

The ARF tumor suppressor is best known for its role in stabilizing p53. This study identifies p53-independent functions of ARF in chromosome segregation and the mitotic checkpoint. Mitotic defects caused by loss of ARF are recapitulated by Aurora B overexpression and rescued by partial depletion of Aurora B.
Abstract
The ARF tumor suppressor is part of the CDKN2A locus and is mutated or undetectable in numerous cancers. The best-characterized role for ARF is in stabilizing p53 in response to cellular stress. However, ARF has tumor suppressive functions outside this pathway that have not been fully defined. Primary mouse embryonic fibroblasts (MEFs) lacking the ARF tumor suppressor contain abnormal numbers of chromosomes. However, no role for ARF in cell division has previously been proposed. Here we demonstrate a novel, p53-independent role for ARF in the mitotic checkpoint. Consistent with this, loss of ARF results in aneuploidy in vitro and in vivo. ARF−/− MEFs exhibit mitotic defects including misaligned and lagging chromosomes, multipolar spindles, and increased tetraploidy. ARF−/− cells exhibit overexpression of Mad2, BubR1, and Aurora B, but only overexpression of Aurora B phenocopies mitotic defects observed in ARF−/− MEFs. Restoring Aurora B to near-normal levels rescues mitotic phenotypes in cells lacking ARF. Our results define an unexpected role for ARF in chromosome segregation and mitotic checkpoint function. They further establish maintenance of chromosomal stability as one of the additional tumor-suppressive functions of ARF and offer a molecular explanation for the common up-regulation of Aurora B in human cancers.
INTRODUCTION
The ARF tumor suppressor is encoded by an alternate reading frame of the INK4a tumor suppressor in the CDKN2a (also known as the INK4a/ARF) locus. ARF is best known for its role in the p53 pathway. In response to cellular stress, ARF is released from the nucleolus and enters the cytoplasm, where it binds MDM2 (Pomerantz et al., 1998; Zhang et al., 1998), an E3 ubiquitin ligase that inhibits the transcriptional activity of p53 (Momand et al., 1992) and targets it for degradation (Honda et al., 1997). Binding of ARF inhibits the ability of MDM2 to ubiquitinate p53 and also promotes MDM2 degradation, thus stabilizing p53 and allowing for cell cycle arrest (Zhang et al., 1998; Honda and Yasuda, 1999). However, it has been shown that ARF has tumor-suppressive roles outside of this pathway (Korgaonkar et al., 2002). Mice lacking ARF and p53 develop a wider tumor spectrum and are more likely to have multiple tumors per animal than mice lacking p53 alone (Weber et al., 2000a). In addition, high levels of ARF expression impede entry into and progress through S phase in cells lacking p53 and MDM2 (Weber et al., 2000a; Yarbrough et al., 2002), suggesting that ARF interacts with distinct partners to mediate cell cycle delay. ARF can also bind and inhibit numerous transcription factors independently of p53, including FoxM1B, E2F, and Myc (Kalinichenko et al., 2004; Ozenne et al., 2010). p53-independent functions of ARF have also been identified in ribosome biogenesis and sumoylation (Ozenne et al., 2010). Thus, ARF has multiple functions outside of the p53 pathway, some of which contribute to its tumor-suppressive activity.
Aneuploidy, an abnormal chromosome complement that deviates from a multiple of the haploid, occurs in ∼85% of human tumors (Weaver and Cleveland, 2006; Zasadil et al., 2013). Aneuploidy is often accompanied by chromosomal instability (CIN), the recurrent gain or loss of chromosomes during multiple divisions. Aneuploidy and CIN serve as markers of poor prognosis in multiple cancer types, including lymphomas, soft tissue sarcomas, and cancers of the lung, breast, ovaries, and colon. Several mouse models that develop aneuploidy and CIN have an elevated rate of spontaneous and/or carcinogen-induced tumorigenesis (Ricke et al., 2008; Holland and Cleveland, 2009; Schvartzman et al., 2010; Zasadil et al., 2013). Together, the data suggest that aneuploidy and a low rate of CIN can be tumor promoting.
Chromosome missegregation during mitosis is a common cause of aneuploidy and CIN in tumor cells (Cimini et al., 2001; Ganem et al., 2009; Silkworth et al., 2009; Ryan et al., 2012). The mitotic checkpoint (also termed the spindle assembly checkpoint) is the major cell cycle regulator acting during mitosis to prevent chromosome missegregation. To produce genetically identical progeny, replicated sister chromatids are sorted and segregated on a bipolar spindle composed of microtubules. Sister chromatids attach to spindle microtubules through their kinetochores—protein structures that assemble at centromeric DNA. The mitotic checkpoint delays the separation of sister chromatids, which occurs at the transition from metaphase to anaphase, until all kinetochores have formed stable attachments to spindle microtubules (Rieder et al., 1994; 1995). Sister separation occurring before formation of stable attachments to opposite poles results in random segregation of chromosomes, increasing the likelihood of producing aneuploid progeny. To prevent this, unattached kinetochores recruit mitotic checkpoint components, including Bub1, BubR1, Mad1, Mad2, and CENP-E, to catalytically generate a diffusible inhibitor of the anaphase-promoting complex/cyclosome (APC/C; Nilsson et al., 2008; Kulukian et al., 2009; Han et al., 2013). Once all kinetochores have stable microtubule attachments, the checkpoint is satisfied and APC/C becomes active, leading to sister chromatid separation and the generation of euploid progeny (Vleugel et al., 2012).
The Aurora B kinase is a member of the chromosomal passenger complex (CPC), which localizes to inner centromeres in early mitosis and relocates to the spindle midzone after anaphase onset (Cooke et al., 1987). During prometaphase, Aurora B functions to release improper kinetochore–microtubule interactions that would impair accurate chromosome segregation (Lan et al., 2004; Cimini et al., 2006). Aurora B overexpression occurs in a variety of human cancers (Chieffi et al., 2006; Vischioni et al., 2006; Chen et al., 2009; Lin et al., 2010), and inhibitors of Aurora B are in clinical trials (Komlodi-Pasztor et al., 2012). In this study we identify novel, p53-independent, tumor-suppressive functions of ARF in chromosome segregation and mitotic checkpoint signaling via regulation of Aurora B protein levels.
RESULTS
Loss of ARF causes aneuploidy in vitro
We and others reported that ARF loss results in abnormal chromosome numbers in MEFs (di Tommaso et al., 2009; Silk et al., 2013). However, the mechanism remained unclear. To determine how loss of ARF resulted in chromosome missegregation, MEFs were prepared from E14.5 ARF−/− embryos and wild-type littermates. Loss of ARF caused a significant increase in aneuploidy, as assessed by chromosome spreads (also known as metaphase spreads; Figure 1, A–C). Although most aneuploid ARF−/− cells had a near-diploid chromosome number, ARF−/− MEFs also exhibited a 2.4-fold increase in near-tetraploidy (Figure 1, D and E), suggestive of cytokinesis failure. Consistent with this, MEFs lacking ARF also had a 1.9-fold increase in the percentage of binucleate cells relative to MEFs with a wild-type complement of ARF (Figure 1, F and G).
FIGURE 1:

ARF loss causes aneuploidy in vitro. (A) Diploid MEF chromosome spread with 40 chromosomes. Scale bar, 10 μm. (B) Average aneuploidy in MEFs. n = 3 experiments of 50 spreads each. (C) Histogram of chromosome spreads from B. Inset, enlarged histogram showing the percentages of near-tetraploid MEFs. (D) Tetraploid MEF chromosome spread with 80 chromosomes. Scale bar, 10 μm. (E) Average tetraploidy in MEFs. n = 3 experiments of 100 spreads each. (F) Binucleate MEF. Red, F-actin stained with phalloidin. Blue, DNA. Scale bar, 20 μm. (G) Average percentage of binucleation in MEFs of the indicated genotypes. n = 250 cells from each of three independent experiments. **p < 0.001.
ARF−/− animals develop aneuploidy in vivo
To test whether ARF loss results in chromosome missegregation in an intact organism, we collected splenocytes from 5-mo-old mice for analysis of aneuploidy using chromosome spreads (Figure 2A). Indeed, ARF−/− splenocytes showed a 3.7-fold increased level of aneuploidy relative to splenocytes from wild-type mice (Figure 2B), demonstrating that loss of ARF is sufficient to induce aneuploidy in vivo. All aneuploid splenocytes had near-diploid numbers of chromosomes (Figure 2C). To further examine this, we analyzed tissue from the small intestine by fluorescent in situ hybridization (FISH) using a probe for chromosome 11 (Figure 2D). Loss of ARF resulted in a significant increase in abnormal numbers of chromosome 11 in intestinal tissue (Figure 2, E and F). Overall, these results indicate that ARF is required to maintain chromosomal stability in vitro and in vivo.
FIGURE 2:
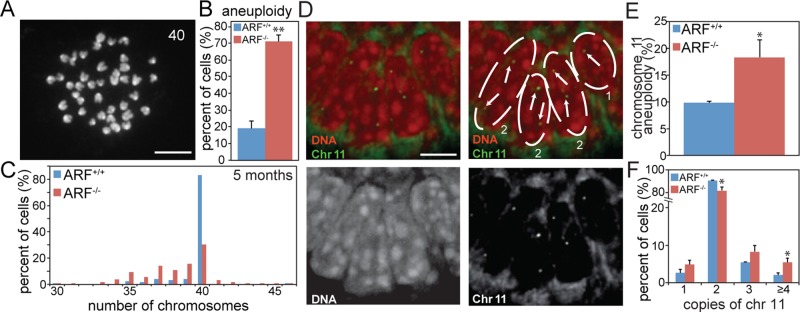
ARF is required to maintain chromosomal stability in vivo. (A) Chromosome spread from a mouse splenocyte with 40 chromosomes. Scale bar, 10 μm. (B) Average aneuploidy in 5-mo-old mouse splenocytes. n = 3 independent experiments of 50 spreads each. (C) Histogram of chromosome spreads from B. No tetraploid spreads were observed in splenocytes. Mitotic indices of splenocytes were comparable in wild-type (0.52 ± 0.18%) and ARF-null (0.62 ± 0.03%) animals. n ≥ 930 cells from three animals. (D) Murine intestine labeled with FISH probe to chromosome 11. Top right is labeled to indicate nuclear boundaries and number of chromosome 11 signals in each cell. (E) Percentage of cells of the indicated genotypes with greater or less than two copies of chromosome 11. (F) Percentage of cells with the indicated number of copies of chromosome 11. *p < 0.05; **p < 0.001.
Loss of ARF causes mitotic defects
The increased rate of aneuploidy in ARF−/− cells suggested that passage through mitosis in the absence of ARF would result in mitotic defects. Indeed, examination of asynchronously cycling MEFs in mitosis revealed that loss of ARF resulted in a 5.2-fold increased frequency of misaligned chromosomes when the majority of chromosomes were at the metaphase plate (Figure 3, A and B), suggesting a deficit in chromosome congression. To test this directly, we treated cells with the proteasome inhibitor MG132 to prevent anaphase onset and permit additional time for chromosome alignment to occur. ARF−/− cells showed a substantial decrease in the percentage of cells that successfully aligned their chromosomes as compared with wild type. This was true whether MG132 was added to asynchronous cells or to those that had formerly established monopolar spindles due to treatment with the Eg5/KSP inhibitor monastrol (Figure 3, C and D). ARF−/− MEFs also showed an increase in chromosomes that lag behind the main bodies of segregating DNA during anaphase or telophase (lagging chromosomes; Figure 3, E and F), which are commonly used as a marker of chromosome missegregation (Thompson and Compton, 2011).
FIGURE 3:

ARF−/− cells exhibit mitotic defects. (A, B) ARF−/− MEFs have increased levels of misaligned chromosomes. (A) Left, normal metaphase. Right, cell with metaphase plate and misaligned chromosome (arrow). (B) Percentage of MEFs of the indicated genotypes containing visible metaphase plates and misaligned chromosomes. n > 50 metaphases from three independent experiments. (C, D) ARF loss causes congression defects. (C) Left, image of MG132-treated cell with fully congressed chromosomes. Right, image of MG132-treated cell with incompletely aligned chromosomes. (D) Quantitation of the percentage of wild-type and ARF−/− cells that successfully aligned their chromosomes after 3 h treatment with 10 μM MG132 (left) or after 18 h of 100 μM monastrol to induce monopolar spindles, followed by washout into 3 h of MG132 (mon→MG; right). (E, F) MEFs lacking ARF have increased levels of lagging chromosomes in anaphase and telophase. (E) Left, normal anaphase. Right, abnormal anaphase cell with lagging chromosome indicated by arrow. (F) Percentage of cells of the indicated genotype with lagging chromosomes. n >100 anaphases and telophases from thee independent experiments. (G, H) ARF−/− MEFs have an elevated frequency of supernumery centrosomes. (G) Interphase cells with two (left) or four centrosomes (right), denoted by arrows. (H) Percentage of interphase cells with abnormal numbers of centrosomes. n > 250 cells from each of three independent experiments. (I–K) ARF loss results in multipolar spindles. (I) Example of a multipolar spindle in an ARF−/− MEF. (J) Percentage of MEFs with abnormal spindles. (K) Histogram showing number of poles per spindle. n > 100 mitotic cells from each of three independent experiments. Scale bars, 5 μm. *p < 0.05.
Cytokinesis failure produces tetraploid G1 cells with two centrosomes. After centriole replication in S phase, tetraploid cells can develop supernumery centrosomes. Consistent with the 2.4-fold increase in tetraploidy in cells lacking ARF expression (Figure 1E), ARF loss in MEFs resulted in a twofold increase in cells with abnormal numbers of centrosomes in interphase (Figure 3, G and H). Because centrosomes are the primary nucleating sites of microtubules, these extra centrosomes can result in formation of multipolar spindles in the subsequent mitosis. Further analysis of mitotic cells showed that MEFs lacking ARF have a threefold increased incidence of multipolar spindles (Figure 3, I– K). Together, these mitotic defects would be expected to result in increased rates of chromosome missegregation and aneuploidy in ARF−/− cells.
Loss of ARF results in a weakened mitotic checkpoint
To determine whether mitotic checkpoint function is compromised in ARF−/− cells, we challenged MEFs with the microtubule poison colcemid. Colcemid treatment results in loss of spindle microtubules, producing unattached kinetochores and an active mitotic checkpoint. Whereas wild-type MEFs accumulated in mitosis in response to colcemid, ARF−/− cells had a significantly reduced mitotic index (Figure 4A). Consistent with this, time-lapse analysis revealed that the duration of mitosis was significantly shorter in cells lacking ARF (Figure 4B). To confirm that this was an ARF-specific effect, we tested whether mitotic arrest in response to colcemid could be rescued by transfection with a yellow fluorescent protein (YFP)–tagged version of the ARF protein. Like endogenous ARF, ARF-YFP localizes to nucleoli during interphase (Weber et al., 2000b; Supplemental Figure S1, A and B). High levels of ARF overexpression were reported to cause an interphase arrest because they result in an increase in cells with a 2n, G0/G1 DNA content on flow cytometry profiles. In certain cell types, a substantial increase (20–30%) in the percentage of cells with a 2n content of DNA is observed (Quelle et al., 1995; Weber et al., 2000a; Yarbrough et al., 2002). However, in other cases, the increase in the G0/G1 population is ≤10% (Quelle et al., 1995; Yarbrough et al., 2002). In most cases, the percentage of ARF-overexpressing cells in G2/M is similar, or somewhat increased, compared to the percentage in control cells (Quelle et al., 1995; Weber et al., 2000a; Yarbrough et al., 2002). Indeed, when treated with the microtubule poison nocodazole, cells overexpressing ARF retain their ability to enter mitosis (Zhang et al., 1998). Consistent with these data, the mitotic index in cells expressing ARF-YFP was indistinguishable from that in cells transfected with empty vector in the absence of microtubule poisons (Figure 4C, left). However, expression of ARF-YFP was sufficient to restore mitotic checkpoint activity in ARF−/− cells. The mitotic index of ARF−/- cells expressing ARF-YFP was indistinguishable from the mitotic index of wild-type cells transfected with empty vector after 16 h of colcemid treatment (Figure 4C, right). This rescue experiment confirms that the mitotic checkpoint defect in ARF−/− cells is due to an acute requirement for ARF in mitotic checkpoint signaling as opposed to one or more compensatory changes that occurred during development in response to loss of ARF.
FIGURE 4:

ARF−/− cells have a weakened mitotic checkpoint. (A) Mitotic index of MEFs treated with colcemid for the specified number of hours. n = 250 cells at each time point from each of three independent experiments. (B) Duration of mitosis in MEFs treated with colcemid, as assessed by time-lapse microscopy. n = 100 cells from three independent experiments. (C) ARF transfection rescues mitotic checkpoint activity in ARF−/− MEFs. After 32 h of transfection with ARF-YFP or empty vector, MEFs were treated with colcemid or vehicle for 16 h before analysis of mitotic index. n = 250 cells from each of three independent experiments. (D) Mitotic index of MEFs infected with retroviruses expressing empty vector or wild-type or V24E p14ARF. Green fluorescent protein (GFP) was also expressed from an internal ribosomal entry site. After 32 h of infection, cells were treated with colcemid for 16 h before analysis of mitotic index. n > 250 cells from each of three independent experiments. (E) Percentage of metaphase cells with misaligned chromosomes after 72 h of infection with retroviruses expressing empty vector or wild-type or V24E p14ARF. Both wild-type p14ARF and the MDM2-binding-domain mutant V24E rescue the occurrence of misaligned chromosomes in ARF−/− MEFs. n > 50 metaphases from three independent experiments. (F) Percentage of anaphase or telophase MEFs with lagging chromosomes after infection with empty vector or wild-type or V24E p14ARF. Both wild-type and V24E p14ARF reduce the incidence of lagging chromosomes in ARF−/− cells. n > 100 anaphases and telophases from three independent experiments. (G) Levels of the mitotic checkpoint components Mad2 and BubR1 are elevated in ARF−/− MEFs, whereas levels of Bub1 and CENP-E remain unchanged. Tubulin, loading control. *p < 0.05.
To test whether a version of ARF containing a mutation in the MDM2-binding site encoded by exon 1β is capable of rescuing the mitotic checkpoint defect caused by ARF loss, we infected ARF−/− MEFs with a retrovirus expressing either wild-type or V24E p14ARF. Valine 24 occurs in a 15–amino acid peptide (amino acids 16–30) that binds MDM2 (Bothner et al., 2001), and cells expressing the V24E ARF mutant are impaired in their ability to stabilize p53 and activate its transcriptional activity (Korgaonkar et al., 2002; di Tommaso et al., 2009). Interestingly, the V24E mutant was as effective as the wild type in rescuing the mitotic checkpoint defect in ARF−/− cells (Figure 4D), suggesting that this function is independent of p53. Furthermore, both wild-type and V24E ARF rescued the occurrence of misaligned and lagging chromosomes in ARF−/− MEFs (Figure 4, E and F).
ARF−/− cells exhibit p53-independent mitotic defects
Aneuploidy has also been reported in p53−/− MEFs (Harvey et al., 1993). To formally test whether the mitotic effects of ARF loss were p53 dependent, we bred mice deficient in ARF and p53 to produce animals heterozygous for both genes. Doubly heterozygous mice were then interbred to produce wild-type, ARF−/−, p53−/−, and ARF−/−;p53−/− MEFs, which were examined for mitotic defects. Both ARF−/− and p53−/− cells displayed misaligned chromosomes in the presence of a metaphase plate, but removal of ARF in cells lacking p53 further increased the percentage of cells with misaligned chromosomes (Figure 5A), suggesting that ARF promotes chromosome congression independently of p53. Lagging chromosomes were also observed in both ARF−/− and p53−/− cells. However, cells lacking both tumor suppressors had a higher level of lagging chromosomes than cells lacking either tumor suppressor alone (Figure 5B), indicating that ARF also uses p53-independent mechanisms to prevent lagging chromosomes.
FIGURE 5:
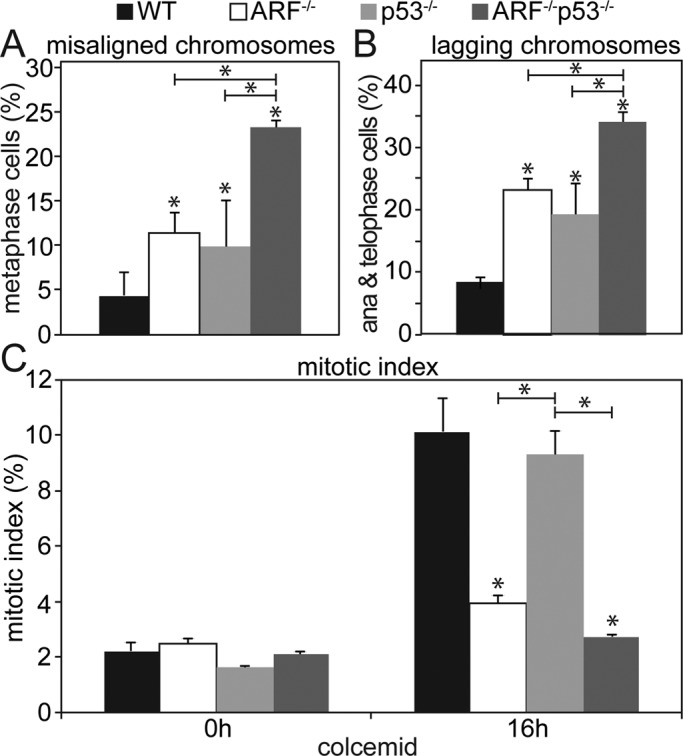
Mitotic functions of ARF are p53-independent. (A) Percentage of MEFs with misaligned chromosomes in metaphase. Loss of ARF increases the incidence of misaligned chromosomes in p53−/− cells. n > 80 metaphases from three independent experiments. (B) Percentage of MEFs with lagging chromosomes in anaphase or telophase. Although loss of either ARF or p53 increases the percentage of cells containing lagging chromosomes, reduction of ARF in p53−/− cells causes a further increase, consistent with a p53-independent role for ARF in preventing chromosome lagging. n > 100 anaphases and telophases from each of three independent experiments. (C) Mitotic index of MEFs of the indicated genotypes ± 16 h of colcemid treatment. ARF, but not p53, is required for the mitotic checkpoint–mediated increase in mitotic index in response to colcemid. n > 250 cells from each of three independent experiments. *p < 0.05.
To assess mitotic checkpoint function, we examined the mitotic index of MEFs with normal and reduced levels of p53 and/or ARF. No differences in mitotic index were observed in the absence of microtubule poisons (Figure 5C, left). After 16 h of colcemid treatment, wild-type cells accumulated in mitosis, as expected (Figure 5C, right). ARF−/− MEFs exhibited a decreased mitotic index relative to wild-type cells, consistent with the results in Figure 4, A– D. However, p53−/− MEFs accumulated in mitosis to the same extent as wild-type MEFs, indicating that p53 is not required for the mitotic checkpoint. Thus, the function of ARF in mitotic checkpoint signaling is independent of the p53 pathway.
Expression of BubR1 and Mad2 is elevated in ARF−/− cells
To determine the mechanism by which ARF functions in the mitotic checkpoint, we first examined ARF localization during mitosis. Although it is well established that ARF localizes to nucleoli during interphase (Weber et al., 2000b), localization of ARF in mitosis has not previously been reported. We found that endogenous ARF had a punctate localization during mitosis. A small portion of the puncta frequently colocalized with spindle poles (Supplemental Figure S1, C and D). However, ARF did not localize to kinetochores in asynchronous or colcemid-treated mitotic cells (Supplemental Figure S1, C and E).
To further understand the effects of ARF on mitotic checkpoint signaling, we compared the expression levels of the mitotic checkpoint proteins Bub1, CENP-E, BubR1, and Mad2 in cells expressing and lacking ARF. Immunoblot analysis of protein extracts from these cells showed that whereas Bub1 and CENP-E levels remained unchanged between wild-type and ARF−/− cells, both BubR1 and Mad2 levels were increased (Figure 4G). Despite the increase in overall protein levels of BubR1 and Mad2, only Mad2 showed enhanced kinetochore recruitment as assessed by quantitative immunofluorescence (Supplemental Figure S2). This could be due to more significant up-regulation of Mad2 or because of saturation of available BubR1-binding sites.
Overexpression of BubR1 does not cause mitotic defects
BubR1 is overexpressed in a variety of human cancers, including those of the breast, lung, and stomach (Weaver and Cleveland, 2006). BubR1 levels are also elevated in ARF−/− splenocytes (Supplemental Figure S3A). To determine whether overexpression of BubR1 caused the mitotic defects seen in ARF−/− MEFs, we incorporated a tetracycline-inducible, YFP-tagged human BubR1 expression vector into chromosomally stable colorectal cancer cells (DLD1) expressing the Tet repressor. Upon treatment with tetracycline, YFP-BubR1 was expressed (Supplemental Figure S3B) and localized to kinetochores (Supplemental Figure S3C). However, expression of YFP-BubR1 did not cause a significant increase in either misaligned or lagging chromosomes (Supplemental Figure S3, D and E). Nor did YFP-BubR1 expression induce supernumery centrosomes (Supplemental Figure S3F) or multipolar spindles (Supplemental Figure S3G) or alter mitotic checkpoint fidelity (Supplemental Figure S3H). Together, these data indicate that increased BubR1 expression is not responsible for the mitotic defects observed in ARF−/− MEFs, consistent with recent findings in BubR1-overexpressing mice (Baker et al., 2013).
Overexpression of Mad2 does not recapitulate mitotic phenotypes of ARF loss
Because overexpression of BubR1 was not sufficient to mimic the mitotic defects in ARF−/− cells, we next examined whether overexpression of Mad2 could do so. We overexpressed human Mad2 using the same tetracycline-inducible system used for BubR1. Overexpression of Mad2 (Supplemental Figure S4A) resulted in an increase in lagging chromosomes during anaphase and telophase (Supplemental Figure S4B), whereas misaligned chromosomes in metaphase were unaffected (Supplemental Figure S4C). Overexpression of Mad2 also failed to induce abnormal numbers of centrosomes in interphase or multipolar spindles in mitosis (Supplemental Figure S4, D and E). We next examined whether overexpression of Mad2 would weaken the mitotic checkpoint, as did ARF loss. However, overexpression of Mad2 did not have a significant effect on mitotic index (Supplemental Figure S4F), consistent with previous results (Sironi et al., 2001). It has been reported that reduction of p53 causes overexpression of Mad2 (Schvartzman et al., 2011), a finding that we were able to recapitulate (Supplemental Figure S4G). Thus, it is unsurprising that up-regulation of Mad2, which is p53-dependent, in not responsible for the p53-independent effects of ARF loss.
Aurora B kinase is overexpressed in ARF−/− cells in vitro and in vivo
Having found that up-regulation of neither Mad2 nor BubR1 could replicate the set of mitotic defects that occur due to ARF loss, we examined other ARF binding partners for evidence of effects on known mitotic proteins. Two transcription factors inhibited by ARF, FoxM1B and Myc, have been found to regulate mitotic genes, including the kinases Aurora A and B, as well as CENP-F (Laoukili et al., 2005; Courapied et al., 2010; den Hollander et al., 2010). We found that levels of Aurora A and CENP-F were unchanged in ARF−/− versus wild-type MEFs (Supplemental Figure S5). However, levels of Aurora B were elevated in ARF−/− MEFs in comparison to wild type (Figure 6A). Of importance, Aurora B expression was also increased in splenocytes of mice lacking ARF (Figure 6B), indicating that levels of Aurora B are increased in vivo as well as in vitro.
FIGURE 6:
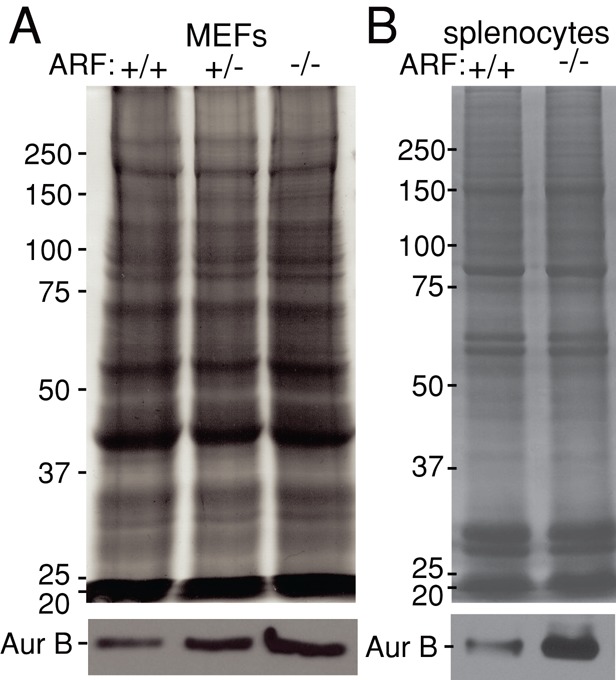
Aurora B expression is elevated in ARF−/− cells in vitro and in vivo. (A) Immunoblot showing increased levels of Aurora B in MEFs with reduced expression of ARF. (B) Splenocytes from ARF−/− mice express heightened amounts of Aurora B relative to splenocytes from wild-type animals. Coomassie is used as a loading control.
The half-life of Aurora B is increased in ARF−/− cells
Expression of Myc-GFP increases Aurora B promoter activity ∼30-fold (den Hollander et al., 2010). ARF binds and inhibits the transcriptional activity of Myc (Qi et al., 2004), suggesting that transcription of Aurora B is elevated in ARF−/− cells. However, quantitative PCR analysis using two distinct primer sets revealed that Aurora B mRNA levels are only modestly increased in ARF−/− cells compared with wild type (Figure 7A). To determine whether the elevated levels of Aurora B in ARF−/− cells were due to increased protein stability, we treated cells with the protein synthesis inhibitor cycloheximide and followed them over 12 h to determine the half-life of Aurora B protein. The Aurora B half-life in wild-type MEFs was similar to the half-life reported for human Aurora B in HeLa cells (Nguyen et al., 2005). However, the Aurora B half-life was increased approximately threefold in ARF null cells compared with wild type (Figure 7, B and C), revealing that the overexpression of Aurora B that occurs in the absence of ARF is likely due to effects on protein stability rather than transcription.
FIGURE 7:
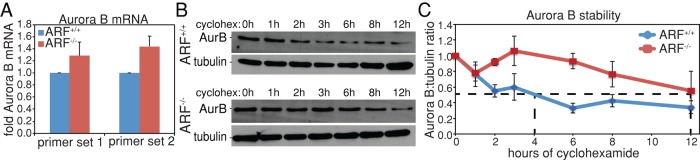
Elevated levels of Aurora B in ARF−/− cells are due to increased protein stability. (A) Quantitative real-time PCR examining relative Aurora B mRNA levels in ARF+/+ and ARF−/− cells. Two different primer sets show a modest, nonsignificant increase in Aurora B transcript levels in ARF−/− primary MEFs. p = 0.285 and 0.1088 for primer sets 1 and 2, respectively, as assessed by Wilcoxon signed rank test. n = 3. (B) Immunoblots showing Aurora B levels after the indicated number of hours of cyclohexamide treatment to prevent new protein synthesis. (C) Quantitation of Aurora B protein levels in ARF+/+ and ARF−/− cells after cycloheximide treatment. Aurora B is substantially more stable in ARF−/− than in ARF+/+ cells. n = 3 independent experiments. Mean values for Aurora B protein levels are as follows (90% mean confidence intervals are shown in parentheses): ARF+/+, 1 h, 0.7615 (0.5816, 0.9414); 2 h, 0.5503 (0.4635, 0.6371); 3 h, 0.6008 (0.4052, 0.7964); 6 h, 0.3308 (0.2618, 0.3997); 8 h, 0.4255 (0.3474, 0.5037); 12 h, 0.3417 (0.2518, 0.4315); ARF−/−,1 h, 0.7777 (0.6665, 0.8889); 2 h, 0.9211 (0.8913, 0.951); 3 h, 1.061 (0.8448, 1.277); 6 h, 0.9289 (0.8282, 1.03); 8 h, 0.7647 (0.5965, 0.933); and 12 h, 0.5516 (0.2819, 0.8213).
Overexpression of Aurora B phenocopies ARF loss
To determine whether overexpression of Aurora B was sufficient to induce the mitotic defects seen in ARF−/− cells, YFP-tagged Aurora B was transfected into wild-type MEFs. Aurora B-YFP was expressed (Figure 8A) and localized appropriately to inner centromeres in prometaphase (Figure 8B) and the midbody in telophase (Figure 8C). When challenged with colcemid to activate the mitotic checkpoint and arrest cells in mitosis, wild-type cells transfected with Aurora B exhibited a significantly lower mitotic index than cells transfected with empty vector (Figure 8D), indicating that overexpression of Aurora B alone is sufficient to weaken the mitotic checkpoint.
FIGURE 8:

Overexpression of Aurora B phenocopies ARF loss. (A) Immunoblot showing expression of Aurora B-YFP in primary MEFs. Tubulin is shown as a loading control. (B, C) Aurora B-YFP localizes appropriately. Aurora B-YFP transiently transfected into wild-type MEFs localizes to (B) inner centromeres in prometaphase and (C) the midbody in telophase. Scale bars, 5 μm. (D) Reduced mitotic index in wild-type cells expressing Aurora B-YFP after 16 h of treatment with 100 ng/ml colcemid, showing that overexpression of Aurora B is sufficient to weaken mitotic checkpoint signaling. n > 250 cells from each of three independent experiments. (E–I) Wild-type MEFs transfected with Aurora B-YFP have elevated levels of (E) misaligned chromosomes in metaphase (n ≥ 50 cells with metaphase plates from three independent experiments), (F) lagging chromosomes in anaphase and telophase (n > 100 anaphases and telophases from three independent experiments), (G) binucleate interphase cells (n > 250 cells from each of three independent experiments), (H) supernumery (>2) centrosomes in interphase (n > 250 cells from each of three independent experiments), and (I) multipolar spindles in mitosis (n > 100 mitotic cells from each of three independent experiments), as compared with wild-type MEFs transfected with empty vector (EV). *p < 0.05.
Elevated expression of Aurora B also caused other mitotic defects. Wild-type MEFs transfected with Aurora B had increased frequencies of misaligned chromosomes in metaphase (Figure 8E) and lagging chromosomes during anaphase and telophase (Figure 8F), similar to what was observed in ARF−/− MEFs (Figure 3, A– F). Increased Aurora B expression was also sufficient to cause elevated levels of binucleate cells (Figure 8G), consistent with cytokinesis failure and the formation of tetraploid cells. As would be expected after tetraploidization, expression of Aurora B-YFP resulted in supernumery centrosomes during interphase (Figure 8H) and multipolar spindles during mitosis (Figure 8I). Together, these results demonstrate that overexpression of Aurora B is sufficient to phenocopy ARF loss.
Partial depletion of Aurora B rescues mitotic defects in ARF−/− cells
To further confirm that elevated levels of Aurora B were responsible for the mitotic phenotypes observed in ARF−/− cells, Aurora B was depleted to near wild-type levels by transfection of two distinct small interfering RNA (siRNA) sequences. Transfection of either siRNA or a combination of both depleted Aurora B in ARF−/− cells to a level similar to that in wild-type MEFs (Figure 9A). Partial depletion of Aurora B rescued mitotic checkpoint function in ARF−/− MEFs and restored their ability to accumulate in mitosis in response to microtubule depolymerization (Figure 9B). Reducing levels of Aurora B in ARF−/− MEFs also rescued their chromosome congression defect (Figure 9C) and significantly reduced their incidence of lagging chromosomes (Figure 9D). These experiments show that normalization of Aurora B levels is sufficient to ameliorate the mitotic defects characteristic of ARF−/− cells.
FIGURE 9:

Partial knockdown of Aurora B to near wild-type levels rescues mitotic defects in ARF−/− cells. (A) Immunoblot demonstrating partial knockdown of Aurora B in ARF−/− primary MEFs to levels similar to those found in wild-type MEFs. Tubulin is shown as a loading control. (B) Partial knockdown of Aurora B rescues the mitotic checkpoint defect in ARF−/− cells. n > 250 cells from each of three independent experiments. (C) Partial depletion of Aurora B by siRNA rescues the chromosome alignment defect in ARF−/− cells. n > 50 cells with visible metaphase plates from each of three independent experiments. (D) Knockdown of Aurora B by siRNA to near-wild-type levels in ARF−/− cells partially rescues the occurrence of lagging chromosomes. n > 100 anaphases and telophases from each of three independent experiments. *p < 0.05.
DISCUSSION
Evidence for p53-independent tumor-suppressive functions of ARF has existed for more than a decade (Weber et al., 2000a), but a full understanding of these numerous effects has remained elusive. At high levels, ARF can impede progression through interphase (Weber et al., 2000a; Yarbrough et al., 2002; Eymin et al., 2003; Datta et al., 2005) and sequester—and thereby inhibit—the potentially oncogenic transcription factors Myc and E2F in a p53-independent manner (Ozenne et al., 2010). There is also evidence that ARF participates in the DNA damage response in a p53-independent, as well as a p53-dependent, manner (Eymin et al., 2006). In this context, we provide evidence that ARF maintains chromosomal stability in a p53-independent manner. Because CIN is a hallmark of human cancers, this is likely to be an additional mechanism by which ARF enacts tumor suppression independently of p53.
It was previously proposed that p53 is essential for the mitotic checkpoint after p53−/− MEFs showed a decreased mitotic index and increased ploidy in response to exposure to the microtubule poison nocodazole (Cross et al., 1995). However, subsequent studies found that p53−/− cells delay in mitosis for an equivalent amount of time as wild-type cells after treatment with microtubule poisons in both fixed (Kienitz et al., 2005) and live (Lanni and Jacks, 1998) assays. The increased ploidy of the nocodazole-treated p53−/− MEFs was the result of a G1 function of p53 in preventing rereplication of DNA after mitotic slippage (Di Leonardo et al., 1997). Thus, p53 is not required for an intact mitotic checkpoint response. Consistent with these previous studies, we show that the mitotic checkpoint defect observed in ARF−/− cells is independent of p53 (Figure 5C).
The role of Aurora B in the mitotic checkpoint has also been a matter of some debate. Budding yeast contain a single Aurora family homologue, Ipl1. Ipl1 and Aurora B play well-established roles in destabilizing improper kinetochore–microtubule attachments that do not place sister chromatids under tension (Biggins et al., 1999; Biggins and Murray, 2001; Ditchfield et al., 2003; Hauf et al., 2003; Lan et al., 2004). This error-correction activity results in the production of unattached kinetochores, which activate the mitotic checkpoint. Whether Ipl1/Aurora B activates the mitotic checkpoint merely by generating unattached kinetochores or whether additional functions are required has been disputed. In budding yeast, Ipl1 is required for a mitotic checkpoint arrest caused by lack of tension but not by lack of attachment (Biggins et al., 1999; Biggins and Murray, 2001; King et al., 2007). However, the fission yeast homologue, ark1, is required for the attachment-sensing checkpoint as well (Petersen and Hagan, 2003). Experiments in human cells with the Aurora B inhibitors Hesperadin and ZM447439 demonstrated that Aurora B–inhibited cells were unable to mount a robust, long-term mitotic checkpoint response to the microtubule-stabilizing drug Taxol, which produces attached kinetochores that are not under tension. However, the checkpoint response was maintained in the absence of kinetochore–microtubule attachments caused by the microtubule-destabilizing drug nocodazole (Ditchfield et al., 2003; Hauf et al., 2003). This was consistent with the findings in budding yeast. Because Aurora B–inhibited cells were capable of maintaining arrest in nocodazole, when all kinetochores are unattached, these data were often interpreted as indicating that Aurora B kinase activity is not required for mitotic checkpoint signaling. However, more recent evidence indicates that Aurora B kinase activity is incompletely inhibited by standard drug concentrations. Higher concentrations of Aurora B inhibitors anticipated to inhibit >95% of kinase activity prevent mitotic arrest in the absence of microtubules, demonstrating a requirement for Aurora B, and its associated kinase activity, in sensing attachment as well as tension (Santaguida et al., 2011). Consistent with this, a mutant of the Aurora B binding partner INCENP that is sufficient for correction of erroneous kinetochore–microtubule interactions is defective in supporting mitotic checkpoint activity (Vader et al., 2007). In addition, function-blocking Aurora B antibodies abrogate both the tension- and attachment-based mitotic checkpoints (Kallio et al., 2002), as does RNA interference–mediated depletion of Aurora B (Ditchfield et al., 2003). Thus, the weight of the evidence favors the interpretation that Aurora B functions in the mitotic checkpoint response to unattached kinetochores in higher eukaryotes. Intriguingly, our data show that overexpression of Aurora B also impairs mitotic checkpoint signaling.
Up-regulation of Mad2 and BubR1 in ARF−/− cells is likely to be a p53-dependent phenotype. Mad2 levels are elevated in p53−/− MEFs (Schvartzman et al., 2011; Supplemental Figure S4G) and in cells lacking the p53 transcriptional target and CDK inhibitor p21 (Schvartzman et al., 2011). Conversely, ectopic expression of p21 in wild-type MEFs results in lower levels of Mad2 protein. This reduction in Mad2 does not occur in cells triply negative for Rb and the pocket proteins p107 and p130, indicating that p21 is acting through the Rb pathway. In wild-type cells, p21 inhibits cyclin D/CDK4, which hyperphosphorylates and inactivates Rb, leading to activation of E2F transcription factors and S-phase entry. Consistent with the known pathway of cyclin D/CDK4–mediated inactivation of Rb, p21 mutants that cannot bind CDKs were unable to repress Mad2 promoter activity (Schvartzman et al., 2011). Thus, reduced stability of p53 is likely to contribute to elevated levels of Mad2 in ARF−/− cells. A similar mechanism is likely to explain up-regulation of BubR1 in p53−/− (Schvartzman et al., 2011) and ARF−/− cells, although data supporting the opposite conclusion—that p53 activates BubR1 transcription—have also been reported (Oikawa et al., 2005).
Interestingly, both reduced and elevated expression of mitotic checkpoint components, including Mad2, BubR1, and Aurora B, can result in similar phenotypes. Reduction of each of these components results in chromosome missegregation (Dobles et al., 2000; Michel et al., 2001; Ditchfield et al., 2003; Hauf et al., 2003; Honda et al., 2003; Baker et al., 2004; Meraldi et al., 2004). Overexpression of Mad2 (Sotillo et al., 2007; Supplemental Figure S4B) or Aurora B (Figure 8, E and F), but not BubR1 (Baker et al., 2013; Supplemental Figure S3), also causes segregation defects. With respect to the mitotic checkpoint, a decrease of Mad2, BubR1, or Aurora B impairs mitotic checkpoint signaling (Hoyt et al., 1991; Li and Murray, 1991; Kallio et al., 2002; Ditchfield et al., 2003; Hauf et al., 2003; Santaguida et al., 2011). Elevated expression of Mad2 delays fission yeast (He et al., 1997; Kim et al., 1998), Xenopus extracts (Chen et al., 1998; Fang et al., 1998), and certain tissue culture cells (Howell et al., 2000; Sotillo et al., 2007) in metaphase. In contrast, overexpression of BubR1 does not hyperactivate mitotic checkpoint signaling. Elevated levels of BubR1 have no notable effect on mitotic index in asynchronously cycling cells (Baker et al., 2013; Supplemental Figure S3H) and can actually rescue a checkpoint defect caused by heterozygous loss of Rae1 (Baker et al., 2013). Here we show that Aurora B overexpression results in a third outcome—reduction of mitotic checkpoint activity (Figure 8D). With respect to their tumor phenotypes, mice heterozygous for Mad2 or BubR1 have a relatively subtle increase in spontaneous (Mad2; Michel et al., 2001) or carcinogen-induced (BubR1; Dai et al., 2004; Baker et al., 2006) tumors. Overexpression of Mad2 causes a substantially more severe tumor phenotype than reduction of Mad2, in which tumors occur with increased penetrance and earlier onset (Sotillo et al., 2007). BubR1 overexpression, on the other hand, actually decreases tumor incidence and extends lifespan (Baker et al., 2013). Animal models with ubiquitously elevated levels of Aurora B have not been reported, but Aurora B overexpression is common in human tumors, in which it correlates with poor prognosis (Bischoff et al., 1998; Ehara et al., 2003; Araki et al., 2004; Sorrentino et al., 2005; Chieffi et al., 2006; Vischioni et al., 2006). Thus, both increased and decreased expression of these genes sometimes results in similar phenotypes. Whereas previous work showed that reduction of Aurora B protein or kinase activity results in mitotic defects, the data reported here reveal that overexpression of Aurora B causes similar defects, indicating that levels of Aurora B must be tightly controlled to prevent aneuploidy.
It is interesting to note that the duration of the mitotic checkpoint response to microtubule poisons is substantially longer in human than in rodent cells (Rieder and Maiato, 2004). It has long been known that rodent cells are less sensitive than human cells to perturbations of the microtubule cytoskeleton (Gupta, 1985). Rodent cells are also more likely to exit mitosis and resynthesize their DNA in the presence of microtubule poisons than are human cells (Kung et al., 1990). In both cases, Chinese hamster cells are the least responsive to microtubule perturbation, whereas mouse cells exhibit an intermediate phenotype between hamster and human cells. Rodent cells exhibit reduced uptake of vinblastine, colchicine, and Taxol compared with human cells, suggesting that lower internal concentration may account for the decrease in mitotic arrest and cell death (Gupta, 1985; Parekh and Simpkins, 1996). However, more recent experiments suggested that differences in phosphoregulation of the N-terminus of Mad1 confer species-specific differences to the stringency of the mitotic checkpoint (Haller et al., 2006). It will now be of interest to determine the consequences of ARF loss on the longer mitotic arrest observed in human cells.
Although independent of p53, our results demonstrate that the mitotic checkpoint defect caused by loss of ARF is a result of overexpression of Aurora B. Elevated Aurora B levels have been observed in a number of human malignancies, including non–small cell lung cancer (Vischioni et al., 2006), hepatocellular carcinoma (Lin et al., 2010), epithelial ovarian cancer (Chen et al., 2009), and prostate cancer (Chieffi et al., 2006). Further, high expression of Aurora B was correlated with poor prognosis in these cancers, and inhibitors of Aurora B are in clinical trials (Komlodi-Pasztor et al., 2012). Our results show that overexpression of Aurora B is sufficient to weaken the mitotic checkpoint and results in chromosome missegregation and CIN. ARF loss, by gene deletion, mutation, or promoter methylation, is likely to be one mechanism responsible for Aurora B overexpression in tumors.
MATERIALS AND METHODS
Animals, cell culture, and treatments
Animals were maintained in a C57BL/6 background and handled in accordance with the policies of the Institutional Animal Care and Use Committee of the University of Wisconsin–Madison. Primary MEFs were generated from embryonic day 14.5 (E14.5) embryos in 6-cm dishes with 3 ml of chilled 0.05% trypsin/EDTA. After the head, liver, and tail (for genotyping) were removed, embryos were minced using scissors, pipetted up and down using a 10-ml pipette, and incubated at 37°C for 15 min. Embryos were pipetted up and down with an additional 2 ml of trypsin/EDTA and a 5-ml pipette before further incubation at 37°C for 10 min. The solution was transferred to a 15-ml conical tube with 5 ml of primary MEF media. After permitting debris to settle for 1–2 min, we transferred the supernatant to a second 15-ml conical tube. MEFs in the supernatant were pelleted and resuspended in high-glucose DMEM (Invitrogen, Carlsbad, CA) containing 15% fetal bovine serum (FBS; Sigma-Aldrich, St. Louis, MO), 0.1 mM nonessential amino acids (Invitrogen), 1 mM sodium pyruvate (Invitrogen), 1 μM 2-mercaptoethanol (Acros Biological/Thermo Fisher, Waltham, MA), 2 mM l-glutamine (Invitrogen), and 50 μg/ml penicillin/streptomycin (Invitrogen) and cultured in 3% O2 and 10% CO2 at 37°C. Experiments were performed on MEFs between passages 3 and 20. Retroviruses (a kind gift from Dawn Quelle, University of Iowa) were added along with 8 μg/ml Polybrene 48 h before harvest of cells.
Flp-In TRex DLD1 cells (Invitrogen) were grown in high-glucose DMEM, 10% FBS, 2 mM l-glutamine, and 50 μg/ml penicillin/streptomycin and cultured in 5% CO2 at 37°C. Unless otherwise indicated, colcemid (Enzo, Farmingdale, NY) was used at 100 ng/ml, and mitotic indices were collected after 16 h of treatment. To induce expression of Mad2 and BubR1 transgenes, DLD1 cells were treated with 0.25 μM tetracycline for 48 h before analysis.
siRNA, 40 nM, directed against luciferase GL2 (control) or Aurora B (SASI_Mm01 00056605 and 00056608; Sigma-Aldrich) was transfected using Dharmafect 4. Analysis was performed 48 h posttransfection.
Chromosome (metaphase) spreads
Chromosome spreads were performed as in Weaver et al. (2007). Briefly, cultured cells were grown to 80% confluence in 6-cm dishes before being treated with 100 ng/ml colcemid for ∼4 h. Cells were then harvested, pelleted, and resuspended with 5 ml of room temperature 75 mM KCl for 11 min. Cells were treated with 1 ml of fresh fix (3:1 methanol:acetic acid) and pelleted before being placed in 4 ml of fresh fix overnight at 4°C.
For splenocyte spreads, spleens were shredded using two pairs of forceps. Single cells were obtained by pipetting using a P1000 and a Pasteur pipette and transferred to 15-ml tubes. After fragments settled, cells in the supernatant were pelleted and resuspended in 4 ml of RPMI 1640 (HyClone) with 100 μg/ml gentamicin (Life Technologies, Grand Island, NY), 11 μg/ml phytohemagglutinin (Sigma-Aldrich), 75 μg/ml lipopolysaccharide (Invitrogen), 10% FBS, and 5 μg/ml colchicine and cultured in 15-ml tubes at 37°C and 10% CO2 for 12 h. Splenocytes were pelleted, resuspended in 5 ml of 75 mM KCl prewarmed to 37°C, and incubated at 37°C for 45 min. One milliliter of 3:1 methanol and acetic acid fix was added before centrifugation. Cells were resuspended in 4 ml of fixative and stored overnight at 4°C.
Cultured cells and splenocytes were pelleted and resuspended in fresh fix twice before being dropped onto precleaned microscope slides and dried at 75°C. DNA was visualized with 4′,6-diamidino-2-phenylindole (DAPI).
Immunoblots
Cells from ∼90% confluent 6-cm dishes were trypsinized, washed with phosphate-buffered saline (PBS), resuspended in 100 μl of ELB lysis buffer (250 mM NaCl, 0.1% NP-40, 50 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid [HEPES], pH 7, 5 mM EDTA) and 25 μl of 5× sample buffer, boiled for 10 min, and stored at −80°C. We ran 30 μg of samples on 12% acrylamide gels and transferred them to nitrocellulose. Primary and secondary antibodies were diluted in 5% milk in Tris-buffered saline plus 0.1% Tween 20. Primary antibody dilutions were as follows: Bub1, 1:500; BubR1, 1:200 (Taylor et al., 2001); CENP-E, 1:200 (Brown et al., 1996); Mad2, 1:200 (Kops et al., 2005); Aurora B, 1:500 (Cell Signaling); α-tubulin, 1:250 (DM1a; Sigma-Aldrich), and ARF, 1:250 (ab80; Abcam).
Immunofluorescence microscopy
Cells were grown in 12-well plates with 18-mm round coverslips until ∼80% confluent. Coverslips were washed with microtubule stabilizing buffer (MTSB; 100 mM 1,4-piperazinediethanesulfonic acid, pH 6.9, 30% glycerol, 1 mM ethylene glycol tetraacetic acid, and 1 mM MgSO4) and fixed with precooled methanol at −20°C for 5 min or 4% formaldehyde or 0.5% glutaraldehyde (Tousimis, Rockville, MD) in MTSB at room temperature for 10 min. Glutaraldehyde was quenched with 0.2% NaBH4 for 20 min. Coverslips were washed twice with PBS and blocked in triton block (0.2 M glycine, 2.5% FBS, and 0.1% Triton X-100 in PBS) at 4°C. For formaldehyde and glutaraldehyde fixations, cells were preextracted with 0.5% Triton X-100 in MTSB for 1–5 min at 37°C. Primary antibody dilutions were as for immunoblots except for Bub1 (1:200), Aurora B (1:2500), and α-tubulin (YL1/2; 1:500). Images were acquired on a Nikon Eclipse Ti-E inverted fluorescence microscope using a CoolSNAP HQ2 camera and a 100×/1.4 numerical aperture (NA) oil objective. Chromosome spread images are from a single z. Other images are maximum projections of 0.2-μm z-stacks deconvolved using the AQI module in Nikon Elements unless otherwise indicated.
Fluorescence in situ hybridization
Small intestine was dissected, washed with PBS, and fixed in 10% buffered Formalin (Fisher, PIttsburgh, PA), followed by three washes in 70% ethanol every 24 h. Paraffin sections, 8 μm, were dewaxed in xylene and hydrated in an ethanol series before cross-linking reversal in 8% sodium thiocyanate (Fisher) for 30 min at 80°C. Tissue was digested with 0.5 mg/ml proteinase K for 5 min at 37°C and washed in 2× SSC buffer (300 mM NaCl, 30 mM sodium citrate) for 2 min before dehydration in an ethanol series. A 200-kb FISH probe recognizing the thymidine kinase locus on chromosome 11 (Kreatech KBI-30501, Buffalo Grove, IL) was added to each tissue before coverslipping and sealing with rubber cement. Slides were placed on an 80°C metal plate for 5 min for denaturation before hybridizing 48 h at 37°C. After removal of coverslips and rubber cement, tissues were washed in 0.4× SSC plus 0.3% NP40 at 72°C for 2 min before a second wash in 2× SSC plus 0.1% NP40. After dehydration in an ethanol series, 0.3 μg/ml DAPI in Vectashield was used to mount the samples and counterstain. Images were acquired using a Nikon Eclipse Ti-E fluorescence microscope using a 100×/1.4 NA objective and a Hamamatsu Orca Flash 4.0 camera.
Quantitative real-time PCR
RNA was isolated from MEFs using TRIzol. One microgram of the resulting RNA was reverse transcribed (iScript; Bio-Rad, Hercules, CA). cDNA was resuspended in nuclease-free water in a total volume 100 μl. For real-time PCR analysis, 1 μl of resuspended cDNA was used in a total reaction volume of 25 μl containing 300 nM forward and reverse primers and 1× IQ SYBR Green Supermix (Bio-Rad). An annealing temperature of 55°C was used during thermocycling for all primers. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as an internal control, and melting curves of products were performed at the end of each experiment to confirm the amplification of a single product. Relative mRNA levels were calculated using the ΔΔCt method. Primer sequences: Aurora B set 1, forward, 5′-CAGAAGGAGAACGCCTACCC; Aurora B set 1, reverse, 5′-GAGAGCAAGCGCAGATGTC-3′; Aurora B set 2, forward, 5′-TCAGAAGGAGAACGCCTACCC-3′; Aurora B set 2, reverse, 5′-GACTCTCTGGGACAACGTGTT-3′. GAPDH forward, 5′-CCA ATG TGT CCG TCG TGG ATC-3′; reverse, 5′-GTT GAA GTC GCA GGA GAC AA-3′.
Statistical analysis
Error bars represent mean ± SE unless otherwise specified. Statistical significance was concluded at p < 0.05.
Supplementary Material
Acknowledgments
We thank Dawn Quelle for providing p14ARF viral constructs and Scott Nelson for assistance with microscopy. This work was supported in part by grants from the National Institutes of Health (R01CA140458) and the American Cancer Society (IRG-58-011-48). Additional support was provided by National Institutes of Health T32 GM008688 and T32 CA009135 (E.B. and L.Z.).
Abbreviations used:
- ARF
alternative reading frame
- CIN
chromosomal instability
- MEF
murine embryonic fibroblast
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E14-05-0966) on July 23, 2014.
REFERENCES
- Araki K, Nozaki K, Ueba T, Tatsuka M, Hashimoto N. High expression of Aurora-B/Aurora and Ipll-like midbody-associated protein (AIM-1) in astrocytomas. J Neurooncol. 2004;67:53–64. doi: 10.1023/b:neon.0000021784.33421.05. [DOI] [PubMed] [Google Scholar]
- Baker DJ, Dawlaty MM, Wijshake T, Jeganathan KB, Malureanu L, van Ree JH, Crespo-Diaz R, Reyes S, Seaburg L, Shapiro V, et al. Increased expression of BubR1 protects against aneuploidy and cancer and extends healthy lifespan. Nat Cell Biol. 2013;15:96–102. doi: 10.1038/ncb2643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker DJ, Jeganathan KB, Cameron JD, Thompson M, Juneja S, Kopecka A, Kumar R, Jenkins RB, de Groen PC, Roche P, et al. BubR1 insufficiency causes early onset of aging-associated phenotypes and infertility in mice. Nat Genet. 2004;36:744–749. doi: 10.1038/ng1382. [DOI] [PubMed] [Google Scholar]
- Baker DJ, Jeganathan KB, Malureanu L, Perez-Terzic C, Terzic A, van Deursen JM. Early aging-associated phenotypes in Bub3/Rae1 haploinsufficient mice. J Cell Biol. 2006;172:529–540. doi: 10.1083/jcb.200507081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biggins S, Murray AW. The budding yeast protein kinase Ipl1/Aurora allows the absence of tension to activate the spindle checkpoint. Genes Dev. 2001;15:3118–3129. doi: 10.1101/gad.934801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biggins S, Severin FF, Bhalla N, Sassoon I, Hyman AA, Murray AW. The conserved protein kinase Ipl1 regulates microtubule binding to kinetochores in budding yeast. Genes Dev. 1999;13:532–544. doi: 10.1101/gad.13.5.532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bischoff JR, Anderson L, Zhu Y, Mossie K, Ng L, Souza B, Schryver B, Flanagan P, Clairvoyant F, Ginther C, et al. A homologue of Drosophila aurora kinase is oncogenic and amplified in human colorectal cancers. EMBO J. 1998;17:3052–3065. doi: 10.1093/emboj/17.11.3052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bothner B, Lewis WS, DiGiammarino EL, Weber JD, Bothner SJ, Kriwacki RW. Defining the molecular basis of Arf and Hdm2 interactions. J Mol Biol. 2001;314:263–277. doi: 10.1006/jmbi.2001.5110. [DOI] [PubMed] [Google Scholar]
- Brown KD, Wood KW, Cleveland DW. The kinesin-like protein CENP-E is kinetochore-associated throughout poleward chromosome segregation during anaphase-A. J Cell Sci. 1996;109:961–969. doi: 10.1242/jcs.109.5.961. [DOI] [PubMed] [Google Scholar]
- Chen RH, Shevchenko A, Mann M, Murray AW. Spindle checkpoint protein Xmad1 recruits Xmad2 to unattached kinetochores. J Cell Biol. 1998;143:283–295. doi: 10.1083/jcb.143.2.283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YJ, Chen CM, Twu NF, Yen MS, Lai CR, Wu HH, Wang PH, Yuan CC. Overexpression of Aurora B is associated with poor prognosis in epithelial ovarian cancer patients. Virchows Arch. 2009;455:431–440. doi: 10.1007/s00428-009-0838-3. [DOI] [PubMed] [Google Scholar]
- Chieffi P, Cozzolino L, Kisslinger A, Libertini S, Staibano S, Mansueto G, De Rosa G, Villacci A, Vitale M, Linardopoulos S, et al. Aurora B expression directly correlates with prostate cancer malignancy and influence prostate cell proliferation. Prostate. 2006;66:326–333. doi: 10.1002/pros.20345. [DOI] [PubMed] [Google Scholar]
- Cimini D, Howell B, Maddox P, Khodjakov A, Degrassi F, Salmon ED. Merotelic kinetochore orientation is a major mechanism of aneuploidy in mitotic mammalian tissue cells. J Cell Biol. 2001;153:517–527. doi: 10.1083/jcb.153.3.517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cimini D, Wan X, Hirel CB, Salmon ED. Aurora kinase promotes turnover of kinetochore microtubules to reduce chromosome segregation errors. Curr Biol. 2006;16:1711–1718. doi: 10.1016/j.cub.2006.07.022. [DOI] [PubMed] [Google Scholar]
- Cooke CA, Heck MM, Earnshaw WC. The inner centromere protein (INCENP) antigens: movement from inner centromere to midbody during mitosis. J Cell Biol. 1987;105:2053–2067. doi: 10.1083/jcb.105.5.2053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Courapied S, Cherier J, Vigneron A, Troadec MB, Giraud S, Valo I, Prigent C, Gamelin E, Coqueret O, Barré B. Regulation of the Aurora-A gene following topoisomerase I inhibition: implication of the Myc transcription factor. Mol Cancer. 2010;9:205. doi: 10.1186/1476-4598-9-205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cross SM, Sanchez CA, Morgan CA, Schimke MK, Ramel S, Idzerda RL, Raskind WH, Reid BJ. A p53-dependent mouse spindle checkpoint. Science. 1995;267:1353–1356. doi: 10.1126/science.7871434. [DOI] [PubMed] [Google Scholar]
- Dai W, Wang Q, Liu T, Swamy M, Fang Y, Xie S, Mahmood R, Yang YM, Xu M, Rao CV. Slippage of mitotic arrest and enhanced tumor development in mice with BubR1 haploinsufficiency. Cancer Res. 2004;64:440–445. doi: 10.1158/0008-5472.can-03-3119. [DOI] [PubMed] [Google Scholar]
- Datta A, Sen J, Hagen J, Korgaonkar CK, Caffrey M, Quelle DE, Hughes DE, Ackerson TJ, Costa RH, Raychaudhuri P. ARF directly binds DP1: interaction with DP1 coincides with the G1 arrest function of ARF. Mol Cell Biol. 2005;25:8024–8036. doi: 10.1128/MCB.25.18.8024-8036.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- den Hollander J, Rimpi S, Doherty JR, Rudelius M, Buck A, Hoellein A, Kremer M, Graf N, Scheerer M, Hall MA, et al. Aurora kinases A and B are up-regulated by Myc and are essential for maintenance of the malignant state. Blood. 2010;116:1498–1505. doi: 10.1182/blood-2009-11-251074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Leonardo A, Khan SH, Linke SP, Greco V, Seidita G, Wahl GM. DNA rereplication in the presence of mitotic spindle inhibitors in human and mouse fibroblasts lacking either p53 or pRb function. Cancer Res. 1997;57:1013–1019. [PubMed] [Google Scholar]
- Ditchfield C, Johnson VL, Tighe A, Ellston R, Haworth C, Johnson T, Mortlock A, Keen N, Taylor SS. Aurora B couples chromosome alignment with anaphase by targeting BubR1, Mad2, and Cenp-E to kinetochores. J Cell Biol. 2003;161:267–280. doi: 10.1083/jcb.200208091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- di Tommaso A, Hagen J, Tompkins V, Muniz V, Dudakovic A, Kitzis A, Ladeveze V, Quelle DE. Residues in the alternative reading frame tumor suppressor that influence its stability and p53-independent activities. Exp Cell Res. 2009;315:1326–1335. doi: 10.1016/j.yexcr.2009.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobles M, Liberal V, Scott ML, Benezra R, Sorger PK. Chromosome missegregation and apoptosis in mice lacking the mitotic checkpoint protein Mad2. Cell. 2000;101:635–645. doi: 10.1016/s0092-8674(00)80875-2. [DOI] [PubMed] [Google Scholar]
- Ehara H, Yokoi S, Tamaki M, Nishino Y, Takahashi Y, Deguchi T, Kimura M, Yoshioka T, Okano Y. Expression of mitotic Aurora/Ipl1p-related kinases in renal cell carcinomas: an immunohistochemical study. Urol Res. 2003;31:382–386. doi: 10.1007/s00240-003-0354-x. [DOI] [PubMed] [Google Scholar]
- Eymin B, Claverie P, Salon C, Leduc C, Col E, Brambilla E, Khochbin S, Gazzeri S. p14ARF activates a Tip60-dependent and p53-independent ATM/ATR/CHK pathway in response to genotoxic stress. Mol Cell Biol. 2006;26:4339–4350. doi: 10.1128/MCB.02240-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eymin B, Leduc C, Coll JL, Brambilla E, Gazzeri S. p14ARF induces G2 arrest and apoptosis independently of p53 leading to regression of tumours established in nude mice. Oncogene. 2003;22:1822–1835. doi: 10.1038/sj.onc.1206303. [DOI] [PubMed] [Google Scholar]
- Fang G, Yu H, Kirschner MW. The checkpoint protein MAD2 and the mitotic regulator CDC20 form a ternary complex with the anaphase-promoting complex to control anaphase initiation. Genes Dev. 1998;12:1871–1883. doi: 10.1101/gad.12.12.1871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganem NJ, Godinho SA, Pellman D. A mechanism linking extra centrosomes to chromosomal instability. Nature. 2009;460:278–282. doi: 10.1038/nature08136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta RS. Species-specific differences in toxicity of antimitotic agents toward cultured mammalian cells. J Natl Cancer Inst. 1985;74:159–164. [PubMed] [Google Scholar]
- Haller K, Kibler KV, Kasai T, Chi YH, Peloponese JM, Yedavalli VS, Jeang KT. The N-terminus of rodent and human MAD1 confers species-specific stringency to spindle assembly checkpoint. Oncogene. 2006;25:2137–2147. doi: 10.1038/sj.onc.1209259. [DOI] [PubMed] [Google Scholar]
- Han J-S, Holland AJ, Fachinetti D, Kulukian A, Cetin B, Cleveland DW. Catalytic Assembly of the mitotic checkpoint inhibitor BubR1-Cdc20 by Mad2-induced functional switch in Cdc20. Mol Cell. 2013;51:92–104. doi: 10.1016/j.molcel.2013.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvey M, Sands AT, Weiss RS, Hegi ME, Wiseman RW, Pantazis P, Giovanella BC, Tainsky MA, Bradley A, Donehower LA. In vitro growth characteristics of embryo fibroblasts isolated from p53-deficient mice. Oncogene. 1993;8:2457–2467. [PubMed] [Google Scholar]
- Hauf S, Cole RW, LaTerra S, Zimmer C, Schnapp G, Walter R, Heckel A, van Meel J, Rieder CL, Peters JM. The small molecule hesperadin reveals a role for Aurora B in correcting kinetochore-microtubule attachment and in maintaining the spindle assembly checkpoint. J Cell Biol. 2003;161:281–294. doi: 10.1083/jcb.200208092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He X, Patterson TE, Sazer S. The Schizosaccharomyces pombe spindle checkpoint protein mad2p blocks anaphase and genetically interacts with the anaphase-promoting complex. Proc Natl Acad Sci USA. 1997;94:7965–7970. doi: 10.1073/pnas.94.15.7965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holland AJ, Cleveland DW. Boveri revisited: chromosomal instability, aneuploidy and tumorigenesis. Nat Rev Mol Cell Biol. 2009;10:478–487. doi: 10.1038/nrm2718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honda R, Körner R, Nigg EA. Exploring the functional interactions between Aurora B, INCENP, and survivin in mitosis. Mol Biol Cell. 2003;14:3325–3341. doi: 10.1091/mbc.E02-11-0769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honda R, Tanaka H, Yasuda H. Oncoprotein MDM2 is a ubiquitin ligase E3 for tumor suppressor p53. FEBS Lett. 1997;420:25–27. doi: 10.1016/s0014-5793(97)01480-4. [DOI] [PubMed] [Google Scholar]
- Honda R, Yasuda H. Association of p19(ARF) with Mdm2 inhibits ubiquitin ligase activity of Mdm2 for tumor suppressor p53. EMBO J. 1999;18:22–27. doi: 10.1093/emboj/18.1.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howell BJ, Hoffman DB, Fang G, Murray AW, Salmon ED. Visualization of Mad2 dynamics at kinetochores, along spindle fibers, and at spindle poles in living cells. J Cell Biol. 2000;150:1233–1250. doi: 10.1083/jcb.150.6.1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoyt MA, Totis L, Roberts BT. S. cerevisiae genes required for cell cycle arrest in response to loss of microtubule function. Cell. 1991;66:507–517. doi: 10.1016/0092-8674(81)90014-3. [DOI] [PubMed] [Google Scholar]
- Kalinichenko VV, Major ML, Wang X, Petrovic V, Kuechle J, Yoder HM, Dennewitz MB, Shin B, Datta A, Raychaudhuri P, et al. Foxm1b transcription factor is essential for development of hepatocellular carcinomas and is negatively regulated by the p19ARF tumor suppressor. Genes Dev. 2004;18:830–850. doi: 10.1101/gad.1200704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kallio MJ, McCleland ML, Stukenberg PT, Gorbsky GJ. Inhibition of Aurora B kinase blocks chromosome segregation, overrides the spindle checkpoint, and perturbs microtubule dynamics in mitosis. Curr Biol. 2002;12:900–905. doi: 10.1016/s0960-9822(02)00887-4. [DOI] [PubMed] [Google Scholar]
- Kienitz A, Vogel C, Morales I, Muller R, Bastians H. Partial downregulation of MAD1 causes spindle checkpoint inactivation and aneuploidy, but does not confer resistance towards Taxol. Oncogene. 2005;24:4301–4310. doi: 10.1038/sj.onc.1208589. [DOI] [PubMed] [Google Scholar]
- Kim SH, Lin DP, Matsumoto S, Kitazono A, Matsumoto T. Fission yeast Slp1: an effector of the Mad2-dependent spindle checkpoint. Science. 1998;279:1045–1047. doi: 10.1126/science.279.5353.1045. [DOI] [PubMed] [Google Scholar]
- King EM, Rachidi N, Morrice N, Hardwick KG, Stark MJ. Ipl1p-dependent phosphorylation of Mad3p is required for the spindle checkpoint response to lack of tension at kinetochores. Genes Dev. 2007;21:1163–1168. doi: 10.1101/gad.431507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komlodi-Pasztor E, Sackett DL, Fojo AT. Inhibitors targeting mitosis: tales of how great drugs against a promising target were brought down by a flawed rationale. Clin Cancer Res. 2012;18:51–63. doi: 10.1158/1078-0432.CCR-11-0999. [DOI] [PubMed] [Google Scholar]
- Kops GJ, Kim Y, Weaver BA, Mao Y, McLeod I, Yates JR, 3rd, Tagaya M, Cleveland DW. ZW10 links mitotic checkpoint signaling to the structural kinetochore. J Cell Biol. 2005;169:49–60. doi: 10.1083/jcb.200411118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korgaonkar C, Zhao L, Modestou M, Quelle DE. ARF function does not require p53 stabilization or Mdm2 relocalization. Mol Cell Biol. 2002;22:196–206. doi: 10.1128/MCB.22.1.196-206.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulukian A, Han JS, Cleveland DW. Unattached kinetochores catalyze production of an anaphase inhibitor that requires a Mad2 template to prime Cdc20 for BubR1 binding. Dev Cell. 2009;16:105–117. doi: 10.1016/j.devcel.2008.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kung AL, Sherwood SW, Schimke RT. Cell line-specific differences in the control of cell cycle progression in the absence of mitosis. Proc Natl Acad Sci USA. 1990;87:9553–9557. doi: 10.1073/pnas.87.24.9553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lan W, Zhang X, Kline-Smith SL, Rosasco SE, Barrett-Wilt GA, Shabanowitz J, Hunt DF, Walczak CE, Stukenberg PT. Aurora B phosphorylates centromeric MCAK and regulates its localization and microtubule depolymerization activity. Curr Biol. 2004;14:273–286. doi: 10.1016/j.cub.2004.01.055. [DOI] [PubMed] [Google Scholar]
- Lanni JS, Jacks T. Characterization of the p53-dependent postmitotic checkpoint following spindle disruption. Mol Cell Biol. 1998;18:1055–1064. doi: 10.1128/mcb.18.2.1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laoukili J, Kooistra MR, Brás A, Kauw J, Kerkhoven RM, Morrison A, Clevers H, Medema RH. FoxM1 is required for execution of the mitotic programme and chromosome stability. Nat Cell Biol. 2005;7:126–136. doi: 10.1038/ncb1217. [DOI] [PubMed] [Google Scholar]
- Li R, Murray AW. Feedback control of mitosis in budding yeast. Cell. 1991;66:519–531. doi: 10.1016/0092-8674(81)90015-5. [DOI] [PubMed] [Google Scholar]
- Lin ZZ, Jeng YM, Hu FC, Pan HW, Tsao HW, Lai PL, Lee PH, Cheng AL, Hsu HC. Significance of Aurora B overexpression in hepatocellular carcinoma. Aurora B overexpression in HCC. BMC Cancer. 2010;10:461. doi: 10.1186/1471-2407-10-461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meraldi P, Draviam VM, Sorger PK. Timing and checkpoints in the regulation of mitotic progression. Dev Cell. 2004;7:45–60. doi: 10.1016/j.devcel.2004.06.006. [DOI] [PubMed] [Google Scholar]
- Michel LS, Liberal V, Chatterjee A, Kirchwegger R, Pasche B, Gerald W, Dobles M, Sorger PK, Murty VV, Benezra R. MAD2 haplo-insufficiency causes premature anaphase and chromosome instability in mammalian cells. Nature. 2001;409:355–359. doi: 10.1038/35053094. [DOI] [PubMed] [Google Scholar]
- Momand J, Zambetti GP, Olson DC, George D, Levine AJ. The mdm-2 oncogene product forms a complex with the p53 protein and inhibits p53-mediated transactivation. Cell. 1992;69:1237–1245. doi: 10.1016/0092-8674(92)90644-r. [DOI] [PubMed] [Google Scholar]
- Nguyen HG, Chinnappan D, Urano T, Ravid K. Mechanism of Aurora-B degradation and its dependency on intact KEN and A-boxes: identification of an aneuploidy-promoting property. Mol Cell Biol. 2005;25:4977–4992. doi: 10.1128/MCB.25.12.4977-4992.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsson J, Yekezare M, Minshull J, Pines J. The APC/C maintains the spindle assembly checkpoint by targeting Cdc20 for destruction. Nat Cell Biol. 2008;10:1411–1420. doi: 10.1038/ncb1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oikawa T, Okuda M, Ma Z, Goorha R, Tsujimoto H, Inokuma H, Fukasawa K. Transcriptional control of BubR1 by p53 and suppression of centrosome amplification by BubR1. Mol Cell Biol. 2005;25:4046–4061. doi: 10.1128/MCB.25.10.4046-4061.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozenne P, Eymin B, Brambilla E, Gazzeri S. The ARF tumor suppressor: structure, functions and status in cancer. Int J Cancer. 2010;127:2239–2247. doi: 10.1002/ijc.25511. [DOI] [PubMed] [Google Scholar]
- Parekh H, Simpkins H. Species-specific differences in taxol transport and cytotoxicity against human and rodent tumor cells. Evidence for an alternate transport system. Biochem Pharmacol. 1996;51:301–311. doi: 10.1016/0006-2952(95)02176-0. [DOI] [PubMed] [Google Scholar]
- Petersen J, Hagan IM. S. pombe aurora kinase/survivin is required for chromosome condensation and the spindle checkpoint attachment response. Curr Biol. 2003;13:590–597. doi: 10.1016/s0960-9822(03)00205-7. [DOI] [PubMed] [Google Scholar]
- Pomerantz J, Schreiber-Agus N, Liégeois NJ, Silverman A, Alland L, Chin L, Potes J, Chen K, Orlow I, Lee HW, et al. The Ink4a tumor suppressor gene product, p19Arf, interacts with MDM2 and neutralizes MDM2’s inhibition of p53. Cell. 1998;92:713–723. doi: 10.1016/s0092-8674(00)81400-2. [DOI] [PubMed] [Google Scholar]
- Qi Y, Gregory MA, Li Z, Brousal JP, West K, Hann SR. p19ARF directly and differentially controls the functions of c-Myc independently of p53. Nature. 2004;431:712–717. doi: 10.1038/nature02958. [DOI] [PubMed] [Google Scholar]
- Quelle DE, Zindy F, Ashmun RA, Sherr CJ. Alternative reading frames of the INK4a tumor suppressor gene encode two unrelated proteins capable of inducing cell cycle arrest. Cell. 1995;83:993–1000. doi: 10.1016/0092-8674(95)90214-7. [DOI] [PubMed] [Google Scholar]
- Ricke RM, van Ree JH, van Deursen JM. Whole chromosome instability and cancer: a complex relationship. Trends Genet. 2008;24:457–466. doi: 10.1016/j.tig.2008.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rieder CL, Cole RW, Khodjakov A, Sluder G. The checkpoint delaying anaphase in response to chromosome monoorientation is mediated by an inhibitory signal produced by unattached kinetochores. J Cell Biol. 1995;130:941–948. doi: 10.1083/jcb.130.4.941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rieder CL, Maiato H. Stuck in division or passing through: what happens when cells cannot satisfy the spindle assembly checkpoint. Dev Cell. 2004;7:637–651. doi: 10.1016/j.devcel.2004.09.002. [DOI] [PubMed] [Google Scholar]
- Rieder CL, Schultz A, Cole R, Sluder G. Anaphase onset in vertebrate somatic cells is controlled by a checkpoint that monitors sister kinetochore attachment to the spindle. J Cell Biol. 1994;127:1301–1310. doi: 10.1083/jcb.127.5.1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan SD, Britigan EM, Zasadil LM, Witte K, Audhya A, Roopra A, Weaver BA. Up-regulation of the mitotic checkpoint component Mad1 causes chromosomal instability and resistance to microtubule poisons. Proc Natl Acad Sci USA. 2012;109:E2205–E2214. doi: 10.1073/pnas.1201911109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santaguida S, Vernieri C, Villa F, Ciliberto A, Musacchio A. Evidence that Aurora B is implicated in spindle checkpoint signalling independently of error correction. EMBO J. 2011;30:1508–1519. doi: 10.1038/emboj.2011.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schvartzman JM, Duijf PH, Sotillo R, Coker C, Benezra R. Mad2 is a critical mediator of the chromosome instability observed upon Rb and p53 pathway inhibition. Cancer Cell. 2011;19:701–714. doi: 10.1016/j.ccr.2011.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schvartzman JM, Sotillo R, Benezra R. Mitotic chromosomal instability and cancer: mouse modelling of the human disease. Nat Rev Cancer. 2010;10:102–115. doi: 10.1038/nrc2781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silk AD, Zasadil LM, Holland AJ, Vitre B, Cleveland DW, Weaver BA. Chromosome missegregation rate predicts whether aneuploidy will promote or suppress tumors. Proc Natl Acad Sci USA. 2013;110:E4134–E4141. doi: 10.1073/pnas.1317042110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silkworth WT, Nardi IK, Scholl LM, Cimini D. Multipolar spindle pole coalescence is a major source of kinetochore mis-attachment and chromosome mis-segregation in cancer cells. PLoS One. 2009;4:e6564. doi: 10.1371/journal.pone.0006564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sironi L, Melixetian M, Faretta M, Prosperini E, Helin K, Musacchio A. Mad2 binding to Mad1 and Cdc20, rather than oligomerization, is required for the spindle checkpoint. EMBO J. 2001;20:6371–6382. doi: 10.1093/emboj/20.22.6371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorrentino R, Libertini S, Pallante PL, Troncone G, Palombini L, Bavetsias V, Spalletti-Cernia D, Laccetti P, Linardopoulos S, Chieffi P, et al. Aurora B overexpression associates with the thyroid carcinoma undifferentiated phenotype and is required for thyroid carcinoma cell proliferation. J Clin Endocrinol Metab. 2005;90:928–935. doi: 10.1210/jc.2004-1518. [DOI] [PubMed] [Google Scholar]
- Sotillo R, Hernando E, Diaz-Rodriguez E, Teruya-Feldstein J, Cordon-Cardo C, Lowe SW, Benezra R. Mad2 overexpression promotes aneuploidy and tumorigenesis in mice. Cancer Cell. 2007;11:9–23. doi: 10.1016/j.ccr.2006.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor SS, Hussein D, Wang Y, Elderkin S, Morrow CJ. Kinetochore localisation and phosphorylation of the mitotic checkpoint components Bub1 and BubR1 are differentially regulated by spindle events in human cells. J Cell Sci. 2001;114:4385–4395. doi: 10.1242/jcs.114.24.4385. [DOI] [PubMed] [Google Scholar]
- Thompson SL, Compton DA. Chromosome missegregation in human cells arises through specific types of kinetochore-microtubule attachment errors. Proc Natl Acad Sci USA. 2011;108:17974–17978. doi: 10.1073/pnas.1109720108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vader G, Cruijsen CW, van Harn T, Vromans MJ, Medema RH, Lens SM. The chromosomal passenger complex controls spindle checkpoint function independent from its role in correcting microtubule kinetochore interactions. Mol Biol Cell. 2007;18:4553–4564. doi: 10.1091/mbc.E07-04-0328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vischioni B, Oudejans JJ, Vos W, Rodriguez JA, Giaccone G. Frequent overexpression of aurora B kinase, a novel drug target, in non-small cell lung carcinoma patients. Mol Cancer Ther. 2006;5:2905–2913. doi: 10.1158/1535-7163.MCT-06-0301. [DOI] [PubMed] [Google Scholar]
- Vleugel M, Hoogendoorn E, Snel B, Kops GJ. Evolution and function of the mitotic checkpoint. Dev Cell. 2012;23:239–250. doi: 10.1016/j.devcel.2012.06.013. [DOI] [PubMed] [Google Scholar]
- Weaver BA, Cleveland DW. Does aneuploidy cause cancer. Curr Opin Cell Biol. 2006;18:658–667. doi: 10.1016/j.ceb.2006.10.002. [DOI] [PubMed] [Google Scholar]
- Weaver BA, Silk AD, Montagna C, Verdier-Pinard P, Cleveland DW. Aneuploidy acts both oncogenically and as a tumor suppressor. Cancer Cell. 2007;11:25–36. doi: 10.1016/j.ccr.2006.12.003. [DOI] [PubMed] [Google Scholar]
- Weber JD, Jeffers JR, Rehg JE, Randle DH, Lozano G, Roussel MF, Sherr CJ, Zambetti GP. p53-independent functions of the p19(ARF) tumor suppressor. Genes Dev. 2000a;14:2358–2365. doi: 10.1101/gad.827300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber JD, Kuo ML, Bothner B, DiGiammarino EL, Kriwacki RW, Roussel MF, Sherr CJ. Cooperative signals governing ARF-mdm2 interaction and nucleolar localization of the complex. Mol Cell Biol. 2000b;20:2517–2528. doi: 10.1128/mcb.20.7.2517-2528.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yarbrough WG, Bessho M, Zanation A, Bisi JE, Xiong Y. Human tumor suppressor ARF impedes S-phase progression independent of p53. Cancer Res. 2002;62:1171–1177. [PubMed] [Google Scholar]
- Zasadil LM, Britigan EM, Weaver BA. 2n or not 2n: Aneuploidy, polyploidy and chromosomal instability in primary and tumor cells. Semin Cell Dev Biol. 2013;24:370–379. doi: 10.1016/j.semcdb.2013.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Xiong Y, Yarbrough WG. ARF promotes MDM2 degradation and stabilizes p53: ARF-INK4a locus deletion impairs both the Rb and p53 tumor suppression pathways. Cell. 1998;92:725–734. doi: 10.1016/s0092-8674(00)81401-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.