement amplification (MDA) increased the amount of nucleic acids obtained from pooled cellulose DNA-SIP incubations prior to the isolation of 25- to 75-kb DNA fragments via pulsed-field gel electrophoresis (PFGE). The cellulose-SIP metagenomic library contained ~83,000 clones with an average insert size of 31 kb based on restriction digestion of a subset of 40 random clones (data not shown). These results compare favorably to a library of ~10,500 clones generated from MDA-amplified SIP-enriched seawater DNA, which had an average insert size of 27 kb, ranging from 17 to 40 kb (26).
We used a combined parallel approach for functional screening of 2,876 randomly selected clones from the cellulose-enriched metagenomic library. Growth of colonies on LB supplemented with carboxymethyl cellulose (CMC), followed by poststaining with Congo red (59), facilitated identification of clones expressing either endoglucanase (EC 3.2.1.X) or glucosidase (EC 3.2.1.X) activities (60). From the 2,876 clones screened, we identified eight positive clones, two of which (C2380 and C2044) were capable of hydrolyzing CMC (Table 2). Restriction mapping showed that these two clones were distinct (Fig. 5). Clones C122 and C2194 carried dissimilar DNAs encoding β-N-acetyl-galactosaminidases (EC 3.2.1.53). β-Glucosidase activities (EC 3.2.1.21) were detected in clones C424, C762, and C1088. Clones C424 and C1088 contained overlapping DNA—probably from the same organism—consistent with the substrate activity profiles. Restriction pattern of clone C1024 was similar to C1088 and C424 (Fig. 5), but C1024 had both α-l-arabinofuranosidase (EC 3.2.1.55) and β-glucosidase (EC 3.2.1.21). The open reading frame (ORF) encoding the β-glucosidase was likely located in the overlapping region.
TABLE 2 .
Substrate-specific activities of positive metagenomic clones from the [13C]cellulose DNA-SIP library
| Clone | Insert size (kb) |
Activity (μM MU released)a |
CMC activityb |
||||
|---|---|---|---|---|---|---|---|
| α-l-Arabinofuranoside pyranoside |
β-d- Cellobiopyranoside |
β-d- Glucopyranoside |
β-d- Xylopyranoside |
N-Acetyl-β-d- galactosaminide |
|||
| C122 | 21.6 | 0.4 | 0.2 | 0.6 | 0.7 | 124.2 | − |
| C424 | 8.2 | 0.9 | 57.6 | 109.4 | 1.6 | 0.7 | − |
| C762 | 13.5 | 2.4 | 5.4 | 21.2 | 0.7 | 0.4 | − |
| C1024 | 16.8 | 123.8 | 6.5 | 35.8 | 1.7 | 0.5 | − |
| C1088 | 11.9 | 0.5 | 25.6 | 79.2 | 1.2 | 0.6 | − |
| C2194 | 12.9 | 0.5 | 0.3 | 0.6 | 0.4 | 39.6 | − |
| C2380 | 14.9 | 0.38 | 0.46 | 0.53 | 0.41 | 0.40 | +++ |
| C2044 | 14.7 | 0.40 | 0.40 | 0.52 | 0.39 | 0.36 | ++ |
Cellulase activity was scored by Congo red staining of clones on the LB-CMC plate. Other activities were measured in cell-free extracts using methylumbelliferone-based substrates. MU, methylumbelliferone units based on equal volumes of sample for each assay.
CMC, carboxymethyl cellulose. Plate-based clearing (high, +++; medium, ++; negative, −) was detected by Congo red stain and activity based on comparison to those of positive and negative controls.
FIG 5 .
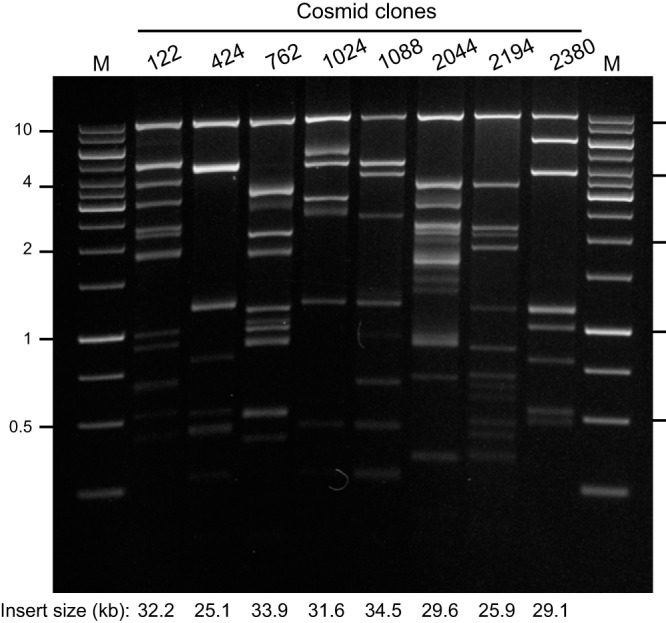
Restriction of cosmid DNA with EcoRI-HindIII-BamHI. DNA sizes in kb are marked on the left and right. M, molecular size markers. The sizes of digested DNA fragments except for the cosmid backbone (the very top band) were added up to obtain the insert sizes of the cloned metagenomic DNA.
End sequencing of the positive isolates demonstrated that most clones had at least one end sequence matching the known cellulolytic member of the Gammaproteobacteria, Cellvibrio sp. (61), with 69 to 95% identity (Table 3). Other top BLAST matches included Saccharophagus degradans, Dyadobacter fermentans, Alicyclobacillus acidocaldarius, and Chthoniobacter flavus (Table 3), with 29 to 97% identity. Although these bacteria are not well characterized to date, other researchers have reported that they use cellulose and other carbohydrates as a carbon source and/or they contain GHs encoded in their genome (62–65). As predicted, the end sequence identities for C424 and C1088 were very similar taxonomically (i.e., Cellvibrio sp.). On the other hand, end sequence data for C122 and C2194 did not suggest a similar genomic origin (Table 3), consistent with the restriction pattern of these cosmids (Fig. 5).
TABLE 3 .
Analysis of cosmid insert end sequences
| Clone | BLASTx result fora: |
|||
|---|---|---|---|---|
| Forward read |
Reverse read |
|||
| Description | E value (% identity [no. positive/total]) |
Description | E value (% identity [no. positive/total]) |
|
| C122 |
Porphyromonas gingivalis (4-amino-4-deoxy-l-arabinose transferase) |
4e–5 (29 [40/139]) |
Cellvibrio japonicus Ueda107 (β-xylosidase) |
8e–136 (82 [131/162]) |
| C424 |
Cellvibrio sp. strain BR (DNA-directed DNA polymerase) |
1e–28 (69 [66/80]) |
Cellvibrio sp. strain BR (Glucuronate isomerase) |
2e–103 (91 [157/163]) |
| C762 |
Chthoniobacter flavus (putative PAS/PAC sensor protein) |
1e–86 (78 [151/171]) |
Sorangium cellulosum (hypothetical protein) |
2e–28 (54 [83/125]) |
| C1024 |
Cellvibrio sp. strain BR (glucuronate isomerase) |
2e–17 (95 [34/40]) |
Cellvibrio sp. strain BR (gluconolaconase) |
2e–46 (80 [85/96]) |
| C1088 |
Saccharophagus degradans (SSS sodium solute transporter superfamily) |
6e–61 (68 [123/150]) |
Cellvibrio sp. strain BR (auxin efflux carrier) |
5e–44 (75 [101/114]) |
| C2194 |
Dyadobacter fermentans (ROK family protein) |
1e–91 (95% [140/142]) | Failed sequencing reaction |
|
| C2380 |
Alicyclobacillus acidocaldarius (Glyoxalase/bleomycin resistance protein/dioxygenase) |
2e–15 (52 [51/69]) |
Cellvibrio sp. strain BR (glucosamine fructose-6-phosphate aminotransferase, isomerizing) |
3e–105 (96 [162/163]) |
| C2044 |
Cellvibrio sp. strain BR (DNA polymerase III subunit delta) |
1e–71 (96 [116/118]) |
Dyadobacter fermentans (hypothetical protein) |
9e–129 (97 [181/184]) |
Cosmids were end sequenced with M13 forward and reverse primers flanking the site of metagenomic DNA insertion. For each clone, two end sequences were obtained and are referred to as “reverse” and “forward” reads. Top matches for BLASTx analyses are shown. Positive results are the number of amino acids from the query that match the amino acids from the subject sequence. The total number of amino acids from the subject is shown.
Posterior analysis of reverse and forward end sequences of the positive clones was done by comparing end sequences to Illumina forward and reverse reads from whole-genome sequencing of the three SIP libraries (see Table S8 in the supplemental material). The results showed that the majority of end sequences were represented in the cellulose library, as expected, and only a few sequence matches were found in other libraries using the selected threshold.
The high frequency of positive clones after screening of DNA-SIP-derived clones compares favorably to those from previous soil functional metagenomic studies reporting the recovery of single positive cellulose hits from screening of tens of thousands of clones. For example, a single cellulose-encoding clone and two xylanase-encoding clones were recovered from functional screening of 13,800 clones from three fosmid metagenomic libraries derived from grassland in Germany, with an insert size range of between 19 and 30 kb (11). Also, one cellulase-encoding clone was retrieved from the functional screening of 3,024 clones from a bacterial artificial chromosome metagenomic library derived from red soil in China, with insert sizes ranging from 25 to 165 kb (12). In another study, one cellulase-encoding clone was recovered from functional screening of 14,000 clones with an average insert size of 5 kb from a metagenomic phagemid library from a forest soil in China (13). Finally, a CMC-positive clone was retrieved from a metagenomic fosmid library derived from wetland soil in South Korea, after screening of 70,000 clones with an average insert of 40 kb (14). Although not conducted here, a well-replicated direct comparison of GH gene recovery from metagenomic libraries prepared from SIP-derived heavy DNA, light DNA, and the original soil DNA would be necessary to confirm the effectiveness of DNA-SIP. In addition, the ability to recover GH genes in high proportions using cultivation-based enrichment approaches is a well-established alternative to direct metagenomics (15). DNA-SIP incubations are designed to be less dependent on rapid growth of a readily cultivated subset of the microbial community (40). Indeed, our labeled DNA contained many OTUs that were classified poorly within described bacterial taxonomies (see Tables S1 to S6 in the supplemental material). Direct DNA-SIP and enrichment culture comparisons would be valuable but have not yet been conducted to our knowledge.
In summary, the combination of DNA-SIP and metagenomics helped recover soil GHs in higher proportions than all previously reported efforts via direct metagenomics, which demonstrates the power of using DNA-SIP as an activity-based prefilter for targeted metagenomic approaches. Our study demonstrated the capability of scaling DNA-SIP analysis for the interrogation of multiple environmental samples with multiple substrates, with sampling at multiple time points. A high-quality cosmid library with >31-kb inserts was constructed from heavy DNA originating from a 13C-cellulose-incubated sample, and highly efficient screening of GHs from a small set of clones (0.3% positive hits) showed strong potential of the techniques combined in this study for functional metagenomics. Identification of the genes encoding GHs and characterization of these enzymes are ongoing and further functional screening of the 13C-cellulose DNA-SIP library clones in other surrogate hosts will be assessed to identify additional GH representation.
MATERIALS AND METHODS
Soil samples.
Three soil samples from the Canadian MetaMicroBiome Library (http://www.cm2bl.org/) were used: Arctic tundra 1 (1AT), temperate rainforest (7TR), and agricultural soil-wheat (11AW). Triplicate surface soils from the top 10 cm below the litter layer were combined to prepare a single composite for each site. Composite soil samples were sieved (2 mm), and subsamples were sent to the Agriculture and Food Laboratory (University of Guelph, Guelph, Ontario, Canada) for analysis of physicochemical properties (Table 1).
SIP.
d-Glucose was obtained from Bio Basic (Markham, Ontario, Canada). (U-13C6)-d-glucose (99%) was supplied by Cambridge Isotope Laboratories (Cambridge, Ontario, Canada). d-(+)-cellobiose, d-(–)-arabinose, and d-(+)-xylose were purchased from Sigma-Aldrich. d-(UL-13C5)-arabinose, d-(UL-13C5)-xylose, and (UL-13C12)-cellobiose were obtained from Omicron Biochemicals (South Bend, IN).
To minimize carbon available for competition with labeled substrates, composite soil samples were preincubated for 2 weeks in darkness at 15°C for 1AT and at 24°C for 7TR and 11AW. Ten grams of soil samples was added to 120-ml serum vials, which were sealed with butyl septa. Incubations were conducted with stable-isotope (13C) and native (12C) substrates, as well as no-substrate controls, for each of the three soils. Finely shredded cellulose was prepared from Gluconacetobacter xylinus grown with 13C- or 12C-glucose (30) as the sole carbon source. Purified bacterial cellulose (200 mg, 6.6 mmol C) was mixed into serum vials in a single dose. Labeled (13C) and unlabeled (12C) substrates were added to soil samples in multiple dosages over periods of 1 week and 3 weeks for glucose, cellobiose, xylose, and arabinose incubations or 3 weeks and 6 weeks for the cellulose incubations. Serum vials were aerated once per week for 1 h in a fume hood. The weight of incubation vials was assessed weekly, and water-filled pore space (WFPS) was maintained between 50 and 60% by adding distilled water and/or substrate for each incubation according to the following formula (34): WFPS = w [ρb ρs/ρs − ρb], where w is the gravimetric water content (%), ρb is the soil bulk density (g/cm3), and ρs is the soil particle density (2.65 g/cm3).
GC.
CO2 accumulation in the headspaces of serum vials was determined using a GC-2014 gas chromatograph (Shimadzu) equipped with a thermal conductivity detector (TCD), methanizer, and a flame ionization detector (FID). The gas chromatography (GC) temperatures were maintained for the oven (80°C), TCD (280°C), methanizer (380°C), and FID (250°C). No-carbon control incubations and separate serum vials amended with 12C-glucose were used as surrogates for experimental vials because an N2-free headspace was required for measurement of O2 with the gas chromatograph. The headspaces of these separate vials were flushed with helium and supplemented with oxygen (20%) at the start of the experiment. Headspace CO2 and O2 were measured every 3 days by direct injection of 0.5 ml of headspace gas through a packed Poropak Q column with a helium flow of 20 ml/min.
DNA extraction and isopycnic centrifugation.
Two grams of soil was sampled from each vial at the time points described above. DNA was extracted with a PowerSoil DNA Isolation kit (MO BIO Laboratories, Carlsbad, CA) according to the manufacturer’s instructions. Extracted DNA was quantified using a NanoDrop 2000 UV-Vis spectrophotometer (Thermo Scientific; Montreal, Quebec, Canada) and a 1% agarose gel with a 1-kb DNA ladder (Invitrogen) for comparison. Cesium chloride (CsCl) gradients were processed by ultracentrifugation, and 12 fractions were collected for each sample as described previously (16, 66).
DGGE.
The V3 regions of bacterial 16S rRNA genes were PCR amplified using primers 341f-GC and 518r (67). Each reaction mixture contained 19.75 µl of UV-treated water, 2.5 µl of 10× ThermoPol reaction buffer (New England BioLabs), 0.05 µl of deoxynucleoside triphosphates (dNTPs) (100 mM), 0.05 µl of forward primer 341f-GC (100 µM), 0.05 µl of reverse primer 518r (100 µM), 1.5 µl of bovine serum albumin (BSA) (10 mg/ml), 0.25 µl of Taq DNA polymerase (5 U/µl) (New England BioLabs), and 1 µl of DNA template purified from each gradient fraction. The PCR conditions were initial denaturation at 95°C for 5 min, followed by 30 cycles of denaturation at 95°C for 1 min, annealing at 55°C for 1 min, and extension at 72°C for 1 min, followed by a final extension at 72°C for 7 min. All PCR products were analyzed on 1% agarose gels prior to DGGE.
Five microliters of each PCR product was loaded onto a 10% polyacrylamide gel with a denaturing gradient of 30 to 70%. Gels were run at 60° C for 14 h at 85 V (DGGEK-2001-110; C.B.S. Scientific, San Diego, CA) as described previously (43). A custom DGGE ladder was loaded into the two outside wells of the gel for subsequent normalization. Gels were stained for 45 min with SYBR green I nucleic acid gel stain (Thermo Fisher) and rinsed once in water prior to imaging. Gel images were taken with a Pharos Plus molecular imager system (Bio-Rad).
Next-generation sequencing.
High-throughput sequencing of the 16S rRNA gene (V3 region) and paired-end-read assembly were conducted as described previously (68, 69). Based on DGGE data, we sequenced gradient fractions 6 (heavy) and 10 (light) for 1AT and fractions 5 (heavy) and 10 (light) for 7TR and 11AW (60 samples in total). Three 25-µl PCR amplifications per sample were conducted, each containing 5 µl of the 5× Phusion HF buffer (Finnzyme, Finland), 0.125 µl of the V3F-modified primer (100 µM), 1.25 µl of an indexed reverse primer (10 µM) (V3-1R to V3-60R), 0.2 µl of dNTPs (100 mM), 0.25 µl of the Phusion high-fidelity DNA polymerase (2 U/µl) (Finnzyme), and 1 µl of DNA template (1 to 10 ng). The PCR conditions were as follows: initial denaturation at 98°C for 2 min, followed by 20 cycles of denaturation at 98°C for 10 s, annealing at 50°C for 30 s, and extension at 72°C for 15 s. A final extension was performed at 72°C for 7 min. The triplicate 330-bp PCR products were pooled and analyzed on a 2% agarose gel. Individually indexed composites were combined in equal nanogram amounts and then resolved on a 2% agarose gel. The amplicon fragment was excised and purified using Wizard SV gel and PCR cleanup system (Promega, Madison, WI). Libraries were subjected to 108-bp end sequencing on the Genome Analyzer IIx (Illumina, Inc., San Diego, CA) at the Plant Biotechnology Institute (Saskatoon, Saskatchewan, Canada).
Shotgun metagenomic sequencing was performed on DNA from three pooled fractions of the 13C-labeled DNA from each treatment. Pooling of heavy DNA resulted in three composite samples for sequencing: (i) “low pH” (fractions 5, 6, and 7 of 1AT and fractions 4, 5, and 6 of 7TR) for week 3 incubations with glucose, cellobiose, arabinose, and xylose; (ii) “agricultural” (fractions 4, 5, and 6 for 11AW) for week 3 incubations with glucose, cellobiose, arabinose, and xylose; and (iii) “cellulose” (fractions 5, 6, and 7 for 1AT and fractions 4, 5, and 6 for 7TR and 11AW) for week 6 incubations with cellulose. Shotgun sequencing samples of metagenomic DNA were prepared using the Nextera DNA sample preparation kit (Illumina). Pooled heavy DNA (25 to 50 ng) was fragmented using the tagmentation reaction (~200 to 5,000 bp), according to the manufacturer’s instructions and purified using the DNA Clean & Concentrator kit (Zymo Research Corporation, Irvine, CA). Purified fragments were used as the template for a five-cycle PCR amplification; indexed sequencing adapters (Epicenter, Madison, WI) were used for the PCR. Each amplified sample was purified and subjected to size selection (400 to 800 bp) using a Pippin Prep device (Sage Science, Beverly, MA). Afterward, each library was quantified using the KAPA library quantification kit (KAPA Biosystems Woburn, MA). Equimolar samples were pooled, concentrated, and quantified. Final concentrations were adjusted to 10 nM. Libraries were sequenced using the HiSeq2000 sequencing system (Illumina) by the Institute for Genomic Biology Core Facility (University of Illinois). Sequencing was performed using a TruSeq SBS kit (version 3), and data were analyzed using the Cassava 1.8 pipeline. Error rates were estimated at below 0.3%. Each sample yielded 42 to 90 million 100-bp end reads of 62 to 63% average GC content.
Statistical analysis.
Taxonomic classification with RDP v2.2 (confidence 0.8 and GreenGenes Oct 2012 revision), principal coordinates analysis (PCoA) with weighted UniFrac distances, multiresponse permutation procedures (MRPP), and indicator species (IS) analyses of 16S rRNA gene sequences generated by assembled paired-end reads were performed using automated exploration of microbial diversity (AXIOME) automation of PANDAseq (69), the QIIME pipeline (70), and custom AXIOME analyses (71).
MG-RAST analysis and CAZy annotation.
Paired-end shotgun sequences from the pooled heavy DNA samples were analyzed for GHs using the MG-RAST pipeline (72). Reads were annotated by comparison to sequences in the UniProt database (73), with no maximum E value cutoff, a 54% minimum percentage identity cutoff, and a 30-bp minimum-alignment-length cutoff. Using custom Perl scripts (see Algorithms S1 and S2 in the supplemental material), Swissprot and Trembl database (UniProt release 2012 to 2014) hits were paired with matching GH family CAZy identifiers by comparing an extracted database of accession numbers to CAZy identifiers (see Texts S1 and S2 in the supplemental material).
Cellulose-enriched metagenomic library construction.
High-molecular-weight DNA was extracted from all three soil samples that were amended with 13C-labeled bacterial cellulose (week 6 time point), using a gentle enzymatic lysis (74). Humic acids were removed from crude DNA as described previously (75), using the SCODA device (Aurora, Boreal Genomics; Vancouver, BC, Canada) with one wash cycle (70 V/cm, 10°C, 90 min) and two concentration cycles (70 V/cm, 10°C, 60 min). DNA was analyzed using a 1% agarose gel and quantified with the NanoDrop 2000 spectrophotometer. Samples were subjected to cesium chloride density gradient ultracentrifugation and fraction collection as described previously with minor modifications. Gradient fractions were diluted with 1 volume of water and then, following addition of 2 volumes of ethanol, the DNA was precipitated overnight at −20°C. DNA was collected by centrifugation for 30 min at 13,000 × g. The DNA was air dried, dissolved in 300 µl of water, and then precipitated by adding 1/10 vol of 3 M sodium acetate (pH 5.3) and 2 volumes of ethanol. After confirming that the fingerprints generated from an alternative lysis protocol were the same as those observed by DGGE, pooled samples and fractions for large-insert cosmid cloning were mixed in the same equal nanogram ratio used to prepare template for sequence-based metagenomics.
To obtain a sufficient amount of DNA for 13C-cellulose-enriched metagenomic library construction, triplicate multiple displacement amplification (MDA) reactions were conducted using the illustra GenomiPhi V2 DNA amplification kit (GE Healthcare, Mississauga, Ontario, Canada), according to the manufacturer’s instructions. Each reaction mixture consisted of ~7 ng of DNA template in order to minimize potential amplification bias (26, 30, 76), yielding 3 to 4 µg of amplified DNA. Positive-control DNA from the kit and negative controls without DNA were run in parallel. MDA products were quantified on a 1% agarose gel and then pooled.
To inactivate ϕ29 DNA polymerase, MDA-amplified DNA (100 µl) was mixed with 613 µl of Tris-EDTA (TE), 73 µl of 10× gel loading buffer, and 6.8 µl of 20% SDS. After being heated at 65°C for 10 min, the sample was left on ice for 5 min and then centrifuged at 15,900 × g for 5 min. The DNA-containing supernatant was loaded onto a 1% pulsed-field agarose gel (with Tris-acetate-EDTA [TAE] buffer) in order to size select DNA. Pulsed-field gel electrophoresis (PFGE) (CHEF Mapper; Bio-Rad) was run at 14°C, 5.5 V/cm, 120° angle, and an initial 1.0-s to final 6.0-s switch time for 20 h. The outer lanes were loaded with a size marker, and following electrophoresis, these lanes were sliced off, poststained with SYBR green I nucleic acid gel stain, and visualized with a Clare Chemical Research Dark Reader. After reassembly of the gel, a gel slice corresponding to 25 to 75 kb of sample DNA was excised, electroeluted, and concentrated as described previously (77). DNA end repair, ligation with cosmid pJC8, packaging, and transduction into E. coli HB101 were performed as reported previously (77). Resulting recombinant cosmid clones were pooled and saved in 7% dimethyl sulfoxide (DMSO) in 1-ml aliquots at −75°C. Prior to pooling, 40 random E. coli clones from the plates were selected for analysis of cosmid DNA restriction patterns. The average sizes of cloned metagenomic DNA and coverage of bacterial genomes were calculated based on sizes of EcoRI-HindIII-BamHI fragments and the number of recombinant library clones. Additionally, 2,876 random clones were inoculated into LB-Tc in 96-well plates and then grown overnight at 37°C for functional screening.
Functional screening.
Clones were randomly selected and subjected to activity-based screening of GHs in E. coli HB101. These clones were grown in 96-well microtiter plates and were replicated onto 150-mm LB-Tc agar plates supplemented with carboxymethyl cellulose (CMC) (0.2%). The plates were incubated at 37°C for 1 week. Following removal of colonies from the plates by washing with water, 0.1% Congo red was used for poststaining.
These clones were also tested for activity on a host of methylumbelliferyl-based fluorogenic proxy substrates. Clones were first grown in LB broth containing 15 µg/ml tetracycline at 37°C in microtiter plates. Each well contained one glass bead, and plates were incubated with orbital shaking. After 24 h, 70 µl of preculture was transferred to a deep-well plate (96 wells) and cultured in Terrific Broth containing 15 µg/ml tetracycline for a further 24 h at 37° C with a glass bead and orbital shaking. Cells were collected by centrifugation and frozen. For lysis, cell pellets were thawed and chemically lysed using the BugBuster protein extraction reagent (Novagen). GH activities in cell-free extracts were measured using α-l-arabino-furanoside/pyranoside, β-d-cellobiopyranoside, β-d-glucopyranoside, β-d-xylopyranoside, and N-acetyl-β-d-galactosaminide. Reactions were carried out in 384-well microplates. Library lysates were incubated with 0.1 mM each substrate for 1 h at 50° C in a 40-µl sodium citrate-buffered (50 mM, pH 5) reaction mixture. Reactions were stopped by the addition of 40 µl of 0.2 M glycine (pH 10). Fluorescence was detected at 445 nm following excitation at 370 nm. Clones that demonstrated activity on one or more substrates were subcultured and rescreened on appropriate substrates to eliminate false-positive reactions. Protein concentrations were measured by the Bradford method with bovine serum albumin (BSA) used as a standard.
End sequences of positive cosmid clones were obtained by Sanger sequencing using M13 forward and reverse primers at TCAG (Toronto, Ontario, Canada). We used BLASTx searches of translated nucleotide sequences against the NCBI protein database. End sequences were deposited in GenBank. Posterior BLAST analysis was done searching for sequence similarities in the three libraries: low pH, agricultural, and cellulose (forward and reverse). Sequences with >95% similarity and >30 bp were recorded as positive matches.
Nucleotide sequence accession numbers.
Paired-end reads have been deposited in MG-RAST under identification no. 4482593.3 (low-pH forward), 4483544.3 (low-pH reverse), 4482599.3 (cellulose forward), 4483820.3 (cellulose reverse), 4482600.3 (agricultural forward), and 4483819.3 (agricultural reverse). End sequences of cosmid clones have been deposited in GenBank under accession no. KG771718 to KG771732.
SUPPLEMENTAL MATERIAL
This program will add CAZy IDs to a list of UniProt accession numbers. It requires the input list of ACCESSION to CAZy IDs for Swissprot and Trembl and a target file in Windows .csv format. Download
This supporting file was used by Algorithm S2 for Trembl to CAZy conversions. Download
This supporting file was used by Algorithm S2 for Swissprot to CAZy conversions. Download
This program will extract all Swissprot IDs with CAZy annotations from a target database. Download
(Figure S1) Oxygen concentrations in soil incubations, with and without added glucose. Headspace was flushed with helium and amended with oxygen at weekly intervals for Arctic tundra (1AT) (A), temperate rainforest (7TR) (B), and agricultural (11AW) (C) soils. (Figure S2) Bacterial denaturing gradient gel electrophoresis fingerprints of density gradient fractions recovered after glucose DNA-SIP incubation of Arctic tundra, temperate rainforest, and agricultural soils (3 weeks of incubation). Fractions are arranged from 1 (heaviest) to 12 (lightest). Both 12C (unlabeled) incubations and 13C (labeled) incubations are shown. The gel strips shown beneath each DGGE are 1% agarose gels stained with ethidium bromide to demonstrate isopycnic separation of DNA within the cesium chloride density gradient. (Figure S3) Bacterial denaturing gradient gel electrophoresis fingerprints of density gradient fractions recovered after cellobiose DNA-SIP incubation of Arctic tundra, temperate rainforest, and agricultural soils (3 weeks of incubation). Fractions are arranged from 1 (heaviest) to 12 (lightest). Both 12C (unlabeled) incubations and 13C (labeled) incubations are shown. The gel strips shown beneath each DGGE are 1% agarose gels stained with ethidium bromide to demonstrate isopycnic separation of DNA within the cesium chloride density gradient. (Figure S4) Bacterial denaturing gradient gel electrophoresis fingerprints of density gradient fractions recovered after arabinose DNA-SIP incubation of Arctic tundra, temperate rainforest, and agricultural soils (3 weeks of incubation). Fractions are arranged from 1 (heaviest) to 12 (lightest). Both 12C (unlabeled) incubations and 13C (labeled) incubations are shown. The gel strips shown beneath each DGGE are 1% agarose gels stained with ethidium bromide to demonstrate isopycnic separation of DNA within the cesium chloride density gradient. (Figure S5) Bacterial denaturing gradient gel electrophoresis fingerprints of density gradient fractions recovered after xylose DNA-SIP incubation of Arctic tundra, temperate rainforest, and agricultural soils (3 weeks of incubation). Fractions are arranged from 1 (heaviest) to 12 (lightest). Both 12C (unlabeled) incubations and 13C (labeled) incubations are shown. The gel strips shown beneath each DGGE are 1% agarose gels stained with ethidium bromide to demonstrate isopycnic separation of DNA within the cesium chloride density gradient. (Figure S6) Bacterial denaturing gradient gel electrophoresis fingerprints of density gradient fractions recovered after cellulose DNA-SIP incubation of Arctic tundra, temperate rainforest, and agricultural soils (6 weeks of incubation). Fractions are arranged from 1 (heaviest) to 12 (lightest). Both 12C (unlabeled) incubations and 13C (labeled) incubations are shown. The gel strips shown beneath each DGGE are 1% agarose gels stained with ethidium bromide to demonstrate isopycnic separation of DNA within the cesium chloride density gradient. Download
(Table S1) Indicator species analysis comparing heavy DNA samples versus light DNA samples from all three soils combined (tundra [1AT], temperate rainforest [7TR], and agricultural soil-wheat [11AW]), using a threshold of a 0.7 indicator value (IV) cutoff and a 250 minimum sequence sum. The number of OTUs is represented per treatment and as the sum of all treatments. Representative sequences are also shown. Colors are intended to assist with table interpretation. (Table S2) Indicator species analysis comparing heavy DNA samples versus light DNA samples from all three soils considered separately (tundra [1AT], temperate rainforest [7TR], and agricultural soil-wheat [11AW]), using a threshold of a 0.7 indicator value (IV) cutoff and 250 minimum sequence sum. The number of OTUs is represented per each treatment and as the sum of all treatments. Representative sequences are also shown. Dark shading corresponds to the heavy DNA samples. Note that this table contains multiple spreadsheets. (Table S3) Indicator species analysis comparing heavy DNA samples versus light DNA samples for each substrate from all three soils combined (tundra [1AT], temperate rainforest [7TR], and agricultural soil-wheat [11AW]) using a threshold of a 0.7 indicator value (IV) cutoff and 250 minimum sequence sum. The number of OTUs is represented per treatment and as the sum of all treatments. Representative sequences are also shown. Dark shading corresponds to the heavy DNA samples of interest. Note that this table contains multiple spreadsheets. (Table S4) Indicator species analysis comparing heavy DNA samples versus light DNA samples from agricultural soil-wheat (11AW), using a threshold of a 0.7 indicator value (IV) cutoff and 250 minimum sequence sum. The number of OTUs is represented per treatment and as the sum of all treatments. Representative sequences are also shown. Dark shading corresponds to the heavy DNA samples of interest. Note that this table contains multiple spreadsheets. (Table S5) Indicator species analysis comparing heavy DNA samples versus light DNA samples from temperate rainforest (7TR), using a threshold of a 0.7 indicator value (IV) cutoff and 250 minimum sequence sum. The number of OTUs is represented per treatment and as the sum of all treatments. Representative sequences are also shown. Dark shading corresponds to the heavy DNA samples of interest. Note that this table contains multiple spreadsheets. (Table S6) Indicator species analysis comparing heavy DNA samples versus light DNA samples from Arctic tundra (1AT), using a threshold of a 0.7 indicator value (IV) cutoff and 250 minimum sequence sum. The number of OTUs is represented per treatment and as the sum of all treatments. Representative sequences are also shown. Dark shading corresponds to the heavy DNA samples of interest. Note that this table contains multiple spreadsheets.
Sequence counts by glycoside hydrolase family annotation in pooled paired-end DNA sequences, based on Swiss-Prot annotations that were managed by MG-RAST.
Sequence matches for forward and reverse cellulose SIP cosmid end sequences against whole-genome paired-end Illumina data generated from the three SIP heavy DNA pools.
ACKNOWLEDGMENT
This work was supported by a Strategic Projects Grant from the Natural Sciences and Engineering Research Council of Canada (NSERC).
Footnotes
Citation Verastegui Y, Cheng J, Engel K, Kolczynski D, Mortimer S, Lavigne J, Montalibet J, Romantsov T, Hall M, McConkey BJ, Rose DR, Tomashek JJ, Scott BR, Charles TC, Neufeld JD. 2014. Multisubstrate isotope labeling and metagenomic analysis of active soil bacterial communities. mBio 5(4):e01157-14. doi:10.1128/mBio.01157-14.
REFERENCES
- 1. Nannipieri P, Ascher J, Ceccherini MT, Landi L, Pietramellara G, Renella G. 2003. Microbial diversity and soil functions. Eur. J. Soil Sci. 54:655–670. 10.1046/j.1351-0754.2003.0556.x [DOI] [Google Scholar]
- 2. Amann RI, Ludwig W, Schleifer KH. 1995. Phylogenetic identification and in situ detection of individual microbial cells without cultivation. Microbiol. Rev. 59:143–169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Torsvik V, Øvreås L. 2002. Microbial diversity and function in soil: from genes to ecosystems. Curr. Opin. Microbiol. 5:240–245. 10.1016/S1369-5274(02)00324-7 [DOI] [PubMed] [Google Scholar]
- 4. Tkacz JS, Lange L. 2004. Advances in fungal biotechnology for industry, agriculture and medicine. Springer Verlag, Berlin, Germany [Google Scholar]
- 5. Bernard L, Mougel C, Maron PA, Nowak V, Lévêque J, Henault C, Haichar FZ, Berge O, Marol C, Balesdent J, Gibiat F, Lemanceau P, Ranjard L. 2007. Dynamics and identification of soil microbial populations actively assimilating carbon from 13C-labelled wheat residue as estimated by DNA- and RNA-SIP techniques. Environ. Microbiol. 9:752–764. 10.1111/j.1462-2920.2006.01197.x [DOI] [PubMed] [Google Scholar]
- 6. Haichar FZ, Achouak W, Christen R, Heulin T, Marol C, Marais MF, Mougel C, Ranjard L, Balesdent J, Berge O. 2007. Identification of cellulolytic bacteria in soil by stable isotope probing. Environ. Microbiol. 9:625–634. 10.1111/j.1462-2920.2006.01182.x [DOI] [PubMed] [Google Scholar]
- 7. Bernard L, Maron PA, Mougel C, Nowak V, Lévêque J, Marol C, Balesdent J, Gibiat F, Ranjard L. 2009. Contamination of soil by copper affects the dynamics, diversity, and activity of soil bacterial communities involved in wheat decomposition and carbon storage. Appl. Environ. Microbiol. 75:7565–7569. 10.1128/AEM.00616-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Schellenberger S, Kolb S, Drake HL. 2010. Metabolic responses of novel cellulolytic and saccharolytic agricultural soil bacteria to oxygen. Environ. Microbiol. 12:845–861. 10.1111/j.1462-2920.2009.02128.x [DOI] [PubMed] [Google Scholar]
- 9. Simon C, Daniel R. 2009. Achievements and new knowledge unraveled by metagenomic approaches. Appl. Microbiol. Biotechnol. 85:265–276. 10.1007/s00253-009-2233-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ferrer M, Beloqui A, Timmis KN, Golyshin PN. 2009. Metagenomics for mining new genetic resources of microbial communities. J. Mol. Microbiol. Biotechnol. 16:109–123. 10.1159/000142898 [DOI] [PubMed] [Google Scholar]
- 11. Nacke H, Engelhaupt M, Brady S, Fischer C, Tautzt J, Daniel R. 2012. Identification and characterization of novel cellulolytic and hemicellulolytic genes and enzymes derived from German grassland soil metagenomes. Biotechnol. Lett. 34:663–675. 10.1007/s10529-011-0830-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Liu J, Liu WD, Zhao XL, Shen WJ, Cao H, Cui ZL. 2011. Cloning and functional characterization of a novel endo-beta-1,4-glucanase gene from a soil-derived metagenomic library. Appl. Microbiol. Biotechnol. 89:1083–1092. 10.1007/s00253-010-2828-4 [DOI] [PubMed] [Google Scholar]
- 13. Wang F, Li F, Chen G, Liu W. 2009. Isolation and characterization of novel cellulase genes from uncultured microorganisms in different environmental niches. Microbiol. Res. 164:650–657. 10.1016/j.micres.2008.12.002 [DOI] [PubMed] [Google Scholar]
- 14. Kim SJ, Lee CM, Han BR, Kim MY, Yeo YS, Yoon SH, Koo BS, Jun HK. 2008. Characterization of a gene encoding cellulase from uncultured soil bacteria. FEMS Microbiol. Lett. 282:44–51. 10.1111/j.1574-6968.2008.01097.x [DOI] [PubMed] [Google Scholar]
- 15. Steele HL, Jaeger KE, Daniel R, Streit WR. 2009. Advances in recovery of novel biocatalysts from metagenomes. J. Mol. Microbiol. Biotechnol. 16:25–37. 10.1159/000142892 [DOI] [PubMed] [Google Scholar]
- 16. Neufeld JD, Vohra J, Dumont MG, Lueders T, Manefield M, Friedrich MW, Murrell JC. 2007. DNA stable-isotope probing. Nat. Protoc. 2:860–866. 10.1038/nprot.2007.109 [DOI] [PubMed] [Google Scholar]
- 17. Dumont MG, Murrell JC. 2005. Stable isotope probing—linking microbial identity to function. Nat. Rev. Microbiol. 3:499–504. 10.1038/nrmicro1162 [DOI] [PubMed] [Google Scholar]
- 18. Radajewski S, Ineson P, Parekh NR, Murrell JC. 2000. Stable-isotope probing as a tool in microbial ecology. Nature 403:646–649. 10.1038/35001054 [DOI] [PubMed] [Google Scholar]
- 19. Wackett LP. 2004. Stable isotope probing in biodegradation research. Trends Biotechnol. 22:153–154. 10.1016/j.tibtech.2004.01.013 [DOI] [PubMed] [Google Scholar]
- 20. Chen Y, Murrell JC. 2010. When metagenomics meets stable-isotope probing: progress and perspectives. Trends Microbiol. 18:157–163. 10.1016/j.tim.2010.02.002 [DOI] [PubMed] [Google Scholar]
- 21. Pinnell LJ, Charles TC, Neufeld JD. 2011. Stable-isotope probing and metagenomics, p 97–114 In Murrell JC, Whiteley AS. (ed), Stable isotopes in microbial molecular ecology. ASM Press, Washington, DC [Google Scholar]
- 22. Schwarz S, Waschkowitz T, Daniel R. 2006. Enhancement of gene detection frequencies by combining DNA-based stable-isotope probing with the construction of metagenomic DNA libraries. World J. Microbiol. Biotechnol. 22:363–367. 10.1007/s11274-005-9042-z [DOI] [Google Scholar]
- 23. Dumont MG, Radajewski SM, Miguez CB, McDonald IR, Murrell JC. 2006. Identification of a complete methane monooxygenase operon from soil by combining stable isotope probing and metagenomic analysis. Environ. Microbiol. 8:1240–1250. 10.1111/j.1462-2920.2006.01018.x [DOI] [PubMed] [Google Scholar]
- 24. Kalyuzhnaya MG, Lapidus A, Ivanova N, Copeland AC, McHardy AC, Szeto E, Salamov A, Grigoriev IV, Suciu D, Levine SR, Markowitz VM, Rigoutsos I, Tringe SG, Bruce DC, Richardson PM, Lidstrom ME, Chistoserdova L. 2008. High-resolution metagenomics targets specific functional types in complex microbial communities. Nat. Biotechnol. 26:1029–1034. 10.1038/nbt.1488 [DOI] [PubMed] [Google Scholar]
- 25. Chen Y, Dumont MG, Neufeld JD, Bodrossy L, Stralis-Pavese N, McNamara NP, Ostle N, Briones MJ, Murrell JC. 2008. Revealing the uncultivated majority: combining DNA stable-isotope probing, multiple displacement amplification and metagenomic analyses of uncultivated Methylocystis in acidic peatlands. Environ. Microbiol. 10:2609–2622. 10.1111/j.1462-2920.2008.01683.x [DOI] [PubMed] [Google Scholar]
- 26. Neufeld JD, Chen Y, Dumont MG, Murrell JC. 2008. Marine methylotrophs revealed by stable-isotope probing, multiple displacement amplification and metagenomics. Environ. Microbiol. 10:1526–1535. 10.1111/j.1462-2920.2008.01568.x [DOI] [PubMed] [Google Scholar]
- 27. Sul WJ, Park J, Quensen JF, Rodrigues JL, Seliger L, Tsoi TV, Zylstra GJ, Tiedje JM. 2009. DNA-stable isotope probing integrated with metagenomics for retrieval of biphenyl dioxygenase genes from polychlorinated biphenyl-contaminated river sediment. Appl. Environ. Microbiol. 75:5501–5506. 10.1128/AEM.00121-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lee TK, Lee J, Sul WJ, Iwai S, Chai B, Tiedje JM, Park J. 2011. Novel biphenyl-oxidizing bacteria and dioxygenase genes from a Korean tidal mudflat. Appl. Environ. Microbiol. 77:3888–3891. 10.1128/AEM.00023-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Eichorst SA, Kuske CR. 2012. Identification of cellulose-responsive bacterial and fungal communities in geographically and edaphically different soils by using stable isotope probing. Appl. Environ. Microbiol. 78:2316–2327. 10.1128/AEM.07313-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Pinnell LJ, Dunford E, Ronan P, Hausner M, Neufeld JD. 11 June 2014. Recovering glycoside hydrolase genes from active tundra cellulolytic bacteria Can. J. Microbiol. 10.1139/cjm-2014-0193 . [DOI] [PubMed] [Google Scholar]
- 31. Neufeld JD, Engel K, Cheng J, Moreno-Hagelsieb G, Rose DR, Charles TC. 2011. Open resource metagenomics: a model for sharing metagenomic libraries. Stand. Genomic Sci. 5:203–210. 10.4056/sigs.1974654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Fierer N, Jackson RB. 2006. The diversity and biogeography of soil bacterial communities. Proc. Natl. Acad. Sci. U. S. A. 103:626–631. 10.1073/pnas.0507535103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Stotzky G. 1997. Soil as an environment for microbial life, p 1–20 In Elsas JD, Modern soil microbiology. Dekker, New York, NY. [Google Scholar]
- 34. Franzluebbers AJ. 1999. Microbial activity in response to water-filled pore space of variably eroded southern Piedmont soils. Appl. Soil Ecol. 11:91–101. 10.1016/S0929-1393(98)00128-0 [DOI] [Google Scholar]
- 35. Linn DM, Doran JW. 1984. Effect of water-filled pore space on carbon dioxide and nitrous oxide production in tilled and nontilled soils. Soils Sci. Soc. J. 48:1267–1272. 10.2136/sssaj1984.03615995004800060013x [DOI] [Google Scholar]
- 36. Koizumi S, Yue Z, Tomita Y, Kondo T, Iwase H, Yamaguchi D, Hashimoto T. 2008. Bacterium organizes hierarchical amorphous structure in microbial cellulose. Eur. Phys. J. E Soft Matter 26:137–142. 10.1140/epje/i2007-10259-3 [DOI] [PubMed] [Google Scholar]
- 37. Hall M, Bansal P, Lee JH, Realff MJ, Bommarius AS. 2010. Cellulose crystallinity—a key predictor of the enzymatic hydrolysis rate. FEBS J 277:1571–1582. 10.1111/j.1742-4658.2010.07585.x [DOI] [PubMed] [Google Scholar]
- 38. Medeiros PM, Fernandes MF, Dick RP, Simoneit BR. 2006. Seasonal variations in sugar contents and microbial community in a ryegrass soil. Chemosphere 65:832–839. 10.1016/j.chemosphere.2006.03.025 [DOI] [PubMed] [Google Scholar]
- 39. Hill PW, Farrar JF, Jones DL. 2008. Decoupling of microbial glucose uptake and mineralization in soil. Soil Biol. Biochem. 40:616–624. 10.1016/j.soilbio.2007.09.008 [DOI] [Google Scholar]
- 40. Neufeld JD, Dumont MG, Vohra J, Murrell JC. 2007. Methodological considerations for the use of stable isotope probing in microbial ecology. Microb. Ecol. 53:435–442. 10.1007/s00248-006-9125-x [DOI] [PubMed] [Google Scholar]
- 41. Leschine SB. 1995. Cellulose degradation in anaerobic environments. Annu. Rev. Microbiol. 49:399–426. 10.1146/annurev.mi.49.100195.002151 [DOI] [PubMed] [Google Scholar]
- 42. Ronan P, Yeung CW, Schellenberg J, Sparling R, Wolfaardt GM, Hausner M. 2013. A versatile and robust aerotolerant microbial community capable of cellulosic ethanol production. Bioresour. Technol. 129:156–163. 10.1016/j.biortech.2012.10.164 [DOI] [PubMed] [Google Scholar]
- 43. Green SJ, Leigh MB, Neufeld JD. 2010. Denaturing gradient gel electrophoresis (DGGE) for microbial community analysis, p 4137–4158 In Timmis KN, Microbiology of hydrocarbon and lipid microbiology. Springer-Verlag, Berlin, Germany [Google Scholar]
- 44. Good IJ. 1953. The population frequencies of species and the estimation of population parameters Biometrika 40:237–264. 10.2307/2333344 [DOI] [Google Scholar]
- 45. Bartram AK, Jiang X, Lynch MD, Masella AP, Nicol GW, Dushoff J, Neufeld JD. 2014. Exploring links between pH and bacterial community composition in soils from the Craibstone Experimental Farm. FEMS Microbiol. Ecol. 87:403–415 [DOI] [PubMed] [Google Scholar]
- 46. Lauber CL, Hamady M, Knight R, Fierer N. 2009. Pyrosequencing-based assessment of soil pH as a predictor of soil bacterial community structure at the continental scale. Appl. Environ. Microbiol. 75:5111–5120. 10.1128/AEM.00335-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Dufrêne M, Legendre P. 1997. Species assemblages and indicator species: the need for a flexible asymmetrical approach. Ecol. Monogr. 67:345–366. 10.1890/0012-9615(1997)067[0345:SAAIST]2.0.CO;2 [DOI] [Google Scholar]
- 48. Zhang DC, Liu HC, Xin YH, Yu Y, Zhou PJ, Zhou YG. 2008. Salinibacterium xinjiangense sp. nov., a psychrophilic bacterium isolated from the China No. 1 glacier. Int. J. Syst. Evol. Microbiol. 58:2739–2742. 10.1099/ijs.0.65802-0 [DOI] [PubMed] [Google Scholar]
- 49. Shin SC, Kim SJ, Lee JK, Lee H, Lee J, Hong SG, Lee YM, Park H, Park H. 2012. Genome sequence of a Salinibacterium sp. isolated from Antarctic soil. J. Bacteriol. 194:2404. 10.1128/JB.00235-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Han SK, Nedashkovskaya OI, Mikhailov VV, Kim SB, Bae KS. 2003. Salinibacterium amurskyense gen. nov., sp. nov., a novel genus of the family Microbacteriaceae from the marine environment. Int. J. Syst. Evol. Microbiol. 53:2061–2066. 10.1099/ijs.0.02627-0 [DOI] [PubMed] [Google Scholar]
- 51. Yoo SH, Weon HY, Kim BY, Hong SB, Kwon SW, Cho YH, Go SJ, Stackebrandt E. 2006. Devosia soli sp. nov., isolated from greenhouse soil in Korea. Int. J. Syst. Evol. Microbiol. 56:2689–2692. 10.1099/ijs.0.64214-0 [DOI] [PubMed] [Google Scholar]
- 52. Lee SD. 2007. Devosia subaequoris sp. nov., isolated from beach sediment. Int. J. Syst. Evol. Microbiol. 57:2212–2215. 10.1099/ijs.0.65185-0 [DOI] [PubMed] [Google Scholar]
- 53. Štursová M, Žifčáková L, Leigh MB, Burgess R, Baldrian P. 2012. Cellulose utilization in forest litter and soil: identification of bacterial and fungal decomposers. FEMS Microbiol. Ecol. 80:735–746. 10.1111/j.1574-6941.2012.01343.x [DOI] [PubMed] [Google Scholar]
- 54. Vasilyeva LV, Omelchenko MV, Berestovskaya YY, Lysenko AM, Abraham WR, Dedysh SN, Zavarzin GA. 2006. Asticcacaulis benevestitus sp. nov., a psychrotolerant, dimorphic, prosthecate bacterium from tundra wetland soil. Int. J. Syst. Evol. Microbiol. 56:2083–2088. 10.1099/ijs.0.64122-0 [DOI] [PubMed] [Google Scholar]
- 55. Angelov A, Loderer C, Pompei S, Liebl W. 2011. Novel family of carbohydrate-binding modules revealed by the genome sequence of Spirochaeta thermophila DSM 6192. Appl. Environ. Microbiol. 77:5483–5489. 10.1128/AEM.00523-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Hoover RB, Pikuta EV, Bej AK, Marsic D, Whitman WB, Tang J, Krader P. 2003. Spirochaeta americana sp. nov., a new haloalkaliphilic, obligately anaerobic spirochaete isolated from soda Mono Lake in California. Int. J. Syst. Evol. Microbiol. 53:815–821. 10.1099/ijs.0.02535-0 [DOI] [PubMed] [Google Scholar]
- 57. Wang TY, Chen HL, Lu MY, Chen YC, Sung HM, Mao CT, Cho HY, Ke HM, Hwa TY, Ruan SK, Hung KY, Chen CK, Li JY, Wu YC, Chen YH, Chou SP, Tsai YW, Chu TC, Shih CC, Li WH, Shih MC. 2011. Functional characterization of cellulases identified from the cow rumen fungus Neocallimastix patriciarum W5 by transcriptomic and secretomic analyses. Biotechnol. Biofuels 4:24. 10.1186/1754-6834-4-24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Lombard V, Ramulu HG, Drula E, Coutinho PM, Henrissat B. 2014. The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 42:D490–D495. 10.1093/nar/gkt1178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Teather RM, Wood PJ. 1982. Use of Congo red-polysaccharide interactions in enumeration and characterization of cellulolytic bacteria from the bovine rumen. Appl. Environ. Microbiol. 43:777–780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Enebro J, Momcilovic D, Siika-Aho M, Karlsson S. 2009. Investigation of endoglucanase selectivity on carboxymethyl cellulose by mass spectrometric techniques. Cellulose 16:271–280. 10.1007/s10570-008-9254-0 [DOI] [PubMed] [Google Scholar]
- 61. Mergaert J, Lednická D, Goris J, Cnockaert MC, De Vos P, Swings J. 2003. Taxonomic study of Cellvibrio strains and description of Cellvibrio ostraviensis sp. nov., Cellvibrio fibrivorans sp. nov. and Cellvibrio gandavensis sp. nov. Int. J. Syst. Evol. Microbiol. 53:465–471. 10.1099/ijs.0.02316-0 [DOI] [PubMed] [Google Scholar]
- 62. Taylor LE, Henrissat B, Coutinho PM, Ekborg NA, Hutcheson SW, Weiner RM. 2006. Complete cellulase system in the marine bacterium Saccharophagus degradans strain 2-40T. J. Bacteriol. 188:3849–3861. 10.1128/JB.01348-05 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Lang E, Lapidus A, Chertkov O, Brettin T, Detter JC, Han C, Copeland A, del Rio TG, Nolan M, Chen F, Lucas S, Tice T, Cheng JF, Land M, Hauser L, Chang YJ, Jeffries CD, Kopitz M, Bruce D, Goodwin L, Pitluck S, Ovchinnikova G, Pati A, Ivanova N, Mavrommatis K, Chen A, Palaniappan K, Chain P, Bristow J, Eisen JA, Markowitz V, Hugenholtz P, Göker M, Rohde M, Kyrpides NC, Klenk HP. 2009. Complete genome sequence of Dyadobacter fermentans type strain (NS114T). Stand. Genomic Sci. 1:133–140. 10.4056/sigs.19262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Chen Y, He Y, Zhang B, Yang J, Li W, Dong Z, Hu S. 2011. Complete genome sequence of Alicyclobacillus acidocaldarius strain Tc-4-1. J. Bacteriol. 193:5602–5603. 10.1128/JB.05709-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Kant R, van Passel MW, Palva A, Lucas S, Lapidus A, Glavina del Rio TG, Dalin E, Tice H, Bruce D, Goodwin L, Pitluck S, Larimer FW, Land ML, Hauser L, Sangwan P, de Vos WM, Janssen PH, Smidt H. 2011. Genome sequence of Chthoniobacter flavus Ellin 428, an aerobic heterotrophic soil bacterium. J. Bacteriol. 193:2902–2903. 10.1128/JB.00295-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Dunford EA, Neufeld JD. 2010. DNA stable-isotope probing (DNA-SIP). J. Vis. Exp. 42:e2027. 10.3791/202.7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Muyzer G, de Waal EC, Uitterlinden AG. 1993. Profiling of complex microbial populations by denaturing gradient gel electrophoresis analysis of polymerase chain reaction-amplified genes coding for 16S rRNA. Appl. Environ. Microbiol. 59:695–700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Bartram AK, Lynch MD, Stearns JC, Moreno-Hagelsieb G, Neufeld JD. 2011. Generation of multimillion 16S rRNA gene libraries from complex microbial communities by assembling paired-end Illumina reads. Appl. Environ. Microbiol. 77:3846–3852. 10.1128/AEM.02772-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Masella AP, Bartram AK, Truszkowski JM, Brown DG, Neufeld JD. 2012. PANDAseq: paired-end assembler for Illumina sequences. BMC Bioinformatics 13:31. 10.1186/1471-2105-13-31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Peña AG, Goodrich JK, Gordon JI, Huttley GA, Kelley ST, Knights D, Koenig JE, Ley RE, Lozupone CA, McDonald D, Muegge BD, Pirrung M, Reeder J, Sevinsky JR, Turnbaugh PJ, Walters WA, Widmann J, Yatsunenko T, Zaneveld J, Knight R. 2010. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7:335–336. 10.1038/nmeth.f.303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Lynch MDJ, Masella AP, Hall MW, Bartram AK, Neufeld JD. 2013. AXIOME: automated exploration of microbial diversity. GigaScience 2:3. 10.1186/2047-217X-2-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Meyer F, Paarmann D, D’Souza M, Olson R, Glass EM, Kubal M, Paczian T, Rodriguez A, Stevens R, Wilke A, Wilkening J, Edwards RA. 2008. The metagenomics RAST server—a public resource for the automatic phylogenetic and functional analysis of metagenomes. BMC Bioinformatics 9:386. 10.1186/1471-2105-9-386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Apweiler R, Bairoch A, Wu CH, Barker WC, Boeckmann B, Ferro S, Gasteiger E, Huang H, Lopez R, Magrane M, Martin MJ, Natale DA, O’Donovan C, Redaschi N, Yeh LS. 2004. UniProt: the universal protein knowledgebase. Nucleic Acids Res. 32:D115–D119. 10.1093/nar/gnh110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Zhou J, Bruns MA, Tiedje JM. 1996. DNA recovery from soils of diverse composition. Appl. Environ. Microbiol. 62:316–322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Engel K, Pinnell L, Cheng J, Charles TC, Neufeld JD. 2012. Nonlinear electrophoresis for purification of soil DNA for metagenomics. J. Microbiol. Methods 88:35–40. 10.1016/j.mimet.2011.10.007 [DOI] [PubMed] [Google Scholar]
- 76. Binga EK, Lasken RS, Neufeld JD. 2008. Something from (almost) nothing: the impact of multiple displacement amplification on microbial ecology. ISME J 2:233–241. 10.1038/ismej.2008.10 [DOI] [PubMed] [Google Scholar]
- 77. Cheng J, Pinnell L, Engel K, Neufeld JD, Charles TC. 2014. Construction of broad-host-range cosmids and metagenomic libraries from Canadian agricultural soils. J. Microbiol. Methods 99:27–34. 10.1016/j.mimet.2014.01.015 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
This program will add CAZy IDs to a list of UniProt accession numbers. It requires the input list of ACCESSION to CAZy IDs for Swissprot and Trembl and a target file in Windows .csv format. Download
This supporting file was used by Algorithm S2 for Trembl to CAZy conversions. Download
This supporting file was used by Algorithm S2 for Swissprot to CAZy conversions. Download
This program will extract all Swissprot IDs with CAZy annotations from a target database. Download
(Figure S1) Oxygen concentrations in soil incubations, with and without added glucose. Headspace was flushed with helium and amended with oxygen at weekly intervals for Arctic tundra (1AT) (A), temperate rainforest (7TR) (B), and agricultural (11AW) (C) soils. (Figure S2) Bacterial denaturing gradient gel electrophoresis fingerprints of density gradient fractions recovered after glucose DNA-SIP incubation of Arctic tundra, temperate rainforest, and agricultural soils (3 weeks of incubation). Fractions are arranged from 1 (heaviest) to 12 (lightest). Both 12C (unlabeled) incubations and 13C (labeled) incubations are shown. The gel strips shown beneath each DGGE are 1% agarose gels stained with ethidium bromide to demonstrate isopycnic separation of DNA within the cesium chloride density gradient. (Figure S3) Bacterial denaturing gradient gel electrophoresis fingerprints of density gradient fractions recovered after cellobiose DNA-SIP incubation of Arctic tundra, temperate rainforest, and agricultural soils (3 weeks of incubation). Fractions are arranged from 1 (heaviest) to 12 (lightest). Both 12C (unlabeled) incubations and 13C (labeled) incubations are shown. The gel strips shown beneath each DGGE are 1% agarose gels stained with ethidium bromide to demonstrate isopycnic separation of DNA within the cesium chloride density gradient. (Figure S4) Bacterial denaturing gradient gel electrophoresis fingerprints of density gradient fractions recovered after arabinose DNA-SIP incubation of Arctic tundra, temperate rainforest, and agricultural soils (3 weeks of incubation). Fractions are arranged from 1 (heaviest) to 12 (lightest). Both 12C (unlabeled) incubations and 13C (labeled) incubations are shown. The gel strips shown beneath each DGGE are 1% agarose gels stained with ethidium bromide to demonstrate isopycnic separation of DNA within the cesium chloride density gradient. (Figure S5) Bacterial denaturing gradient gel electrophoresis fingerprints of density gradient fractions recovered after xylose DNA-SIP incubation of Arctic tundra, temperate rainforest, and agricultural soils (3 weeks of incubation). Fractions are arranged from 1 (heaviest) to 12 (lightest). Both 12C (unlabeled) incubations and 13C (labeled) incubations are shown. The gel strips shown beneath each DGGE are 1% agarose gels stained with ethidium bromide to demonstrate isopycnic separation of DNA within the cesium chloride density gradient. (Figure S6) Bacterial denaturing gradient gel electrophoresis fingerprints of density gradient fractions recovered after cellulose DNA-SIP incubation of Arctic tundra, temperate rainforest, and agricultural soils (6 weeks of incubation). Fractions are arranged from 1 (heaviest) to 12 (lightest). Both 12C (unlabeled) incubations and 13C (labeled) incubations are shown. The gel strips shown beneath each DGGE are 1% agarose gels stained with ethidium bromide to demonstrate isopycnic separation of DNA within the cesium chloride density gradient. Download
(Table S1) Indicator species analysis comparing heavy DNA samples versus light DNA samples from all three soils combined (tundra [1AT], temperate rainforest [7TR], and agricultural soil-wheat [11AW]), using a threshold of a 0.7 indicator value (IV) cutoff and a 250 minimum sequence sum. The number of OTUs is represented per treatment and as the sum of all treatments. Representative sequences are also shown. Colors are intended to assist with table interpretation. (Table S2) Indicator species analysis comparing heavy DNA samples versus light DNA samples from all three soils considered separately (tundra [1AT], temperate rainforest [7TR], and agricultural soil-wheat [11AW]), using a threshold of a 0.7 indicator value (IV) cutoff and 250 minimum sequence sum. The number of OTUs is represented per each treatment and as the sum of all treatments. Representative sequences are also shown. Dark shading corresponds to the heavy DNA samples. Note that this table contains multiple spreadsheets. (Table S3) Indicator species analysis comparing heavy DNA samples versus light DNA samples for each substrate from all three soils combined (tundra [1AT], temperate rainforest [7TR], and agricultural soil-wheat [11AW]) using a threshold of a 0.7 indicator value (IV) cutoff and 250 minimum sequence sum. The number of OTUs is represented per treatment and as the sum of all treatments. Representative sequences are also shown. Dark shading corresponds to the heavy DNA samples of interest. Note that this table contains multiple spreadsheets. (Table S4) Indicator species analysis comparing heavy DNA samples versus light DNA samples from agricultural soil-wheat (11AW), using a threshold of a 0.7 indicator value (IV) cutoff and 250 minimum sequence sum. The number of OTUs is represented per treatment and as the sum of all treatments. Representative sequences are also shown. Dark shading corresponds to the heavy DNA samples of interest. Note that this table contains multiple spreadsheets. (Table S5) Indicator species analysis comparing heavy DNA samples versus light DNA samples from temperate rainforest (7TR), using a threshold of a 0.7 indicator value (IV) cutoff and 250 minimum sequence sum. The number of OTUs is represented per treatment and as the sum of all treatments. Representative sequences are also shown. Dark shading corresponds to the heavy DNA samples of interest. Note that this table contains multiple spreadsheets. (Table S6) Indicator species analysis comparing heavy DNA samples versus light DNA samples from Arctic tundra (1AT), using a threshold of a 0.7 indicator value (IV) cutoff and 250 minimum sequence sum. The number of OTUs is represented per treatment and as the sum of all treatments. Representative sequences are also shown. Dark shading corresponds to the heavy DNA samples of interest. Note that this table contains multiple spreadsheets.
Sequence counts by glycoside hydrolase family annotation in pooled paired-end DNA sequences, based on Swiss-Prot annotations that were managed by MG-RAST.
Sequence matches for forward and reverse cellulose SIP cosmid end sequences against whole-genome paired-end Illumina data generated from the three SIP heavy DNA pools.