

Abstract
Regulated endocytic trafficking is the central mechanism facilitating a variety of neuromodulatory events, by dynamically controlling receptor, ion channel, and transporter cell surface presentation on a minutes time scale. There is a broad diversity of mechanisms that control endocytic trafficking of individual proteins. Studies investigating the molecular underpinnings of trafficking have primarily relied upon surface biotinylation to quantitatively measure changes in membrane protein surface expression in response to exogenous stimuli and gene manipulation. However, this approach has been mainly limited to cultured cells, which may not faithfully reflect the physiologically relevant mechanisms at play in adult neurons. Moreover, cultured cell approaches may underestimate region-specific differences in trafficking mechanisms. Here, we describe an approach that extends cell surface biotinylation to the acute brain slice preparation. We demonstrate that this method provides a high-fidelity approach to measure rapid changes in membrane protein surface levels in adult neurons. This approach is likely to have broad utility in the field of neuronal endocytic trafficking.
Keywords: Neuroscience, Issue 86, Trafficking, endocytosis, internalization, biotinylation, brain, neurons, transporter, protein kinase C
Introduction
Endocytic trafficking is a ubiquitous cellular mechanism that fine-tunes the plasma membrane presentation of a variety of integral membrane proteins. Endocytosis delivers vital nutrients to the intracellular milieu1 and desensitizes receptor signaling in response to receptor activation2. Endocytic recycling back to the plasma membrane can additionally enhance cellular signaling by increasing protein expression levels at the cell surface3. Moreover, membrane trafficking perturbations are implicated in numerous disease and pathological conditions4,5, stressing the need to investigate the molecular mechanisms that govern protein endocytic trafficking. While many proteins utilize classic clathrin-dependent internalization mechanisms, mounting evidence over the past several years demonstrates that multiple clathrin-independent endocytic mechanisms govern the endocytic potential of an increasing array of proteins6,7. Thus, the need to investigate endocytic mechanisms facilitating trafficking in physiological relevant systems has grown considerably.
In the brain, endocytic trafficking of receptors, ion channels and neurotransmitter transporters has a primary role in establishing synaptic plasticity8-11 and response to drugs of abuse12-15, ultimately impacting neuronal excitability and synaptic responses. To date the majority of neuronal trafficking studies rely on either heterologous expression systems or cultured primary neurons, neither of which may reliably reflect mechanisms at play in adult neurons. Here, we report an approach that uses surface biotinylation to quantitatively measure surface protein levels in acute brain slices derived from adult rodents. Using this approach, we present data that demonstrate that the mouse striatal dopamine transporter rapidly internalizes in response to phorbol ester-mediated protein kinase C (PKC) activation.
Protocol
All animal handling and tissue harvesting was performed in accordance with the guidelines of the University of Massachusetts Medical School Institutional Animal Care Use Committee (IACUC), following the approved protocol #A1506 (Melikian, P.I.).
Required solutions
Artificial cerebrospinal fluid (ACSF) - Make fresh daily
125 mM NaCl, 2.5 mM KCl, 1.2 mM NaH2PO4, 1.2 mM MgCl2, 2.4 mM CaCl2, 26 mM NaHCO3, and 11 mM glucose
Note: Prepare ACSF as a 10x stock solution, excluding NaHCO3 and glucose. Make 1x working solutions daily from the 10x stock, supplementing with fresh NaHCO3 and glucose.
Sucrose-supplemented ACSF (SACSF) – Make fresh daily
250 mM sucrose, 2.5 mM KCl, 1.2 mM NaH2PO4, 1.2 mM MgCl2, 2.4 mM CaCl2, 26 mM NaHCO3, and 11 mM glucose
Note: Prepare SACSF as a 10x stock solution, excluding NaHCO3 and glucose. Make 1x working solutions daily from the 10x stock, supplementing with fresh NaHCO3 and glucose.
Sulfo-N-hydroxysuccinyl-SS-Biotin (sulfo-NHS-SS-biotin, Pierce Chemical Company)
Stock solutions should be 200 mg/ml in DMSO and are resistant to multiple freeze/thaw cycles. Aliquots are stored at -20 °C. The succinyl ester is rapidly hydrolyzed in aqueous solution, so working solutions should be prepared immediately prior to applying to slices.
Slice Quench Solution
ACSF supplemented with 100 mM glycine
RIPA Lysis Buffer
10 mM Tris, pH 7.4, 150 mM NaCl, 1.0 mM EDTA, 1% Triton-X-100, 0.1% SDS, 1% Na deoxycholate
RIPA with Protease Inhibitors (RIPA/PI) – Make fresh daily
RIPA supplemented with 1 μM leupeptin, 1 μM pepstatin, 1 μM aprotinin, and 1 mM phenylmethyl sulfonyl fluoride.
1. Prepare Brain Slices
Make fresh 1x SACSF and 1x ACSF
Chill SACSF on ice in a small beaker. This will be used to hold the freshly harvested mouse brain.
Saturate the ACSF and SACSF with oxygen by bubbling with 95%/5% O2/CO2, 20 min on ice.
P30-38 mice should be used for optimal tissue viability. Sacrifice animals by cervical dislocation and decapitation, and rapidly remove brains into prechilled, oxygenated SACSF.
Using a vibrating microtome, make 300 μm brain sections in region of interest.
If desired, slices can be further dissected prior to recovery to enrich for particular brain regions or separate right and left hemispheres to use as control and experimental slices, respectively.
Using a fire-polished Pasteur pipette, transfer slices to mesh-bottomed chambers set into 24-well plates.
Allow slices to recover for 40 min, 31 °C in oxygenated ACSF, with continual, gentle bubbling.
2. Drug Treatment (if appropriate) and Slice Biotinylation
Following recovery, wash slices 3x in prewarmed (37 °C), oxygenated ACSF bubbling constantly with 95%/5% O2/CO2.
- Add test compounds and incubate with continuous oxygenation.
- For convenience, add a 1/10th volume of 10x concentrated drug, and mix by gently inverting.
- Shake plates gently in a water bath at the desired temperature.
Following drug treatment, rapidly chill slices by washing 3x in ice cold ACSF.
3. Biotinylate Surface Proteins
Prepare 1.0 mg/ml sulfo-NHS-SS-biotin in ice cold ACSF immediately prior to labeling.
Add 0.75 ml sulfo-NHS-SS-biotin to slices and incubate slices on ice, 45 min.
Wash slices three times quickly with ice cold ACSF, then incubate for 10 min in ice cold ACSF on ice.
Wash slices three times with ice cold slice quench buffer and incubate with 0.75 ml slice quench buffer two times, 25 min, on ice to quench free sulfo-NHS-SS-biotin.
4. Prepare Tissue Lysates
Wash slices three times in ice cold ACSF and transfer each slice to a microcentrifuge tube using a fire-polished Pasteur pipette.
Gently pellet slice by centrifuging 200 x g, 1 min and carefully aspirate remaining ACSF.
Add 400 µl ice cold RIPA/PI and break up tissue by pipetting up and down once through a P200 pipette.
Transfer dissociated slice/RIPA to a fresh tube and incubate 30 min, 4 °C, rotating, to complete lysis.
Pellet cellular debris by centrifuging, 18,000 x g, 15 min, 4 °C.
Determine lysate protein concentrations using the BCA protein assay, with bovine serum albumin (BSA) as a standard.
5. Isolate Biotinylated Proteins
- Optimize Bead/total protein ratio. Note: Individual protein expression levels can vary widely across different brain regions. Unless all of the biotinylated protein in a given amount of tissue lysate is captured, it is not possible to accurately detect any potential changes in surface expression. Therefore, it is imperative to empirically determine the optimal bead/total protein ratio for a particular protein/brain region prior to embarking upon a new slice biotinylation study.
- Incubate 25 µl streptavidin agarose beads with increasing amounts of tissue lysate (approximate range 25-200 µg), and then proceed as described below for binding, washing and elution steps.
- Quantify the resulting immunoreactive bands and choose a bead/lysate ratio in the linear range of binding that will permit accurate quantification of either increased or decreased protein surface expression.
- Prepare Streptavidin-Agarose Beads
- Determine total bead volume needed for all samples. Prepare a sufficient bead volume for that amount plus one extra (i.e. 4 samples at 25 µl/sample = 100 µl beads + one extra = 125 µl beads.
- Vortex Streptavidin bead stock and pipette out desired volume. If using a P200 pipette, cut off the end of the tip to prevent blockage or bead damage.
- Wash beads 3 times in 0.5-1.0 ml RIPA/PI to remove preservatives, vortexing after each RIPA addition and collecting beads between washes by centrifuging 18,000 x g, 1 min, at room temperature.
- Aspirate off buffer between washes using a glass Pasteur pipette attached to a vacuum flask. A plastic P200 tip attached to the end of the glass Pasteur pipette provides finer control when aspirating. It is not necessary to remove absolutely all the buffer for the first two washes, as it risks aspirating beads into the pipette. After the final wash, remove as much excess buffer as possible without aspirating the beads.
- Add RIPA/PI to bring beads back to their original volume, pipetting up and down to resuspend. Avoid vortexing at this point, as beads will stick to the tube wall.
- Aliquot beads into microcentrifuge tubes using a P200 with the tip cut off. Pipette up and down several times between sampling to assure beads remain evenly suspended and dispersed among the tubes.
- Bind biotinylated proteins to streptavidin beads
- Distribute cell lysates to the tubes containing the agarose beads. For experiments where multiple samples are being compared, make sure to use the same amount of protein for each sample for accurate comparisons.
- Add additional RIPA/PI to bring to samples to a 200 µl minimal volume. This assures that samples will mix adequately during the incubation and also unifies the protein concentrations across samples.
- Place tubes in a tube rotator and mix overnight at 4 °C.
- In separate tubes, dispense an equivalent of the total lysate volume used for each sample. Alternatively, if the sample volumes are high, a fraction of total lysate volume can be reserved instead, to accommodate maximal load volumes on SDS-PAGE gels. These will be used to normalize the surface expression to the total protein amount.
- Add either an equal volume of 2x or 1/5 volume of 6x SDS-PAGE sample buffer and either incubate at 4 °C, in parallel with bead samples, or store at -20 °C until samples are analyzed.
6. Elute and Analyze Samples
Pellet beads by centrifuging 18,000 x g, 2 min, at room temperature.
Aspirate supernatant and wash beads three times with 0.75 ml RIPA. To minimize bead loss, leave a small head volume of buffer above beads between washes. After the final wash, remove as much RIPA as possible, without disrupting the bead pellet.
Elute biotinylated proteins from streptavidin beads by reducing the disulfide linkage. Add 25 µl 2x SDS-PAGE reducing sample buffer, vortex well and rotate samples 30 min, room temperature. Note: Many membrane proteins have a high tendency to aggregate when boiled in SDS-PAGE sample buffer, severely impairing their electrophoretic mobility. If this is the case for the protein being investigated, avoid heating samples and, instead, elute slowly at room temperature. If boiling is absolutely necessary (e.g. if another protein assessed in parallel requires boiling), sample buffer can be supplemented with 2 M urea (final concentration) to minimize aggregation. While effective in reducing aggregation in some cases, urea often compromises band appearances.
Analyze samples by immunoblot.
Thaw total lysate samples and rotate in parallel with bead samples, 30 min, room temperature.
Separate proteins on SDS-PAGE gels.
- Identify protein(s) of interest by immunoblotting.
- Be certain that bands are detected in the linear range of detection for proper quantification.
- To assure that the biotinylation reagent has not gained access to intracellular proteins via damaged/compromised cells. This is best accomplished by immunoblotting in parallel for an intracellular protein specific to the cell type being investigated.
- Quantify band densities and calculate relative protein surface density as a percent of the total protein expression level.
Representative Results
The neuronal dopamine transporter is internalized in response to PKC activation in cell lines16-20. Despite many reports demonstrating PKC-induced DAT surface losses in a variety of cell lines and expression systems, it has been challenging to confirm this finding in cultured dopaminergic neurons21-23. We used mouse striatal slices to directly test whether DAT internalizes in response to PKC activation in adult dopaminergic neurons. Following slice preparation, slices were hemisected along the midline and slices from identical planes were treated ±1 µM phorbol myristate-13 acetate (PMA) for 30 min, 37 °C. Slices were rapidly chilled and surface proteins were biotinylated and isolated as described in the Protocol. Immunoblots were probed for DAT, and also for tyrosine hydroxylase (TH), in parallel, to measure whether the biotinylation reagent gained any intracellular access in dopaminergic neurons. As seen in Figure 1 (Top), we detected robust DAT surface expression in mouse striatum under basal (vehicle-treated) conditions, with 81.4±5.8% of total DAT at the cell surface. PKC activation with 1 µM PMA, 30 min, 37 °C significantly decreased DAT surface expression to 60.8±5.2% total DAT, which corresponds to ~30% loss of DAT from the plasma membrane. In contrast, only 1.6±0.4% of total TH was biotinylated (Figure 1, bottom), consistent with its intracellular localization and confirming that the biotinylation reagent was excluded from the cell interior of dopaminergic neurons.
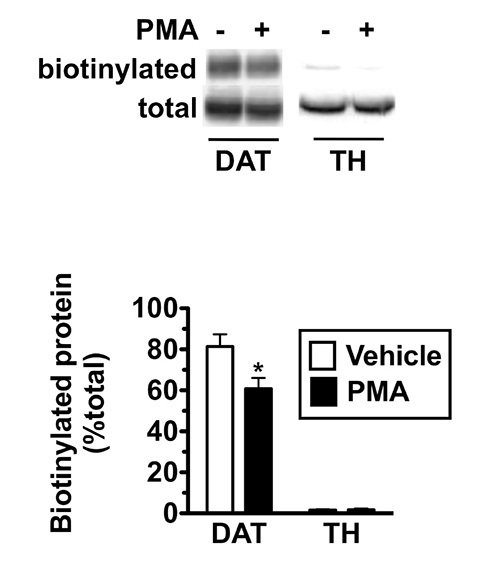 Figure 1. Acute PKC activation decreases dopamine transporter surface levels in adult striatal neurons. Mouse Striatal Slice Biotinylation. Acute mouse striatal slices were prepared as described in Protocol and were treated ±1 µM PMA, 30 min, 37 °C. Surface proteins were covalently coupled to biotin and were isolated by batch streptavidin chromatography. Samples underwent SDS-PAGE and immunoblotting with rat anti-DAT and mouse anti-TH antibodies. Immunoreactive bands were captured with a VersaDoc CCD camera imaging station and densities from nonsaturating bands were quantified using Quantity One software. Top: Representative immunoblot displaying biotinylated and total DAT and TH following the indicated treatments. Bottom: Average data. Biotinylated protein signal expressed as % total protein ± S.E.M. *Significantly different from control, Student’s t test, p < 0.03, n = 6.
Figure 1. Acute PKC activation decreases dopamine transporter surface levels in adult striatal neurons. Mouse Striatal Slice Biotinylation. Acute mouse striatal slices were prepared as described in Protocol and were treated ±1 µM PMA, 30 min, 37 °C. Surface proteins were covalently coupled to biotin and were isolated by batch streptavidin chromatography. Samples underwent SDS-PAGE and immunoblotting with rat anti-DAT and mouse anti-TH antibodies. Immunoreactive bands were captured with a VersaDoc CCD camera imaging station and densities from nonsaturating bands were quantified using Quantity One software. Top: Representative immunoblot displaying biotinylated and total DAT and TH following the indicated treatments. Bottom: Average data. Biotinylated protein signal expressed as % total protein ± S.E.M. *Significantly different from control, Student’s t test, p < 0.03, n = 6.
Discussion
Despite longstanding knowledge that endocytic trafficking critically impacts synaptic signaling in the brain, it has proved challenging to quantitatively measure changes in protein surface expression in adult neurons. In this work, we report a reliable approach to label surface protein ex vivo in acute brain slices. Brain slice preparations have a longstanding history of utility for electrophysiological recordings, as they maintain synaptic connections and cell viability up to hours after their preparation. Moreover, slicing strategies can be optimized to preserve specific synaptic connections between various brain regions of interest.
Much of the prior work investigating neuronal protein trafficking in brain-derived preparations has depended primarily upon synaptosomes and primary cultured neurons. The acute slice biotinylation approach method boasts several advantages over either of these: synaptosomes are physically removed from the axon and may not contain the necessary molecular factors to faithfully recapitulate intact neuronal protein trafficking. Additionally, synaptosomal preparations are often contaminated with membrane fragments that may skew experimental results. Primary neuronal cultures are typically derived from developmentally immature neurons that may not have appropriately differentiated into their mature neuronal phenotypes expressing cell-specific trafficking mechanisms. In contrast, acute slices exhibit high degree of cell viability and are derived from adult animals. Moreover, surface trafficking can be compared following in vivo molecular manipulations, such as gene delivery/knockdown, optogenetic neuronal stimulation/inhibition, or following in vivo drug treatments or behavioral adaptations.
Although there are many advantages to the ex vivo slice approach, there are several limitations as well. Acute (noncultured) brain slices have limited viability and are therefore not suitable for chronic drug treatments. Moreover, we observed that they do not remain viable during temperature shift experiments, where slices are rapidly chilled and rewarmed. Further, slices are most viable when prepared from P21-P35 mice, potentially limiting the amount of time that in vivo treatments can be undertaken prior to performing experiments. Indeed, using a high-sucrose cutting solution, we find that DAT trafficking is less reproducibly observed in slices prepared from P35-P42 mice (Gabriel and Melikian, unpublished data). However, we observe markedly improved slice viability in older animals using the modified slice preparation as described by Zhao et al.24 This approach is an excellent alternative where methodological parameters require using older animals (i.e. following either viral-mediated protein/RNA expression, establishing behaviors, or chronic in vivo drug treatments).
There are several additional technical factors that may influence the experimental outcome. It is imperative to empirically determine the optimal bead/total protein ratio needed to capture all of the biotinylated protein in a given amount of total lysate protein prior to attempting quantitative experiments. This issue is frequently overlooked, but can be the deciding factor in being able to detect changes in protein surface expression. If all of the biotinylated protein is not quantitatively recovered, changes in surface levels will be either undetectable or inaccurately measured. Another variable that could influence outcomes is the volume of lysis buffer used to dissociate the tissue slices. The absolute tissue mass used will depend on the region of interest being investigated, and will likely require more or less lysis buffer in order to achieve complete tissue solubilization. As such, final lysate protein concentrations may also vary across brain regions. Therefore, lysis conditions should be empirically determined for a given brain region and kept constant between independent experiments.
In summary, we provide a detailed protocol for measuring neuronal protein trafficking in adult neurons using ex vivo acute brain slices. Utilization of this approach is likely to lead to a more in-depth understanding of endocytic trafficking mechanisms that underlie neuronal function.
Disclosures
The authors have no financial interests that conflict with this work.
Acknowledgments
This work was funded by NIH grants DA15169 and DA035224 to H.E.M.
References
- Conner SD, Schmid SL. Regulated portals of entry into the cell. Nature. 2003;422:37–44. doi: 10.1038/nature01451. [DOI] [PubMed] [Google Scholar]
- Zastrow M, Williams JT. Modulating neuromodulation by receptor membrane traffic in the endocytic pathway. Neuron. 2012;76:22–32. doi: 10.1016/j.neuron.2012.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leto D, Saltiel AR. Regulation of glucose transport by insulin: traffic control of GLUT4. Nat. Rev. Mol. Cell Biol. 2012;13:383–396. doi: 10.1038/nrm3351. [DOI] [PubMed] [Google Scholar]
- Liu YW, Lukiyanchuk V, Schmid SL. Common membrane trafficking defects of disease-associated dynamin 2 mutations. Traffic. 2011;12:1620–1633. doi: 10.1111/j.1600-0854.2011.01250.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, DiFiglia M. The recycling endosome and its role in neurological disorders. Prog. Neurobiol. 2012. pp. 127–141. [DOI] [PubMed]
- Sandvig K, Pust S, Skotland T, van Deurs B. Clathrin-independent endocytosis: mechanisms and function. Curr. Opin. Cell Biol. 2011;23:413–420. doi: 10.1016/j.ceb.2011.03.007. [DOI] [PubMed] [Google Scholar]
- Kumari S, Mg S, Mayor S. Endocytosis unplugged: multiple ways to enter the cell. Cell Res. 2010;20:256–275. doi: 10.1038/cr.2010.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barry MF, Ziff EB. Receptor trafficking and the plasticity of excitatory synapses. Curr. Opin. Neurobiol. 2002;12:279–286. doi: 10.1016/s0959-4388(02)00329-x. [DOI] [PubMed] [Google Scholar]
- Bredt DS, Nicoll RA. AMPA receptor trafficking at excitatory synapses. Neuron. 2003;40:361–379. doi: 10.1016/s0896-6273(03)00640-8. [DOI] [PubMed] [Google Scholar]
- Kerchner GA, Nicoll RA. Silent synapses and the emergence of a postsynaptic mechanism for LTP. Nat. Rev. Neurosci. 2008;9:813–825. doi: 10.1038/nrn2501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malinow R, Malenka RC. AMPA receptor trafficking and synaptic plasticity. Annu. Rev. Neurosci. 2002;25:103–126. doi: 10.1146/annurev.neuro.25.112701.142758. [DOI] [PubMed] [Google Scholar]
- Borgland SL, Malenka RC, Bonci A. Acute and chronic cocaine-induced potentiation of synaptic strength in the ventral tegmental area: electrophysiological and behavioral correlates in individual rats. J. Neurosci. 2004;24:7482–7490. doi: 10.1523/JNEUROSCI.1312-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong Y, et al. Cocaine-induced potentiation of synaptic strength in dopamine neurons: Behavioral correlates in GluRA(-/-) mice. PNAS. 2004;101:14282–14287. doi: 10.1073/pnas.0401553101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyman SE, Malenka RC, Nestler EJ. Neural mechanisms of addiction: the role of reward-related learning and memory. Annu. Rev. Neurosci. 2006;29:565–598. doi: 10.1146/annurev.neuro.29.051605.113009. [DOI] [PubMed] [Google Scholar]
- Thomas MJ, Malenka RC. Synaptic plasticity in the mesolimbic dopamine system. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 2003;358:815–819. doi: 10.1098/rstb.2002.1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorkina T, Hoover BR, Zahniser NR, Sorkin A. Constitutive and protein kinase C-induced internalization of the dopamine transporter is mediated by a clathrin-dependent mechanism. Traffic. 2005;6:157–170. doi: 10.1111/j.1600-0854.2005.00259.x. [DOI] [PubMed] [Google Scholar]
- Holton KL, Loder MK, Melikian HE. Nonclassical, distinct endocytic signals dictate constitutive and PKC-regulated neurotransmitter transporter internalization. Nat. Neurosci. 2005;8:881–888. doi: 10.1038/nn1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loder MK, Melikian HE. The dopamine transporter constitutively internalizes and recycles in a protein kinase C-regulated manner in stably transfected PC12 cell lines. J. Biol. Chem. 2003;278:22168–22174. doi: 10.1074/jbc.M301845200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melikian HE, Buckley KM. Membrane trafficking regulates the activity of the human dopamine transporter. J. Neurosci. 1999;19:7699–7710. doi: 10.1523/JNEUROSCI.19-18-07699.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniels GM, Amara SG. Regulated trafficking of the human dopamine transporter. Clathrin-mediated internalization and lysosomal degradation in response to phorbol esters. J. Biol. Chem. 1999;274:35794–35801. doi: 10.1074/jbc.274.50.35794. [DOI] [PubMed] [Google Scholar]
- Sorkina T, et al. RNA interference screen reveals an essential role of Nedd4-2 in dopamine transporter ubiquitination and endocytosis. J. Neurosci. 2006;26:8195–8205. doi: 10.1523/JNEUROSCI.1301-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eriksen J, et al. Visualization of dopamine transporter trafficking in live neurons by use of fluorescent cocaine analogs. J. Neurosci. 2009;29:6794–6808. doi: 10.1523/JNEUROSCI.4177-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao A, Simmons D, Sorkin A. Differential subcellular distribution of endosomal compartments and the dopamine transporter in dopaminergic neurons. Mol. Cell Neurosci. 2011;46:148–158. doi: 10.1016/j.mcn.2010.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao S, et al. Cell type-specific channelrhodopsin-2 transgenic mice for optogenetic dissection of neural circuitry function. Nat. Methods. 2011;8:745–752. doi: 10.1038/nmeth.1668. [DOI] [PMC free article] [PubMed] [Google Scholar]