

In response to oxidative stress, cyclin C translocates from the nucleus to the cytoplasm, where it interacts with the mitochondrial fission machinery and induces extensive fragmentation of this organelle. Med13p is identified as the anchor protein that retains cyclin C in the nucleus.
Abstract
The yeast cyclin C-Cdk8 kinase forms a complex with Med13p to repress the transcription of genes involved in the stress response and meiosis. In response to oxidative stress, cyclin C displays nuclear to cytoplasmic relocalization that triggers mitochondrial fission and promotes programmed cell death. In this report, we demonstrate that Med13p mediates cyclin C nuclear retention in unstressed cells. Deleting MED13 allows aberrant cytoplasmic cyclin C localization and extensive mitochondrial fragmentation. Loss of Med13p function resulted in mitochondrial dysfunction and hypersensitivity to oxidative stress–induced programmed cell death that were dependent on cyclin C. The regulatory system controlling cyclin C-Med13p interaction is complex. First, a previous study found that cyclin C phosphorylation by the stress-activated MAP kinase Slt2p is required for nuclear to cytoplasmic translocation. This study found that cyclin C-Med13p association is impaired when the Slt2p target residue is substituted with a phosphomimetic amino acid. The second step involves Med13p destruction mediated by the 26S proteasome and cyclin C-Cdk8p kinase activity. In conclusion, Med13p maintains mitochondrial structure, function, and normal oxidative stress sensitivity through cyclin C nuclear retention. Releasing cyclin C from the nucleus involves both its phosphorylation by Slt2p coupled with Med13p destruction.
INTRODUCTION
Elevated levels of reactive oxygen species (ROS) are commonly observed during aging or in response to environmental stress. High ROS levels cause lipid oxidation, protein aggregation, and DNA damage (Morano et al., 2012), leading to activation of stress-response pathways (Estruch, 2000). If the damage is too extensive, the cell will initiate the programmed cell death (PCD) pathway (Mazzoni and Falcone, 2008). An early step in the stress-response pathway is extensive mitochondrial fragmentation or fission (Chan, 2012). Several studies support a model that hyperfission helps facilitate mitochondrial outer membrane permeability, leading to release of proapoptotic factors and caspase activation (Youle and Karbowski, 2005).
In yeast and mammalian cells, cyclin C-Cdk8 regulates transcription through association with the RNA polymerase II holoenzyme (for a review, see Nemet et al., 2014). The cyclin C-Cdk8 kinase controls transcription though modification of the basal transcriptional machinery (Akoulitchev et al., 2000), chromatin (Meyer et al., 2008; Knuesel et al., 2009b), or transcription factors (Hirst et al., 1999; Nelson et al., 2003). In addition to Cdk8p, cyclin C also associates with two additional proteins (Med12p and Med13p) in a complex termed the Cdk8 module (Borggrefe et al., 2002). This module is highly conserved and is found either free (Knuesel et al., 2009a) or associated (Conaway and Conaway, 2011) with the Mediator, a 25–30 protein complex that associates with the RNA polymerase II holoenzyme (Ansari and Morse, 2013). Expression profiling revealed that components of this module control the expression of a similar subset of genes (Zhu et al., 2006). However, the individual components have also been shown to have varying roles in transcriptional control during Drosophila development (Gobert et al., 2010). In addition, Med12p, but not the other components of the module, is required for induction of the multidrug transporter PDR5 in mitochondrial DNA–deficient (rho0) cells (Shahi et al., 2010), indicating an important exchange of information occurs between these two organelles.
Recent studies have revealed an important second role for cyclin C that is independent of Cdk8p. In response to oxidative or ethanol stress, cyclin C, but not Cdk8p, translocates from the nucleus to the cytoplasm (Cooper et al., 2012), where it interacts with the fission machinery to induce mitochondrial hyperfission (Cooper et al., 2014). Consistent with a role for mitochondrial fission and PCD execution, loss of cyclin C function restricts fission (Cooper et al., 2014) and enhances cell viability following stress (Krasley et al., 2006). Conversely, aberrant localization of cyclin C in the cytoplasm induces stress-independent hyperfission and sensitizes the cell to oxidative stress (Cooper et al., 2014). These results indicate that the decision to retain cyclin C in the nucleus or release it into the cytoplasm is an important regulator of PCD initiation. A previous study revealed that Cdk8p is required for normal cytoplasmic translocation of cyclin C (Cooper et al., 2012), but the mechanism was unknown. Here, we provide evidence that Med13p plays the opposite role to Cdk8p by retaining cyclin C in the nucleus in unstressed cells. In response to stress, cyclin C release from Med13p requires the stress-activated MAP kinase Slt2p and Cdk8p activity. Aberrant cyclin C relocalization to the cytoplasm results in continuous mitochondrial fragmentation and dysfunction. These results indicate that Med13-cyclin C interaction is controlled by multiple signals to insure the proper subcellular localization of cyclin C in stressed and unstressed cells.
RESULTS
Med13p is required for nuclear retention of cyclin C in unstressed cultures
In response to several types of stress, the transcription factor cyclin C translocates from the nucleus to the cytoplasm through a mechanism that requires Cdk8p (Cooper et al., 2012, 2014). Therefore we next determined whether the two remaining components of the Cdk8 module, Med12p and Med13p, also regulate cyclin C relocalization. Using fluorescence microscopy, we monitored the localization of a functional cyclin C–yellow fluorescent protein (cyclin C-YFP) reporter protein in med12∆ or med13∆ mutants before and following H2O2 stress application. In wild-type cells, H2O2 induces cyclin C translocation from the nucleus to the cytoplasm, where it interacts with the mitochondria to induce fission (see Cooper et al., 2014; and Figure 1A). Deleting MED12 did not affect cyclin C nuclear localization in unstressed cells or stress-induced cytoplasmic relocalization (Supplemental Figure S1A). However, cyclin C-YFP formed multiple cytoplasmic foci in the unstressed med13∆ strain (Figure 1B) similar to those observed in oxidatively stressed wild-type cells. In addition, these foci colocalized with fragmented mitochondria. These results indicate that Med13p is required for cyclin C retention in the nucleus of unstressed cells.
FIGURE 1:

Med13p retains cyclin C in the nucleus. (A) Subcellular localization of cyclin C (cyclin C-YFP), mitochondria (mt-DsRed), and nuclei (DAPI) were monitored by fluorescence microscopy in a wild-type cell (RSY10) before and following H2O2 treatment (1 mM) as indicated. (B) An unstressed med13∆ culture (RSY1701) was examined as in A. (C) Representative image of mitochondrial morphology in med13∆ cnc1∆ mutant (RSY1712, left panel) and quantitation of mitochondrial fission in cells with the indicated genotype (mean ± SEM, n ≥ 3). (D) Experiment described in B was repeated in unstressed dnm1∆ (RSY1750) or dnm1∆ med13∆ (RSY1894) mutant strains. DAPI staining was omitted in these experiments. Scale bar: 5 μM; h = hours.
In addition to cyclin C mislocalization, we also observed that the mitochondria were highly fragmented in the med13∆ mutant similar to what is observed in stressed wild-type cells (compare mt-DsRed panels in Figure 1, A and B, quantified in C). We previously reported that the stress-induced cytoplasmic relocalization of cyclin C triggers extensive mitochondrial fission (Cooper et al., 2014). Therefore we next determined whether the fragmented mitochondrial phenotype observed in the med13∆ mutant was dependent on cyclin C. A med13∆ cnc1∆ double mutant was constructed, and mitochondrial morphology was monitored in unstressed cultures. These experiments indicated that cyclin C is required for the hyperfission phenotype associated with the med13∆ allele (Figure 1C). Finally, we determined whether this fragmentation was dependent on Dnm1p, the dynamin-like GTPase required for fission (Sesaki and Jensen, 1999). Similar to the med13∆ strain, cyclin C-YFP exhibited cytoplasmic localization in the unstressed dnm1∆ med13∆ double mutant (Figure 1D). Although cyclin C could be observed associated with the mitochondria in the double mutant, the mitochondria retained their aggregated or net-like phenotype similar to the dnm1∆ single mutant. These results are consistent with a model that the extensive mitochondrial fragmentation observed in a med13∆ mutant requires cyclin C relocalization to the cytoplasm and Dnm1p activity.
Our previous work revealed that stress-induced relocalization to the cytoplasm triggers cyclin C proteolysis (Cooper et al., 2012). Consistent with these results, cyclin C was destroyed more rapidly in med13∆ mutants exposed to H2O2 stress (Figure S1B). This instability required the oxidative stress response, as glucose-repressible shutoff experiments revealed no significant difference in cyclin C stability in unstressed cells (Figure S1C). These results indicate that precocious cytoplasmic localization of cyclin C in itself is insufficient to induce its destruction. However, cytoplasmic cyclin C appears more rapidly targeted by a stress-activated destruction pathway.
Med13p regulates Cdk8p nucleolar localization
Our previous studies (Cooper et al., 2012) found that Cdk8p does not relocate to the cytoplasm in H2O2-treated cells. Rather, it colocalizes with the nucleolar marker Nop1p–red fluorescent protein (Nop1p-RFP) in response to oxidative stress (Figure 2A). Therefore we next determined whether Med13p is required for Cdk8p nuclear localization and/or its stress-induced concentration in the nucleolus. To address this question, we monitored Cdk8p–green fluorescent protein (Cdk8p-GFP) localization in unstressed or stressed med13∆ mutant cultures. In the unstressed med13∆ culture, the Cdk8p-GFP signal exhibited a diffuse nuclear pattern similar to that observed in wild-type cells (Figure 2B). However, Cdk8p-GFP also exhibited nucleolar localization in 78% (SEM ± 8, n = 3) of the unstressed med13∆ cells (Figure 2B, top panels). These observations suggest that Med13p normally prevents Cdk8p entry into the nucleolar compartment in unstressed cells. Following H2O2 treatment, Cdk8p-GFP and Nop1p-RFP colocalization still occurred in the med13∆ cells. However, the overall Cdk8p-GFP signal also remained diffused over the nucleus rather than concentrating in the nucleolus as observed in wild-type cells. These results suggest a complicated role for Med13p in regulating Cdk8p subnuclear localization. In unstressed cells, Med13p prevents Cdk8p nucleolar localization. However, Med13p is also required for normal consolidation of Cdk8p in the nucleolus of stressed cells.
FIGURE 2:

Regulation of GFP-Cdk8p localization by Med13p. GFP-Cdk8p localization was monitored by fluorescence microscopy in the wild-type RSY10 (A) or med13∆ mutant RSY1701 (B) strains before and 2 h after H2O2 addition as indicated. Nucleolar and nuclear locations were followed by Nop1p-RFP and DAPI signals, respectively. Arrows indicate cells that are enlarged in the zoom panels. Scale bar: 5 μM; h = hours.
Med13p protects cells for H2O2-induced cell death
Mitochondrial hyperfission is an early step in the stress response and is associated with PCD induction (Eisenberg et al., 2007). Supporting this model, cnc1∆ mutants fail to undergo stress-induced mitochondrial hyperfission (Cooper et al., 2014) and are resistant to H2O2-induced PCD (Krasley et al., 2006; Cooper et al., 2014). Therefore we next examined the role of Med13p in regulating the cellular response to oxidative stress. Although extensive mitochondrial fragmentation is observed in unstressed med13∆ cells, this event on its own did not induce cell death, as evidenced by the similar plating efficiency of unstressed wild-type and mutant cultures (Figure 3A, top rows). However, med13∆ cells demonstrated a hypersensitivity to H2O2 (second panel) compared with wild-type while cnc1∆ strains (third panel) were resistant to this pro-oxidant. To determine whether this hypersensitivity was due to aberrant cyclin C localization, we repeated the experiment with a med13Δ cnc1Δ double mutant. Loss of cyclin C suppressed the H2O2 hypersensitivity associated with the med13∆ allele (Figure 3A, right panel) similar to levels observed for the cnc1∆ allele alone. These results suggest that Med13p-dependent retention of cyclin C in the nucleus prevents hypersensitivity to H2O2. Finally, terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assays and flow-cytometry analyses indicated that the elevated cell death observed in stressed med13∆ mutants corresponded to an increase in cells exhibiting positive TUNEL signal (Figure 3B). As expected, TUNEL-positive cells were reduced in either the cnc1∆ or cnc1∆ med13∆ strains. Taken together, these results suggest that loss of Med13p function predisposes cells to programmed cell death through a cyclin C–dependent mechanism.
FIGURE 3:
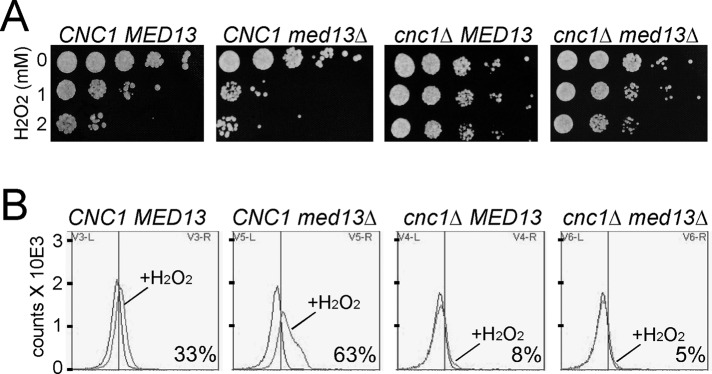
Med13p is required for normal oxidative stress sensitivity. (A) Mid–log wild-type (RSY10), med13∆ (RSY1701), cnc1∆ (RSY391), and cnc1∆ med13∆ (RSY1712) cultures were treated with the H2O2 concentrations as indicated for 2 h and then serially diluted (1:10) before being plated on rich growth medium. The plates were incubated 3 d before image collection. (B) TUNEL assays were performed on strains described in A that were treated with 2 mM H2O2 for 20 h. Typical histograms are shown depicting fluorescence-activated cell sorting analysis of untreated and treated samples as indicated. The numbers in the lower right of each panel indicate the percentage of the population exhibiting a TUNEL-positive signal (SEM < 5%, n = 3).
Med13p maintains mitochondrial DNA integrity
Previous studies have implicated excessive mitochondrial fission with loss of mitochondrial function (Chen et al., 2005). These experiments were accomplished using knockdown or deletion of fusion machinery components. To ask this same question without altering the basic fission or fusion machinery, we monitored mitochondrial function in med13∆ mutants, using growth on a respiratory-necessary carbon source (acetate) as the readout. These studies revealed that the med13∆ mutant was not able to grow on acetate medium but displayed nearly normal growth rate in the presence of the fermentable carbon dextrose (Figure 4A). This result indicates that Med13p is required for mitochondrial function, confirming the results of an earlier study (Shahi et al., 2010). Next we examined whether loss of mitochondrial function was dependent on cyclin C. Double mutant experiments revealed that, similar to the stress hypersensitivity phenotype, deleting CNC1 also suppressed the mitochondrial defect observed in med13∆ mutants. These results suggest that the extensive mitochondrial fragmentation induced by aberrant cyclin C cytoplasmic localization is deleterious to mitochondrial function. For further exploration of this possibility, the med13∆ dnm1∆ double mutant described earlier was assayed for mitochondrial function. Following approximately 90 generations in the presence of a fermentable carbon source, 97% (± 3, n = 4) of the double mutant cells were still respiration competent compared with <5% for med13∆ mutants. These results suggest, as others have reported, that continuous mitochondrial fission is deleterious to long-term maintenance of mitochondrial function.
FIGURE 4:
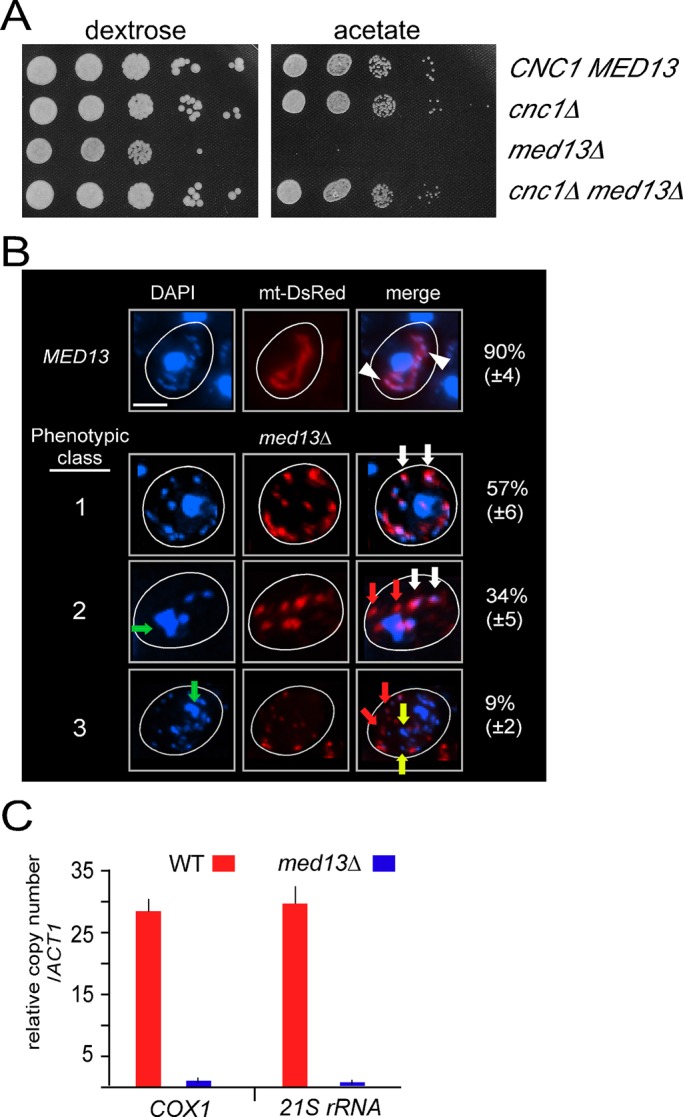
Med13p is required to maintain mitochondrial DNA integrity. (A) Mid–log cultures as described in Figure 3 with the indicated genotype were grown in dextrose medium and then serially diluted (1:10) before being plated on medium containing either dextrose or acetate as the sole carbon source. The plates were incubated 3 d before image collection. (B) Mitochondrial morphology and mtDNA abundance was monitored by mt-DsRed and DAPI staining, respectively, in wild-type (RSY10) and med13∆ mutant (RSY1701) cells. Arrowheads indicate typical mtDNA nucleoids within the mitochondria in wild-type cells. Representative images of the three general phenotypic classes observed in the med13∆ strain are shown with quantification. White arrows indicate normal overlapping mtDNA-mitochondrial signals; green arrows indicate abnormal nuclear morphology; red arrows indicate examples of fragmented mitochondria without a visual mtDNA signal; yellow arrows indicate DAPI staining signals that may represent nuclear fragmentation or aberrant mtDNA signals. (C) qPCR analysis of two mtDNA loci COX1 and 21S rRNA in the strains indicated. Values are depicted relative to the single-copy ACT1 locus. Results shown are the means (± SEM) from three biological replicates. Scale bar: 5 μM.
Loss of mitochondrial function can be the result of mutations in either the mitochondrial or nuclear genome. In yeast, respiration-deficient cells can exhibit total loss of mitochondrial DNA (mtDNA), a condition termed Rho0. To determine whether med13∆ mutants retained their mtDNA, we conducted 4′,6-diamidino-2-phenylindole (DAPI) staining followed by fluorescence microscopy. Wild-type cells exhibited small nucleoids throughout the mitochondrial continuum (Figure 4B, arrowheads). In med13∆ cells, three classes of mtDNA signals were observed. The predominant class 1 phenotype exhibited normal-appearing mtDNA signals associated with the mitochondria (white arrows). However, two additional classes were observed. Class 2 mutants displayed mtDNA-mitochondrial association but also exhibited abnormal nuclear morphology (green arrows). In addition, DAPI-staining signals were absent in a subset of the mitochondrial signals (red arrows). It is important to note that DAPI staining alone is not sufficient to conclude the absence of mtDNA, but rather only that the DNA signal is reduced. Finally, class 3 mutants display a more degraded nuclear DAPI signal with additional DAPI staining bodies not associated with the nucleus or the mitochondria (yellow arrows). Taken together, these results indicated that med13∆ mutants retain mtDNA, although the amount of the DNA may be reduced.
To further characterize mtDNA integrity in med13∆ mutants, we utilized quantitative real-time PCR (qPCR) to test for the presence of COX1 and 21S rRNA alleles. Both alleles were quantitated using qPCR and then compared with a nuclear gene control (ACT1). The primers were chosen to generate relatively small amplicons (102 and 29 base pairs, respectively) to detect retention of discreet regions of the mitochondrial genome. In addition, these loci are on opposite sides of the mitochondrial genome. This experiment produced a calculated wild-type copy number of COX1 and 21S rRNA at 28 and 29, respectively (Figure 4C). This copy number is in the normal range for mtDNA (Williamson and Fennell, 1979). However, less than one copy of either locus was measured in the med13∆ mutant. These results indicate that significant deletions of mtDNA occurred in the med13∆ strain and support the model that excessive fission is deleterious to overall mitochondrial genome maintenance.
Med13p is destroyed in response to H2O2 stress
Our results are consistent with a model that disrupting cyclin C-Med13p association is important to release the cyclin into the cytoplasm in stressed cells. Therefore we next investigated whether Med13p regulation itself provided insight into how this interaction is dissolved. Initially, we monitored Med13p localization in the cell expressing an endogenously tagged functional MED13-YFP allele. As expected, fluorescence microscopy revealed that Med13p-YFP displayed diffuse nuclear staining in unstressed cells (Figure S2). Following 1-h H2O2 treatment, no difference in Med13p-YFP localization was noted, indicating that relocalization of this factor is not a primary regulatory mechanism. Next we examined Med13p levels in an H2O2-stressed culture expressing an endogenously tagged MED13 allele (13-myc). Western blot analysis revealed a dramatic reduction in Med13p levels following stress application (Figure 5A, left panel). Med13p-myc reduction was due to enhanced degradation and not translation inhibition, as Med13p is relatively stable as determined by translation inhibition experiments in unstressed cells (right panel). Thus Med13p is actively targeted for degradation during oxidative stress. To determine whether Med13p destruction required the 26S proteasome, we repeated this experiment in a strain deleted for UMP1, a gene whose product is required for 20S proteasome maturation (Ramos et al., 1998). In response to H2O2 stress, Med13p-myc was protected from destruction in the ump1∆ mutant compared with the wild-type control (Figure 5B). These results indicate that Med13p destruction is most likely directed by a ubiquitin-mediated mechanism.
FIGURE 5:

Med13p is destroyed in response to oxidative stress. (A) Western blot analysis of endogenously tagged Med13p-13myc (RSY17896) during an H2O2 time-course experiment (left panel). Med13p-13myc turnover rate was monitored in an unstressed log phase culture following the addition of cycloheximide (CHX, right panel). For all panels, “con” indicates the untagged parental strain (RSY10) controlling for nonspecific α-myc cross-reactivity. Pgk1p levels were used as loading controls. (B) Med13p-13myc levels were monitored by Western blot analysis during H2O2 stress time course in wild-type (RSY1786) and ump1∆ (RSY1961) strains as described in (A). (C) Med13p-myc levels were monitored by Western blot in extracts prepared from a wild-type (RSY1786) or cnc1∆ (RSY1930) strains before and following H2O2 treatment. (D) Med13p-13myc levels were monitored by Western blot analysis during H2O2 stress time course in cnc1∆ strain (RSY1930) harboring the cyclin CHAD∆ expression plasmid pBK217. h = hours.
To determine whether cyclin C is involved in H2O2-induced Med13p destruction, we monitored Med13p-myc levels in stressed cnc1∆ mid–log cultures. Similar to ump1∆ strains, Med13p-myc levels remained elevated in the cnc1∆ mutant following H2O2 addition. These results indicated that cyclin C is required for Med13p turnover in stressed cells. We have previously demonstrated that a domain at the amino terminal end of cyclin C, the holoenzyme association domain, or HAD (Cooper and Strich, 1999), is required for Med13p association (Cooper et al., 2014). Therefore we next examined whether cyclin C association per se was necessary for Med13p destruction. A cnc1∆ strain was transformed with a plasmid expressing cyclin C with a small internal deletion of 10 amino acids in the HAD (HAD∆). In response to H2O2 exposure, Med13p destruction was also prevented in cells expressing cyclin CHAD∆ (Figure 5D). These results indicate that oxidative stress–induced Med13p destruction required the proteasome and cyclin C association.
Med13p destruction is mediated by Cdk8p activity
The requirement of cyclin C for Med13p destruction suggested a role for Cdk8p in this process. Therefore we next monitored Med13p levels in a cdk8∆ mutant expressing a kinase-dead cdk8 allele (cdk8KD; Surosky et al., 1994). Similar to the cnc1∆ mutant, Med13p was protected from destruction in the strain expressing the kinase-dead allele (Figure 6A). These results indicate that Cdk8p kinase activity is required for Med13p destruction. We next examined the impact that loss of Cdk8p kinase activity had on cyclin C localization. A cdk8∆-null strain expressing either wild-type or the kinase-dead allele of CDK8, along with cyclin C-YFP, was subjected to H2O2 stress (0.8 mM) for 2 h; this was followed by fluorescence microscopy. As expected, the strain expressing wild-type CDK8 exhibited normal cyclin C-YFP relocalization to the cytoplasm (Figure 6B, top panels). Conversely, cyclin C-YFP formed a single focus associated with the nuclear periphery (bottom panels). This observation is similar to our previous study, which found nucleolar targeting of cyclin C-YFP in stressed cdk8∆ cells (Cooper et al., 2014). These results are consistent with a model that Med13p destruction is required for cyclin C translocation from the nucleolus to the cytoplasm.
FIGURE 6:

Med13p destruction requires Cdk8p activity. (A) Med13p-13myc levels were monitored in an H2O2 (0.8 mM) stressed mid–log cdk8∆ strain (RSY1954) expressing the wild-type CDK8 (pPL144-21) or a kinase-dead derivative (pPL144-23). Pgk1p levels were used as a loading control. (B) Localization of cyclin C-YFP in wild-type or Cdk8pKD-expressing cells following 2 h treatment with H2O2 (0.8 mM). Subnuclear localization of cyclin C-YFP in the cdk8KD-expressing cells is indicated by the arrowheads. The Normoski (Nom.) and nuclear (DAPI) images are indicated. (C) Endogenously tagged Med13p-3 hemagglutinin (HA) levels were monitored in an unstressed wild-type strain (RSY1788) harboring constitutively active RHO1 (RHO1G19V or RHO1Q68L) expression plasmids. Protein extracts were immunoprecipitated with HA monoclonal antibodies, and the immunoprecipitates were probed for the presence of Med13p-HA. The parental strain (con) and no antibody controls are shown. Tub1p levels were monitored as a loading control. (D) Cyclin CS266A-YFP localization was monitored in unstressed cnc1∆ (RSY391) or cnc1∆ med13∆ (RSY1712) strains. The Normoski (Nom.) and nuclear (DAPI) images are indicated. (E) Coimmunoprecipitation studies were conducted in extracts prepared from a wild-type strain expressing Med13p-myc (RSY1786) and either cyclin C-YFP (pBK38) or cyclin CS266E-YFP (pBK53). α-myc or α-GFP immunoprecipitates were probed for the presence of cyclin C-YFP (E) or Med13p-myc (F) as indicated. Open brackets indicate the no immunoprecipitation antibody controls. GFP lanes contain extracts prepared from cells expressing only GFP (pUG36) to control for interaction of GFP alone with Med13p-myc. Scale bar: 5 μM; h = hours.
Med13p destruction and cyclin C translocation are controlled by separate signaling pathways
Modification of cyclin C on Ser-266 by the cell wall integrity (CWI) MAP kinase Slt2p/Mpk1p is required for normal cyclin C translocation in response to H2O2 stress (Jin et al., 2014). This MAP kinase module is stimulated by Rho1p through protein kinase C (Pkc1p; for a review, see Levin, 2011). To determine whether Med13p stability is controlled by the CWI pathway, we monitored the levels of Med13p-myc in unstressed cultures harboring plasmids expressing wild-type RHO1 or one of two constitutively active alleles (Q68L or G19V; Sekiya-Kawasaki et al., 2002). Our previous studies found that the presence of activated Rho1p was sufficient to induce cyclin C relocalization and destruction in the absence of stress (Jin et al., 2014). Western blot analysis revealed that Med13p-myc levels were not altered in the presence of the activated RHO1 alleles (Figure 6C). These results indicate that Slt2p activity is not sufficient to drive Med13p destruction. These results raised the question of the relationship between cyclin C phosphorylation and Med13p destruction with respect to cyclin C release from the nucleus. To address this question, we used a Ser-266 to alanine (S266A) mutant form of cyclin C that prevents Slt2p phosphorylation and its subsequent relocalization to the cytoplasm under low-stress conditions (Jin et al., 2014). Localization of cyclin CS266A-YFP was monitored in unstressed cnc1∆ and cnc1∆ med13∆ mutants. Although nuclear in the wild-type strain, we found cyclin CS266A in the cytoplasm in the med13∆ mutant (Figure 6D). These results indicate that med13∆ is epistatic to cyclin CS266A and formally implies that Med13p function is either downstream or independent of Ser-266 phosphorylation.
To further test this model, we conducted coimmunoprecipitation experiments between Med13p and either cyclin C or cyclin CS266E. This substitution mutation mimics cyclin C phosphorylation and allows partial release of cyclin C into the cytoplasm (Jin et al., 2014). Extracts were prepared from mid–log cultures (no stress) expressing Med13p-myc and either cyclin C-YFP or cyclin CS266E-YFP. These samples were immunoprecipitated with either α-GFP or α-myc antibodies, and then the immunoprecipitates were subjected to Western blot analysis probing for the presence of cyclin C-YFP or cyclin CS266E-YFP. This experiment revealed a reduction cyclin CS266E-YFP able to immunoprecipitate with Med13p-myc (compare lanes 6 and 7, Figure 6E). No significant differences were observed in cyclin C or cyclin CS266E levels (lanes 2 and 3). In addition, this interaction was independent of the YFP tag (lane 5) and required the myc antibody (lane 8). When the coimmunoprecipitation was examined by probing for the presence of Med13p-myc, a similar result was obtained. The substitution mutation was again less able to interact with Med13p-myc (compare lanes 2 and 3, Figure 6F). These results indicate that cyclin CS266E reduces but does not eliminate Med13p association. In addition, these results provide a mechanism for the requirement of Slt2p in mediating cyclin C relocalization to the cytoplasm. Taken together, our results support a two-step system controlling cyclin C-Med13p association and, ultimately, cyclin C nuclear retention or release.
DISCUSSION
In unstressed cells, cyclin C and Cdk8p form a complex with two additional proteins (Med12p and Med13p) that associates with the Mediator to control gene transcription. In response to stress, cyclin C translocates from the nucleus to the cytoplasm, where it promotes both mitochondrial fragmentation and PCD. Therefore the switch governing cyclin C retention or release from the nucleus is an important cell fate discriminator. In this paper, we demonstrate that Med13p is responsible for retaining cyclin C in the nucleus in unstressed cells. Deleting MED13 releases cyclin C into the cytoplasm, inducing extensive mitochondrial fission, oxidative stress hypersensitivity, and loss of mtDNA integrity. Our previous studies revealed that cyclin C relocalization requires Cdk8p and activation of the CWI pathway. We now provide mechanisms to explain the requirement of each factor. We found that Med13p is destroyed in response to oxidative stress in a manner dependent on Cdk8p activity. In addition, our data indicate that Slt2p-dependent phosphorylation of cyclin C helps destabilize its interaction with Med13p. Taken together, these data reveal that Med13p maintains mitochondrial function and protects the cells from aberrant PCD execution through retention of cyclin C in the nucleus.
We find that loss of Med13p activity results in extensive mitochondrial fragmentation in unstressed cells. In light of our previous report (Cooper et al., 2014) and results from the present study, release of cyclin C into the cytoplasm is responsible for this dramatic fission phenotype. Two additional phenotypes associated with the med13∆ allele include H2O2 hypersensitivity and loss of mitochondrial function. The respiration dysfunction could be caused by mutation within mitochondrial genome or misexpression of a nuclear gene caused by the med13∆ allele. Several results indicate that the respiration deficiency in med13Δ mutant is due to mtDNA defect and not the transcriptional role of Med13p. First, we quantified a significant loss in two mtDNA loci in med13∆ strains. Second, dissection of MED13/med13 diploids resulted in spores exhibiting both active and inactive mitochondria. However, the mitochondrial defective phenotype did not cosegregate with the med13 mutant allele. Finally, studies in mice (Chen et al., 2003, 2005; An et al., 2013) and yeast (Hermann et al., 1998; Sesaki and Jensen, 2001) have reported that the inability to undergo fusion causes elevated mtDNA damage. These data argue that aberrant mitochondrial fission induced by constitutive cytoplasmic cyclin C localization accelerates loss of active mitochondria. An alternative explanation that we cannot rule out is that a transcriptional defect associated with loss of Med13p function reduces the efficiency of mitochondrial maintenance. Therefore, unlike nuclear petite mutants that display instant loss of mitochondrial activity, med13∆ mutants may undergo an overall degradation of mitochondrial function that is manifested only after many generations. However, our finding that dnm1∆ alleles are able to suppress the loss of mitochondrial function in a med13∆ strain argues against this possibility. Additional studies into the exact role of cyclin C in promoting mitochondrial fission may help distinguish between these possibilities.
The second phenotype we observed is oxidative stress hypersensitivity. This observation may also be related to the impact of constant mitochondrial fragmentation. Many studies have observed that mitochondrial fragmentation is an early step in the stress-response pathway (for reviews, see Scott et al., 2003; Youle and Karbowski, 2005). Consistent with this connection, we have previously demonstrated that cells lacking cyclin C fail to undergo fission and are resistant to oxidative stress (Cooper et al., 2014). Studies in mammalian cells have found that the proapoptotic BH-3 family member Bax is recruited to sites of fission (Karbowski et al., 2002; Yuan et al., 2007; Cassidy-Stone et al., 2008; Brooks et al., 2011). Therefore it is possible that the constitutive recruitment of the fission machinery to the mitochondria by cyclin C may elevate the efficiency by which BH-3 proteins can induce PCD. In yeast, a BH-3 protein (Ybh3p) has been identified (Buttner et al., 2011). Currently, studies are underway to determine the relationship between the stress hypersensitivity associated with med13∆ alleles and Ybh3p activity.
This study and our previous work have identified two domains, the HAD and Ser-266 region, as sites controlling cyclin C nuclear localization. Structural analysis of the Saccharomyces pombe cyclin C provides a clue as to how these domains act together. Cyclins contain a repeat of the cyclin box fold, a five alpha-helix bundle (Hoeppner et al., 2005). The amino cyclin box universally binds its cognate Cdk, while a role for the second cyclin box remains elusive. In addition, all cyclins possess an amino terminal helix of varying length that appears to have different functions. For cyclin A, the amino terminal helix folds back on itself to make contact with Cdk2 (Jeffrey et al., 1995). Conversely, this region in cyclin C contains the HAD (see Figure 7A) and has been described as flexible (Hoeppner et al., 2005) or more rigid (Schneider et al., 2011). Previously, we demonstrated that the hydrophobic residues in this domain (indicated by yellow coloring), as well as its alpha-helical nature, are required for HAD function (Cooper and Strich, 1999). In addition, the cyclin C HAD mutant is less able to coimmunoprecipitate with Med13p, causing partial release from the nucleus and an intermediate, mixed fusion–fission phenotype, in unstressed cells (Cooper et al., 2014). Similarly, another study demonstrated that a phosphomimetic substitution mutation at Ser-266 (S266E) also displays a reduced ability to associate with Med13p and causes a partial release of cyclin C from the nucleus (Jin et al., 2014). These results suggest a common role for these domains. The solved cyclin C structure (Hoeppner et al., 2005) allowed us to model Ser-266 to the loop region between the third and fourth helix of the second cyclin box (Figure 7A). Interestingly, both the HAD and Ser-266 regions are on the same side of cyclin C, raising the possibility that they represent a docking site for Med13p, as previously suggested (Hoeppner et al., 2005). Therefore combining our genetic results with the crystal data suggests that the HAD and Ser-266 regions form a protein-binding domain on cyclin C away from Cdk8p interaction. Consistent with this model, the crystal structure also predicts a hydrophobic pocket (Figure 7A, light blue region) that may facilitate protein:protein interaction.
FIGURE 7:
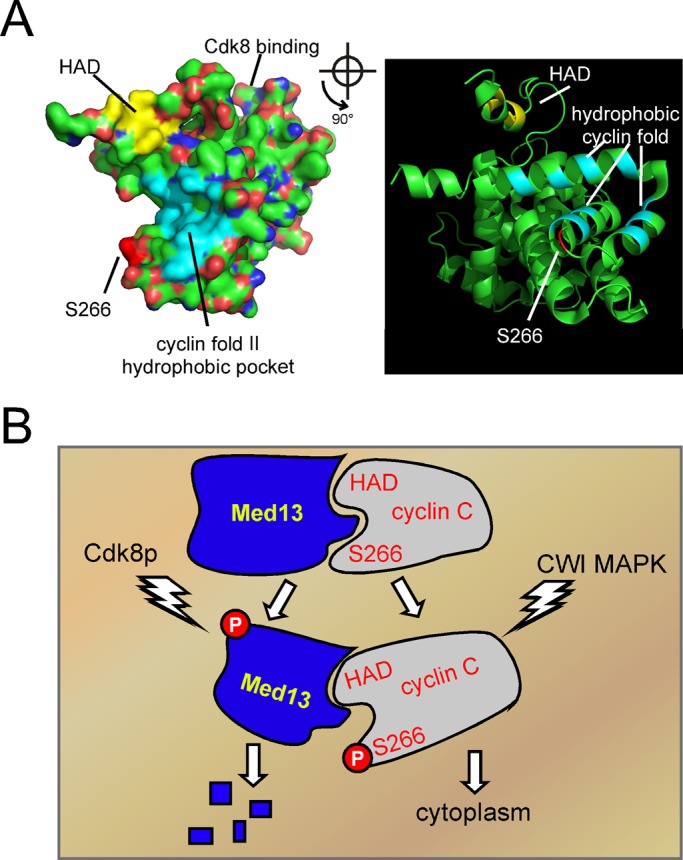
Models for Med13p control of cyclin C subcellular localization. (A) RasMol-generated images of the cyclin C crystal structure solved by Hoeppner et al. (2005). A space-filling (left) and ribbon (right) diagram with the HAD (yellow), Ser-266 (dark red), and the hydrophobic pocket (light blue) imposed on the cyclin C structure. The ribbon diagram is rotated 90° with respect to the space-filling model. The cyclin box region that binds Cdk8p is on the other side of the space-filling model. The Ser-266 region is approximate, as the loop region in which it resides was not modeled. (B) Model for regulation of cyclin C release from Med13p. Proposed interaction of Med13p with the cyclin box II region is depicted with the HAD and Ser-266 region as indicated. Activation of the CWI MAP kinase Slt2p results in phosphorylation of cyclin C on Ser-266, causing partial disruption of this interaction. Phosphorylation by Cdk8p makes Med13p susceptible to ubiquitin-mediated proteolysis, resulting in complete release of cyclin C into the cytoplasm.
Our finding that cyclin C is cytoplasmic in unstressed med13∆ mutants indicates that the system controlling cyclin C translocation does not require a stress signal. Therefore Med13p release appears to be the critical decision point in controlling cyclin C localization. We have recently demonstrated that the CWI MAP kinase Slt2p phosphorylates Ser-266 (Jin et al., 2014) and that this modification is required for efficient cyclin C cytoplasmic translocation. Therefore cyclin C phosphorylation reduces its ability to bind Med13p (Figure 7B). To relieve HAD binding, Med13p is destroyed, which commits the cell to cyclin C release. Interestingly, the CWI signal transduction pathway that mediates cyclin C phosphorylation is not involved in Med13p proteolysis, indicating the existence of another pathway triggering this process. Indeed, Med13p is a known substrate of the protein kinase A signal transduction pathway (Chang et al., 2004). We found that cyclin C and Cdk8p kinase activities are required for Med13p destruction. As a previous study found that mammalian Med13 is a substrate of Cdk8 (Knuesel et al., 2009b), the interaction we observe may be direct. In addition, a recent study found that Cdk8 phosphorylation induced the destruction of the Mediator component Med3 (Gonzalez et al., 2014). Finally, similar to our results, steady-state turnover of mammalian Med13 is mediated by a ubiquitin-mediated process (Davis et al., 2013). Taken together, our findings are consistent with a model that stress-activated destruction of Med13p requires phosphorylation by Cdk8p. This possibility implies that activation of two separate pathways is required for full release of cyclin C. It is reasonable to expect that cyclin C translocation to the cytoplasm is tightly controlled to prevent aberrant mitochondrial fission and/or elevated sensitivity to stress, two outcomes that are deleterious to cell fitness.
MATERIALS AND METHODS
Yeast strains and plasmids
All Saccharomyces cerevisiae strains used in this study are derivatives of a W303-1A variant (Strich et al., 1989) and are listed in Table 1. In accordance with the gene nomenclature standardization efforts (Bourbon et al., 2004), CNC1 (a.k.a. SSN3/SRB11/UME3), MED12 (a.k.a. SRB8/SSN5), MED13 (a.k.a. SSN2/SRB9/UME2), and CDK8 (SSN8/SRB10/UME5) gene designations will be used. Gene deletions were constructed as described previously (Longtine et al., 1998). The med13Δ cnc1Δ strain (RSY1712) was generated by deleting MED13 in the cnc1∆ mutant RSY391. The endogenous MED13-yECitrine::KanMx6 construct was made using pKT140 (Sheff and Thorn, 2004). The strain containing the integrated CNC1-TAP allele (RSY1010) was a gift from Nynke L. van Berkum. Plasmids pKC337, pKC333, and pBK37 were described previously (Cooper et al., 1999, 2012). pBK217 containing the CNC1HAD∆ allele was previously described (Cooper et al., 2014). Mitochondria visualization was achieved using pMt-DsRed (a gift from J. Nunnari; Naylor et al., 2006). Plasmids pUG36 (MET25-GFP control), pBK38 (cyclin C-YFP), and pBK53 (cyclin CS266E-YFP) have been previously described (Niedenthal et al., 1996; Jin et al., 2014). The CDK8/SSN8/SRB10 (pPL144-21) and cdk8KD (pPL144-23) expression plasmids have been previously described (Surosky et al., 1994). The G19V (pYO964) and Q68L (pYO965) constitutively active RHO1 expression plasmids (Sekiya-Kawasaki et al., 2002) were a gift from Y. Ohya. A ump1 mutant strain (Ramos et al., 1998) used to generate RSY1961 was a gift from R. J. Dohmen.
TABLE 1:
S. cerevisiae strains.
| Strain | Genotype | Source |
|---|---|---|
| RSY10 | Strich et al., 1989 | |
| RSY391 | cnc1::LEU2 | Cooper et al., 1999 |
| RSY392 | cnc1::TRP1 | Cooper et al., 1999 |
| RSY1701 | med13::HIS3 | This study |
| RSY1712 | cnc1::LEU2 med13::HIS3 | This study |
| RSY1786 | MED13-13myc::KanMX6 | This study |
| RSY1812 | MED13–yECitrine::KanMX6 | This study |
| RSY1961 | his3∆ leu2-3112 lys2-801 trp1-∆63 ura3-52 ump1::HIS3 MED13-13myc::KanMX6 | This study |
| RSY1930 | cnc1::LEU2 MED13-13myc::KanMX6 | This study |
| RSY1700 | med12::HIS3 | This study |
| RSY1750 | dnm1::KanMX6 | This study |
| RSY1894 | dnm1::KanMX6 med13::HIS3 | This study |
| RSY1954 | cdk8::his5+ cnc1::LEU2 MED13-13myc::KanMX6 | This study |
| RSY1788 | MED13-3HA::KanMX6 | This study |
| RSY1861 | KanMX6-GAL1pro-GFP-CDK8 | This study |
| RSY1863 | KanMX6-GAL1pro-GFP-CDK8 med13::HIS3 | This study |
All strains are derived from the W303 background and contain the genotype MATa ade2 ade6 can1-100 his3-11,15 leu2-3112 trp1-1 ura3-1, except RSY1961.
Growth and stress assays
Cells were grown in either rich, nonselective medium (YPDA) or synthetic minimal medium (SC) allowing plasmid selection as previously described (Cooper et al., 1999). Clonogenic viability studies were conducted with mid–log phase (6 × 106 cells/ml) treated with 1 or 2 mM H2O2 for 2 h and then serially diluted (1:10) and plated on the nonselective medium (YPDA). TUNEL assays were conducted essentially as previously described (Madeo et al., 1997; Krasley et al., 2006). TUNEL-positive cells were measured by fluorescence-activated cell analysis using the Accuri C6 cell analyzer. All statistical analysis was performed using the Student's t test with p < 0.05 considered significant. All analyses were conducted with at least three independent cultures with 300 or more cells counted per time point. Quantitative PCR analysis of mtDNA loci was accomplished using Taqman cybergreen method (Applied Biosystems, Grand Island, NY). The threshold cycle number (CT) values were normalized to the nuclear ACT1 locus. COX1-F-5′-CTACAGATACAGCATTTCCAAGA; COX1-R-5′-GTGCCTG-AATAGATGATAATGGT; 21S-F-5′-AATTGACCCGAAAGCAAACG’ 21S-R-5′-TTGCAACAT-CAACCTGTTCGA; ACT1-F-5′-GTATGTGTAAAGCCGGTTTTG; ACT1-R-5′-CATGATACCT-TGGTGTCTTGG
Microscopy and cell analysis
Intracellular localization studies of chimeric fusion proteins were performed in fixed or living cells as indicated in the figure legends. Cells were fixed in 3.7% paraformaldehyde and stained with DAPI, as previously described (Cooper and Strich, 2002). For all experiments, the cells were grown to mid–log (5 × 106 cells/ml), treated with 1 mM H2O2 for the time indicated in the text, and then analyzed by fluorescence microscopy. Images were obtained using a Nikon microscope (model E800) with a 60× objective (Plan Fluor Oil, NA 1.3) and a CCD camera (RETIGA Exi). Data were collected using Autoquant and processed using Image Pro software. All images were obtained using the same exposures for the course of the experiment.
Western blot analysis
Straight Western blot analysis was performed as previously described (Kushnirov, 2000) using 20 ml of mid–log phase cells per sample. For immunoprecipitations, protein extracts were prepared from mid–log phase cultures using mild RIPA buffer (150 mM NaCl, 1% NP-40, 0.15% doxycycline, 50 mM Tris-HCl, pH 8) and glass bead lysis, as described previously (Cooper et al., 1999). Protein turnover rates were determined in mid–log phase (5 × 106 cell/ml) cultures treated with cycloheximide (10 mg/l). Coimmunoprecipitation analyses were performed using 500 μg whole-cell extracts. Immunoprecipitations were conducted overnight at 4°C with agitation. Then protein complexes were bound to protein G-agarose beads (Roche, Indianapolis, IN) and processed according to the manufacturer's manual. GFP polyclonal antibody (Living Colors; Clontech, Mountain View, CA) or anti–c-myc monoclonal (9E10) antibody (Roche) was used for immunoprecipitations and Western blot analysis. The 12G10 mouse monoclonal anti-Tub1p antibody (Developmental Studies Hybridoma Bank, University of Iowa) and 3-phosphoglycerate K (Pgk1p) mouse immunoglubulin G1 (IgG1) monoclonal (22C5D8) antibody (Invitrogen) were used to detect Tub1p and Pgk1p, respectively, as loading controls in this study. Western blot signal was detected using alkaline phosphatase–conjugated goat anti-mouse IgG (H+L) or anti-rabbit IgG (H+L) (Jackson Laboratories, Bar Harbor, ME) and the CDP-Star chemiluminescence reagent (Tropix, Life Technologies, Grand Island, NY).
Supplementary Material
Acknowledgments
We thank M. Staley for help in strain construction and plasmid preparations; N. L. van Berkum (University of Massachusetts Medical School, Worcester, MA), J. Nunnari (University of California, Davis), Y. Ohya (University of Tokyo, Japan), F. Luca (University of Pennsylvania, Philadelphia), and R. J. Dohmen (University of Cologne, Germany) for strains and plasmids; S. Moye-Rowley (University of Iowa, Iowa City) for critical reading of the manuscript; and A. V. Parshin (RowanSOM) for assistance with the RasMol program to generate the cyclin C model images. This work was supported by grants from the National Institutes of Health (CA099003, GM086788) to R.S. and the W. W. Smith Charitable Trust (#CO604) to K.F.C.
Abbreviations used:
- CWI
cell wall integrity
- DAPI
4′,6-diamidino-2-phenylindole
- GFP
green fluorescent protein
- HAD
holoenzyme association domain
- IgG
immunoglubulin G
- mtDNA
mitochondrial DNA
- PCD
programmed cell death
- RFP
red fluorescent protein
- ROS
reactive oxygen species
- YFP
yellow fluorescent protein
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E14-05-0953) on July 23, 2014.
*Present address: Department of Molecular Physiology and Biophysics, University of Iowa, Iowa City, IA 52242.
REFERENCES
- Akoulitchev S, Chuikov S, Reinberg D. TFIIH is negatively regulated by cdk8-containing mediator complexes. Nature. 2000;407:102–106. doi: 10.1038/35024111. [DOI] [PubMed] [Google Scholar]
- An HJ, Cho G, Lee JO, Paik SG, Kim YS, Lee H. Higd-1a interacts with Opa1 and is required for the morphological and functional integrity of mitochondria. Proc Natl Acad Sci USA. 2013;110:13014–13019. doi: 10.1073/pnas.1307170110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ansari SA, Morse RH. Mechanisms of Mediator complex action in transcriptional activation. Cell Mol Life Sci. 2013;70:2743–2756. doi: 10.1007/s00018-013-1265-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borggrefe T, Davis R, Erdjument-Bromage H, Tempst P, Kornberg RD. A complex of the Srb8, -9, -10, and -11 transcriptional regulatory proteins from yeast. J Biol Chem. 2002;277:44202–44207. doi: 10.1074/jbc.M207195200. [DOI] [PubMed] [Google Scholar]
- Bourbon HM, Aguilera A, Ansari AZ, Asturias FJ, Berk AJ, Bjorklund S, Blackwell TK, Borggrefe T, Carey M, Carlson M, et al. A unified nomenclature for protein subunits of mediator complexes linking transcriptional regulators to RNA polymerase II. Mol Cell. 2004;14:553–557. doi: 10.1016/j.molcel.2004.05.011. [DOI] [PubMed] [Google Scholar]
- Brooks C, Cho SG, Wang CY, Yang T, Dong Z. Fragmented mitochondria are sensitized to Bax insertion and activation during apoptosis. Amer J Physiol. 2011;300:C447–455. doi: 10.1152/ajpcell.00402.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buttner S, Ruli D, Vogtle FN, Galluzzi L, Moitzi B, Eisenberg T, Kepp O, Habernig L, Carmona-Gutierrez D, Rockenfeller P, et al. A yeast BH3-only protein mediates the mitochondrial pathway of apoptosis. EMBO J. 2011;30:2779–2792. doi: 10.1038/emboj.2011.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cassidy-Stone A, Chipuk JE, Ingerman E, Song C, Yoo C, Kuwana T, Kurth MJ, Shaw JT, Hinshaw JE, Green DR, et al. Chemical inhibition of the mitochondrial division dynamin reveals its role in Bax/Bak-dependent mitochondrial outer membrane permeabilization. Dev Cell. 2008;14:193–204. doi: 10.1016/j.devcel.2007.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan DC. Fusion and fission: interlinked processes critical for mitochondrial health. Ann Rev Genet. 2012;46:265–287. doi: 10.1146/annurev-genet-110410-132529. [DOI] [PubMed] [Google Scholar]
- Chang YW, Howard SC, Herman PK. The Ras/PKA signaling pathway directly targets the Srb9 protein, a component of the general RNA polymerase II transcription apparatus. Mol Cell. 2004;15:107–116. doi: 10.1016/j.molcel.2004.05.021. [DOI] [PubMed] [Google Scholar]
- Chen H, Chomyn A, Chan DC. Disruption of fusion results in mitochondrial heterogeneity and dysfunction. J Biol Chem. 2005;280:26185–26192. doi: 10.1074/jbc.M503062200. [DOI] [PubMed] [Google Scholar]
- Chen H, Detmer SA, Ewald AJ, Griffin EE, Fraser SE, Chan DC. Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J Cell Biol. 2003;160:189–200. doi: 10.1083/jcb.200211046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conaway RC, Conaway JW. Function and regulation of the Mediator complex. Curr Opin Genet Dev. 2011;21:225–230. doi: 10.1016/j.gde.2011.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper KF, Khakhina S, Kim SK, Strich R. Stress-induced nuclear-to-cytoplasmic translocation of cyclin c promotes mitochondrial fission in yeast. Dev Cell. 2014;28:161–173. doi: 10.1016/j.devcel.2013.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper KF, Mallory MJ, Strich R. Oxidative stress-induced destruction of the yeast C-type cyclin Ume3p requires phosphatidylinositol-specific phospholipase C and the 26S proteasome. Mol Cell Biol. 1999;19:3338–3348. doi: 10.1128/mcb.19.5.3338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper KF, Scarnati MS, Krasley E, Mallory MJ, Jin C, Law MJ, Strich R. Oxidative-stress-induced nuclear to cytoplasmic relocalization is required for Not4-dependent cyclin C destruction. J Cell Sci. 2012;125:1015–1026. doi: 10.1242/jcs.096479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper KF, Strich R. Functional analysis of the Ume3p/Srb11p-RNA polymerase II holoenzyme interaction. Gene Expr. 1999;8:43–57. [PMC free article] [PubMed] [Google Scholar]
- Cooper KF, Strich R. Saccharomyces cerevisiae C-type cyclin Ume3p/Srb11p is required for efficient induction and execution of meiotic development. Eukaryot Cell. 2002;1:66–74. doi: 10.1128/EC.01.1.66-74.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis MA, Larimore EA, Fissel BM, Swanger J, Taatjes DJ, Clurman BE. The SCF-Fbw7 ubiquitin ligase degrades MED13 and MED13L and regulates CDK8 module association with Mediator. Genes Dev. 2013;27:151–156. doi: 10.1101/gad.207720.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenberg T, Buttner S, Kroemer G, Madeo F. The mitochondrial pathway in yeast apoptosis. Apoptosis. 2007;12:1011–1023. doi: 10.1007/s10495-007-0758-0. [DOI] [PubMed] [Google Scholar]
- Estruch F. Stress-controlled transcription factors, stress-induced genes and stress tolerance in budding yeast. FEMS Microbiol Rev. 2000;24:469–486. doi: 10.1111/j.1574-6976.2000.tb00551.x. [DOI] [PubMed] [Google Scholar]
- Gobert V, Osman D, Bras S, Auge B, Boube M, Bourbon HM, Horn T, Boutros M, Haenlin M, Waltzer L. A genome-wide RNA interference screen identifies a differential role of the mediator CDK8 module subunits for GATA/RUNX-activated transcription in Drosophila. Mol Cell Biol. 2010;30:2837–2848. doi: 10.1128/MCB.01625-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez D, Hamidi N, Del Sol R, Benschop JJ, Nancy T, Li C, Francis L, Tzouros M, Krijgsveld J, Holstege FC, et al. Suppression of Mediator is regulated by Cdk8-dependent Grr1 turnover of the Med3 coactivator. Proc Natl Acad Sci USA. 2014;111:2500–2505. doi: 10.1073/pnas.1307525111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermann GJ, Thatcher JW, Mills JP, Hales KG, Fuller MT, Nunnari J, Shaw JM. Mitochondrial fusion in yeast requires the transmembrane GTPase Fzo1p. J Cell Biol. 1998;143:359–373. doi: 10.1083/jcb.143.2.359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirst M, Kobor MS, Kuriakose N, Greenblatt J, Sadowski I. GAL4 is regulated by the RNA polymerase II holoenzyme-associated cyclin-dependent protein kinase SRB10/CDK8. Mol Cell. 1999;3:673–678. doi: 10.1016/s1097-2765(00)80360-3. [DOI] [PubMed] [Google Scholar]
- Hoeppner S, Baumli S, Cramer P. Structure of the mediator subunit cyclin C and its implications for CDK8 function. J Mol Biol. 2005;350:833–842. doi: 10.1016/j.jmb.2005.05.041. [DOI] [PubMed] [Google Scholar]
- Jeffrey PD, Russo AA, Polyak K, Gibbs E, Hurwitz J, Messague J, Pavletich NP. Mechanism of CDK activation revealed by the structure of cyclin A-CDK2 complex. Nature. 1995;376:3113–3320. doi: 10.1038/376313a0. [DOI] [PubMed] [Google Scholar]
- Jin C, Strich R, Cooper KF. Slt2p phosphorylation induces cyclin C nuclear-to-cytoplasmic translocation in response to oxidative stress. Mol Biol Cell. 2014;25:1396–1407. doi: 10.1091/mbc.E13-09-0550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karbowski M, Lee YJ, Gaume B, Jeong SY, Frank S, Nechushtan A, Santel A, Fuller M, Smith CL, Youle RJ. Spatial and temporal association of Bax with mitochondrial fission sites, Drp1, and Mfn2 during apoptosis. J Cell Biol. 2002;159:931–938. doi: 10.1083/jcb.200209124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knuesel MT, Meyer KD, Bernecky C, Taatjes DJ. The human CDK8 subcomplex is a molecular switch that controls Mediator coactivator function. Genes Dev. 2009a;23:439–451. doi: 10.1101/gad.1767009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knuesel MT, Meyer KD, Donner AJ, Espinosa JM, Taatjes DJ. The human CDK8 subcomplex is a histone kinase that requires Med12 for activity and can function independently of Mediator. Mol Cell Biol. 2009b;29:650–661. doi: 10.1128/MCB.00993-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krasley E, Cooper KF, Mallory MJ, Dunbrack R, Strich R. Regulation of the oxidative stress response through Slt2p-dependent destruction of cyclin C in Saccharomyces cerevisiae. Genetics. 2006;172:1477–1486. doi: 10.1534/genetics.105.052266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kushnirov VV. Rapid and reliable protein extraction from yeast. Yeast. 2000;16:857–860. doi: 10.1002/1097-0061(20000630)16:9<857::AID-YEA561>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- Levin DE. Regulation of cell wall biogenesis in Saccharomyces cerevisiae: the cell wall integrity signaling pathway. Genetics. 2011;189:1145–1175. doi: 10.1534/genetics.111.128264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longtine MS, McKenzie A, 3rd, Demarini DJ, Shah NG, Wach A, Brachat A, Philippsen P, Pringle JR. Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast. 1998;14:953–961. doi: 10.1002/(SICI)1097-0061(199807)14:10<953::AID-YEA293>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- Madeo F, Frohlich E, Frohlich KU. A yeast mutant showing diagnostic markers of early and late apoptosis. J Cell Biol. 1997;139:729–734. doi: 10.1083/jcb.139.3.729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazzoni C, Falcone C. Caspase-dependent apoptosis in yeast. Biochim Biophys Acta. 2008;1783:1320–1327. doi: 10.1016/j.bbamcr.2008.02.015. [DOI] [PubMed] [Google Scholar]
- Meyer KD, Donner AJ, Knuesel MT, York AG, Espinosa JM, Taatjes DJ. Cooperative activity of Cdk8 and GCN5L within Mediator directs tandem phosphoacetylation of histone H3. EMBO J. 2008;27:1447–1457. doi: 10.1038/emboj.2008.78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morano KA, Grant CM, Moye-Rowley WS. The response to heat shock and oxidative stress in Saccharomyces cerevisiae. Genetics. 2012;190:1157–1195. doi: 10.1534/genetics.111.128033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naylor K, Ingerman E, Okreglak V, Marino M, Hinshaw JE, Nunnari J. Mdv1 interacts with assembled Dnm1 to promote mitochondrial division. J Biol Chem. 2006;281:2177–2183. doi: 10.1074/jbc.M507943200. [DOI] [PubMed] [Google Scholar]
- Nelson C, Goto S, Lund K, Hung W, Sadowski I. Srb10/Cdk8 regulates yeast filamentous growth by phosphorylating the transcription factor Ste12. Nature. 2003;421:187–190. doi: 10.1038/nature01243. [DOI] [PubMed] [Google Scholar]
- Nemet J, Jelicic B, Rubelj I, Sopta M. The two faces of Cdk8, a positive/negative regulator of transcription. Biochimie. 2014;97:22–27. doi: 10.1016/j.biochi.2013.10.004. [DOI] [PubMed] [Google Scholar]
- Niedenthal RK, Riles L, Johnston M, Hegemann JH. Green fluorescent protein as a marker for gene expression and subcellular localization in budding yeast. Yeast. 1996;12:773–786. doi: 10.1002/(SICI)1097-0061(19960630)12:8%3C773::AID-YEA972%3E3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- Ramos PC, Hockendorff J, Johnson ES, Varshavsky A, Dohmen RJ. Ump1p is required for proper maturation of the 20S proteasome and becomes its substrate upon completion of the assembly. Cell. 1998;92:489–499. doi: 10.1016/s0092-8674(00)80942-3. [DOI] [PubMed] [Google Scholar]
- Schneider EV, Bottcher J, Blaesse M, Neumann L, Huber R, Maskos K. The structure of CDK8/CycC implicates specificity in the CDK/cyclin family and reveals interaction with a deep pocket binder. J Mol Biol. 2011;412:251–266. doi: 10.1016/j.jmb.2011.07.020. [DOI] [PubMed] [Google Scholar]
- Scott SV, Cassidy-Stone A, Meeusen SL, Nunnari J. Staying in aerobic shape: how the structural integrity of mitochondria and mitochondrial DNA is maintained. Curr Opin Cell Biol. 2003;15:482–488. doi: 10.1016/s0955-0674(03)00070-x. [DOI] [PubMed] [Google Scholar]
- Sekiya-Kawasaki M, Abe M, Saka A, Watanabe D, Kono K, Minemura-Asakawa M, Ishihara S, Watanabe T, Ohya Y. Dissection of upstream regulatory components of the Rho1p effector, 1,3-beta-glucan synthase, in Saccharomyces cerevisiae. Genetics. 2002;162:663–676. doi: 10.1093/genetics/162.2.663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sesaki H, Jensen RE. Division versus fusion: Dnm1p and Fzo1p antagonistically regulate mitochondrial shape. J Cell Biol. 1999;147:699–706. doi: 10.1083/jcb.147.4.699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sesaki H, Jensen RE. UGO1 encodes an outer membrane protein required for mitochondrial fusion. J Cell Biol. 2001;152:1123–1134. doi: 10.1083/jcb.152.6.1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shahi P, Gulshan K, Naar AM, Moye-Rowley WS. Differential roles of transcriptional mediator subunits in regulation of multidrug resistance gene expression in Saccharomyces cerevisiae. Mol Biol Cell. 2010;21:2469–2482. doi: 10.1091/mbc.E09-10-0899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheff MA, Thorn KS. Optimized cassettes for fluorescent protein tagging in Saccharomyces cerevisiae. Yeast. 2004;21:661–670. doi: 10.1002/yea.1130. [DOI] [PubMed] [Google Scholar]
- Strich R, Slater MR, Esposito RE. Identification of negative regulatory genes that govern the expression of early meiotic genes in yeast. Proc Natl Acad Sci USA. 1989;86:10018–10022. doi: 10.1073/pnas.86.24.10018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surosky RT, Strich R, Esposito RE. The yeast UME5 gene regulates the stability of meiotic mRNAs in response to glucose. Mol Cell Biol. 1994;14:3446–3458. doi: 10.1128/mcb.14.5.3446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson DH, Fennell DJ. Visualization of yeast mitochondrial DNA with the fluorescent stain “DAPI.”. Methods Enzymol. 1979;56:728–733. doi: 10.1016/0076-6879(79)56065-0. [DOI] [PubMed] [Google Scholar]
- Youle RJ, Karbowski M. Mitochondrial fission in apoptosis. Nat Rev Mol Cell Biol. 2005;6:657–663. doi: 10.1038/nrm1697. [DOI] [PubMed] [Google Scholar]
- Yuan H, Gerencser AA, Liot G, Lipton SA, Ellisman M, Perkins GA, Bossy-Wetzel E. Mitochondrial fission is an upstream and required event for bax foci formation in response to nitric oxide in cortical neurons. Cell Death Diff. 2007;14:462–471. doi: 10.1038/sj.cdd.4402046. [DOI] [PubMed] [Google Scholar]
- Zhu X, Wiren M, Sinha I, Rasmussen NN, Linder T, Holmberg S, Ekwall K, Gustafsson CM. Genome-wide occupancy profile of mediator and the Srb8-11 module reveals interactions with coding regions. Mol Cell. 2006;22:169–178. doi: 10.1016/j.molcel.2006.03.032. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.