

Abstract
Taxol (generic name paclitaxel) is a microtubule-stabilizing drug that is approved by the Food and Drug Administration for the treatment of ovarian, breast, and lung cancer, as well as Kaposi's sarcoma. It is used off-label to treat gastroesophageal, endometrial, cervical, prostate, and head and neck cancers, in addition to sarcoma, lymphoma, and leukemia. Paclitaxel has long been recognized to induce mitotic arrest, which leads to cell death in a subset of the arrested population. However, recent evidence demonstrates that intratumoral concentrations of paclitaxel are too low to cause mitotic arrest and result in multipolar divisions instead. It is hoped that this insight can now be used to develop a biomarker to identify the ∼50% of patients that will benefit from paclitaxel therapy. Here I discuss the history of paclitaxel and our recently evolved understanding of its mechanism of action.
HOW TAXOL WAS DISCOVERED AND RENAMED PACLITAXEL
Between 1960 and 1981, the National Cancer Institute (NCI) and the U.S. Department of Agriculture (USDA) collaborated on a plant screening program that collected and tested 115,000 extracts from 15,000 species of plants to identify naturally occurring compounds with anticancer activity. Samples from a single Pacific yew tree, Taxus brevifolia, were obtained by USDA botanist Arthur Barclay on the last day of his expedition in 1962. After his return, crude extracts from bark, twigs, needles, and fruit were tested, and bark extract was found to be cytotoxic. Mansukh Wani and Monroe Wall, working under contract with the NCI at the Research Triangle Institute (Research Triangle Park, NC), received T. brevifolia samples in 1964. By 1967, they had isolated and identified the active ingredient from the bark of T. brevifolia and named it taxol, based on its species of origin and the presence of hydroxyl groups (Perdue and Hartwell, 1969; Wall and Wani, 1995). In 1971, they published the structure of taxol (Wani et al., 1971), and it entered the NCI drug development program (Table 1).
TABLE 1:
How taxol became paclitaxel.
| Year | Event |
| 1964 | Sample from Pacific yew tree T. brevifolia found to be cytotoxic in joint program between the NCI and the USDAa,b,c |
| 1967 | Wall lab identifies active ingredient in T. brevifolia and names taxolb |
| 1971 | Taxol structure publishedd |
| 1978 | Taxol shows efficacy against mouse tumor modelsc |
| 1979 | Horwitz lab shows that taxol stabilizes microtubulese |
| 1984 | Taxol enters phase I clinical trialsa,c |
| 1985 | Taxol enters phase II clinical trialsa,c |
| 1991 | The NCI selects BMS to commercialize taxol; taxol transitions from public to private property |
| First congressional hearing on acquisition of taxol by BMSa,c | |
| 1992 | BMS trademarks the name “Taxol” and assigns new generic name of paclitaxel |
| The Food and Drug Administration (FDA) approves Taxol for ovarian cancer | |
| Pacific Yew Act passed to ensure survival of T. brevifoliaa,c,f | |
| 1993 | Second congressional hearing on acquisition of Taxol by BMSa,c |
| 1994 | The FDA approves Taxol for breast cancer |
| The FDA approves semisynthetic manufacture of Taxol by BMSa | |
| 1995 | Protection of T. brevifolia by Pacific Yew Act endsa |
| 1999 | The FDA approves Taxol for non–small cell lung cancer (NSCLC)f |
f Tuma (2003).
Taxol showed mixed results in preclinical trials and was not uniformly considered the most promising plant product. The insolubility of taxol in water necessitated its formulation with polyethoxylated castor oil, which can cause severe anaphylactic reactions and further dampened enthusiasm. However, by 1978 taxol had shown efficacy in a subset of mouse tumor models, including P388 leukemia (Fuchs and Johnson, 1978), and it entered clinical trials in 1984 (Walsh and Goodman, 2002a; Tuma, 2003; Table 1).
Several clinical trials were delayed because of a shortage of taxol, the only source of which at the time was the slow-growing T. brevifolia. Despite the scarcity, a clinical study on ovarian cancer proceeded and eventually concluded that 30% of patients with advanced ovarian cancer responded to taxol therapy (McGuire et al., 1989). High demand for taxol resulted in severe depletion of T. brevifolia, since removing the bark killed the trees. In 1990, the Department of the Interior was petitioned to include T. brevifolia on the list of endangered species, and the Pacific Yew Act was passed in 1992 to safeguard the tree (Walsh and Goodman, 1999).
In 1988, it was estimated that the cost of manufacturing taxol from the existing T. brevifolia was 10 times the budget available for the project at the NCI, and environmental concerns regarding the long-term prospects for T. brevifolia were growing. Owing to the limited accessibility of taxol, as well as its unique structure and cytotoxic potential, at least 30 laboratories worldwide competed to develop a total synthesis. However, because of the complexity of the molecule, these efforts were not successful until 1994 (Holton et al., 1994a, b; Nicolaou et al., 1994). Ultimately, several methods for total synthesis were developed, all of which require roughly 40 steps of reactions, and a more practical, semisynthetic protocol became the standard approach for production. However, in 1989, no immediate solution for obtaining large quantities of taxol was apparent, and the NCI made the decision to transfer taxol to a pharmaceutical company for commercialization. The request for applications received four responses, and Bristol-Myers Squibb (BMS) was selected in 1991. In 1992, BMS trademarked the name “Taxol” and created the new generic name paclitaxel (Walsh and Goodman, 1999, 2002a, b; Table 1). This occurred despite the fact that the term taxol had been used in >600 manuscripts published over the course of 20 years.
Congressional hearings were held in 1991 and 1993 regarding the transition of taxol to Taxol and paclitaxel (Table 1). The hearings questioned granting BMS a monopoly on a natural resource, as well as the higher price that was charged for drugs, like Taxol, that were identified and developed with federal funding rather than private money. The subcommittee staff concluded that the agreements between NCI and BMS were “not sufficient to fully protect the public interest” (United States Congress, House Committee on Small Business, Subcommittee on Regulation, Business Opportunities, and Energy, 1992). However, the agreements were not substantively altered. Taxol is the most profitable chemotherapy drug in history, and the only drug in clinical use identified by the plant screening program (Walsh and Goodman, 1999, 2002a, b).
BASIC EFFECTS OF PACLITAXEL IN VITRO AND IN VIVO
In 1977, the NCI sent samples of paclitaxel (still referred to as taxol at that point) to Susan Horwitz at Albert Einstein College of Medicine (New York, NY). In 1979, she reported that paclitaxel promotes the assembly of microtubules—polymers composed of repeating subunits of α- and β-tubulin heterodimers. Paclitaxel reduces the critical concentration of purified tubulin subunits necessary for polymerization into microtubules in vitro and increases the percentage of tubulin subunits that assemble. Furthermore, microtubules polymerized in the presence of paclitaxel are protected from the disassembly normally induced by cold or calcium treatment (Schiff et al., 1979). These effects were in stark contrast to previously identified microtubule poisons, including colchicine and vinca alkaloids, which prevent microtubule polymerization (Malawista and Bensch, 1967; Bensch and Malawista, 1968; De Brabander et al., 1981).
Similar to its effects on purified tubulin, paclitaxel promotes microtubule polymerization and stabilization in living cells, where it is capable of antagonizing the effects of colchicine and vinca alkaloids (Schiff and Horwitz, 1980; De Brabander et al., 1981). Phenotypically, paclitaxel treatment arrests a diverse array of cell types in mitosis, in both animal tumor models and cell culture (Fuchs and Johnson, 1978; Schiff and Horwitz, 1980; Milas et al., 1995; Jordan and Wilson, 2004; Orth et al., 2008). A large majority of reports indicate that paclitaxel-arrested cells are in metaphase and contain near-normal, bipolar spindles. Canonical images of paclitaxel treatment show cells with chromosomes aligned at the cell equator, although in some cases one or a few chromosomes have been reported to remain misaligned (Jordan et al., 1993, 1996; Waters et al., 1996). A small number of studies have reported that certain concentrations of paclitaxel induce multipolar spindles (Chen and Horwitz, 2002; Hornick et al., 2008). However, until recently, there was no evidence to suggest that these effects were clinically relevant. The dominant perception, by far, has been that the antitumor effects of paclitaxel occur due to its ability to arrest cells in metaphase on bipolar spindles.
Paclitaxel-induced mitotic arrest occurs due to activation of the mitotic checkpoint (also known as the spindle assembly checkpoint), the major cell cycle control mechanism acting during mitosis to prevent chromosome missegregation. The mitotic checkpoint delays separation of the chromosomes, which enter mitosis as replicated pairs of sister chromatids, until each pair has made stable attachments to both poles of the mitotic spindle. This arrangement ensures that each daughter cell will receive one copy of every chromatid. Chromatids connect to spindle microtubules through their kinetochores, protein complexes that assemble on centromeric regions of DNA. Unattached kinetochores, which have not made stable attachments to microtubules, activate a signal transduction cascade that delays mitotic progression by inhibiting the anaphase-promoting complex/cyclosome (Kops et al., 2005; Lara-Gonzalez et al., 2012; Foley and Kapoor, 2013). Paclitaxel treatment arrests cells in mitosis due to the presence of a small number of unattached kinetochores (Waters et al., 1998).
In addition to its utility in cancer therapy, paclitaxel is also widely used in cell biology. In untreated cells, bipolar attachment of sister chromatids places kinetochores under tension, which helps stabilize the interactions between kinetochores and spindle microtubules. Paclitaxel treatment reduces the tension on kinetochores that maintain bipolar attachment (Waters et al., 1998), and is a useful tool both for arresting cells in mitosis and for dissecting the contributions of tension versus attachment in satisfying the mitotic checkpoint (Maresca and Salmon, 2010).
CHALLENGES IN STUDYING THE MECHANISM OF PACLITAXEL
A PubMed search for paclitaxel (or Taxol) returns ∼25,000 articles. Despite this vast literature, until recently, the clinically relevant concentration for use in cell culture studies has been unclear. There are several reasons for this. First, paclitaxel is dosed at various levels and on different schedules, depending on the disease and the chemotherapy regimen selected. Second, the concentration of paclitaxel in the plasma changes over time as the drug is cleared, primarily by the liver. Third, paclitaxel accumulates intracellularly in cancer cell lines by 50- to >1000-fold, depending on cell type and the concentration added (Jordan et al., 1993, 1996; Yvon et al., 1999). Therefore the concentration of paclitaxel is almost certainly higher in the tumor than in the plasma, where it is typically measured, but there is no linear calculation to predict the fold concentration. Fourth, intratumoral measurements require a biopsy after initiation of therapy, which is not readily accessible outside of a clinical trial.
In the absence of data establishing the intratumoral concentration of paclitaxel, it was reasonable to infer that its antitumor effects were due to mitotic arrest. Unfortunately, determination of the fate of mitotically arrested cells is not straightforward. Mitotic arrest results in either death during mitosis or an abnormal exit from mitosis, without chromosome segregation or cytokinesis, to form a tetraploid G1 cell; this exit is known as mitotic slippage. After slippage, cells can die, arrest, or continue cycling. What determines the fate of cells after mitotic arrest remains unknown.
One factor frequently implicated in the response to mitotic arrest is the mitotic checkpoint. A functional mitotic checkpoint has been reported by numerous groups to be required for efficient cell killing in response to mitotic arrest. In contrast, cells in which the mitotic checkpoint is weakened have also been reported to be sensitive to paclitaxel. Still other studies have found that the state of the mitotic checkpoint does not affect this sensitivity (Rieder and Maiato, 2004; Weaver and Cleveland, 2005; Yamada and Gorbsky, 2006; Ryan et al., 2012). Some have hypothesized that a weakened mitotic checkpoint confers only short-term resistance to mitotic arrest (Janssen et al., 2009), whereas others have proposed that activation of the mitotic checkpoint followed by mitotic slippage results in optimal cell killing (Tao et al., 2005). One popular hypothesis was that the duration of mitotic arrest is predictive of cell death, with cells that arrest longer being more likely to die. However, multiple studies observing individual cells have now shown that the length of time a cell spends in mitosis cannot predict whether it will survive (Gascoigne and Taylor, 2008; Orth et al., 2008; Shi et al., 2008).
The difficulty of predicting sensitivity to mitotic arrest was further demonstrated in a study using nontransformed, chromosomally stable cells. Time-lapse microscopy was used to identify sister cells that resulted from a normal bipolar division without chromosome missegregation. Unexpectedly, the fates of the sister cells in response to mitotic arrest were completely unrelated. If one cell died from mitosis, its sister was no more likely to die from mitosis than it was to slip into interphase (and either die or survive). Thus cell fate in response to mitotic arrest is stochastic and not determined genetically (Gascoigne and Taylor, 2008).
ALTERNATE HYPOTHESIS OF INTERPHASE ACTION
The predominant hypothesis for the past several decades has been that paclitaxel kills tumor cells as a consequence of mitotic arrest. However, despite significant effects on mitosis that are sufficient to cause cell death, it has been suggested that paclitaxel causes death in tumors through effects on interphase cells. This proposal is largely based on the idea that the mitotic index in tumors is not sufficient to explain the efficacy of paclitaxel. Human tumors have a slow doubling time, and calculations that predict mitotic index based on tumor doubling rates, without accounting for cell death, suggest that an insufficient number of cells pass through mitosis in the presence of paclitaxel to account for tumor shrinkage rates (Komlodi-Pasztor et al., 2011, 2012; Mitchison, 2012). However, cell death has been observed in a wide array of untreated patient tumors, and directly measured proliferative rates are much higher than those estimated based on tumor doubling rates (Kerr and Searle, 1972; Kerr et al., 1972; Searle et al., 1973; Lowe and Lin, 2000). In addition, paclitaxel is retained in tumors for >5 d (Mori et al., 2006; Koshiba et al., 2009), permitting an extended window of time for cells to undergo one or more rounds of division in the presence of drug.
Mechanistically, it is unclear how paclitaxel might enact cell death in interphase without having affected a prior mitosis. It has been hypothesized that paclitaxel may interfere with cell signaling, trafficking, and microtubule-mediated transport (Herbst and Khuri, 2003; Komlodi-Pasztor et al., 2011). However, in cell culture, clinically relevant levels of paclitaxel do not cause death in interphase cells that have not previously undergone mitosis in the context of drug (Janssen et al., 2013; Zasadil et al., 2014). Of interest, in tumor models observed using intravital microscopy, the mitotic index after treatment with doses of paclitaxel expected to cause mitotic arrest was quite low (Orth et al., 2011; Janssen et al., 2013), leading to the suggestion that the microenvironment allows paclitaxel to exhibit interphase effects not observed in culture. However, no clear cytotoxic mechanism has yet emerged.
CLINICALLY RELEVANT CONCENTRATIONS OF PACLITAXEL CAUSE MULTIPOLAR DIVISIONS
To better mimic the antineoplastic effects of paclitaxel in cell culture, we first collaborated with our physician colleagues to design a clinical trial to measure the intratumoral concentration of paclitaxel in primary breast tumors (Zasadil et al., 2014). To remove as many confounding variables as possible, patients who had not received prior therapy and did not require concurrent therapy were enrolled. At 20 h after the initiation of the first dose of 175 mg/m2 paclitaxel, samples were obtained to measure paclitaxel concentration in both plasma and tumor. The 20-h time point was selected because the mitotic index of breast cancer cells in culture is increased ≥15-fold between 16 and 32 h after paclitaxel administration, and we therefore predicted that mitotic arrest would be evident at this time point. To assess whether tumors responded, measurements were obtained by ultrasound and/or mammogram before treatment and after four standard cycles of paclitaxel.
As predicted by prior cell culture experiments, the intratumoral concentration of paclitaxel (1–9 μM) was higher than the plasma concentration (80–280 nM) in all patients. However, contrary to expectations based on cell culture data, mitotic arrest was neither necessary nor sufficient for tumor shrinkage in response to paclitaxel (Zasadil et al., 2014).
As a second step in determining the appropriate dose of paclitaxel with which to treat our cultured cells, we determined the extent to which the drug was concentrated in breast cancer cell lines. Consistent with previous results (Jordan et al., 1993, 1996; Yvon et al., 1999), we found that paclitaxel accumulated to a differing extent in distinct cell lines. High-performance liquid chromatography analysis determined that treatment with low nanomolar concentrations of paclitaxel (5–10 nM for MDA-MB-231 and 10–50 nM for Cal51) resulted in clinically relevant intracellular concentrations of 1–9 μM.
Of interest, whereas higher concentrations of drug cause a robust mitotic arrest in these breast cancer cell lines, clinically relevant concentrations do not. They do, however, induce multipolar spindle formation. Importantly, a majority of mitotic cells in patient tumors treated with paclitaxel also exhibit multipolar spindles. After a brief delay, cultured cells in clinically relevant concentrations of paclitaxel proceed through mitosis on multipolar spindles and often segregate their chromosomes in three, four, or five different directions. However, a portion of the cytokinetic furrows usually fail, and most divisions in paclitaxel produce two or three daughter cells (Figure 1; Zasadil et al., 2014).
FIGURE 1:
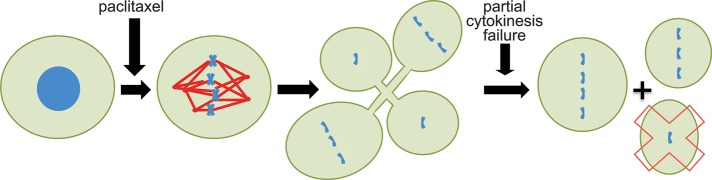
Clinically relevant concentrations of paclitaxel kill tumor cells by inducing multipolar divisions. Cells entering mitosis in the presence of concentrations of paclitaxel equivalent to those in human breast tumors form abnormal spindles that contain additional spindle poles. Rather than mounting a long-term mitotic arrest, these cells enter anaphase and divide their chromosomes in multiple directions. However, a portion of the cytokinetic furrows often fail, and two or three daughter cells are usually produced. Chromosome segregation is randomized due to multipolar division followed by partial cytokinesis failure. The resultant daughter cells are aneuploid, and a portion of these die (red X), presumably due to loss of one or more essential chromosomes.
IMPLICATIONS
Because paclitaxel causes mitotic arrest at concentrations typically used in culture, and was believed to do so in human tumors, numerous other drugs that induce mitotic arrest without affecting microtubule dynamics have entered clinical trials. These include inhibitors of Aurora A, CENP-E, Eg5/KSP, and Plk1. The expectation for these drugs was that they would have the efficacy of paclitaxel without one of its major dose-limiting toxicities—peripheral neuropathy. Peripheral neuropathy after paclitaxel therapy is presumed to result from impaired transport along the longest axons in the body, although data for this are scarce, and some chemotherapy drugs that do not affect microtubules, such as cisplatin, cause the same symptoms. Disappointingly, despite causing an accumulation of mitotic figures, the new classes of antimitotic drugs have yet to match the efficacy of paclitaxel (Chakravarty et al., 2011; Komlodi-Pasztor et al., 2011; Mitchison, 2012). This may be because the cytotoxic effect of paclitaxel in patient tumors is induction of multipolar divisions rather than mitotic arrest.
Numerous screens to identify markers of resistance or sensitivity to paclitaxel have been performed. These have identified a diverse array of candidates, including proteasome subunits, cyclin G1, and solute carrier genes (Rouzier et al., 2005; Swanton et al., 2007; Whitehurst et al., 2007; Pusztai et al., 2009; Juul et al., 2010; Wertz et al., 2011; Njiaju et al., 2012; Russell et al., 2012). However, these discoveries have not yet led to a validated biomarker that predicts which patients will benefit from paclitaxel therapy. This may be due, at least in part, to an emphasis on higher concentrations of drug that cause mitotic arrest and rapid cell death. It is hoped that recognition of the clinically relevant mechanism of paclitaxel will facilitate identification of a biomarker capable of predicting which patients will benefit from its use.
CONCLUSION
Like all drugs, paclitaxel exhibits concentration-dependent effects. It is not surprising that the rapid, dramatic effects of higher paclitaxel concentrations on mitosis and cell death were originally believed to be responsible for its efficacy in cancer therapy. Unfortunately, the barriers to acquisition of patient samples by basic scientists substantially delayed the finding that lower concentrations, which are slower to evoke cell death, are clinically relevant. However, newly available data demonstrate that, rather than causing mitotic arrest, intratumoral concentrations of paclitaxel cause cell death due to chromosome missegregation on multipolar spindles. It is hoped that, in addition to expediting identification of a predictive biomarker for paclitaxel treatment, this insight will also encourage collaboration between basic scientists and clinicians.
Acknowledgments
I thank Mark Burkard, Lauren Zasadil, and Eric Britigan for useful discussions and critiques. This work was supported by the National Institutes of Health (R01CA140458).
Abbreviations used:
- BMS
Bristol-Myers Squibb
- FDA
Food and Drug Administration
- NCI
National Cancer Institute
Footnotes
REFERENCES
- Bensch KG, Malawista SE. Microtubule crystals: a new biophysical phenomenon induced by Vinca alkaloids. Nature. 1968;218:1176–1177. doi: 10.1038/2181176a0. [DOI] [PubMed] [Google Scholar]
- Chakravarty A, Shinde V, Tabernero J, Cervantes A, Cohen RB, Dees EC, Burris H, Infante JR, Macarulla T, Elez E, et al. Phase I assessment of new mechanism-based pharmacodynamic biomarkers for MLN8054, a small-molecule inhibitor of Aurora A kinase. Cancer Res. 2011;71:675–685. doi: 10.1158/0008-5472.CAN-10-1030. [DOI] [PubMed] [Google Scholar]
- Chen JG, Horwitz SB. Differential mitotic responses to microtubule-stabilizing and -destabilizing drugs. Cancer Res. 2002;62:1935–1938. [PubMed] [Google Scholar]
- De Brabander M, Geuens G, Nuydens R, Willebrords R, De Mey J. Taxol induces the assembly of free microtubules in living cells and blocks the organizing capacity of the centrosomes and kinetochores. Proc Natl Acad Sci USA. 1981;78:5608–5612. doi: 10.1073/pnas.78.9.5608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foley EA, Kapoor TM. Microtubule attachment and spindle assembly checkpoint signalling at the kinetochore. Nat Rev Mol Cell Biol. 2013;14:25–37. doi: 10.1038/nrm3494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchs DA, Johnson RK. Cytologic evidence that Taxol, an antineoplastic agent from Taxus brevifolia, acts as a mitotic spindle poison. Cancer Treat Rep. 1978;62:1219–1222. [PubMed] [Google Scholar]
- Gascoigne KE, Taylor SS. Cancer cells display profound intra- and interline variation following prolonged exposure to antimitotic drugs. Cancer Cell. 2008;14:111–122. doi: 10.1016/j.ccr.2008.07.002. [DOI] [PubMed] [Google Scholar]
- Herbst RS, Khuri FR. Mode of action of docetaxel—a basis for combination with novel anticancer agents. Cancer Treat Rev. 2003;29:407–415. doi: 10.1016/s0305-7372(03)00097-5. [DOI] [PubMed] [Google Scholar]
- Holton RA, Kim HB, Somoza C, Liang F, Biediger RJ, Boatman PD, Shindo M, Smith CC, Kim SC, Nadizadeh H, et al. First total synthesis of Taxol. 2. Completion of the C and D rings. J Am Chem Soc. 1994a;116:1599–1600. [Google Scholar]
- Holton RA, Somoza C, Kim HB, Liang F, Biediger RJ, Boatman PD, Shindo M, Smith CC, Kim SC, Nadizadeh H, et al. First total synthesis of Taxol. I. Functionalization of the B ring. J Am Chem Soc. 1994b;116:1597–1598. [Google Scholar]
- Hornick JE, Bader JR, Tribble EK, Trimble K, Breunig JS, Halpin ES, Vaughan KT, Hinchcliffe EH. Live-cell analysis of mitotic spindle formation in Taxol-treated cells. Cell Motil Cytoskeleton. 2008;65:595–613. doi: 10.1002/cm.20283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janssen A, Beerling E, Medema R, van Rheenen J. Intravital FRET imaging of tumor cell viability and mitosis during chemotherapy. PLoS One. 2013;8:e64029. doi: 10.1371/journal.pone.0064029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janssen A, Kops GJ, Medema RH. Elevating the frequency of chromosome mis-segregation as a strategy to kill tumor cells. Proc Natl Acad Sci USA. 2009;106:19108–19113. doi: 10.1073/pnas.0904343106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan MA, Toso RJ, Thrower D, Wilson L. Mechanism of mitotic block and inhibition of cell proliferation by Taxol at low concentrations. Proc Natl Acad Sci USA. 1993;90:9552–9556. doi: 10.1073/pnas.90.20.9552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan MA, Wendell K, Gardiner S, Derry WB, Copp H, Wilson L. Mitotic block induced in HeLa cells by low concentrations of paclitaxel (Taxol) results in abnormal mitotic exit and apoptotic cell death. Cancer Res. 1996;56:816–825. [PubMed] [Google Scholar]
- Jordan MA, Wilson L. Microtubules as a target for anticancer drugs. Nat Rev Cancer. 2004;4:253–265. doi: 10.1038/nrc1317. [DOI] [PubMed] [Google Scholar]
- Juul N, Szallasi Z, Eklund AC, Li Q, Burrell RA, Gerlinger M, Valero V, Andreopoulou E, Esteva FJ, Symmans WF, et al. Assessment of an RNA interference screen-derived mitotic and ceramide pathway metagene as a predictor of response to neoadjuvant paclitaxel for primary triple-negative breast cancer: a retrospective analysis of five clinical trials. Lancet Oncol. 2010;11:358–365. doi: 10.1016/S1470-2045(10)70018-8. [DOI] [PubMed] [Google Scholar]
- Kerr JF, Searle J. A suggested explanation for the paradoxically slow growth rate of basal-cell carcinomas that contain numerous mitotic figures. J Pathol. 1972;107:41–44. doi: 10.1002/path.1711070107. [DOI] [PubMed] [Google Scholar]
- Kerr JF, Wyllie AH, Currie AR. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer. 1972;26:239–257. doi: 10.1038/bjc.1972.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komlodi-Pasztor E, Sackett DL, Fojo AT. Inhibitors targeting mitosis: tales of how great drugs against a promising target were brought down by a flawed rationale. Clin Cancer Res. 2012;18:51–63. doi: 10.1158/1078-0432.CCR-11-0999. [DOI] [PubMed] [Google Scholar]
- Komlodi-Pasztor E, Sackett D, Wilkerson J, Fojo T. Mitosis is not a key target of microtubule agents in patient tumors. Nat Rev Clin Oncol. 2011;8:244–250. doi: 10.1038/nrclinonc.2010.228. [DOI] [PubMed] [Google Scholar]
- Kops GJ, Weaver BA, Cleveland DW. On the road to cancer: aneuploidy and the mitotic checkpoint. Nat Rev Cancer. 2005;5:773–785. doi: 10.1038/nrc1714. [DOI] [PubMed] [Google Scholar]
- Koshiba H, Hosokawa K, Mori T, Kubo A, Watanabe A, Honjo H. Intravenous paclitaxel is specifically retained in human gynecologic carcinoma tissues in vivo. Int J Gynecol Cancer. 2009;19:484–488. doi: 10.1111/IGC.0b013e3181a130db. [DOI] [PubMed] [Google Scholar]
- Lara-Gonzalez P, Westhorpe FG, Taylor SS. The spindle assembly checkpoint. Curr Biol. 2012;22:R966–980. doi: 10.1016/j.cub.2012.10.006. [DOI] [PubMed] [Google Scholar]
- Lowe SW, Lin AW. Apoptosis in cancer. Carcinogenesis. 2000;21:485–495. doi: 10.1093/carcin/21.3.485. [DOI] [PubMed] [Google Scholar]
- Malawista SE, Bensch KG. Human polymorphonuclear leukocytes: demonstration of microtubules and effect of colchicine. Science. 1967;156:521–522. doi: 10.1126/science.156.3774.521. [DOI] [PubMed] [Google Scholar]
- Maresca TJ, Salmon ED. Welcome to a new kind of tension: translating kinetochore mechanics into a wait-anaphase signal. J Cell Sci. 2010;123:825–835. doi: 10.1242/jcs.064790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGuire WP, Rowinsky EK, Rosenshein NB, Grumbine FC, Ettinger DS, Armstrong DK, Donehower RC. Taxol: a unique antineoplastic agent with significant activity in advanced ovarian epithelial neoplasms. Ann Intern Med. 1989;111:273–279. doi: 10.7326/0003-4819-111-4-273. [DOI] [PubMed] [Google Scholar]
- Milas L, Hunter NR, Kurdoglu B, Mason KA, Meyn RE, Stephens LC, Peters LJ. Kinetics of mitotic arrest and apoptosis in murine mammary and ovarian tumors treated with Taxol. Cancer Chemother Pharmacol. 1995;35:297–303. doi: 10.1007/BF00689448. [DOI] [PubMed] [Google Scholar]
- Mitchison TJ. The proliferation rate paradox in antimitotic chemotherapy. Mol Biol Cell. 2012;23:1–6. doi: 10.1091/mbc.E10-04-0335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori T, Kinoshita Y, Watanabe A, Yamaguchi T, Hosokawa K, Honjo H. Retention of paclitaxel in cancer cells for 1 week in vivo and in vitro. Cancer Chemother Pharmacol. 2006;58:665–672. doi: 10.1007/s00280-006-0209-6. [DOI] [PubMed] [Google Scholar]
- Nicolaou KC, Yang Z, Liu JJ, Ueno H, Nantermet PG, Guy RK, Claiborne CF, Renaud J, Couladouros EA, Paulvannan K. Total synthesis of Taxol. Nature. 1994;367:630–634. doi: 10.1038/367630a0. [DOI] [PubMed] [Google Scholar]
- Njiaju UO, Gamazon ER, Gorsic LK, Delaney SM, Wheeler HE, Im HK, Dolan ME. Whole-genome studies identify solute carrier transporters in cellular susceptibility to paclitaxel. Pharmacogenet Genomics. 2012;22:498–507. doi: 10.1097/FPC.0b013e328352f436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orth JD, Kohler RH, Foijer F, Sorger PK, Weissleder R, Mitchison TJ. Analysis of mitosis and antimitotic drug responses in tumors by in vivo microscopy and single-cell pharmacodynamics. Cancer Res. 2011;71:4608–4616. doi: 10.1158/0008-5472.CAN-11-0412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orth JD, Tang Y, Shi J, Loy CT, Amendt C, Wilm C, Zenke FT, Mitchison TJ. Quantitative live imaging of cancer and normal cells treated with kinesin-5 inhibitors indicates significant differences in phenotypic responses and cell fate. Mol Cancer Ther. 2008;7:3480–3489. doi: 10.1158/1535-7163.MCT-08-0684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perdue RE, Jr, Hartwell JL. The search for plant sources of anticancer drugs. Morris Arboretum Bull. 1969;20:35–53. [Google Scholar]
- Pusztai L, Jeong JH, Gong Y, Ross JS, Kim C, Paik S, Rouzier R, Andre F, Hortobagyi GN, Wolmark N, Symmans WF. Evaluation of microtubule-associated protein-Tau expression as a prognostic and predictive marker in the NSABP-B 28 randomized clinical trial. J Clin Oncol. 2009;27:4287–4292. doi: 10.1200/JCO.2008.21.6887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rieder CL, Maiato H. Stuck in division or passing through: what happens when cells cannot satisfy the spindle assembly checkpoint. Dev Cell. 2004;7:637–651. doi: 10.1016/j.devcel.2004.09.002. [DOI] [PubMed] [Google Scholar]
- Rouzier R, Rajan R, Wagner P, Hess KR, Gold DL, Stec J, Ayers M, Ross JS, Zhang P, Buchholz TA, et al. Microtubule-associated protein tau: a marker of paclitaxel sensitivity in breast cancer. Proc Natl Acad Sci USA. 2005;102:8315–8320. doi: 10.1073/pnas.0408974102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell P, Hennessy BT, Li J, Carey MS, Bast RC, Freeman T, Venkitaraman AR. Cyclin G1 regulates the outcome of taxane-induced mitotic checkpoint arrest. Oncogene. 2012;31:2450–2460. doi: 10.1038/onc.2011.431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan SD, Britigan EM, Zasadil LM, Witte K, Audhya A, Roopra A, Weaver BA. Up-regulation of the mitotic checkpoint component Mad1 causes chromosomal instability and resistance to microtubule poisons. Proc Natl Acad Sci USA. 2012;109:E2205–2214. doi: 10.1073/pnas.1201911109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiff PB, Fant J, Horwitz SB. Promotion of microtubule assembly in vitro by Taxol. Nature. 1979;277:665–667. doi: 10.1038/277665a0. [DOI] [PubMed] [Google Scholar]
- Schiff PB, Horwitz SB. Taxol stabilizes microtubules in mouse fibroblast cells. Proc Natl Acad Sci USA. 1980;77:1561–1565. doi: 10.1073/pnas.77.3.1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Searle J, Collins DJ, Harmon B, Kerr JF. The spontaneous occurrence of apoptosis in squamous carcinomas of the uterine cervix. Pathology. 1973;5:163–169. doi: 10.3109/00313027309060831. [DOI] [PubMed] [Google Scholar]
- Shi J, Orth JD, Mitchison T. Cell type variation in responses to antimitotic drugs that target microtubules and kinesin-5. Cancer Res. 2008;68:3269–3276. doi: 10.1158/0008-5472.CAN-07-6699. [DOI] [PubMed] [Google Scholar]
- Swanton C, Marani M, Pardo O, Warne PH, Kelly G, Sahai E, Elustondo F, Chang J, Temple J, Ahmed AA, et al. Regulators of mitotic arrest and ceramide metabolism are determinants of sensitivity to paclitaxel and other chemotherapeutic drugs. Cancer Cell. 2007;11:498–512. doi: 10.1016/j.ccr.2007.04.011. [DOI] [PubMed] [Google Scholar]
- Tao W, South VJ, Zhang Y, Davide JP, Farrell L, Kohl NE, Sepp-Lorenzino L, Lobell RB. Induction of apoptosis by an inhibitor of the mitotic kinesin KSP requires both activation of the spindle assembly checkpoint and mitotic slippage. Cancer Cell. 2005;8:49–59. doi: 10.1016/j.ccr.2005.06.003. [DOI] [PubMed] [Google Scholar]
- Tuma RS. Taxol's journey from discovery to use: lessons and updates. Oncol Times. 2003;25:52–57. [Google Scholar]
- United States Congress, House Committee on Small Business, Subcommittee on Regulation, Business Opportunities, and Energy. Exclusive agreements between federal agencies and Bristol-Myers Squibb Co. for drug development: is the public interest protected. Washington, DC: U.S. Government Printing Office; 1992. [Google Scholar]
- Wall ME, Wani MC. Camptothecin and Taxol: discovery to clinic—thirteenth Bruce F. Cain Memorial Award Lecture. Cancer Res. 1995;55:753–760. [PubMed] [Google Scholar]
- Walsh V, Goodman J. Cancer chemotherapy, biodiversity, public and private property: the case of the anti-cancer drug Taxol. Soc Sci Med. 1999;49:1215–1225. doi: 10.1016/s0277-9536(99)00161-6. [DOI] [PubMed] [Google Scholar]
- Walsh V, Goodman J. From taxol to Taxol: the changing identities and ownership of an anti-cancer drug. Med Anthropol. 2002a;21:307–336. doi: 10.1080/01459740214074. [DOI] [PubMed] [Google Scholar]
- Walsh V, Goodman J. The billion dollar molecule: Taxol in historical and theoretical perspective. Clio Med. 2002b;66:245–267. doi: 10.1163/9789004333499_013. [DOI] [PubMed] [Google Scholar]
- Wani MC, Taylor HL, Wall ME, Coggon P, McPhail AT. Plant antitumor agents. VI. The isolation and structure of Taxol, a novel antileukemic and antitumor agent from Taxus brevifolia. J Am Chem Soc. 1971;93:2325–2327. doi: 10.1021/ja00738a045. [DOI] [PubMed] [Google Scholar]
- Waters JC, Chen RH, Murray AW, Salmon ED. Localization of Mad2 to kinetochores depends on microtubule attachment, not tension. J Cell Biol. 1998;141:1181–1191. doi: 10.1083/jcb.141.5.1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waters JC, Mitchison TJ, Rieder CL, Salmon ED. The kinetochore microtubule minus-end disassembly associated with poleward flux produces a force that can do work. Mol Biol Cell. 1996;7:1547–1558. doi: 10.1091/mbc.7.10.1547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver BA, Cleveland DW. Decoding the links between mitosis, cancer, and chemotherapy: the mitotic checkpoint, adaptation, and cell death. Cancer Cell. 2005;8:7–12. doi: 10.1016/j.ccr.2005.06.011. [DOI] [PubMed] [Google Scholar]
- Wertz IE, Kusam S, Lam C, Okamoto T, Sandoval W, Anderson DJ, Helgason E, Ernst JA, Eby M, Liu J, et al. Sensitivity to antitubulin chemotherapeutics is regulated by MCL1 and FBW7. Nature. 2011;471:110–114. doi: 10.1038/nature09779. [DOI] [PubMed] [Google Scholar]
- Whitehurst AW, Bodemann BO, Cardenas J, Ferguson D, Girard L, Peyton M, Minna JD, Michnoff C, Hao W, Roth MG, et al. Synthetic lethal screen identification of chemosensitizer loci in cancer cells. Nature. 2007;446:815–819. doi: 10.1038/nature05697. [DOI] [PubMed] [Google Scholar]
- Yamada HY, Gorbsky GJ. Spindle checkpoint function and cellular sensitivity to antimitotic drugs. Mol Cancer Ther. 2006;5:2963–2969. doi: 10.1158/1535-7163.MCT-06-0319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yvon AM, Wadsworth P, Jordan MA. Taxol suppresses dynamics of individual microtubules in living human tumor cells. Mol Biol Cell. 1999;10:947–959. doi: 10.1091/mbc.10.4.947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zasadil LM, Andersen KA, Yeum D, Rocque GB, Wilke LG, Tevaarwerk AJ, Raines RT, Burkard ME, Weaver BA. Cytotoxicity of paclitaxel in breast cancer is due to chromosome missegregation on multipolar spindles. Sci Transl Med. 2014;6:229ra243. doi: 10.1126/scitranslmed.3007965. [DOI] [PMC free article] [PubMed] [Google Scholar]