

Abstract
Purpose of Review
This review highlights recently discovered mechanisms that sustain castration-resistant prostate cancer (CRPC) growth and describes advances in CRPC therapeutics.
Recent Findings
Recent reports have shed new light on the molecular processes underlying CRPC survival during androgen deprivation therapy (ADT). This report summarizes recent findings and comments on their clinical relevance. Included in this review is a discussion on molecular mechanisms that regulate AR signaling in normal prostate epithelium and CRPC, biologically significant differences in the androgen-regulated transcriptional programs of androgen-dependent (AD) prostate cancer and CRPC, and recent discoveries involving de novo androgen production and transport. We review the status and results of current clinical trials and finally, discuss the implications of evidence suggesting a declining importance of AR-signaling in prostate cancers with PTEN loss.
Summary
Advances in the understanding of AR signaling in CRPC have identified novel drug targets and improved the rational design of targeted therapy, while illuminating a subset of prostate cancers that may progress to become completely independent of the AR signaling program.
Keywords: Androgen receptor, castration-resistant prostate cancer, PTEN, androgen biosynthesis, androgen receptor splice variants, androgen pathway independent prostate cancer
Introduction
Prostate cancer is the second leading cause of cancer death in American men and mortality remains high despite improvements in therapy [1]. While the majority of primary prostate tumors are treated successfully by radical prostatectomy or external beam radiotherapy, some tumors progress to invasive and disseminated disease. Patients with metastatic prostate cancer are treated with androgen deprivation therapy (ADT) through either chemical or surgical castration. Initial responses to ADT approach 100%, but complete remissions are rare and progressive cancer growth generally resumes after a median of 2-3 years [2]. The emergent castration-resistant prostate cancer (CRPC) is generally incurable with existing treatment approaches.
Because ADT is initially effective and there is copious evidence supporting the re-expression of androgen-regulated signaling in CRPC, most investigational therapies have focused on developing a more potent blockade of androgen receptor (AR) transcriptional activity and the synthesis of androgenic ligands [2]. In this review we report recent advancements in deciphering AR signaling and regulation, identifying mechanisms of intratumoral androgen maintenance, and the development of novel therapies directed toward the AR pathway.
AR Signaling in CRPC
AR signaling is critical to the development of the normal prostate and in the progression from primary to metastatic disease. Continued reliance on AR signaling for survival is a hallmark of CRPC, demonstrated by re-expression of androgen regulated genes (ARGs) after castration [2] and the susceptibility of tumors refractory to conventional ADT to more potent androgen pathway inhibitors [3**-5*]. Therefore, discovering mechanisms of AR expression and AR-driven transcription in tumor cells is imperative.
Regulation of AR Expression
The AR is commonly altered in CRPC with genomic amplification and/or overexpression occurring in up to ∼60% of CRPC metastasis [6*]. In addition to genomic alterations, a recent study identified non-genomic mechanisms leading to AR upregulation in advanced prostate cancers (Figure 1). Analysis of androgen-dependent (AD) prostate cancer cell lines identified an AR-regulated enhancer element located within the second intron of the AR gene itself. Low AR signaling was shown to activate this enhancer and upregulated AR expression. Restoration of AR signaling caused AR to bind the enhancer and recruit lysine-specific demethylase 1 (LSD1) which repressed transcription, demonstrating a negative feedback program that limits endogenous AR expression in normal tissues [7**]. However, when AD prostate cancer cells were grown under castrate-levels of androgen, low AR transcriptional activity resulted in upregulated AR expression, an event that consequently maintains the cellular program of AR-regulated genes. The investigators concluded that the measureable, but low levels of AR ligand in CRPC are sufficient to engage enhancer elements in AR target genes, but insufficient to recruit AR and attendant co-factors to suppressor elements, thus providing a net pro-growth and survival effect without suppressing the AR itself.
Figure 1.
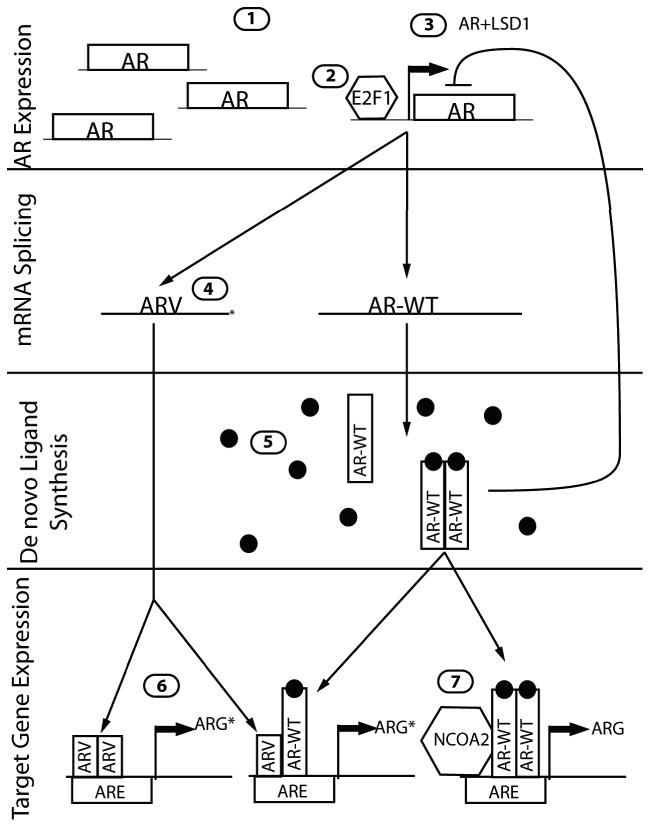
Mechanisms of AR pathway activation in CRPC. 1) AR gene amplification. 2) Rb increases E2F1 activity and AR transcription. 3) Low AR activity leads to increased AR expression via an enhancer element. Increased AR transcriptional activity recruits LSD1 to exert negative feedback on this pathway. 4) mRNA splicing generates LBD-deficient AR variants. 5) Extra-testicular ligand production maintains AR transcriptional activity. 6) ARVs induce ligand-independent expression of ARGs. ARG* represents the differential transcription program activated by some ARVs. 7) AR cofactors, such as NCOA2, amplify the magnitude of AR signaling.
The AR was also recently shown to be regulated through novel functions of the well-characterized retinoblastoma tumor suppression protein. Two independent studies identified widespread genomic loss of retinoblastoma (Rb) in primary tumors (∼5%) and CRPC (35%) [6*], with a loss of Rb protein in up to 74% of CRPC [8*]. Experimentally, loss of normal Rb function enhanced LNCaP xenograft growth in castrate mice. Upon further investigation, this survival benefit was not mediated by losing classical Rb tumor suppressor function, but through an alternate mechanism that involved the consequent upregulated E2F1 expression. In Rb-deficient cells E2F1 was shown to be recruited to the AR regulatory locus resulting in increased AR expression and transcriptional activity, and attendant maintenance of the AR transcriptional program [8*].
Regulation of AR Transcriptional Activity
The action and specificity of nuclear steroid hormone receptors is determined by cofactor binding [9]. A recent large-scale genomic analysis of primary and metastatic tumors found a novel AR cofactor, NCOA2, to be upregulated in a subset of primary (8%) and metastatic tumors (37%). Follow-up experiments in LNCaP cells found that NCOA2 expression correlated with an increased AR transcriptional response to dihydrotestosterone (DHT). NCOA2 overexpression in primary tumors also correlated with a higher risk of recurrence, linking AR activity in primary tumors to future adverse outcomes [6*].
In addition to globally increased AR activity, altered AR signaling in CRPC appears to alter the spectrum of transcriptional targets. A striking example of an alternative AR transcriptional program emerged experiments comparing ARGs in the AD cell model LNCaP and its CR subline LNCaP-abl. Wang et al discovered that the AR binding sites and resulting transcript profile in LNCaP-abl were enriched for genes promoting M-phase progression, notably ubiquitin-conjugating enzyme E2C (UBE2C). UBE2C expression was also found to be upregulated in clinical CRPC [10**]. Other studies have also identified a shift towards pro-mitotic gene expression in conjunction with decreased AR signaling. However, an alternative hypothesis regarding increased mitosis stems from a decrease in the pro-differentiation role of AR, and not a global change in the repertoire of genes regulated by AR [7**, 8*]. None-the-less, these findings necessitate a reassessment of the influence of unliganded AR in mediating key drivers of cell growth.
AR Splice Variants
A substantial number of genes encoded by the genome are transcribed with alternative exon usage and the AR is among this group. Of relevance to CRPC, several AR splice variants (ARVs) encoding ligand-binding-domain (LBD) deficient receptors represent a newly discovered mechanism contributing to ADT resistance. ARVs were initially discovered in the CWR22R xenograft line, and have since been identified in the VCaP cell line, LuCaP xenografts, the Myc-CaP genetically engineered mouse model of prostate cancer and clinical CRPC [10**, 11, 12**, 13*]. While several studies indicate that ARVs are able to function independently of full-length AR (ARFL), work by Watson et al demonstrated that several ARVs remain dependent on ARFL heterodimerization for nuclear translocation and transcriptional activity, and overall AR activity remains sensitive to be LBD-targeted antiandrogens [13*].
Among the recently described ARVs is a transcript that is devoid of exons 5-7 encoding the LBD, designated ARv567es, but retaining the hinge region necessary for nuclear translocation [14]. This ARV is constitutively localized to the nucleus and appears to be less dependent on ARFL for transcriptional activity. ARv567es can upregulate ARFL expression and induce a subset of ARGs in a ligand-independent fashion, supporting a mechanism for CR growth. LNCaP cells engineered to express ARv567es also upregulate a cohort of genes not known to be influenced by the ligand-activated full-length receptor, suggesting this AR variant controls a unique transcriptional program [12**, 15].
To date, two ARVs identified in human cancer (AR-V7, ARv567es) and one identified in a mouse model (mAR-V4) have been shown to significantly enhance ADT survival in prostate cell lines and are upregulated in response to castration [12**, 13*]. Those ARVs dependent on ARFL for transcriptional activity (AR-V7 and mAR-V4) are sensitive to ARFL inhibitors [13*]. There are no published data regarding the sensitivity of ARv567es to inhibition by new potent AR antagonists, however effective blockade seems unlikely since ARv567es translocates to the nucleus and activates ARGs in the absence of ARFL [12**]. Thus, since ARv567es is also commonly found in clinical CRPC [12**], therapeutics that effectively inhibit AR activity through a non-LBD mechanism may be required to successfully treat prostate cancers that express this AR type.
Maintenance of Intratumoral Androgens
In men, extra-testicular androgen production occurs in adrenal glands and recent studies suggest that synthesis may also occur de novo in CRPC [16*, 17*, 18*, 19*]. The efficacy of steroid synthesis inhibitors in clinical trials supports the importance of extra-testicular ligand production in clinical disease [20**] and there is emerging evidence suggesting that active steroid transport also plays a role in ADT resistance.
Androgen Synthesis
Two novel mechanisms of regulating androgen biosynthesis were discovered by studying AD prostate cancer models. In one pathway, Lock et al investigated androgen-stimulated cholesterol-ester production in prostate cancer. They discovered that cholesterol-esters are broken down into cholesterol and fatty acid in response to ADT. Fatty acid is further converted to arachidonic acid (AA), which initiates a transcription program that increases steroid acute regulatory protein (StAR) expression. StAR co-localizes with CYP11A on the mitochondrial membrane and catalyzes the rate-limiting step in steroid biosynthesis: conversion of cholesterol to pregnenelone. Further investigation demonstrated increased expression of steroidogenic enzymes and de novo steroid synthesis in androgen-deprived LNCaP cells that were stimulated with exogenous AA [17*].
Hyperinsulinemia, a clinically-observed side-effect of ADT, can activate an alternative intratumoral steroid biosynthesis pathway. Insulin treatment was sufficient to induce steroid biosynthesis in the three widely-used cell line models of AD prostate cancer. Following insulin stimulation, intracellular and secreted steroids were present in levels sufficient to induce ARG expression. Importantly, insulin upregulated the expression of StAR [18*], similar to the result of AA stimulation [17*], revealing a common, and potentially druggable target in intratumoral androgen synthesis.
Androgen Transport
Steroid hormones enter cells primarily via passive diffusion through the plasma membrane, but several studies now indicate that active transport by organic anion-transport polypeptides (OATP super-family, encoded by the SLCO gene family) contribute to ADT resistance. OATPs transport many known drugs and small molecules, including steroid hormones [21]. The clinical relevance of these transporters in CRPC is supported by recent studies demonstrating that a subgroup of clinical prostate cancers overexpress specific members of the SLCO family and outcome analyses of two independent patient cohorts demonstrated that genomic variants of SLCO2B1 and SLCO1B3 associated with a shorter time to disease progression [22*] and higher rates of prostate specific mortality [23*]. The associations of SLCO2B1 and SLCO1B3 variants with higher mortality is clinically significant because they are both androgen transporters [24, 25], however polymorphisms in each gene are associated with differential rates of steroid transport. Cell-line experiments showed contrasting levels of dihydroepiandosterone (DHEAS) transport between 3 SLCO2B1 SNP variants. SLCO1B3 SNP variants were not associated with shorter patient time to progression in isolation, but interacted with SLCO2B1 variants that confer high-DHEAS transport to decrease patient time to disease progression [22*].
AR Interactions with oncogenic signaling pathways
Inactivation of the PTEN tumor suppressor is one of the most common abnormalities found in prostate cancers [6*]. PTEN inactivation de-regulates PI3K/AKT signaling and engages multiple survival and proliferation pathways. The interaction between the PI3K/AKT pathway and AR activity is complex. While early studies suggested that AKT phosphorylation of AR repressed androgen-mediated transcription [26], other reports suggests that PI3K/ATK activation results in a net increase in proliferation and ARG expression [27**]. However, recent publications suggest a more complex feedback loop between PTEN, PI3K/AKT signaling, and AR activity.
Two independent reports using the PTEN conditionally-null mouse model of prostate cancer incorporated PTEN/AKT-mediated AR-suppression into a feedback loop. While both studies determined that PTEN deletion reduces AR expression, the mechanisms contributing to these events varied. Carver et al proposed a model where AR activity is upregulated by HER kinase activation which in turn is inhibited by AKT activity [28**]. Therefore, PTEN loss and increased AKT signaling inhibit HER kinases and consequently diminish AR activity (Figure 2). Thus, pharmacological inhibition of the AKT pathway resulted in the paradoxical re-activation of AR signaling. However, previously published studies on HER kinase activation in human models of prostate cancer reported HER kinase activity decreased AR expression [29]. Reciprocal interactions whereby active AR signaling suppressed AKT signaling via FKBP5 and PHLPP intermediaries provides a rationale for co-targeting these pathways. Though clinical trials of HER inhibitors as monotherapy in CRPC have not yielded substantial responses, serum PSA declines have been noted in some patients [30, 31]. This may be the result of reduced tumor burden or decreased AR activity. Clearly, the effect of HER kinase activity, the PI3K pathway, and relationships with AR signaling in prostate cancer is incompletely understood and further investigation is needed in order to design clinical studies that exploit their interactions.
Figure 2.
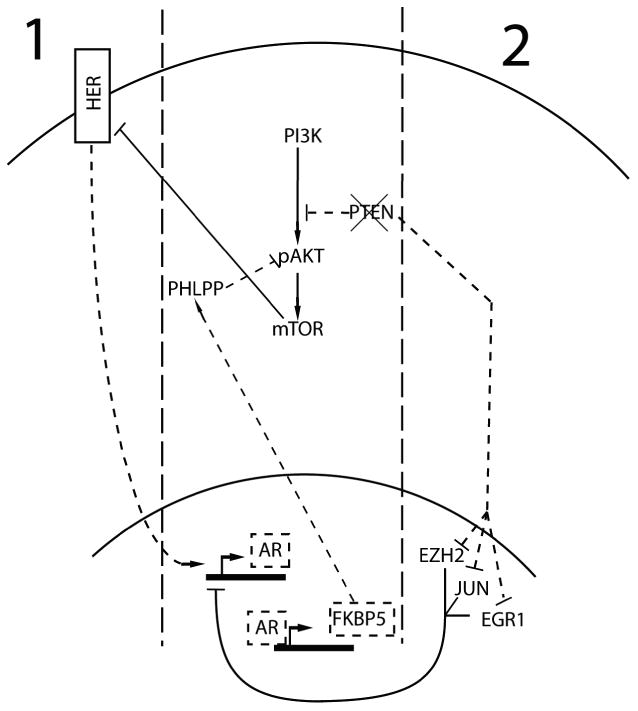
Feedback pathways of AR, PTEN, and PI3K. AR transcriptional activity represses PI3K signaling through the expression of FKBP5. FKBP5 facilitates the dephosphorylation of AKT by PHLPP. PTEN loss, common in prostate cancer, increases PI3K pathway activity and may also repress AR. Two prominent hypotheses explaining the mechanism of PI3K activity and AR repression are presented. 1) Carver et al hypothesize that HER kinases induce expression of AR, and increased PI3K signaling inhibits HER kinases in an mTOR-dependent fashion. This decreases AR expression [28**]. 2) Mullholland et al propose a mechanism by which PTEN negatively regulates the expression and activity of a number of proteins that modulate AR activity (EZH2, JUN, EGR1). PTEN loss deregulates these proteins, repressing AR expression [27**]. Dashed lines indicate pathways where activity is downregulated by PTEN loss.
A second model, proposed by Mullholland et al, suggests that PTEN loss, independent of AKT, represses AR activity [27**]. Further, using elegant genetically engineered mouse models that completely ablated AR signaling in prostate cancers---something not yet achieved in humans---the investigators demonstrated that the resulting AR-null cancers arising in a Pten-null genetic background had substantially increased rates of cell proliferation. While proposing somewhat different mechanisms, the significant contribution of these studies is evidence that AKT activation combined with PTEN loss is sufficient for prostate tumor growth in the absence of robust AR signaling, and may actively repress the AR program. Thus, further reduction in AR activity by more potent ADT might select for tumors with activated PI3K/AKT/MTOR signaling and little or no residual AR activity.
AR Pathway-Directed Therapies
Clinically, the emergence of CRPC is reflected by the progressive growth of metastatic tumors in the absence of serum androgens which is accompanied by the reactivation of the AR program as exemplified by rising serum concentrations of PSA. To continue targeting this reactivated AR pathway, new potent pharmacological inhibitors of ligand synthesis and the AR have been developed and shown to be of clinical benefit.
Antiandrogens
Conventional AR antagonists offer incomplete transcriptional inhibition, and exhibit partial agonist activity in the presence of mutated AR, high levels of the AR, or high concentrations of inhibitor. Thus, more potent antagonists lacking agonist activity are necessary. A promising novel antiandrogen, MDV-3100, recently completed a Phase I/II trial in 140 patients with CRPC. Of those treated, 56% experienced a >50% decrease in serum PSA, and imaging demonstrated that 22% of patients with soft tissue metastasis and 56% of patients with bony metastasis saw a stabilization of disease. Furthermore, there was no evidence of agonist activity and dose-related fatigue was the most common side effect [5*]. Formal reports of ongoing phase 3 clinical trials are eagerly awaited.
Most AR antagonist development has targeted the C-terminal LBD, a domain that is lost in ARVs [12**, 13*]. The AR N-terminal domain contains the DNA-binding elements common to both ARFL and ARVs. Thus, inhibition of all AR forms could potentially be accomplished via N-terminal disruption. Anderson et al recently reported studies of a novel small-molecule N-terminal AR inhibitor, EPI-001. EPI-001 specifically binds the AR N-terminus and disrupts transcription and protein-protein interactions. Preliminary investigation showed that EPI-001 treatment caused decreased ARG expression and inhibited LNCaP xenograft growth in intact mice. Furthermore, EPI-001 inhibited LBD-deficient ARV activity [3**]. While not yet ready for clinical trials, EPI-001 is a significant discovery that will serve as a lead compound to identify other AR N-terminal inhibitors.
Androgen Synthesis Inhibitors
Recently, a Phase III trial was completed that demonstrated improved outcomes in patients treated with abiraterone acetate, an irreversible CYP17A inhibitor. Patients previously treated with docetaxel received a combination of abiraterone/prednisone or placebo/prednisone. Overall survival was significantly increased in patients treated with abiraterone (14.8 months) versus patients treated with placebo (10.9 months). All other secondary endpoints (PSA response, time to PSA progression, progression-free survival) were also improved in the abiraterone treatment group. Furthermore, the side effect profile was generally limited to the effects of mineralocorticoid excess [20**]. Importantly, disease progression on Abiraterone was associated with a rising PSA, once again indicating incomplete suppression of AR signaling.
While abiraterone-resistant tumors have not yet been evaluated from men who have relapsed on abiraterone treatment, mechanisms of resistance have been investigated in vivo using prostate cancer xenografts. Reminiscent of AR-inhibitor trials, in vivo abiraterone resistance appears to occur via upregulation of the drug target, CYP17A, along with higher expression of ARV or mutated AR [16*, 19*]. This underscores a continued reliance on AR-mediated transcriptional pathways, suggesting that combination therapy co-targeting the AR and CYP17A might evoke a more prolonged therapeutic response.
Conclusions
The heterogeneous molecular composition of advanced CRPC makes treatment very challenging. However, a common feature involves the activation and re-expression of the AR program following ADT. Of relevance for therapeutics, the molecular mechanisms resulting in ARG re-expression appear to be diverse. Current literature suggests that tumors re-express ARGs following ADT by upregulation of the targeted pathway (e.g. CYP17), activation of an alternative pathway of resistance—of which there appear to be several, or both. It is important that future work focus on the development of additional ligand and synthesis inhibitors, in addition to investigating combination therapy between different ADT modalities (androgen synthesis inhibitors with more potent AR antagonists, for example). Targeting newly implicated ADT resistance mechanisms – such as ARVs, NCOA2, HER kinases, and AKT/PI3K – in combination with existing therapy may act to further suppress CRPC.
A potential application of recent studies would be to tailor therapy to prostate cancer subtypes. For example, ADT use in primary prostate cancer has produced mixed results and is associated with significant morbidity [32]. Molecular analyses of tumors prior to, or during therapy may assist in rationally selecting patients with specific AR-enhancing pathways for co-targeting strategies. Finally, there is emerging experimental and clinical evidence of prostate tumors that can completely overcome their dependence on AR [27**, 28**, 33], and it is plausible that aggressive ADT may select for tumors that are truly androgen-pathway independent. Mechanisms of androgen-pathway independence should be investigated in more depth to identify potential therapeutic targets in order to anticipate the emergence of this clinically important variant of prostate cancer.
Key Points.
Androgen receptor signaling remains the dominant growth pathway in prostate cancers that progress in the setting of low serum androgens.
There is evidence that PI3K/AKT/mTOR signaling can bypass the need for AR in a PTEN-null tumors.
Emerging anti-AR and anti-androgen biosynthesis pharmaceuticals are an improvement over conventional ADT, although CRPC recurring from second-line ADT suggests that more aggressive ADT is still necessary.
Acknowledgments
The authors' work on the Androgen Receptor pathway is supported by the Pacific Northwest Prostate Cancer SPORE P50CA097186, RC1 CA146849, DOD Synergy Award PC093509, and the Prostate Cancer Foundation. EB was supported through a DOD Predoctoral Fellowship Award, W81XWH-10-1-0133.
References
- 1.Jemal A, Siegel R, Xu J, et al. Cancer statistics, 2010. CA Cancer J Clin. 2010 Sep-Oct;60(5):277–300. doi: 10.3322/caac.20073. [DOI] [PubMed] [Google Scholar]
- 2.Harris WP, Mostaghel EA, Nelson PS, et al. Androgen deprivation therapy: progress in understanding mechanisms of resistance and optimizing androgen depletion. Nat Clin Pract Urol. 2009 Feb;6(2):76–85. doi: 10.1038/ncpuro1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3**.Andersen RJ, Mawji NR, Wang J, et al. Regression of castrate-recurrent prostate cancer by a small-molecule inhibitor of the amino-terminus domain of the androgen receptor. Cancer Cell. 2010 Jun 15;17(6):535–46. doi: 10.1016/j.ccr.2010.04.027. This study documents the development of the first N-terminal AR inhibitor, EPI-001. Epi-001 inhibits the transcriptional activity full-length AR and LBD-deficient AR variants. [DOI] [PubMed] [Google Scholar]
- 4.Attard G, Reid AH, Yap TA, et al. Phase I clinical trial of a selective inhibitor of CYP17, abiraterone acetate, confirms that castration-resistant prostate cancer commonly remains hormone driven. J Clin Oncol. 2008 Oct 1;26(28):4563–71. doi: 10.1200/JCO.2007.15.9749. [DOI] [PubMed] [Google Scholar]
- 5*.Scher HI, Beer TM, Higano CS, et al. Antitumour activity of MDV3100 in castration-resistant prostate cancer: a phase 1-2 study. Lancet. 2010 Apr 24;375(9724):1437–46. doi: 10.1016/S0140-6736(10)60172-9. A clinical trial demonstrating improvement in serum PSA and radiographic progression in patients treated with the novel antiandrogen MDV3100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6*.Taylor BS, Schultz N, Hieronymus H, et al. Integrative genomic profiling of human prostate cancer. Cancer Cell. 2010 Jul 13;18(1):11–22. doi: 10.1016/j.ccr.2010.05.026. This report combines mRNA transcript and CGH data with clinical outcomes data, in addition to identifying a novel AR cofactor. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7**.Cai C, He HH, Chen S, et al. Androgen receptor gene expression in prostate cancer is directly suppressed by the androgen receptor through recruitment of lysine-specific demethylase 1. Cancer Cell. 2011 Oct 18;20(4):457–71. doi: 10.1016/j.ccr.2011.09.001. This article describes how AR expression is controlled by a newly discovered enhancer element in the second intron. Importantly, it provides a mechanism explaining the upregulation of AR observed in CRPC, even in the absence of gene amplification. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8*.Sharma A, Yeow WS, Ertel A, et al. The retinoblastoma tumor suppressor controls androgen signaling and human prostate cancer progression. J Clin Invest. 2010 Dec 1;120(12):4478–92. doi: 10.1172/JCI44239. Rb tumor suppressor inactivation functions as a tumor-progression event in prostate cancer through the deregulation of AR exprssion. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Heinlein CA, Chang C. Androgen receptor (AR) coregulators: an overview. Endocr Rev. 2002 Apr;23(2):175–200. doi: 10.1210/edrv.23.2.0460. [DOI] [PubMed] [Google Scholar]
- 10**.Wang Q, Li W, Zhang Y, et al. Androgen receptor regulates a distinct transcription program in androgen-independent prostate cancer. Cell. 2009 Jul 23;138(2):245–56. doi: 10.1016/j.cell.2009.04.056. This is the first report of a AR-regulated pro-mitotic transcriptional program unique to CRPC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dehm SM, Schmidt LJ, Heemers HV, et al. Splicing of a novel androgen receptor exon generates a constitutively active androgen receptor that mediates prostate cancer therapy resistance. Cancer Res. 2008 Jul 1;68(13):5469–77. doi: 10.1158/0008-5472.CAN-08-0594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12**.Sun S, Sprenger CC, Vessella RL, et al. Castration resistance in human prostate cancer is conferred by a frequently occurring androgen receptor splice variant. J Clin Invest. 2010 Aug 2;120(8):2715–30. doi: 10.1172/JCI41824. By analyzing prostate cancer xenografts, this study identified a LBD-deficient ARV that was constitutively recruited to the nucleus and commonly expressed in clinical CRPC samples. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13*.Watson PA, Chen YF, Balbas MD, et al. Constitutively active androgen receptor splice variants expressed in castration-resistant prostate cancer require full-length androgen receptor. Proc Natl Acad Sci U S A. 2010 Sep 28;107(39):16759–65. doi: 10.1073/pnas.1012443107. Mouse and most human ARVs were characterized for dependence on ARFL for translocation to the nucleus and ability to promote CRPC growth. Full-length AR was required for ARV activity. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Haelens A, Tanner T, Denayer S, et al. The hinge region regulates DNA binding, nuclear translocation, and transactivation of the androgen receptor. Cancer Res. 2007 May 1;67(9):4514–23. doi: 10.1158/0008-5472.CAN-06-1701. [DOI] [PubMed] [Google Scholar]
- 15.Zhang X, Morrissey C, Sun S, et al. Androgen receptor variants occur frequently in castration resistant prostate cancer metastases. PLoS One. 2011;6(11):e27970. doi: 10.1371/journal.pone.0027970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16*.Cai C, Chen S, Ng P, et al. Intratumoral de novo steroid synthesis activates androgen receptor in castration-resistant prostate cancer and is upregulated by treatment with CYP17A1 inhibitors. Cancer Res. 2011 Oct 15;71(20):6503–13. doi: 10.1158/0008-5472.CAN-11-0532. This study demonstrates de novo androgen production in CRPC and showed that CYP17A upregulation can confer resistance to abiraterone treatment. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17*.Locke JA, Guns ES, Lehman ML, et al. Arachidonic acid activation of intratumoral steroid synthesis during prostate cancer progression to castration resistance. Prostate. 2010 Feb 15;70(3):239–51. doi: 10.1002/pros.21057. This study illustrates an AR-mediated mechanism by which tumors can generate de novo androgen production using the byproducts of fatty acid breakdown. [DOI] [PubMed] [Google Scholar]
- 18*.Lubik AA, Gunter JH, Hendy SC, et al. Insulin increases de novo steroidogenesis in prostate cancer cells. Cancer Res. 2011 Sep 1;71(17):5754–64. doi: 10.1158/0008-5472.CAN-10-2470. ADT induces hyperinsulinemia in patients with CRPC. This report provides evidence thathigh insulin levels can contribute to castration resistance by promoting de novo androgen production. [DOI] [PubMed] [Google Scholar]
- 19*.Mostaghel EA, Marck BT, Plymate SR, et al. Resistance to CYP17A1 inhibition with abiraterone in castration-resistant prostate cancer: induction of steroidogenesis and androgen receptor splice variants. Clin Cancer Res. 2011 Sep 15;17(18):5913–25. doi: 10.1158/1078-0432.CCR-11-0728. Using two LuCaP xenografts as models of human CRPC, this article demonstrates that abirateroneresistance can occur through upregulation of ARVs and steroidogenic enzymes. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20**.de Bono JS, Logothetis CJ, Molina A, et al. Abiraterone and increased survival in metastatic prostate cancer. N Engl J Med. 2011 May 26;364(21):1995–2005. doi: 10.1056/NEJMoa1014618. A Phase -III clinical trial in patients with CRPC that demonstrates increased overall survival in abiraterone treated groups, improvements in all secondary endpoints, and minimal drug-related toxicity. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hagenbuch B, Meier PJ. The superfamily of organic anion transporting polypeptides. Biochim Biophys Acta. 2003 Jan 10;1609(1):1–18. doi: 10.1016/s0005-2736(02)00633-8. [DOI] [PubMed] [Google Scholar]
- 22*.Yang M, Xie W, Mostaghel E, et al. SLCO2B1 and SLCO1B3 may determine time to progression for patients receiving androgen deprivation therapy for prostate cancer. J Clin Oncol. 2011 Jun 20;29(18):2565–73. doi: 10.1200/JCO.2010.31.2405. Cell lines were used to show that specific SLCO gene variants could transport androgens at different rates. Androgen transporting variants were associated with decreased time to progression. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23*.Wright JL, Kwon EM, Ostrander EA, et al. Expression of SLCO transport genes in castration-resistant prostate cancer and impact of genetic variation in SLCO1B3 and SLCO2B1 on prostate cancer outcomes. Cancer Epidemiol Biomarkers Prev. 2011 Apr;20(4):619–27. doi: 10.1158/1055-9965.EPI-10-1023. Prostate cancer-specific mortality was associated with the expression of androgen-transporting SLCO gene variants. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hamada A, Sissung T, Price DK, et al. Effect of SLCO1B3 haplotype on testosterone transport and clinical outcome in caucasian patients with androgen-independent prostatic cancer. Clin Cancer Res. 2008 Jun 1;14(11):3312–8. doi: 10.1158/1078-0432.CCR-07-4118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tamai I, Nezu J, Uchino H, et al. Molecular identification and characterization of novel members of the human organic anion transporter (OATP) family. Biochem Biophys Res Commun. 2000 Jun 24;273(1):251–60. doi: 10.1006/bbrc.2000.2922. [DOI] [PubMed] [Google Scholar]
- 26.Lin HK, Yeh S, Kang HY, et al. Akt suppresses androgen-induced apoptosis by phosphorylating and inhibiting androgen receptor. Proc Natl Acad Sci U S A. 2001 Jun 19;98(13):7200–5. doi: 10.1073/pnas.121173298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27**.Mulholland DJ, Tran LM, Li Y, et al. Cell autonomous role of PTEN in regulating castration-resistant prostate cancer growth. Cancer Cell. 2011 Jun 14;19(6):792–804. doi: 10.1016/j.ccr.2011.05.006. By using mice harboring conditional PTEN and AR knockouts, the authors demonstrate that AR function is not necessary to generate prostate epithelial tumors. Furthermore, they show that PTEN loss further suppresses AR expression and transcriptional output, suggesting a mechanism for CRPC progression. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28**.Carver BS, Chapinski C, Wongvipat J, et al. Reciprocal feedback regulation of PI3K and androgen receptor signaling in PTEN-deficient prostate cancer. Cancer Cell. 2011 May 17;19(5):575–86. doi: 10.1016/j.ccr.2011.04.008. This article demonstrates that PI3K kinase pathway activation leads to decreased AR transcriptional output in both human and murine cancers, and suggests that androgen-dependence may be restored by inhibiting PI3K signaling in CRPC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cai C, Portnoy DC, Wang H, et al. Androgen receptor expression in prostate cancer cells is suppressed by activation of epidermal growth factor receptor and ErbB2. Cancer Res. 2009 Jun 15;69(12):5202–9. doi: 10.1158/0008-5472.CAN-09-0026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nabhan C, Lestingi TM, Galvez A, et al. Erlotinib has moderate single-agent activity in chemotherapy-naive castration-resistant prostate cancer: final results of a phase II trial. Urology. 2009 Sep;74(3):665–71. doi: 10.1016/j.urology.2009.05.016. [DOI] [PubMed] [Google Scholar]
- 31.Whang YE, Armstrong AJ, Rathmell WK, et al. A phase II study of lapatinib, a dual EGFR and HER-2 tyrosine kinase inhibitor, in patients with castration-resistant prostate cancer. Urol Oncol. 2011 Mar 9; doi: 10.1016/j.urolonc.2010.09.018. [DOI] [PubMed] [Google Scholar]
- 32.Pagliarulo V, Bracarda S, Eisenberger MA, et al. Contemporary role of androgen deprivation therapy for prostate cancer. Eur Urol. 2012 Jan;61(1):11–25. doi: 10.1016/j.eururo.2011.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Roudier MP, True LD, Higano CS, et al. Phenotypic heterogeneity of end-stage prostate carcinoma metastatic to bone. Hum Pathol. 2003 Jul;34(7):646–53. doi: 10.1016/s0046-8177(03)00190-4. [DOI] [PubMed] [Google Scholar]