

Actin filament formation plays an essential role in cell movement, and posttranslational modifications regulate actin filament assembly. Prolyl hydroxylase 3 (PHD3) interacts with nonmuscle actins in HeLa cells and catalyzes prolyl hydroxylation of actin. Inhibition of PHD3 activity or expression increases F-actin formation and cell motility.
Abstract
Actin filaments play an essential role in cell movement, and many posttranslational modifications regulate actin filament assembly. Here we report that prolyl hydroxylase 3 (PHD3) interacts with nonmuscle actin in human cells and catalyzes hydroxylation of actin at proline residues 307 and 322. Blocking PHD3 expression or catalytic activity by short hairpin RNA knockdown or pharmacological inhibition, respectively, decreased actin prolyl hydroxylation. PHD3 knockdown increased filamentous F-actin assembly, which was reversed by PHD3 overexpression. PHD3 knockdown increased cell velocity and migration distance. Inhibition of PHD3 prolyl hydroxylase activity by dimethyloxalylglycine also increased actin polymerization and cell migration. These data reveal a novel role for PHD3 as a negative regulator of cell motility through posttranslational modification of nonmuscle actins.
INTRODUCTION
Cell movement is a fundamental biological process that is critical for the development and maintenance of multicellular organisms. Dysregulation of cell movement is associated with disease processes, most notably cancer (Ridley et al., 2003). Actin is a highly conserved cytoskeletal protein that plays an essential role in eukaryotic cell movement (Pollard and Copper, 2009). It consists of six isoforms in vertebrates: three α isoforms and one γ isoform are selectively expressed in skeletal, cardiac, and smooth muscle cells, whereas the nonmuscle β- and γ-actin isoforms are present in most cell types (Herman, 1993). Actin isoforms differ by only a few amino acids, and their protein structures are similar, but their biological functions are distinct in different tissues and cellular compartments (Herman, 1993).
Actin exists in monomeric (G-actin) and filamentous (F-actin) forms in cells (Dominguez and Homes, 2011). Polymerization of G-actin into double-stranded F-actin provides intracellular mechanical support that facilitates cell migration (Pollard and Cooper, 2009). Actin polymerization is highly dynamic, and many actin-binding proteins regulate this process (Insall and Machesky, 2009). A large body of data indicates that posttranslational modifications also regulate the balance between G-actin and F-actin (Terman and Kashina, 2013). Phosphorylation of Tyr-53, ADP-ribosylation of Arg-177 and Arg-206, oxidation of Cys-374, Cys-285, Cys-272, and Met-44, covalent cross-linking between Glu-270 and Lys-50, and glutathionylation, carbonylation, or nitrosylation of Cys-374 inhibit actin polymerization (Terashima et al., 1995; Dalle-Donne et al., 2000, 2003; Liu et al., 2006; Aldini et al., 2007; Lassing et al., 2007; Kudryashov et al., 2008; Visschedyk et al., 2010; Farah et al., 2011; Hung et al., 2011). Transglutamination of Lys-50, cross-linking between Gln-41 and Lys-50, methylation of His-73, ADP-ribosylation of Thr-148, and arginylation stabilize F-actin (Hegyi et al., 1992; Nyman et al., 2002; Lang et al., 2010; Saha et al., 2010).
Prolyl hydroxylases (PHDs) are α-ketoglutarate–dependent dioxygenases, which were originally identified as negative regulators of hypoxia-inducible factors (HIFs) and are present in cells of all metazoan species (Epstein et al., 2001; Hampton-Smith and Peet, 2009; Leonarz et al., 2011). PHDs transfer one oxygen atom from O2 to the carbon-4 position of a prolyl residue (Pro) to form (2S, 4R)-4-hydroxyproline, and the other oxygen atom reacts with α-ketoglutarate to form succinate and CO2; in addition to utilizing α-ketoglutarate and O2 as substrates, the enzymatic reaction requires iron (II) and ascorbate as cofactors (Gorres and Raines, 2010; Rose et al., 2011). Prolyl hydroxylated HIF-1α is bound by the von Hippel–Lindau tumor suppressor protein (VHL), which is a key component of the ubiquitin E3 ligase complex that targets HIF-1α for degradation in the 26S proteasome (Kaelin and Ratcliffe, 2008). Three PHD family members (PHD1–3) have been identified. PHD2 is the primary prolyl hydroxylase that regulates HIF-1α protein stability in nonhypoxic cells, whereas knockdown of PHD1 or PHD3 does not affect HIF-1α protein levels in many cancer cell lines (Berra et al., 2003).
PHD3-dependent hydroxylation promotes the degradation of several other proteins, including ATF-4, β2-adrenergic receptor, Paired box 2, and Sprouty 2 (Köditz et al., 2007; Xie et al., 2009; Anderson et al., 2011; Yan et al., 2011). In contrast, PHD3 blocks VHL-mediated ubiquitination of myogenin, thereby stabilizing myogenin in differentiated C2C12 cells (Fu and Taubman, 2010).
In addition to its role in regulating protein stability, PHD3 also exerts other cellular functions. PHD3 induces prolyl hydroxylation of pyruvate kinase M2 (PKM2) to stimulate interaction with HIF-1α, leading to increased HIF-1 transactivation (Luo et al., 2011). PHD3 mediates HCLK2 prolyl hydroxylation to activate the ATR/CHK1 signaling pathway in response to DNA damage (Xie et al., 2012). PHD3 has also been reported to promote apoptosis (Tennant et al., 2010; Xie et al., 2012). Recent studies demonstrated that overexpression of PHD3 inhibits invasion of pancreatic cancer cells (Su et al., 2010). PHD3 knockdown or knockout increases migration of pancreatic cancer cells and macrophages (Kiss et al., 2012; Place et al., 2013). However, the molecular mechanism underlying PHD3-mediated inhibition of cell migration has not been delineated.
In the present study, we find that nonmuscle β- and γ-actin are subject to hydroxylation at Pro-307 and Pro-322, which is catalyzed by PHD3. Inhibition of prolyl hydroxylation increases actin polymerization and cell motility. Thus a novel posttranslational modification of nonmuscle actin provides a novel mechanism for regulating actin polymerization and cell motility.
RESULTS
Nonmuscle β- and γ-actin are subject to PHD3-mediated prolyl hydroxylation
To discover novel prolyl hydroxylated proteins, we performed a quantitative stable isotope labeling by amino acids in cell culture (SILAC) proteomic screen (Figure 1A). HeLa human cervical carcinoma cells were cultured in standard culture medium (light isotope) or medium supplemented with heavy isotope–labeled lysine/arginine and treated with the proteasome inhibitor MG132 and either dimethyloxalylglycine (DMOG), which inhibits PHDs by competing with α-ketoglutarate, or vehicle (dimethyl sulfoxide [DMSO]) for 6 h (Figure 1A). Tryptic peptide digests of cell lysates were immunoprecipitated by anti-hydroxyproline antibody, and the precipitated peptides were analyzed by reverse-phase liquid chromatography interfaced with an LTQ Orbitrap Velos mass spectrometer, which identified a prolyl hydroxylated peptide (KDLYANTVLSGGTTMYP307GIADR) that is present in both of the nonmuscle actins, that is, β- and γ-actin, with a heavy-to-light ratio of <0.8 (Figure 1, B and C). Mass spectrometry also detected another prolyl hydroxylated peptide (EITALAP322STMK), whose sequence is conserved among all six actin isoforms (Figure 1, B and D). A third nonmuscle actin peptide (MTQIMFETFNTP130AMYVAIQAVLSLYASGR) was hydroxylated with a heavy-to-light ratio of <0.8, but mass spectrometry could not distinguish hydroxylation on either the Pro or one of three Met residues of this peptide (Supplemental Figure S1, A and B). Pro-307 and Pro-322 are both located within subdomain 3 of actin (Figure 1E; Kabsch et al., 1990). Taken together, the mass spectrometry data indicate that nonmuscle β- and γ-actin are novel prolyl hydroxylated proteins and that Pro-307 and Pro-322 of nonmuscle actins are hydroxylated in human cells.
FIGURE 1:

Nonmuscle β- and γ-actin are novel prolyl hydroxylated proteins. (A) Schematic representation of the SILAC proteomic screening strategy. (B) Two prolyl hydroxylated peptides of the nonmuscle actin were identified by mass spectrometry. Hydroxyproline residues are underlined. (C, D) MS/MS spectrum of the prolyl hydroxylated nonmuscle actin peptide. Hydroxyproline residues are shown in lowercase. (E) Mapping hydroxylated proline residues Pro-307 and Pro-322 (red) in the protein structure of actin. Hydroxylated Pro-307 and Pro-322 were mapped by SWISS-MODEL (Arnold et al., 2006) using the protein structure of human β-actin (PDB ID 3BYH) as template (Galkin et al., 2008) and visualized using PyMOL (Schrödinger LLC, 2008).
β-Actin was chosen as the representative nonmuscle actin for further evaluation. To identify the prolyl hydroxylase that modifies β-actin, we performed coimmunoprecipitation (co-IP) assays with HeLa cells. Endogenous β-actin was pulled down from whole-cell lysates (WCLs) by an anti-PHD3 antibody but not by control immunoglobulin G (IgG; Figure 2A). In contrast, IP of either overexpressed FLAG-PHD1 or endogenous PHD2 failed to provide evidence of interaction with β-actin under either nonhypoxic (20% O2) or hypoxic (1% O2) conditions (Supplemental Figure S2). Endogenous HIF-1α was coimmunoprecipitated with FLAG-PHD1 or PHD2 from lysates of hypoxic cells, validating the IP assays (Supplemental Figure S2). Thus β-actin selectively interacts with PHD3 in HeLa cells.
FIGURE 2:

PHD3 mediates hydroxylation of actin on Pro-307 and Pro-322. (A) HeLa cells were exposed to 1% O2 for 24 h. IP assays were performed using IgG or anti-PHD3 antibody, followed by immunoblot assays using antibodies against β-actin or PHD3. WCL, whole-cell lysate. (B) In vitro hydroxylation assays were performed using purified WT or mutant GST–β-actin and PHD3, followed by immunoblot assays with anti-hydroxyproline antibody. The immunoblot bands were quantified by densitometry and normalized to WT (mean ± SEM, n = 3). **p < 0.01 vs. WT. (C) HeLa cells were transfected with vector encoding WT β-actin-V5 or β-actin (P307/322A)-V5 and exposed to 1% O2 for 24 h. IP of WCLs was performed using anti-hydroxyproline antibody (Pro-OH), followed by immunoblot assays with anti-V5 antibody. The immunoblot bands were quantified by densitometry and normalized to WT. Representative blots from two independent experiments. (D) HeLa-shSC or HeLa-shPHD3 cells were exposed to 20% or 1% O2 for 24 h. IP of WCLs was performed using anti-hydroxyproline antibody (Pro-OH), followed by immunoblot assays with antibodies against the indicated proteins. The immunoblot bands were quantified by densitometry and normalized to shSC-20% O2. Representative blots from two independent experiments. (E) HeLa cells were treated with desferrioxamine (DFX, 100 μM) for 6 h. IP of WCLs was performed using anti-hydroxyproline antibody (Pro-OH), followed by immunoblot assays with anti–β-actin antibody. The immunoblot bands were quantified by densitometry and normalized to control (CON). Data shown are mean ± SEM, n = 3.
We next performed in vitro hydroxylation assays to determine whether PHD3 directly hydroxylates β-actin. Wild-type (WT) glutathione S-transferase (GST)–β-actin fusion protein was expressed in Escherichia coli, purified, and incubated for 30 min with or without recombinant PHD3 in the presence of O2, α-ketoglutarate, FeSO4, and ascorbate. The anti-hydroxyproline antibody detected WT GST–β-actin when PHD3 was coincubated, indicating that WT GST–β-actin is prolyl hydroxylated by PHD3 in vitro (Figure 2B). To further determine whether PHD3 hydroxylates Pro-130, Pro-307, or Pro-322 of β-actin, we generated mutant GST-β-actin constructs in which Pro-130, Pro-307, or Pro-322 was mutated to alanine. Mutant GST-β-actin fusion proteins were purified from bacteria and incubated with or without recombinant PHD3 in an in vitro hydroxylation assay. PHD3 induced prolyl hydroxylation of GST–β-actin (P322A), GST–β-actin (P307A), or GST–β-actin (P130A), comparable to WT GST–β-actin (Figure 2B). In contrast, double-mutant GST–β-actin (P307/322A) had significantly reduced levels of prolyl hydroxylation (Figure 2B). Triple mutation of Pro-130, Pro-307, and Pro-322 to alanine reduced prolyl hydroxylation of GST–β-actin by 30%, comparable to double-mutant GST–β-actin (P307/322A; Figure 2B). Next, WT and mutant β-actin were expressed in HeLa cells as V5 epitope–tagged proteins. IP of double-mutant β-actin (P307/322A) by anti-hydroxyproline antibody was decreased compared with WT β-actin despite comparable expression levels (Figure 2C). Taken together, these in vitro and cell-based assays indicate that PHD3 hydroxylates Pro-307 and Pro-322 of human β-actin.
To determine whether PHD3 hydroxylates endogenous β-actin, we analyzed the effect of PHD3 loss of function mediated by short hairpin RNA (shRNA). HeLa cells were transduced with a retrovirus encoding a scrambled control shRNA (shSC) or PHD3 shRNA (shPHD3). PHD3 protein levels in WCLs were markedly increased when HeLa-shSC cells were exposed to hypoxic conditions (1% O2). Expression of shPHD3 reduced endogenous PHD3 protein levels in HeLa WCLs (Figure 2D). We previously demonstrated that exposure of HeLa cells to 1% O2 for 24 h induces PHD3 expression, which compensates for reduced catalytic activity under hypoxic conditions, thereby maintaining constant levels of prolyl hydroxylated PKM2 (Luo et al., 2011). Analysis of anti-hydroxyproline immunoprecipitates revealed that levels of prolyl hydroxylated β-actin were reduced in HeLa-shPHD3 cells as compared with HeLa-shSC cells at both 20 and 1% O2 (Figure 2D). Because prolyl hydroxylated β-actin levels were comparable at both 20 and 1% O2, we analyzed cells exposed to 20% O2 in subsequent experiments. Similar results were observed for PKM2, again validating our assay (Figure 2D).
To determine whether PHD3 hydroxylase activity is required for endogenous β-actin hydroxylation, we treated HeLa cells for 6 h with desferrioxamine (DFX), which inhibits PHDs by chelating iron. IP assays using anti-hydroxyproline antibody demonstrated that DFX treatment reduced prolyl hydroxylation of β-actin in HeLa WCLs without changing total β-actin levels (Figure 2E). Taken together, these data indicate that PHD3 directly mediates prolyl hydroxylation of β-actin in HeLa cells.
PHD3-mediated prolyl hydroxylation impairs actin polymerization
To determine whether prolyl hydroxylation regulates F-actin assembly, we stained HeLa-shSC and HeLa-shPHD3 cells with Alexa Fluor 555–conjugated phalloidin, which selectively binds filamentous F-actin but not monomeric G-actin (Lengsfeld et al., 1974). Fluorescence microscopy revealed increased F-actin in HeLa-shPHD3 cells as compared with HeLa-shSC cells (Figure 3A). Increased F-actin was also observed in HeLa-shPHD3-2244 cells, which were transduced with lentivirus encoding a second independent shRNA targeting PHD3 (Supplemental Figure S3), indicating a specific effect of PHD3 knockdown on F-actin formation. To determine whether PHD3 overexpression can reverse PHD3 knockdown-induced F-actin formation, we introduced an expression vector encoding shRNA-resistant PHD3R into HeLa-shPHD3 cells. Phalloidin staining demonstrated that expression of PHD3R reduced F-actin in HeLa-shPHD3 cells to baseline levels (Figure 3, B and C). We further analyzed G-actin and F-actin fractions from HeLa-shSC and HeLa-shPHD3 cells by sedimentation assays, which demonstrated that PHD3 knockdown significantly increased F-actin levels by 1.8-fold, whereas G-actin levels were not altered (Figure 3, D and E). Taken together, these data indicate that PHD3 inhibits actin polymerization in HeLa cells.
FIGURE 3:

PHD3 inhibits actin polymerization in HeLa cells. (A) HeLa-shSC and HeLa-shPHD3 cells were fixed, permeabilized, stained with Alexa Fluor 555–conjugated phalloidin, and imaged by fluorescence microscopy. The boxed areas are enlarged and shown at the bottom. Representative images from at least three independent experiments. Scale bar, 100 μm. (B, C) HeLa-shPHD3 cells were transduced with lentivirus encoding shRNA-resistant PHD3 (PHD3R) or empty vector (EV). The cells were fixed, permeabilized, stained, and imaged by confocal microscopy. F-actin (red) was visualized by Alexa Fluor 555–conjugated phalloidin. PHD3 (green) was visualized by rabbit anti-PHD3 antibody and Alexa Fluor 647–conjugated goat anti-rabbit IgG. Representative images from at least three independent experiments (B). Scale bar, 100 μm. The fluorescence intensity of 12–15 images/group were quantified and normalized to shSC (C; mean ± SEM). ***p < 0.001 vs. shSC;###p < 0.001 vs. EV. (D, E) Actin sedimentation assays were performed, followed by immunoblot assays with antibodies against β-actin or PHD3. (D) F-actin bands were quantified by densitometry and normalized to shSC (E; mean ± SEM, n = 4). *p < 0.05 vs. shSC.
To determine whether the prolyl hydroxylase activity of PHD3 is required to inhibit β-actin polymerization, we treated HeLa cells with the hydroxylase inhibitor DMOG for 72 h. Compared to treatment with vehicle (DMSO), DMOG treatment increased F-actin levels, as shown by phalloidin staining (Figure 4A) and actin sedimentation assays (Figure 4B). These data indicate that the prolyl hydroxylase activity of PHD3 promotes the actin monomeric state.
FIGURE 4:
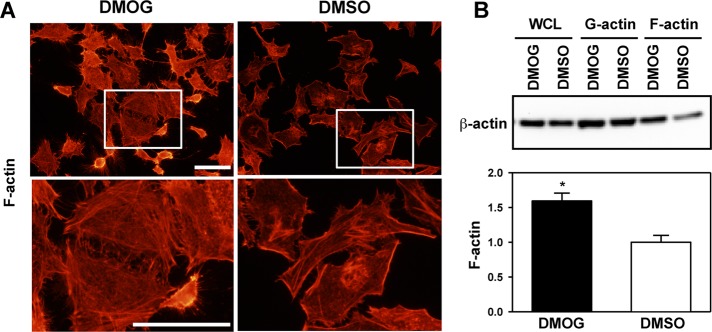
PHD3 inhibitor DMOG increases β-actin polymerization. HeLa cells were treated with DMSO or DMOG (500 μM) for 72 h. (A) Cells were fixed, permeabilized, stained with Alexa Fluor 555–conjugated phalloidin, and imaged by fluorescence microscopy. The boxed areas are enlarged and shown below. Representative images from at least three independent experiments. Scale bar, 100 μm. (B) Actin sedimentation assays were performed, followed by immunoblot assays with anti–β-actin antibody. F-actin bands were quantified by densitometry and normalized to DMSO (mean ± SEM, n = 3). *p < 0.05 vs. DMSO.
PHD3 inhibits cell motility through its prolyl hydroxylase activity
To determine whether PHD3 regulates cell migration, we performed microfluidic assays with HeLa-shSC and HeLa-shPHD3 cells. Cells were seeded onto a multiple-channel microchip, and chemotaxis driven by a serum gradient was monitored for 10 h. The chemotactic migration of HeLa-shPHD3 cells was significantly increased 2.2-fold compared with that of HeLa-shSC cells (Figure 5, A and B, and Supplemental Videos S1 and S2). The mean velocity of HeLa-shPHD3 cells was 3.1-fold greater than that of HeLa-shSC cells (Figure 5C). Consistent with the microfluidic assays, scratch assays demonstrated that the cell-free area was much greater in cultures of HeLa-shSC cells compared with HeLa-shPHD3 cells after 48 h (Figure 5D). PHD3 knockdown did not alter the rate of cell proliferation (Supplemental Figure S4). PHD3-knockdown HeLa cells assumed a spindle-shaped morphology that was distinct from that of HeLa-shSC cells (Supplemental Figure S5).
FIGURE 5:

PHD3 knockdown increases HeLa cell motility. (A–C) Microfluidic assays were performed using HeLa-shSC and HeLa-shPHD3 cells. (A) Representative images from three independent experiments. The yellow asterisks indicate the position of cells after 600 min of migration, and the red asterisks indicate the initial starting position of cells at 0 min. Scale bar, 20 μm. (B) Quantification of chemotactic migration. Mean ± SEM, n = 297 (HeLa-shSC) or 266 (HeLa-shPHD3) cells. ***p < 0.001 vs. shSC. (C) Quantification of cell velocity. Mean ± SEM, n = 305 (HeLa-shSC) or 327 (HeLa-shPHD3) cells. ***p < 0.001 vs. shSC. (D) Scratch assays were performed with HeLa-shSC and HeLa-shPHD3 cells. Representative images at indicated time points from two independent experiments. Scale bar, 100 μm.
To determine whether the prolyl hydroxylase activity of PHD3 is required for inhibition of cell migration, HeLa cells were treated with DMOG for 72 h. Microfluidic assays demonstrated that DMOG significantly increased cell velocity 1.9-fold and chemotactic migration 2.0-fold compared with DMSO (Figure 6 and Supplemental Videos S3 and S4). These data indicate that PHD3 expression and prolyl hydroxylase activity are required for inhibition of cell motility.
FIGURE 6:

DMOG treatment increases HeLa cell migration. (A–C) HeLa cells were pretreated with DMOG (500 μM) or DMSO for 72 h, and microfluidic assays were performed. (A) Representative images from three independent experiments. The yellow asterisks indicate the position of cells after 600 min of migration, and the red asterisks indicate the initial starting position of cells at 0 min. Scale bar, 20 μm. (B) Quantification of cell velocity. Mean ± SEM, n = 412 (DMSO) or 539 (DMOG) cells. ***p < 0.001 vs. DMSO. (C) Quantification of chemotactic migration. Mean ± SEM, n = 360 (DMSO) or 417 (DMOG) cells. ***p < 0.001 vs. DMSO.
DISCUSSION
A growing body of data indicates that PHD3 has a wider range of substrates than PHD1 or PHD2 (Jaakkola and Rantanen, 2013). Several proteins, including ATF-4, β2-adrenergic receptor, HCLK2, paired box 2, and PKM2, are prolyl hydroxylated specifically by PHD3 (Köditz et al., 2007; Xie et al., 2009, 2012; Luo et al., 2011; Yan et al., 2011). In the present study, we demonstrate that nonmuscle β- and γ-actin are also specific substrates of PHD3. Hydroxylation of nonmuscle actin by PHD3 negatively regulated actin polymerization and cell motility. Previous and current studies on β2-adrenergic receptor, HCLK2, PKM2, and actin revealed that there is no strong consensus sequence surrounding proline residues that are hydroxylated by PHD3 (Xie et al., 2009, 2012; Luo et al., 2011). PHD3 can hydroxylate proline residues outside of the putative motif LXXLAP that was originally identified in HIF-1α (Epstein et al., 2001).
In vitro hydroxylation assays and mass spectrometry identified Pro-307 and Pro-322 of human β-actin as two target residues for hydroxylation by PHD3. Double mutation of Pro-307 and Pro-322 of β-actin significantly but incompletely reduced prolyl hydroxylation, and thus additional Pro residues of β-actin may be subject to hydroxylation. Structural analysis of actin demonstrated that Pro-322 is located within a structural hinge (residues 321–324 in subdomain 3) that rotates as a rigid unit, leading to a conformational change (Page et al., 1998). Electron cryomicroscopy also revealed that interaction of Pro-322 with Gly-245 of a neighboring actin molecule is important for actin polymerization (Fujii et al., 2010), suggesting that prolyl hydroxylation may interfere with this interaction to block polymerization. Further studies are required to determine how prolyl hydroxylation impairs actin polymerization and cell motility.
A previous study showed that PHD2 knockdown increased F-actin formation in cancer cells (Vogel et al., 2010). PHD2 knockdown led to increased cofilin-1 phosphorylation, which inhibits F-actin disassembly. Our data demonstrate that PHD2 does not bind to β-actin, which excludes the possibility that PHD2 hydroxylates β-actin in HeLa cells.
In human cancers, metastasis is responsible for ∼90% of cancer deaths. Invasion and metastasis require cancer cell motility, and actin plays a critical role in metastasis (Pollard and Cooper, 2009; Carnell and Insall, 2011). Inhibition of PHD3 markedly increased cell motility. PHD3-knockdown cells also assumed a more spindle-shaped morphology. Thus PHD3 may alter the physical properties of cells by disrupting F-actin formation, thereby impairing cell migration. Expression of PHD3 is down-regulated in pancreatic cancer, colon cancer, and metastatic melanoma (Qi et al., 2008; Xue et al., 2010; Place et al., 2011). PHD3 expression is also negatively correlated with breast cancer grade and stage (Peurala et al., 2012). These findings suggest that PHD3 may inhibit metastasis of multiple human cancers by mediating prolyl hydroxylation of nonmuscle actins to block F-actin assembly. Further studies are required to determine the effect of modulating PHD3 activity on invasion and metastasis in murine cancer models.
MATERIALS AND METHODS
Plasmid constructs
Human β-actin or PHD3 cDNA was amplified by reverse transcription PCR and cloned into pcDNA3.1-V5-His or pGEX-6P-1 vector (GE Healthcare, Piscataway, NJ). Pro–mutant β-actin cDNA was generated using QuikChange Site-directed Mutagenesis Kit (Stratagene, La Jolla, CA) and subcloned to pcDNA3.1-V5-His or pGEX-6P-1 vector. shRNA-resistant PHD3R cDNA was generated using QuikChange Site-directed Mutagenesis Kit (Stratagene) and subcloned to lentiviral vector EF.v-CMV.GFP. Other constructs have been described previously (Luo et al., 2011).
Lentivirus production
HEK293T cells were cotransfected with transducing vector encoding PHD3R or empty vector and packaging vectors pMD.G and pCMVR8.91. After 48 h, lentivirus particles in the medium were harvested, filtered, and transduced into HeLa-shPHD3 cells.
Cell culture
HeLa cells (Scherer et al., 1953) were cultured in DMEM with 10% heat-inactivated fetal bovine serum (FBS) at 37ºC in a 5% CO2/95% air incubator. For hypoxia exposure, cells were placed in a modular incubator chamber (Billups-Rothenberg, Del Mar, CA) that was flushed with a gas mixture containing 1% O2, 5% CO2, and balance N2 and incubated for 24 h at 37°C.
SILAC screen
SILAC (Ong et al., 2002) was performed with HeLa cells that were cultured in DMEM containing 12C6-14N2-lysine/12C6-14N4-arginine (light isotope medium) or 13C6-15N2-lysine/13C6-15N4-arginine (heavy isotope medium) for at least six doublings and treated with DMSO or DMOG (100 μM) for 6 h in the presence of MG132 (10 μM). Cells were lysed in modified radioimmunoprecipitation assay buffer, and equal amounts of lysates were mixed, reduced with 4.1 mM dithiothreitol (DTT), alkylated with 8.3 mM iodoacetamide, and digested overnight with trypsin. The digested peptides were lyophilized, resuspended in buffer (containing 50 mM 3-(N-morpholino)propanesulfonic acid, pH 7.2, 10 mM sodium phosphate, and 50 mM NaCl), and subjected to IP with the anti-hydroxyproline antibody prebound to protein A–agarose beads. The bound peptides were eluted and analyzed by a reversed-phase liquid chromatography system (nanoACQUITY UPLC; Waters, Milford, MA) interfaced with an LTQ Orbitrap Velos for mass spectrometry (MS; Thermo Scientific, Waltham, MA). The MS spectra were acquired in a Fourier transform–Fourier transform data-dependent manner targeting the nine most abundant precursor ions in the m/z range 350–1700 at a resolution of 30,000. Each precursor ion was isolated within a 1.90-m/z window and fragmented with 35% normalized collision energy to form product ions analyzed at 7500 resolution. The tandem mass spectra were searched using MASCOT (version 2.2.0) and SEQUEST search algorithms against the NCBI_GB_167 human protein database through the Proteome Discoverer platform (version 1.3; Thermo Scientific). The MS/MS spectrum of the prolyl hydroxylated peptides was obtained through the PEAKS software and manually examined.
In vitro prolyl hydroxylation assays
WT or mutant GST–β-actin and GST-PHD3 fusion proteins were expressed in E. coli BL21-Gold (DE3) and purified by binding to glutathione-Sepharose beads (GE Healthcare). WT or mutant GST–β-actin was eluted from beads with 20 mM reduced glutathione. Recombinant PHD3 was obtained by removal of GST with PreScission protease at 4°C. WT or mutant GST-β-actin fusion protein was incubated at 30°C for 30 min with or without recombinant PHD3 protein supplemented with 50 mM Tris/HCl (pH 8.0), 100 mM NaCl, 100 μM DTT, 100 μM FeSO4, 5 mM ascorbate, and 1 mM α-ketoglutarate. The prolyl hydroxylation reaction was stopped by adding Laemmli sample buffer and analyzed by immunoblot assays using anti-hydroxyproline antibody (Abcam, Cambridge, MA).
Co-IP assays
Cells were lysed in modified radioimmunoprecipitation assay buffer, and WCLs were incubated overnight with anti-FLAG (Sigma-Aldrich, St. Louis, MO), anti-PHD2 (Novus Biologicals, Littleton, CO), anti-PHD3 (Novus Biologicals), or anti-hydroxyproline (Abcam) antibody in the presence of protein A–agarose beads (Novus Biologicals). After three washes, the bound proteins were fractionated by SDS–PAGE and analyzed by immunoblot assays using antibodies against the following proteins or epitope tag: PHD2, PHD3, β-actin, PKM2 (Novus Biologicals), V5 (Invitrogen, Carlsbad, CA), or FLAG (Sigma-Aldrich).
Microfluidic assays
Microfluidic experiments were performed as previously described (Lin et al., 2012). Briefly, cells were seeded onto microfluidic chips in DMEM containing 0.5% FBS and were allowed to attach for 8-12 h. A gradient was formed across the microchannels by flowing DMEM containing 10% FBS in one of the flanking flow arms and DMEM containing 0.5% FBS in the other arm. During experiments, cells were incubated under 37°C and 95% air/5% CO2. Images were acquired under brightfield illumination every 10 min with a Zeiss Axiovert 200M microscope coupled to a Cascade II:1024 electron-multiplying charge-coupled device camera (Photometrics, Tucson, AZ) using a 40×/1.3 numerical aperture (NA) oil immersion objective. Cells were tracked manually using custom scripts written in Matlab 2007b (MathWorks, Natick, MA). Velocity data were generated based on coordinates obtained from cell tracking. Chemotactic migration measurements were defined as the final displacement of cells from their initial positions. Positive values were assigned to distances toward the gradient, whereas negative values were assigned to distances away from the gradient. Cells migrating within microfluidic channels for <10 h were excluded from these measurements.
Scratch assays
Cells were seeded onto 10-cm dishes and cultured overnight to reach 80–90% confluence. The cell layer was scratched with a sterile 1-ml pipette tip, labeled, and photographed at the indicated time points with an inverted IX71 microscope (Olympus, Center Valley, PA) that was equipped with a digital camera (Olympus DP70) using a 10×/0.3 NA objective.
Fluorescence microscopy
After rinsing with phosphate-buffered saline (PBS), cells were fixed with 3.7% methanol-free formaldehyde for 10 min at room temperature, permeabilized with 0.1% Triton X-100 in PBS for 5 min, and incubated with 1% bovine serum albumin for 60 min. For phalloidin staining, cells were incubated for 120 min with Alexa Fluor 555–conjugated phalloidin at room temperature in the dark. For PHD3 staining, cells were incubated overnight with rabbit anti-PHD3 antibody (5 μg/ml) at 4°C, rinsed, and incubated with Alexa Fluor 647–conjugated goat anti-rabbit IgG antibody (4 μg/ml; Molecular Probes, Eugene, OR) for 120 min at room temperature in the dark. Mounted slides were imaged with an upright Olympus BX51 fluorescence microscope equipped with a digital camera (Olympus DP70) using a 60×/1.25 NA oil objective or a Zeiss LSM 710 confocal laser scanning microscope using a 63×/1.4 NA oil objective. The fluorescence intensity of the image was calculated using ZEN2012 software (Zeiss, Jena, Germany).
Actin sedimentation assays
Cells were homogenized in 0.5 ml of prewarmed lysis buffer containing 50 mM 1,4-piperazinediethanesulfonic acid (pH 6.9), 50 mM NaCl, 5 mM MgCl2, 5 mM ethylene glycol tetraacetic acid, 5% glycerol, 0.1% Igepal CA-630, 0.1% Triton X-100, 0.1% Tween 20, 0.1% β-mercaptoethanol, 0.001% Antifoam C, 100 mM ATP, and protease inhibitor cocktail and centrifuged at 2000 rpm for 5 min to remove unbroken cells. The supernatant (0.4 ml) was centrifuged at 100,000 × g for 60 min at 37°C. The supernatants, which contained G-actin, were collected, and the pellets, which contained F-actin, were resuspended in 0.4 ml of lysis buffer and sonicated. Equal volumes of G-actin and F-actin fractions were analyzed by immunoblot assays using an anti–β-actin antibody (Novus Biologicals).
Statistical analysis
Data are expressed as mean ± SEM. Differences were analyzed by Student's t test; p < 0.05 was considered significant.
Supplementary Material
Acknowledgments
We thank Karen Padgett (Novus Biologicals, Littleton, CO) for providing antibodies against PHD2, PHD3, PKM2, and β-actin; rabbit IgG; and protein A–agarose beads. We are grateful to Linzhao Cheng (Johns Hopkins University School of Medicine) for the EF.v-CMV.GFP vector and Vickram Srinivas (Thomas Jefferson University, Philadelphia, PA) for the FLAG-PHD1 vector. This work was supported by National Institutes of Health Contract N01-HV28180. W.L. is supported by National Institutes of Health Grant K99-CA168746. Y.W. is supported by National Institutes of Health Grant K99-NS078049 and American Heart Association NCRP Scientist Development Grant 12SDG11900071. G.L.S. is the C. Michael Armstrong Professor at the Johns Hopkins University School of Medicine.
Abbreviations used:
- DFX
desferrioxamine
- DMOG
dimethyloxalylglycine
- DMSO
dimethyl sulfoxide
- DTT
dithiothreitol
- FBS
fetal bovine serum
- GST
glutathione S-transferase
- HIF
hypoxia-inducible factor
- IgG
immnoglobulin G
- IP
immunoprecipitation
- NA
numerical aperture
- PBS
phosphate-buffered saline
- PHD
prolyl hydroxylase
- PKM2
pyruvate kinase M2
- SC
scrambled control
- shRNA
short hairpin RNA
- SILAC
stable isotope labeling by amino acids in cell culture
- VHL
von Hippel–Lindau tumor suppressor protein
- WCL
whole-cell lysate
- WT
wild type
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E14-02-0775) on July 30, 2014.
REFERENCES
- Aldini G, Carini M, Vistoli G, Shibata T, Kusano Y, Gamberoni L, Dalle-Donne I, Milzani A, Uchida K. Identification of actin as a 15-deoxy-delta12,14-prostaglandin J2 target in neuroblastoma cells: mass spectrometric, computational, and functional approaches to investigate the effect on cytoskeletal derangement. Biochemistry. 2007;46:2707–2718. doi: 10.1021/bi0618565. [DOI] [PubMed] [Google Scholar]
- Anderson K, Nordquist KA, Gao X, Hicks KC, Zhai B, Gygi SP, Patel TB. Regulation of cellular levels of Sprouty2 protein by prolyl hydroxylase domain and von Hippel-Lindau proteins. J Biol Chem. 2011;286:42027–42036. doi: 10.1074/jbc.M111.303222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold K, Bordoli L, Kopp J, Schwede T. The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling. Bioinformatics. 2006;22:195–201. doi: 10.1093/bioinformatics/bti770. [DOI] [PubMed] [Google Scholar]
- Berra E, Benizri E, Ginouvès A, Volmat V, Roux D, Pouysségur J. HIF prolyl- hydroxylase 2 is the key oxygen sensor setting low steady-state levels of HIF-1a in normoxia. EMBO J. 2003;22:4082–4090. doi: 10.1093/emboj/cdg392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carnell MJ, Insall RH. Actin on disease—studying the pathobiology of cell motility using Dictyostelium discoideum. Semin Cell Dev Biol. 2011;22:82–88. doi: 10.1016/j.semcdb.2010.12.003. [DOI] [PubMed] [Google Scholar]
- Dalle-Donne I, Giustarini D, Rossi R, Colombo R, Milzani A. Reversible S-glutathionylation of Cys 374 regulates actin filament formation by inducing structural changes in the actin molecule. Free Radic Biol Med. 2003;34:23–32. doi: 10.1016/s0891-5849(02)01182-6. [DOI] [PubMed] [Google Scholar]
- Dalle-Donne I, Milzani A, Giustarini D, Di Simplicio P, Colombo R, Rossi R. S-NO-actin: S-nitrosylation kinetics and the effect on isolated vascular smooth muscle. J Muscle Res Cell Motil. 2000;21:171–181. doi: 10.1023/a:1005671319604. [DOI] [PubMed] [Google Scholar]
- Dominguez R, Holmes KC. Actin structure and function. Annu Rev Biophys. 2011;40:169–186. doi: 10.1146/annurev-biophys-042910-155359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epstein AC, Gleadle JM, McNeill LA, Hewitson KS, O'Rourke J, Mole DR, Mukherji M, Metzen E, Wilson MI, Dhanda A, et al. C. elegans EGL-9 and mammalian homologs define a family of dioxygenases that regulate HIF by prolyl hydroxylation. Cell. 2001;107:43–54. doi: 10.1016/s0092-8674(01)00507-4. [DOI] [PubMed] [Google Scholar]
- Farah ME, Sirotkin V, Haarer B, Kakhniashvili D, Amberg DC. Diverse protective roles of the actin cytoskeleton during oxidative stress. Cytoskeleton (Hoboken) 2011;68:340–354. doi: 10.1002/cm.20516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu J, Taubman MB. Prolyl hydroxylase EGLN3 regulates skeletal myoblast differentiation through an NF-kB-dependent pathway. J Biol Chem. 2010;285:8927–8935. doi: 10.1074/jbc.M109.078600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujii T, Iwane AH, Yanagida T, Namba K. Direct visualization of secondary structure of F-actin by electron cryomicroscopy. Nature. 2010;467:724–728. doi: 10.1038/nature09372. [DOI] [PubMed] [Google Scholar]
- Galkin VE, Orlova A, Cherepanova O, Lebart M, Egelman EH. High-resolution cryo-EM structure of the F-actin-fimbrin/plastin ABD2 complex. Proc Natl Acad Sci USA. 2008;105:1494–1498. doi: 10.1073/pnas.0708667105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorres KL, Raines RT. Prolyl 4-hydroxylase. Crit Rev Biochem Mol Biol. 2010;45:106–124. doi: 10.3109/10409231003627991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hampton-Smith RJ, Peet DJ. From polyps to people: a highly familiar response to hypoxia. Ann N Y Acad Sci. 2009;1177:19–29. doi: 10.1111/j.1749-6632.2009.05035.x. [DOI] [PubMed] [Google Scholar]
- Hegyi G, Michel H, Shabanowitz J, Hunt DF, Chatterjie N, Healy-Louie G, Elzinga M. Gln-41 is intermolecularly cross-linked to Lys-113 in F-actin by N-(4-azidobenzoyl)-putrescine. Protein Sci. 1992;1:132–144. doi: 10.1002/pro.5560010113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herman IM. Actin isoforms. Curr Opin Cell Biol. 1993;5:48–55. doi: 10.1016/s0955-0674(05)80007-9. [DOI] [PubMed] [Google Scholar]
- Hung RJ, Pak CW, Terman JR. Direct redox regulation of F-actin assembly and disassembly by Mical. Science. 2011;334:1710–1713. doi: 10.1126/science.1211956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Insall RH, Machesky LM. Actin dynamics at the leading edge: from simple machinery to complex networks. Dev Cell. 2009;17:310–322. doi: 10.1016/j.devcel.2009.08.012. [DOI] [PubMed] [Google Scholar]
- Jaakkola PM, Rantanen K. The regulation, localization, and functions of oxygen-sensing prolyl hydroxylase PHD3. Biol Chem. 2013;394:449–457. doi: 10.1515/hsz-2012-0330. [DOI] [PubMed] [Google Scholar]
- Kabsch W, Mannherz HG, Suck D, Pai EF, Holmes KC. Atomic structure of the actin: DNase I complex. Nature. 1990;347:37–44. doi: 10.1038/347037a0. [DOI] [PubMed] [Google Scholar]
- Kaelin WG, Jr, Ratcliffe PJ. Oxygen sensing by metazoans: the central role of the HIF hydroxylase pathway. Mol Cell. 2008;30:393–402. doi: 10.1016/j.molcel.2008.04.009. [DOI] [PubMed] [Google Scholar]
- Kiss J, Mollenhauer M, Walmsley SR, Kirchberg J, Radhakrishnan P, Niemietz T, Dudda J, Steinert G, Whyte MK, Carmeliet P, et al. Loss of the oxygen sensor PHD3 enhances the innate immune response to abdominal sepsis. J Immunol. 2012;189:1955–1965. doi: 10.4049/jimmunol.1103471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kudryashov DS, Durer ZA, Ytterberg AJ, Sawaya MR, Pashkov I, Prochazkova K, Yeates TO, Loo RR, Loo JA, Satchell KJ, et al. Connecting actin monomers by iso-peptide bond is a toxicity mechanism of the Vibrio cholerae MARTX toxin. Proc Natl Acad Sci USA. 2008;105:18537–18542. doi: 10.1073/pnas.0808082105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Köditz J, Nesper J, Wottawa M, Stiehl DP, Camenisch G, Franke C, Myllyharju J, Wenger RH, Katschinski DM. Oxygen-dependent ATF-4 stability is mediated by the PHD3 oxygen sensor. Blood. 2007;110:3610–3617. doi: 10.1182/blood-2007-06-094441. [DOI] [PubMed] [Google Scholar]
- Lang AE, Schmidt G, Schlosser A, Hey TD, Larrinua IM, Sheets JJ, Mannherz HG, Aktories K. Photorhabdus luminescens toxins ADP-ribosylate actin and RhoA to force actin clustering. Science. 2010;327:1139–1142. doi: 10.1126/science.1184557. [DOI] [PubMed] [Google Scholar]
- Lassing I, Schmitzberger F, Bjornstedt M, Holmgren A, Nordlund P, Schutt CE, Lindberg U. Molecular and structural basis for redox regulation of b-actin. J Mol Biol. 2007;370:331–348. doi: 10.1016/j.jmb.2007.04.056. [DOI] [PubMed] [Google Scholar]
- Lengsfeld AM, Löw I, Wieland T, Dancker P, Hasselbach W. Interaction of phalloidin with actin. Proc Natl Acad Sci USA. 1974;71:2803–2807. doi: 10.1073/pnas.71.7.2803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonarz C, Coleman ML, Boleininger A, Schierwater B, Ratcliffe PJ, Schofield CJ. The hypoxia-inducible transcription factor pathway regulates oxygen sensing in the simplest animal, Trichoplax adherens. EMBO Rep. 2011;12:63–70. doi: 10.1038/embor.2010.170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin B, Holmes WR, Wang CJ, Ueno T, Harwell A, Edelstein-Keshet L, Inoue T, Levchenko A. Synthetic spatially graded Rac activation drives cell polarization and movement. Proc Natl Acad Sci USA. 2012;109:E3668–E3677. doi: 10.1073/pnas.1210295109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Shu S, Hong MS, Levine RL, Korn ED. Phosphorylation of actin Tyr-53 inhibits filament nucleation and elongation and destabilizes filaments. Proc Natl Acad Sci USA. 2006;103:13694–13699. doi: 10.1073/pnas.0606321103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo W, Hu H, Chang R, Zhong J, Knabel M, O'Meally R, Cole RN, Pandey A, Semenza GL. Pyruvate kinase M2 is a PHD3-stimulated coactivator for hypoxia- inducible factor 1. Cell. 2011;145:732–744. doi: 10.1016/j.cell.2011.03.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nyman T, Schuler H, Korenbaum E, Schutt CE, Karlsson R, Lindberg U. The role of MeH73 in actin polymerization and ATP hydrolysis. J Mol Biol. 2002;317:577–589. doi: 10.1006/jmbi.2002.5436. [DOI] [PubMed] [Google Scholar]
- Ong SE, Blagoev B, Kratchmarova I, Kristensen DB, Steen H, Pandey A, Mann M. Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol Cell Proteomics. 2002;1:376–386. doi: 10.1074/mcp.m200025-mcp200. [DOI] [PubMed] [Google Scholar]
- Page R, Lindberg U, Schutt CE. Domain motions in actin. J Mol Biol. 1998;280:463–474. doi: 10.1006/jmbi.1998.1879. [DOI] [PubMed] [Google Scholar]
- Peurala E, Koivunen P, Bloigu R, Haapasaari KM, Jukkola-Vuorinen A. Expressions of individual PHDs associate with good prognostic factors and increased proliferation in breast cancer patients. Breast Cancer Res Treat. 2012;133:179–188. doi: 10.1007/s10549-011-1750-5. [DOI] [PubMed] [Google Scholar]
- Place TL, Fitzgerald MP, Venkataraman S, Vorrink SU, Case AJ, Teoh ML, Domann FE. Aberrant promoter CpG methylation is a mechanism for impaired PHD3 expression in a diverse set of malignant cells. PLoS One. 2011;6:e14617. doi: 10.1371/journal.pone.0014617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Place TL, Nauseef JT, Peterson MK, Henry MD, Mezhir JJ, Domann FE. Prolyl-4-hydroxylase 3 (PHD3) expression is downregulated during epithelial-to-mesenchymal transition. PLoS One. 2013;8:e83021. doi: 10.1371/journal.pone.0083021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollard TD, Cooper JA. Actin, a central player in cell shape and movement. Science. 2009;326:1208–1212. doi: 10.1126/science.1175862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi J, Nakayama K, Gaitonde S, Goydos JS, Krajewski S, Eroshkin A, Bar-Sagi D, Bowtell D, Ronai Z. The ubiquitin ligase Siah2 regulates tumorigenesis and metastasis by HIF-dependent and -independent pathways. Proc Natl Acad Sci USA. 2008;105:16713–16718. doi: 10.1073/pnas.0804063105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridley AJ, Schwartz MA, Burridge K, Firtel RA, Ginsberg MH, Borisy G, Parsons JT, Horwitz AR. Cell migration: integrating signals from front to back. Science. 2003;302:1704–1709. doi: 10.1126/science.1092053. [DOI] [PubMed] [Google Scholar]
- Rose NR, McDonough MA, King ON, Kawamura A, Schofield CJ. Inhibition of 2-oxoglutarate dependent oxygenases. Chem Soc Rev. 2011;40:4364–4397. doi: 10.1039/c0cs00203h. [DOI] [PubMed] [Google Scholar]
- Saha S, Mundia MM, Zhang F, Demers RW, Korobova F, Svitkina T, Perieteanu AA, Dawson JF, Kashina A. Arginylation regulates intracellular actin polymer level by modulating actin properties and binding of capping and severing proteins. Mol Biol Cell. 2010;21:1350–1361. doi: 10.1091/mbc.E09-09-0829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scherer WF, Syverton JT, Gey GO. Studies on the propagation in vitro of poliomyelitis viruses. IV. Viral multiplication in a stable strain of human malignant epithelial cells (strain HeLa) from an epidermoid carcinoma of the cervix. J Exp Med. 1953;97:695–710. doi: 10.1084/jem.97.5.695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrödinger LLC. The PyMOL molecular graphics system, version 1.1. 2008 Available at www.schrodinger.com/pymol/ (accessed 8 July 2014) [Google Scholar]
- Su Y, Loos M, Giese N, Hines OJ, Diebold I, Görlach A, Metzen E, Pastorekova S, Friess H, Büchler P. PHD3 regulates differentiation, tumour growth and angiogenesis in pancreatic cancer. Br J Cancer. 2010;103:1571–1579. doi: 10.1038/sj.bjc.6605936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tennant DA, Gottlieb E. HIF prolyl hydroxylase-3 mediates a-ketoglutarate- induced apoptosis and tumor suppression. J Mol Med (Berl) 2010;88:839–849. doi: 10.1007/s00109-010-0627-0. [DOI] [PubMed] [Google Scholar]
- Terashima M, Yamamori C, Shimoyama M. ADP-ribosylation of Arg28 and Arg206 on the actin molecule by chicken arginine-specific ADP-ribosyltransferase. Eur J Biochem. 1995;231:242–249. [PubMed] [Google Scholar]
- Terman JR, Kashina A. Post-translational modification and regulation of actin. Curr Opin Cell Biol. 2013;25:30–38. doi: 10.1016/j.ceb.2012.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visschedyk DD, Perieteanu AA, Turgeon ZJ, Fieldhouse RJ, Dawson JF, Merrill AR. Photox, a novel actin-targeting mono-ADP-ribosyltransferase from Photorhabdus luminescens. J Biol Chem. 2010;285:13525–13534. doi: 10.1074/jbc.M109.077339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel S, Wottawa M, Farhat K, Zieseniss A, Schnelle M, Le-Huu S, von Ahlen M, Malz C, Camenisch G, Katschinski DM. Prolyl hydroxylase domain (PHD) 2 affects cell migration and F-actin formation via RhoA/Rho-associated kinase-dependent cofilin phosphorylation. J Biol Chem. 2010;285:33756–33763. doi: 10.1074/jbc.M110.132985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie L, Pi X, Mishra A, Fong G, Peng J, Patterson C. PHD3-dependent hydroxylation of HCLK2 promotes the DNA damage response. J Clin Invest. 2012;122:2827–2836. doi: 10.1172/JCI62374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie L, Xiao K, Whalen EJ, Forrester MT, Freeman RS, Fong G, Gygi SP, Lefkowitz RJ, Stamler JS. Oxygen-regulated b2-adrenergic receptor hydroxylation by EGLN3 and ubiquitylation by pVHL. Sci Signal. 2009;2 doi: 10.1126/scisignal.2000444. ra33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue J, Li X, Jiao S, Wei Y, Wu G, Fang J. Prolyl hydroxylase-3 is down-regulated in colorectal cancer cells and inhibits IKKb independent of hydroxylase activity. Gastroenterology. 2010;138:606–615. doi: 10.1053/j.gastro.2009.09.049. [DOI] [PubMed] [Google Scholar]
- Yan B, Jiao S, Zhang HS, Lv DD, Xue J, Fan L, Wu GH, Fang J. Prolyl hydroxylase domain protein 3 targets Pax2 for destruction. Biochem Biophys Res Commun. 2011;409:315–320. doi: 10.1016/j.bbrc.2011.05.012. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.