

Abstract
Adherent cells in culture maintain a polarized state to support movement and intercellular interactions. Nanopodia are thin, elongated, largely F-actin-negative membrane projections in endothelial and cancer cells that can be visualized through TM4SF1 (Transmembrane-4-L-six-family-1) immunofluorescence staining. TM4SF1 clusters in 100-300 μm diameter TMED (TM4SF1 enriched microdomains) containing 3 to as many as 14 individual TM4SF1 molecules. TMED are arranged intermittently along nanopodia at a regular spacing of 1 to 3 TMED per μm and firmly anchor nanopodia to matrix. This enables nanopodia to extend more than 100 μm from the leading front or trailing rear of polarized endothelial or tumor cells, and causes membrane residues to be left behind on matrix when the cell moves away. TMED and nanopodia have been overlooked because of their extreme fragility and sensitivity to temperature. Routine washing and fixation disrupt the structure. Nanopodia are preserved by direct fixation in paraformaldehyde (PFA) at 37 °C, followed by brief exposure to 0.01% Triton X-100 before staining. Nanopodia open new vistas in cell biology: they promise to reshape our understanding of how cells sense their environment, detect and identify other cells at a distance, initiate intercellular interactions at close contact, and of the signaling mechanisms involved in movement, proliferation, and cell-cell communications. The methods that are developed for studying TM4SF1-derived nanopodia may be useful for studies of nanopodia that form in other cell types through the agency of classic tetraspanins, notably the ubiquitously expressed CD9, CD81, and CD151.
Keywords: Cellular Biology, Issue 86, nanopodia, TM4SF1, endothelial cell, tumor cell, F-actin, immunofluorescence staining, tetraspanin
Introduction
During polarization for movement, animal cells extend a variety of dynamic, membrane protrusions from their surfaces, including filopodia, lamellipodia, retraction fibers, and ruffles1. Recently added to this list were nanopodia, a newly recognized type of thin (100-300 μm in diameter), elongated (up to 50-100 μm long) membrane projection that provide membrane channels for the extension of F-actin structures such as filopodia and retraction fiber, and that stain positively with TM4SF1 (Transmembrane-4-L-six-family-1) in cultured endothelial and tumor cells2,3.
TM4SF1 is a protein with tetraspanin-like topology that was originally known as a tumor cell antigen4 before the discovery that the molecule is an endothelial cell biomarker that plays an essential role in endothelial cell proliferation and migration2,3. Immunofluorescence staining revealed that TM4SF1 is localized to perinuclear vesicles and to the plasma membrane, and is enriched in TM4SF1 enriched microdomains (TMED). TMED anchor nanopodia to matrix and present in a regularly spaced banded pattern of 1-3 TMED/µm length of nanopodia. Nanopodia typically extend from a cell's leading front and trailing rear during cell polarization for movement. Due to the firm adherent nature of TMED, nanopodia are unable to retract back into the cell as it moves away; abandoned nanopodia residues thus trace out the path of cellular movement. Nanopodia provide membrane channels for F-actin extension and retraction, and are sites of intercellular interactions and communications2,3. These characteristics mean that nanopodia provide a unique opportunity to study the mechanisms underlying F-actin assembly during cellular polarization, cellular sensing of the environment, determination of the path and direction of cell movement, and intercellular interactions and communications.
Due to the highly hydrophobic nature of TMED and the thin and fragile membranous nature of nanopodia, special care needs to be taken in order to preserve TMED and nanopodia. The destruction of nanopodia and removal of TMED by common laboratory methods is a possible reason why only sixty-three publications have appeared on TM4SF1 since its first discovery in 19861 and for the complete lack of knowledge of TM4SF1 enrichment in 100-300 μm microdomains on the cell surface until the report of TM4SF1 in endothelial cells in 20092.
Conventional immunostaining methods commonly use organic solvents like ethanol, methanol, or acetone to fix cells and use 0.1% or higher Triton X-100 concentration to permeabilize cells5. Studies described here implemented three major changes to the conventional method to reveal TMED and nanopodia: (i) use 37 °C 4% PFA and fix cells in a 37 °C incubator, (ii) apply gentle room temperature PBS washing, and (iii) use less than 0.03% Triton X-100 only briefly to permeabilize cells before addition of primary antibody, as Triton X-100 higher than 0.03% will extract TM4SF1.
All tetraspanins form microdomains on the cell membrane6 and some colocalize with TMED in endothelial and tumor cells2,3. As tetraspanins like CD9, CD81, and CD151 are ubiquitously expressed, the staining protocol described here can be extended to many different cell types that lack TM4SF1 for the studies of nanopodia function.
Protocol
1. Cell Culture on Collagen Coated Glass Disk
Place glass disks (12 mm in diameter) in a glass jar (4 oz) and autoclave to sterilize the discs.
Put 25 ml 70% ethanol in a 50 ml Falcon tube and place it in a cell culture hood. This solution can be reused multiple times until the level of the solution drops to 20 ml.
Place a sharp forceps with an extra fine point in the 70% ethanol for 5 min before using it to handle the glass disk.
Carefully remove the forceps out of the tube and close the cap, then gently place the ethanol treated forceps on top of the tube to air dry for 2 min. Do not allow the tip of the forceps to touch anything in the hood.
Use the sterile forceps to grab one sterile glass disk out of the glass jar and place it in a well of the 24-well cell culture plate. Repeat until the desired number of wells has been populated with discs.
Put 500 μl of Bovine collagen solution (50 ng/ml) into each well that contains a glass disk and place it in a 37 °C, 5% CO2 cell culture incubator for at least 30 min. There is no harm in a longer incubation.
Harvest HUVEC (Human Umbilical Vein Endothelial Cells) or PC3 (prostate tumor cells) that were already grown in 150 mm cell culture plate through trypsinization and block the trypsin activity using its complete culture medium. Collect cells into a 15 ml Falcon tube.
Count the cell number and make sure the viability is greater than 90%.
Pellet the cells by centrifuging at 200 x g for 10 min at 4 °C. Remove the supernatant.
Use the culture medium to dilute the cells to 105 cells/ml. Place cells on ice.
Aspirate the collagen in the cell culture hood and gently place 500 μl of cell suspension to each well in which glass disks were precoated with collagen. 5 x 104 cells/well in the 24-well plate will give 60% confluency for HUVEC and 30% confluency for PC3 cells. Note: add more cell suspension for higher cell density.
Culture the cells in a 37 °C, 5% CO2 incubator for 1 hr, 2 hr, 4 hr, 6 hr, or 24 hr to track nanopodia activities and cell passage. Typically, cells will attach to the glass disk in the first 30 min, then start to polarize and migrate in the first hour, and perform intercellular interactions and cell division in the following hours.
2. Cell Fixation
Each well will need 500 μl 4% PFA (paraformaldehyde) to fix the cells. Either commercially manufactured or freshly made 4% PFA can be used. Calculate the amount of PFA needed based on the number of wells prepared, and place PFA in a 15-ml falcon tube. CAUTION: paraformaldehyde is a suspected carcinogen.
Put the falcon tube in a Styrofoam stand that was already in place in a 37 °C incubator. Leave the tube in the incubator for 30 min to warm up the PFA to 37 °C.
Place a heating pad in the culture hood and turn it on.
Take out the 24-well cell culture plate and the prewarmed PFA out of the incubator and place it on top of the heating pad.
Use one hand to aspirate the medium from a well, and immediately after use the other hand to gently slide 500 μl PFA into the well through the well edge. For easier aspiration, tilt the culture plate at about 45°, leaning it against the Styrofoam stand so that it is stable at that angle. This will facilitate the aspiration and adding PFA solution to the well without disturbing the cells in culture. This is the most crucial step to preserve the original cellular structure of cell activities.
Repeat the process until all wells with a glass disk have received PFA. Note: all steps that need to change pre-existing solution from the well will apply same aspiration procedures.
Place the plate in 37 °C for 5 min.
Take the plate back to the culture hood and tilt against Styrofoam as described in step 2.5. Use one hand to gently transfer the PFA from the well into a collection tube; use the other hand to immediately afterward place at least 500 μl room temperature PBS in the well. After all wells have been washed, return and repeat the PBS washing once more in every well. Empty the collected PFA into a hazardous waste container.
Prepare ICC blocking buffer by adding 2% Fetal Bovine Serum to PBS. 0.04% sodium azide (dilute from 20% stock solution) is added as a preservative. CAUTION: sodium azide is a toxic compound. The buffer can be stored in 4 °C for a long period of time without bacterial contamination.
With the culture plate still tilted against a stand, once again cycle through each well aspirating to remove PBS and immediately afterward adding 500 μl of ICC blocking buffer. Cells are now ready for immunofluorescence staining. If necessary, the fixed cells can be stored in a 4 °C refrigerator for a week without significant loss of TM4SF1 protein.
3. TM4SF1 Immunofluorescence Staining of Nanopodia
In each well, replace the 500 μl ICC blocking buffer with a new blocking buffer containing 0.01% Triton X-100 and let it sit at room temperature for 1 hr. Do not use higher than 0.03% Triton X-100 as it will remove TM4SF1 from cell membranes. PBS washed cells from step 2.10 can be directly moved to the blocking buffer containing Triton if the staining is ready to be started right away.
Dilute primary anti-TM4SF1 (or anti-CD9) antibody to 0.5 μg/ml in ICC blocking buffer.
In each well, remove the ICC/0.01% Triton buffer and replace it with 300 μl of the anti-TM4SF1-antibody solution. Incubate 2 hr at room temperature or leave overnight at 4 °C.
Wash each well with no less than 500 μl PBS. Repeat 3x; each time give at least 5 min of incubation time.
Prepare a secondary antibody and phalloidin solution in ICC blocking buffer, using a 1,000 times dilution of Alexa 488 labeled donkey anti-mouse secondary antibody (2 μg/ml final concentration) and 1,000x dilution of phalloidin (50 ng/ml final concentration).
Remove PBS and add 300 μl of the secondary antibody and phalloidin solution to each well and incubate for 2 hr or leave it overnight at 4 °C.
Repeat PBS washing steps with at least a 5 min incubation per wash. In the last wash, leave each well in PBS for 1 hr.
Drop a single drop (~10 μl) of anti-fade mounting media on a glass slide.
Bend the tip of a 19 G 1 ½ in syringe needle 90° by pressing gently on a hard surface. Use one hand to hold bended needle and gently lift up a glass disk from the well, and use the other hand to grasp the disc using the sharp forceps.
Turn the glass disk face down letting the cell side contact the mounting media on the glass slide. Gently place the glass disk and let it dry overnight in a dark place at room temperature.
Proceed to image the immunofluorescence stained nanopodia, or store the slides in a slide box at -20 °C. The slides will remain viewable for a few months in -20 °C.
Representative Results
For Step 1:
If cells (such as HUVEC and PC3 used in this study) are able to grow normally, cells will attach to the collagen-coated disc within 30 min after they are seeded, polarize and become mobile soon afterward, and extend nanopodia ahead of their path of movement. Figures 1A and 1B respectively shows a polarized and proliferating HUVEC. Figure 2A shows polarized PC3 cells in a mobile state.
For Step 2:
With prewarming of 4% PFA to 37 °C, fixing cells at 37 °C for 20 min, and gentle washing with room temperature PBS, cellular states should be well preserved. Generally, isolated individual HUVEC (Figure 1A, 1) or PC3 (Figure 2A) cells will show a polarized morphology, unless cells are in the state of cell division (Figure 1B, 1). Intercellular interactions leading to cell junction formation (Figure 1C, 1) are also commonly observed. Nanopodia should appear on the cell periphery, and F-actin will often be present in portions of the nanopodia proximal to the cell. Cold temperatures cause F-actin to retract back into cells leaving behind nanopodia membrane channels. Suboptimal temperature, as shown in examples using 4 °C PFA fixation and cold PBS washing, will disturb and destroy nanopodia structure in polarized cells (Figure 1A, 2), proliferating cells (Figure 1B, 2), and artificially produce gaps at intercellular junctions (Figure 1C, 2).
For Step 3:
Many membrane-bound proteins including TM4SF1 are very sensitive to Triton X-100 and will be removed from the cell surface if concentrations higher than 0.03% are used2,3. In order to preserve most of cell surface TM4SF1, PFA fixed cells should only be treated with 0.01% Triton X-100 containing blocking buffer for an hour before changing to anti-TM4SF1 primary antibody that has been diluted in a regular blocking buffer that does not contain Triton X-100. As nanopodia are thin and long membrane projections and attach to matrix through TMED, TMED intensity will likely need to be enhanced through brightness and contrast functions in image processing software like Photoshop in order to reveal and track the path of cell movement through nanopodia residues (Figure 1, 2insets).
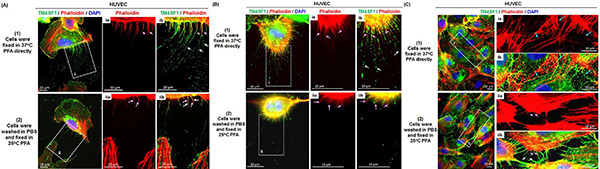 Figure 1. Nanopodia projections during endothelial cell movement, cell division,and junction formation. HUVEC were freshly cultured for 6 hr after plating at 60% confluence on collagen-coated glass disk. Cells were fixed using (1) the modified method where cells were fixed in 37 °C 4% PFA directly without prewashing in PBS, and placed in a 37 °C incubator during the 20 min fixation time, or (2) a conventional staining method where the cells were washed with room temperature PBS twice and fixed in 4% PFA for 20 min at room temperature. Nanopodia, thin and long membrane extensions that are marked by an intermittent, banded pattern of TMED (white arrows) and that often enclose phalloidin-stained F-actin (pink arrows) in portions proximal to the cell, were immunofluorescence stained using anti-TM4SF1 Ab. Three representative HUVEC images were captured to show the appearance of nanopodia during (A) polarization, (B) cell division, and (C) intercellular junction formation. Inset boxes show higher magnifications of nanopodia. During polarized (A) or retracted cellular states (B, C), the nanopodia projections were preserved by directly fixing cells in 37 °C PFA, whereas the structures were largely removed by washing and fixing the cells in room temperature PBS and PFA. (C) 37 °C PFA fixation preserves intercellular junctions (blue arrows), whereas junctions are disturbed by colder temperatures that cause cells to retract, creating a gap between cells bridged by nanopodia of which most contain F-actin (pink arrows) and some do not (white arrows) Click here to view larger image.
Figure 1. Nanopodia projections during endothelial cell movement, cell division,and junction formation. HUVEC were freshly cultured for 6 hr after plating at 60% confluence on collagen-coated glass disk. Cells were fixed using (1) the modified method where cells were fixed in 37 °C 4% PFA directly without prewashing in PBS, and placed in a 37 °C incubator during the 20 min fixation time, or (2) a conventional staining method where the cells were washed with room temperature PBS twice and fixed in 4% PFA for 20 min at room temperature. Nanopodia, thin and long membrane extensions that are marked by an intermittent, banded pattern of TMED (white arrows) and that often enclose phalloidin-stained F-actin (pink arrows) in portions proximal to the cell, were immunofluorescence stained using anti-TM4SF1 Ab. Three representative HUVEC images were captured to show the appearance of nanopodia during (A) polarization, (B) cell division, and (C) intercellular junction formation. Inset boxes show higher magnifications of nanopodia. During polarized (A) or retracted cellular states (B, C), the nanopodia projections were preserved by directly fixing cells in 37 °C PFA, whereas the structures were largely removed by washing and fixing the cells in room temperature PBS and PFA. (C) 37 °C PFA fixation preserves intercellular junctions (blue arrows), whereas junctions are disturbed by colder temperatures that cause cells to retract, creating a gap between cells bridged by nanopodia of which most contain F-actin (pink arrows) and some do not (white arrows) Click here to view larger image.
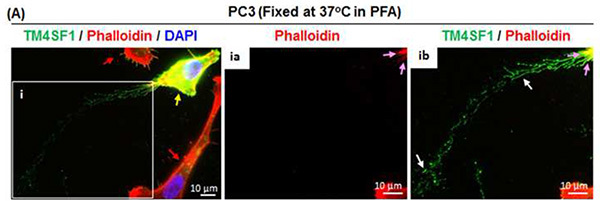 Figure 2. Nanopodia demonstrate the direction of PC3 tumor cell movement. (A) PC3 tumor cells were freshly cultured for 6 hr after plating at 60% confluence on collagen-coated glass disk. Cells were fixed directly in 37 °C 4% PFA and placed in a 37 °C incubator for 20 min to preserve nanopodia. Nanopodia (white arrows) and F-actin (pink arrows) were immunofluorescence stained using anti-TM4SF1 Ab and phalloidin, respectively. The inset box shows nanopodia at higher magnification. This representative image shows that TM4SF1 expression in PC3 cells is heterogeneous; one PC3 cell with strong TM4SF1 staining (yellow arrow) produced long nanopodia and is surrounded by two weakly TM4SF1-stained PC3 cells (red arrows). Nanopodia residues at the trailing edge (ib) indicate the direction of PC3 cell movement during the 6 hr cell culture period. Click here to view larger image.
Figure 2. Nanopodia demonstrate the direction of PC3 tumor cell movement. (A) PC3 tumor cells were freshly cultured for 6 hr after plating at 60% confluence on collagen-coated glass disk. Cells were fixed directly in 37 °C 4% PFA and placed in a 37 °C incubator for 20 min to preserve nanopodia. Nanopodia (white arrows) and F-actin (pink arrows) were immunofluorescence stained using anti-TM4SF1 Ab and phalloidin, respectively. The inset box shows nanopodia at higher magnification. This representative image shows that TM4SF1 expression in PC3 cells is heterogeneous; one PC3 cell with strong TM4SF1 staining (yellow arrow) produced long nanopodia and is surrounded by two weakly TM4SF1-stained PC3 cells (red arrows). Nanopodia residues at the trailing edge (ib) indicate the direction of PC3 cell movement during the 6 hr cell culture period. Click here to view larger image.
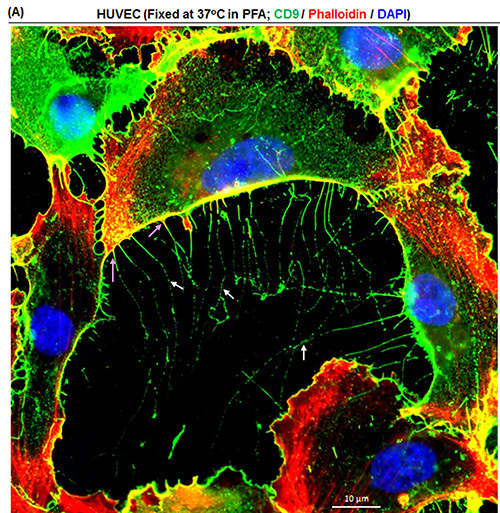 Figure 3. Visualization of nanopodia through tetraspanin CD9. (A)HUVEC were freshly cultured for 6 hr after plating at 60% confluency on a collagen-coated glass disk. Cells were fixed in 37 °C 4% PFA and nanopodia (white arrows) were revealed through anti-CD9 Ab. F-actin (pink arrows) was stained using phalloidin. This representative image shows that CD9 also localizes to nanopodia. Please click here to view a larger version of this figure.
Figure 3. Visualization of nanopodia through tetraspanin CD9. (A)HUVEC were freshly cultured for 6 hr after plating at 60% confluency on a collagen-coated glass disk. Cells were fixed in 37 °C 4% PFA and nanopodia (white arrows) were revealed through anti-CD9 Ab. F-actin (pink arrows) was stained using phalloidin. This representative image shows that CD9 also localizes to nanopodia. Please click here to view a larger version of this figure.
Discussion
Nanopodia are thin cellular membrane channels that firmly attach to matrix through TMED and can extend more than 100 μm from a polarized mobile cell to sense the environment and mediate intercellular interactions2,3. Nanopodia adhere to matrix so firmly that residues are left behind as the cell moves away (Figures 1 and 2). Nanopodia thus allow us to study how cells sense their environment and determine the path of cell movement, and how gene expression or drug treatment changes movement. However, due to the fragility of these thin (100-300 μm width) membrane structures, caution needs to be taken in order to preserve them.
In order to faithfully record cellular activities without disturbing nanopodia, and to study how experimental conditions affect nanopodia activities and the path of cell movement, one should first verify that cells grow effectively in complete medium before pursuing the experiment. Normal healthy primary endothelial cells, such as HUVEC, in fresh EGM2-MV complete culture medium will normally divide approximately every 18 hr during their log phase of growth within six passages of their culture. One should avoid the use of HUVEC that are greater than passage-6 as higher passage numbers will lead to increased numbers of senescent or binuclear cells in culture7 and will affect cells' ability to polarize and produce nanopodia2. PC3 prostate tumor cells are more stable to cultures (unpublished observation), but one should still avoid the use of cells more than ten passages from their removal from the frozen state.
Healthy adherent cells will typically attach to collagen-coated discs within the first 30 min after they are seeded, followed soon afterward by cell polarization. Thus one should expect to see most endothelial cells in a polarized state at the end of the first hour of incubation. If a majority of endothelial cells do not attach to the glass surface, then check whether (a) collagen has passed its expiration date, (b) trypsinization during cell harvest was too stringent, or (c) cells are unhealthy. PC3 cells polarize slower than endothelial cells, but should demonstrate a polarized morphology within 2 hr after seeding. If contamination occurs in culture, then examine whether glass disks or forceps were properly sterilized. Alternatively, one can consider using chamber slides; the protocol described here suggests the use of glass disks in 24-well culture plates in order to save cost and to more easily handle a large number of samples.
Conventional immunostaining methods, use of PBS to wash cells before adding room temperature fixative, easily disturb fine cellular structures like nanopodia and will destroy them during the staining process. This is largely due to the fact that physical stresses like temperature fluctuations during cell fixation or rigorous PBS washing during the staining process are the main causes of fragmentation or removal of nanopodial structures. With three simple methodological changes - (i) do not disturb cell polarization state by washing with PBS before fixation, (ii) fix cells directly in 37 °C prewarmed PFA and incubate cells at 37 °C during fixation, and (ii) gently wash cells in room temperature PBS - nanopodia are largely preserved. Figure 1 shows how PFA and PBS temperatures affect preservation of nanopodia in cultured endothelial cells; Figure 2A shows that TM4SF1 expression is heterogeneous in PC3 cells and that cells with the strongest TM4SF1 staining display the longest nanopodia. Temperature is known to affect F-actin activities and tubulin integrity8,9. Thus fixing cells at the same temperature in which cells were grown (this is 37 °C in the protocol described here) is likely to better preserve the polarized cellular state (Figures 1A and 1B) and intercellular junctions (Figure 1C), permitting more faithful recording of cellular activities at the time when cells are removed for experimental studies. Unlike on the retracting side, nanopodia projections at the cell's leading front are short lived, in part because the cells are constantly moving and overrun leading edge nanopodia as they move forward2,3. Thus the best time to observe nanopodia at the leading front is not long after cells are plated, ideally at around 30-60 min after seeding, a time when cells have attached to matrix and are only beginning to initiate movement. Live cell imaging is an alternative approach which enables a precise record of nanopodia activities at the leading front.
PFA is not the only reagent used in fixation; organic solvents like methanol, ethanol, and acetone are also frequently used in cell fixation for immunostaining10. The best fixative is often decided by the epitope location of a specific antibody used in the staining (unpublished data). For the mouse anti-human TM4SF1 antibody that recognizes extracellular domains of the TM4SF1 (Millipore; used in this study), PFA is the only fixative which reveals TMED in immune-staining, indicating that other organic solvents destroy the epitope. In contrast, rabbit anti-human TM4SF1 polyclonal antibody from Acris (data not shown), whose epitope is located in eighteen amino acids near the N-terminal region, works also with ethanol fixed cells. However, the Acris antibody does not yield good TM4SF1 staining, probably because the N-terminal epitope is preoccupied by TM4SF1 interactions with cytosolic molecule(s) which partially cover the epitope.
Triton X-100 is a nonionic surfactant and is often used in immune-staining with the aim of permeabilizing cell membrane and reducing surface tension of aqueous solutions11,12. However, this capability also brings the risk of solubilizing membrane proteins. TM4SF1 is completely removed from the cell surface by Triton X-100 concentrations greater than 0.05% 2,3. For this reason, only a 1-hr treatment with 0.01% Triton X-100 before addition of the primary antibody is used in the method described here. This treatment permeabilizes the membrane enough to enable peri-nuclear TM4SF1 to be well assessed by the antibodies, while preserving most TM4SF1 on the cell membrane. For double staining of TM4SF1 with proteins located in the nucleus, use 0.03% Triton X-100 in the preincubation stage if 0.01% Triton does not work. This will facilitate Triton X-100 puncturing of the nuclear membrane.
TM4SF1 is highly specific to endothelial cells and tumor cells; most other cell types either do not express TM4SF1 or express it at a very low level2,3. However, classic tetraspanins - a family that encompasses thirty-three members including CD9, CD81, and CD151 - are ubiquitously expressed6, and can also be found in membrane domains in nanopodia2,3 (Figure 3A). Thus, staining classic tetraspanins through the same immune-staining method used for TM4SF1 can enable the studies of nanopodia in cell types that do not express TM4SF1. PC3 tumor cells that express TM4SF1 highly tend to move continuously in a single direction, whereas PC3 cells that weakly express TM4SF1 do not have a clearly defined direction of movement (Figure 2A and unpublished data). TM4SF1 expression level in tumor cells is known to be correlated with their metastatic potential13, and investigations are currently underway linking tumor cells' ability to express TM4SF1 and produce nanopodia to their metastatic potential.
In summary, mastery of effective methods for staining nanopodia enables investigators to track the path of cell movement through nanopodia residues, study how cells sense their environment for movement, how cell movement is modified by alteration of the expression levels of genes (either overexpression or knockdown of genes of interest). Nanopodia represent a new tool for investigation of cell polarization and cell movement.
Disclosures
No conflicts of interest declared.
Acknowledgments
We acknowledge Dr. Harold Dvorak for helpful discussions including the suggestion to try fixation in 37 °C PFA. This work was supported by NIH grant P01 CA92644 and by a contract from the National Foundation for Cancer Research.
References
- Chhabra ES, Higgs HN. The many faces of actin: matching assembly factors with cellular structures. Nat. Cell Biol. 2007;9:1110–1121. doi: 10.1038/ncb1007-1110. [DOI] [PubMed] [Google Scholar]
- Shih SC, et al. The L6 protein TM4SF1 is critical for endothelial cell function and tumor angiogenesis. Cancer Res. 2009;69:3272–3277. doi: 10.1158/0008-5472.CAN-08-4886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zukauskas A, et al. TM4SF1: a tetraspanin-like protein necessary for nanopodia formation and endothelial cell migration. Angiogenesis. 2011;14:345–354. doi: 10.1007/s10456-011-9218-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellstrom I, Beaumier PL, Hellstrom KE. Antitumor effects of L6, an IgG2a antibody that reacts with most human carcinomas. Proc. Natl. Acad. Sci. U.S.A. 1986;83:7059–7063. doi: 10.1073/pnas.83.18.7059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang YY, Ye Z, Cheng L. Molecular imaging and stem cell research. Mol. Imag. 2011;10:111–122. [PubMed] [Google Scholar]
- Hemler ME. Tetraspanin functions and associated microdomains. Nat. Rev. Mol. Cell Biol. 2005;6:801–811. doi: 10.1038/nrm1736. [DOI] [PubMed] [Google Scholar]
- Vasile E, Tomita Y, Brown LF, Kocher O, Dvorak HF. Differential expression of thymosin beta-10 by early passage and senescent vascular endothelium is modulated by VPF/VEGF: evidence for senescent endothelial cells in vivo at sites of atherosclerosis. FASEB. 2001;15:458–466. doi: 10.1096/fj.00-0051com. [DOI] [PubMed] [Google Scholar]
- Itoh TJ, Hotani H. Microtubule dynamics and the regulation by microtubule-associated proteins (MAPs) Uchu Seibutsu Kagaku. 2004;18:116–117. [PubMed] [Google Scholar]
- Caplow M, Shanks J, Ruhlen RL. Temperature-jump studies of microtubule dynamic instability. J. Biol. Chem. 1988;263:10344–10352. [PubMed] [Google Scholar]
- Pollice AA, et al. Sequential paraformaldehyde and methanol fixation for simultaneous flow cytometric analysis of DNA, cell surface proteins, and intracellular proteins. Cytometry. 1992;13:432–444. doi: 10.1002/cyto.990130414. [DOI] [PubMed] [Google Scholar]
- Macarulla JM, et al. Membrane solubilization by the non-ionic detergent triton X-100. A comparative study including model and cell membranes. Revista Espanola de Fisiologia. 1989;45 Suppl:1–8. [PubMed] [Google Scholar]
- Koley D, Bard AJ. Triton X-100 concentration effects on membrane permeability of a single HeLa cell by scanning electrochemical microscopy (SECM) Proc. Natl. Acad. Sci. U.S.A. 2010;107:16783–16787. doi: 10.1073/pnas.1011614107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang YW, et al. CD13 (aminopeptidase N) can associate with tumor-associated antigen L6 and enhance the motility of human lung cancer cells. Int. J. Cancer. 2005;116:243–252. doi: 10.1002/ijc.21089. [DOI] [PubMed] [Google Scholar]