

Abstract
We describe a new platform to identify structure-switching DNA beacon aptamers, which detect small molecules in a specific manner. By clonally amplifying a DNA library designed to fluoresce in response to binding events onto microbeads, aptamer beacons can be selected by stringent fluorescence-assisted sorting. We validated this method by isolating known and novel anti-steroid aptamers from two separate DNA libraries that were structurally enriched with three-way junctions. Importantly, aptamers were retrieved in only a few (three) rounds of selection by this approach and did not require further optimization, significantly streamlining the process of beacon development.
Keywords: aptamers, DNA structures, emulsion PCR, molecular beacons, SELEX
The detection of biological and chemical agents of consequence for human health is an ongoing goal for various biotechnology platforms.[1] Towards this end, nucleic acids that bind to small molecules in a highly specific manner, termed aptamers, have garnered interest for their ease of synthesis and quality control. A special class of aptamers, structure-switching aptamer beacons, contain intra- or intermolecular duplexes that are covalently linked to quencher and fluorescent probes. Upon binding of a target molecule, the proximity of the probes is disrupted, rendering the device fluorescent.[2, 3]
Aptamer beacons have been generated via one of two general strategies. In the first approach, known aptamers are rationally modified by the addition of short sequence extensions. These additional bases serve to disrupt the aptamer’s native fold and to introduce a fluorophore and quencher pair that can be attached covalently[4–7] or by hybridization[8] (Scheme 1 A). Both two-piece and three-piece aptamer beacons have been developed by using this latter method (Scheme 1 B and 1 C).
Scheme 1.
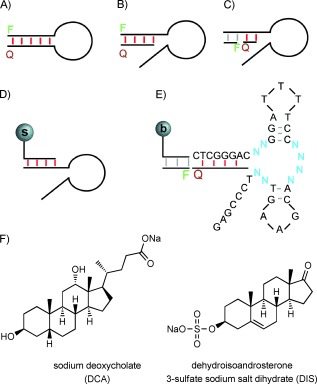
Aptamers can be modified to generate fluorescent detection devices by A) covalent attachment or B) single- or C) dual- hybridization of quencher (Q) and fluorophore (F) dyes. Disruption of a duplex (red) results in a strong fluorescent signal. D) Classical support-based structure-switching aptamer in vitro selection scheme. The support used to immobilize the library is indicated by the letter “s”. E) Fluorescent structure-switching aptamer in vitro selection scheme for the LN8 library used to identify three-piece aptamer beacon devices. The microbead is indicated by the letter “b”. F) Small molecules used in these experiments.
Second, structural-switching aptamers have been directly acquired through in vitro selection.[2, 9–11] For this method, random nucleic acid libraries are typically immobilized on a support via hybridization (Scheme 1 D). Following target addition, sequences that selectively bind a given target are released from the matrix if the binding event induces a conformational rearrangement wherein an internal complementary sequence of the aptamer is preferentially bound. DNA that is eluted in this fashion is PCR-amplified to seed a new, enriched library for use in subsequent rounds of selection.
Recently, several steroid-binding aptamers were isolated from a DNA library that included a designed three-way junction containing just eight variable bases[12]—a surprising result, given that the library contained fewer than 105 unique sequences. However, we note that, even when using a small library such as this, the identification of functional aptamer beacons required 9–13 rounds of selection. Additionally, the selected molecules were not in and of themselves fluorescent sensors and had to be adapted and optimized for this purpose, further complicating the development process.
To streamline the identification of beacon aptamers, we combined the goals of these two approaches to devise an in vitro selection scheme that directly identifies aptamer beacons that fluoresce in the presence of analyte. We demonstrated that this was possible by combining emulsion PCR (ePCR) and fluorescence-activated cell sorting (FACS; Scheme 2). Thus, our approach enabled the de novo selection of functional aptamer beacons without the need for further optimization and modification. Furthermore, because selection pressure in this approach is based on FACS gating, molecules with distinctly different functional properties could potentially be identified.
Scheme 2.
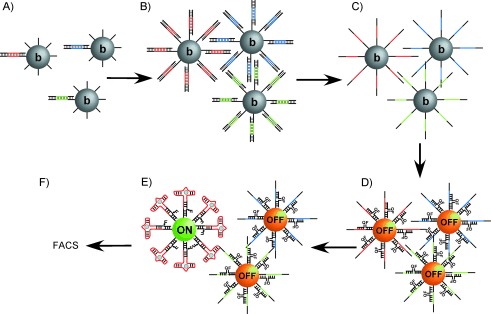
FACS-based selection. A) Microbeads bearing multiple copies (∼105) of a forward primer and a single dsDNA member of the library are amplified by emulsion PCR (ePCR), generating B) beads bearing ∼40 000 copies of a single sequence. The emulsion is broken, and the beads are recovered and C) rendered single-stranded by treatment with NaOH. Single-stranded DNA is hybridized to fluorophore (F) and quencher (Q) oligonucleotides, generating libraries of particles in the “OFF” state. D) Following incubation with target, E) particles which turn “ON” in the presence of ligand can be recovered by F) FACS.
We initially sought to confirm this method by selecting for aptamers that bind DHEA-sulfate (dehydroisoandrosterone 3-sulfate; DIS), a diagnostic of adrenal function. To guarantee the presence of selectable aptamers in the starting pool of sequences, we designed a library based on a previously-explored DIS aptamer beacon library (denoted “LN8” in this work).[12]
To begin, streptavidin-coated microbeads were loaded with 5′ double-biotinylated library molecules at a ratio of one library molecule to one bead, such that probabilistic interactions ensured a large population of beads (∼40 %) bound to a single DNA sequence, a function confirmed in preliminary control experiments (data not shown). The beads were subsequently saturated with 5′ double-biotinylated forward primers, emulsified with the appropriate components to perform PCR, and amplified by ePCR.[13, 14] Once in the emulsion, each bead is protected from contamination from beads in adjacent compartments such that each bead bears a clonal population of DNA. DNA-loaded beads (Supporting Information) were recovered from the emulsion and subsequently treated with strong base to generate ssDNA. The ssDNA-covered beads were neutralized and annealed to FITC- and DABCYL-labeled oligonucleotides before equilibration in binding buffer.
Prior to each sort, a subset of beads bearing ssDNA was incubated with only the FITC-labeled oligonucleotide and analyzed by flow cytometry. This allowed us to identify where the fully “ON” positive events lay in the FACS histogram (Figure 1 A; ON). It is important to note that this control also allowed us to confirm that the ePCR had been conducted successfully without mixing, as indicated by the presence of a negative bead population, corresponding to beads that did not get compartmentalized with library molecules (Figure 1 A; blank).
Figure 1.
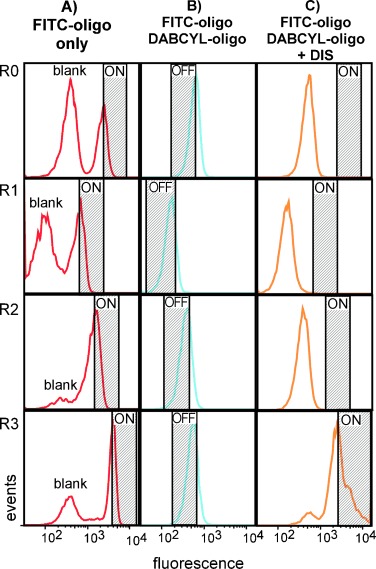
Progress of the LN8 DIS selection by flow cytometry. A) Bead-bound LN8 library annealed to the FITC oligonucleotide to determine the positive gates (“ON′′) and to confirm the presence of unlabeled beads indicating that the emulsion remained intact during PCR (”Blank“). B) Following hybridization of the FITC- and DABCYL-oligonucleotides, beads were quenched and sorted by using the hashed gate (”OFF“) to remove false-positive events. C) Positive selection was initiated by the addition of 200 μm DIS, and beads were sorted for positive events falling in the hashed gate (”ON′′), which was set based on the data collected in A). R0 data is derived from the initial LN8 library, R1 from the first enriched library, etc. Variations in the position of both the “ON” and “OFF” gates were due to day-to-day differences encountered on the flow cytometer and highlight the need for running portions of each individual round of selection prior to each sort in order to accurately define both of these states on any given day.
Following this analysis, the remaining pool was subsequently incubated with both FITC- and DABCYL-labeled oligonucleotides, and library molecules in the “OFF” state were sorted for fully-quenched beads (negative sort). This sorting gate was typically set to collect the ∼75 % darkest population and was required to purge the starting population of false positives—sequences that bound the FITC- but not the DABCYL-labeled oligonucleotides (Figure 1 B, “OFF”). Beads in the “OFF” state were collected, concentrated by centrifugation, and then incubated in the presence of the target (DIS, 200 μm) for ∼25–45 min. The beads were then sorted a second time, and fluorescent events that occurred within the “ON” gate (defined by the FITC-oligonucleotide-only analysis, Figure 1 A) were collected. To impose stringency, we only collected the brightest ∼50 % of the positive control-gated population. DNA from these sorted beads was recovered by PCR to generate an enriched library and used in subsequent rounds of selection (Figure 1; R1–R3).
Following the third round of selection, the LN8 population displayed marked enrichment for functional binders, as evidenced by a substantial fraction of positively fluorescent events (Figure 1 C; R3). To identify these sequences, DNA recovered from the last round of selection was cloned and sequenced. Among 90 sequences, 85 showed high identity to the previously selected “diss. 1” aptamer[12] (Table S1 in the Supporting Information). These sequences differed from the diss. 1 aptamer in only one position, where a G is replaced with an A: we designated this aptamer variant “diss. 1 A”. In the previously hypothesized structure for the diss. 1 aptamer,[12] this G-to-A transition might replace a G–T wobble with an A–T pair.
To identify any effects this transition might have on aptamer activity, we assayed both sequences by a cytometry-based activity assay. Beads loaded with ssDNA were annealed to 5′-FITC- and 3′-DABCYL-labeled oligonucleotides in binding buffer. The beads were then incubated in the presence of increasing concentrations of DIS or a structurally related compound, deoxycholate salt (DCA), for ∼30 min and then analyzed by flow cytometry (Figure 2). In contrast to aptamer beacons isolated from previous selection schemes,[2, 9–12] these assays were simplified, as our aptamer beacons are already capable of fluorescence-based detection and can be utilized to detect analyte without any additional modification.
Figure 2.
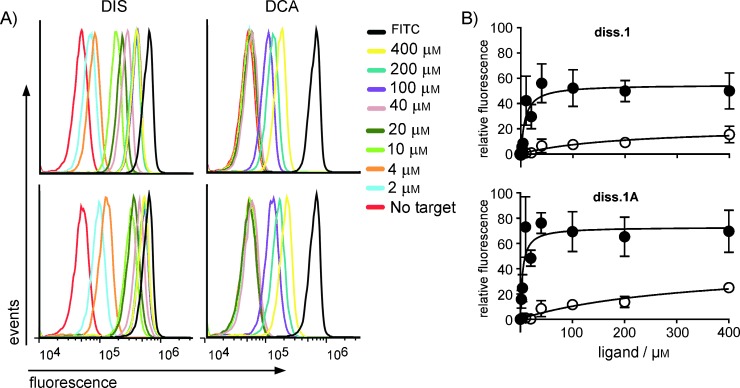
Binding activity of the DIS-responsive aptamer beacons diss. 1 and diss. 1 A. A) Representative flow cytometry histograms of bead-bound aptamer beacon devices incubated in the presence of increasing concentrations of DIS (left) or DCA (right); note scale. All experiments contained a sample reaction performed in the absence of target (no target) or bearing only the FITC-labeled oligonucleotide (FITC), representing the signal expected from a construct which was fully turned “ON′′. B) Binding curves were determined (n=3, SE shown) from the histograms by plotting the mode of relative fluorescence of singlet bead populations in the presence of DIS (filled circles) or DCA (open circles). The calculated apparent binding constants for diss. 1 and diss. 1 A were 7.4±3.5 and 4.1±1.8 μm, respectively. Due to poor binding, the apparent binding constants for DCA could not be determined accurately.
As previously reported, a re-engineered variant of the diss. 1 aptamer optimized for fluorescence signaling provided specific ligand detection with an apparent KD of ∼5 µm, with no apparent signaling when experiments were performed in the presence of DCA.[12] Accordingly, our flow-cytometry-based assay was in qualitative agreement across these two assay formats, yielding apparent binding constants of 7.4±3.5 μm for diss. 1 and 4.1±1.8 μm for diss. 1 A. Interestingly, although a comparison of the diss. 1 and diss. 1 A aptamer binding activity profiles suggested that the difference in their sequences had little consequence on their apparent KD values and specificity, the relative fluorescence of the diss. 1 A aptamer was greater at all tested concentrations of DIS (Figure 2 B). Further, the fluorescence of diss. 1 A-loaded beads increased more rapidly (Figure S1).
We hypothesized that these differences in sensitivity might have allowed the diss. 1 A variant to out compete the diss. 1 aptamer, which was not identified in sequences recovered from the final round of this selection (Table S1). To test this possibility, we ran a single round of ePCR/FACS selection on a 1:1 mixture of diss. 1 and diss. 1 A DNA-loaded beads. In this experiment, the bottom and top 5 % of the observed population were sorted separately in the presence of DIS (170 μm for 45 min), recovered by PCR, and sequenced along with the initial starting beads (not shown). In the pre-sorted population, both diss. 1 and diss. 1 A were present in similar proportions—47 and 53 %, respectively (of 57 sequences analyzed). However, DNA amplified from the 5 % least-fluorescent population was enriched for diss. 1 aptamers (60 % of 57 sequences analyzed), whereas the 5 % highest-fluorescent population was enriched for diss. 1 A aptamers (69 % of 59 sequences analyzed). Thus, the diss. 1 aptamer might have accumulated enough of a selective advantage over multiple rounds of our original LN8 selection to comprise the majority of sequences. More importantly, this suggests that our selection approach can identify aptamers with different properties based solely on gating parameters.
To test whether other designed libraries could be used by this method, we carried out a second selection for DIS-responsive aptamer beacons in which four additional variable positions were introduced into the three-way junction structure. This library, LN12, contains a complexity of ∼107 variants and is more likely to adopt novel folds that deviate from the designed three-way junction because of the additional positions (Table S1 and Figure S2). Also, we reasoned that a more complex library might contain aptamers with more sensitive, improved device responses.
This selection was carried out in a similar manner as the LN8 selection, except that the negative sort was omitted in the first round (Figure 3). This was necessary to maximize the diversity of recovered functional sequences, as a large fraction of beads (∼50 %) were lost between negative and positive sorts, due to stringent FACS logic gates and physical manipulation. This is a valid concern when working with libraries that contain twelve or more random base positions which are characterized by a complexity (∼107) that is near the number of beads used here (∼108). As a consequence of the reduced stringency in the initial round, a “false positive” population was observed in every round, though it was largely selected against by the final round (Figure 3, R1–R3).
Figure 3.
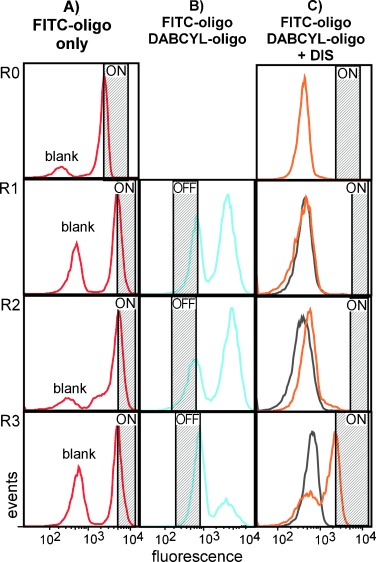
Progress of the LN12 DIS selection by flow cytometry. Bead-bound LN12 library annealed to A) the FITC oligonucleotide to determine the positive gates (“ON′′), or B) both the FITC and DABCYL oligonucleotides to sort quenched beads (”OFF“). C) DIS (200 μm) was added to quenched, sorted beads (gray) and re-sorted for positive events (”ON′′; orange). The negative sort was omitted in the first round. R0 data was derived from the initial LN12 library, R1 from the first enriched library, etc. Variations in the position of both the “ON” and “OFF” gates were due to day-to-day differences encountered on the flow cytometer.
As in the LN8 selection, we observed a small positive fraction of events with ligand-induced fluorescence from the third round of enriched DNA library. We cloned and sequenced 93 sequences from the final (fourth) round recovered DNA library. The three most frequently occurring sequences displayed a high level of similarity to sequences identified in the previous DIS-responsive aptamer beacon selection[12] (Table S1). The most frequent sequence, diss. 1 A.12 (28 copies), bears a high resemblance to the diss. 1 A aptamer, but the presence of additional bases demonstrates that it was clearly derived from the LN12 library. Consistent with this, cytometric analysis of diss. 1 A.12 revealed similar (albeit slightly less sensitive) activation parameters than the corresponding clone from the LN8 library (Figure 4, 26.2±4.1 μm). In addition to sequences (diss. 3.12, diss. 4.12) similar to those reported by Yang and colleagues,[12] we also identified two novel sequences, diss. 5.12 and diss. 6.12. These aptamer beacons displayed DIS-dependent activation, with apparent KD values of 27.1±11.4 and 19.4±6.3 μm, respectively (Figure 4). Although these sequences were isolated from a higher-complexity library, these aptamer beacons did not yield functional sequences with better signaling capabilities. In fact, diss. 5.12 showed higher levels of cross-reactivity with DCA, although the apparent KD for activation was about 20 times lower than that of DIS (Figure 4 and Table S1).
Figure 4.
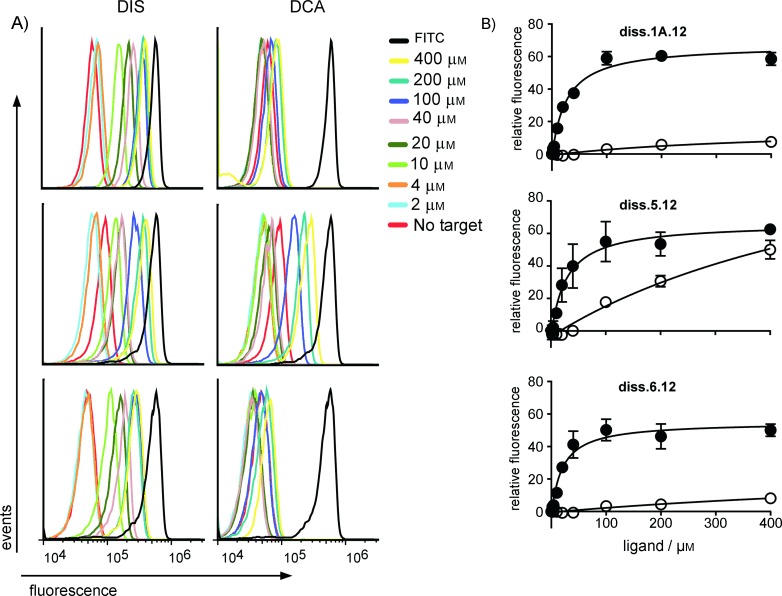
Binding activity of sequences isolated from the LN12 selection. A) Representative flow cytometry histograms of bead-bound aptamer beacon devices incubated in the presence of increasing concentrations of DIS (left) or DCA (right); note scale. All experiments contained a sample reaction performed in the absence of target or bearing only the FITC-labeled oligonucleotide, representing the signal expected from a construct which had fully turned “ON′′. B) Binding curves were determined (n=3, SE shown) from the histograms by plotting the mode of relative fluorescence of singlet bead populations in the presence of DIS (•) or DCA (○). The apparent binding constants for diss. 1 A.12, diss. 5.12, and diss. 6.12 for DIS were 26.2±4.1, 27.1±11.4, and 19.4±6.3 μm, respectively. Due to poor binding, the apparent binding constants for DCA could not be determined accurately.
We take these results to suggest that the increased number of random positions introduced into this library allow for greater flexibility in the designed binding pocket. Thus, the LN8 library might be restricted in its ability to form hydrophobic pockets capable of accommodating different ligands, whereas the LN12 library, which is significantly more diverse, is not. Additionally, it is interesting to note that the responsiveness of selected variants from the LN12 library was approximately fivefold worse than those of the LN8 library, thus suggesting that the additional bases might perturb the responsiveness of library, a consideration for future structured library designs.
Bead-based libraries of DNA generated by ePCR have previously been employed for the identification of genetic variations[15] as well as transcription factors.[16] Bead-based RNA libraries have also previously been used with FACS to identify RNA molecules capable of binding the small molecule dye FlAsH and enhancing its fluorescence.[17] In that report, the RNA libraries were generated chemically on beads by using a traditional split pool approach, which prohibited performing multiple rounds of selection. Here, we employed the use of ePCR and a DNA library to develop a system which allowed us to turn multiple rounds of selection for the direct selection of aptamer beacons. Although our current work is limited to DNA, the use of novel DNA polymerase variants should allow for the generation of libraries composed of alternate materials, including 2′-fluoro RNA, 2′-O-methyl RNA, or other synthetic genetic polymers.[18]
The ability to use FACS to screen libraries of oligonucleotides for aptamers that switch structure and signal the presence of analytes presents a number of significant advantages over previous selection technologies. In particular, unlike almost all other approaches, our selection approach allows for the ability to directly identify signaling molecules. Thus, selected molecules can be readily synthesized and used to generate detection agents with little or no additional engineering or optimization. This led to the identification of a more sensitive DIS aptamer variant that eluded discovery by traditional selection and optimization techniques. Additionally, using a larger library, we identified two novel aptamers with similar detection capabilities. Second, this selection protocol identified sequences within three rounds of selection, with each round taking only a single day of work. This is a marked improvement over the number of rounds required in most selections and the time taken for further conversion of structure-switching aptamers into fluorescent devices. Finally, because the selective pressure is imposed by FACS gating, the selection can potentially be tuned to identify molecules with particular properties (e.g., maximal response, activation kinetics).
Although we have used this approach to search small libraries (∼105–107), as many as 108 events can be screened per hour on most high-throughput sorters (∼30 000 events per second), allowing for selection from larger libraries. This number could be further increased by including multiple sequences per beads during the early rounds of selection and then decreasing the number of variants per bead for the later round as described for other compartmentalization-based selection schemes.[19] For example, initial rounds of selection with 1000 sequences per bead could increase the library complexity to ∼1011, although such a selection would require significant increases in the sensitivity of detection. Finally, our approach could be combined with other, more traditional, selection schemes that allow for pre-selection of structural-switching aptamers from much larger libraries (∼1014–16), or even extended to the selection of other oligonucleotide-based signaling molecules, such as allosteric deoxyribozymes.
Acknowledgments
We would like to thank Amos Yan, Paul Gianella, and Sonald Duclair for helpful suggestions. This work was supported by the National Institutes of Health (grant numbers R21 CA157366 and R21 CA182330).
Supporting Information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re-organized for online delivery, but are not copy-edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
miscellaneous_information
References
- 1.Wang K, Tang Z, Yang CJ, Kim Y, Fang X, Li W, Wu Y, Medley CD, Cao Z, Li J, Colon P, Lin H, Tan W. Angew. Chem. Int. Ed. 2009;48:856. doi: 10.1002/anie.200800370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angew. Chem. 2009;121 [Google Scholar]
- 2.Liu J, Cao Z, Lu Y. Chem. Rev. 2009;109:1948. doi: 10.1021/cr030183i. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nutiu R, Li Y. Methods. 2005;37:16. doi: 10.1016/j.ymeth.2005.07.001. [DOI] [PubMed] [Google Scholar]
- 4.Fang X, Sen A, Vicens M, Tan W. ChemBioChem. 2003;4:829. doi: 10.1002/cbic.200300615. [DOI] [PubMed] [Google Scholar]
- 5.Hamaguchi N, Ellington A, Stanton M. Anal. Biochem. 2001;294:126. doi: 10.1006/abio.2001.5169. [DOI] [PubMed] [Google Scholar]
- 6.Stojanovic MN, de Prada P, Landry DW. J. Am. Chem. Soc. 2001;123:4928. doi: 10.1021/ja0038171. [DOI] [PubMed] [Google Scholar]
- 7.Yamamoto R, Baba T, Kumar PK. Genes Cells. 2000;5:389. doi: 10.1046/j.1365-2443.2000.00331.x. [DOI] [PubMed] [Google Scholar]
- 8.Nutiu R, Li Y. J. Am. Chem. Soc. 2003;125:4771. doi: 10.1021/ja028962o. [DOI] [PubMed] [Google Scholar]
- 9.Morse DP. Biochem. Biophys. Res. Commun. 2007;359:94. doi: 10.1016/j.bbrc.2007.05.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Oh SS, Plakos K, Lou X, Xiao Y, Soh HT. Proc. Natl. Acad. Sci. USA. 2010;107:14053. doi: 10.1073/pnas.1009172107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rajendran M, Ellington AD. Anal. Bioanal. Chem. 2008;390:1067. doi: 10.1007/s00216-007-1735-8. [DOI] [PubMed] [Google Scholar]
- 12.Yang KA, Pei R, Stefanovic D, Stojanovic MN. J. Am. Chem. Soc. 2012;134:1642. doi: 10.1021/ja2084256. [DOI] [PubMed] [Google Scholar]
- 13.Kojima T, Takei Y, Ohtsuka M, Kawarasaki Y, Yamane T, Nakano H. Nucleic Acids Res. 2005;33:e150. doi: 10.1093/nar/gni143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Diehl F, Li M, He Y, Kinzler KW, Vogelstein B, Dressman D. Nat. Methods. 2006;3:551. doi: 10.1038/nmeth898. [DOI] [PubMed] [Google Scholar]
- 15.Dressman D, Yan H, Traverso G, Kinzler KW, Vogelstein B. Proc. Natl. Acad. Sci. USA. 2003;100:8817. doi: 10.1073/pnas.1133470100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schütze T, Rubelt F, Repkow J, Greiner N, Erdmann VA, Lehrach H, Konthur Z, Glökler J. Anal. Biochem. 2011;410:155. doi: 10.1016/j.ab.2010.11.029. [DOI] [PubMed] [Google Scholar]
- 17.Wiesmayr A, Jäschke A. Bioorg. Med. Chem. 2011;19:1041. doi: 10.1016/j.bmc.2010.08.006. [DOI] [PubMed] [Google Scholar]
- 18.Pinheiro VB, Taylor AI, Cozens C, Abramov M, Renders M, Zhang S, Chaput JC, Wengel J, Peak-Chew SY, McLaughlin SH, Herdewijn P, Holliger P. Science. 2012;336:341. doi: 10.1126/science.1217622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zheng Y, Roberts RJ. Nucleic Acids Res. 2007;35:e83. doi: 10.1093/nar/gkm410. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
miscellaneous_information