

Abstract
This study was to evaluate patterns of gene expression and promoter methylation of DR4 from peripheral circulating blood lymphocytes of AD patients and folate-deficiency medium cultured neuroblast cells, and also expression levels of DNMT1, DNMT3a, and MECP2. Blood samples of 25 pairs of AD patients and age- and sex-matched controls were collected. SH-SY5Y cells were cultured with folate-deficiency medium. Bisulfite cloning coupled with sequencing was employed to analyze methylation levels of DR4 gene promoters, and quantitative real time PCR (qRT-PCR) was used to detect gene expression levels of DR4, and also DNA methyltransferase 1 (DNMT1), DNA methyltransferase 3a (DNMT3a) and methyl CpG binding protein 2 (MECP2). Folate concentration was calculated in serum of blood samples. 3-(4,5-di-methylthiazol-2-yl)-2,5-diphenyl tetrazolium (MTT) assay was used to analyze cell viability. The results showed that, the promoter of DR4 was hypomethylated in AD patients and cells cultured in folate-deficiency medium and had site-specific changes (P < 0.05), and these sites were mostly at or nearby some key transcription factor binding sites. Accordance with the hypomethylation, increased expression level of DR4 was observed (P < 0.05). DNMT1 and DNMT3a mRNA level were elevated (P < 0.05) in AD patients and folate deficient medium cultured cells compared with controls (P < 0.05), together with lower folate concentration in AD. MTT assay showed that folate deficiency inhibited cell growth. In summary, folate deficiency can induce apoptosis by increasing DR4 expression with DNA promoter hypomethylation in AD, together with upregulating DNMTs expression, which may be associated with folate deficiency-induced DNA damage.
Keywords: Alzheimer’s disease, DNA methylation, DR4, apoptosis, DNMT
Introduction
Alzheimer’s disease (AD) is a debilitating neurodegenerative disease, which brings heavy burden for the society. The pathology of AD is characterized by senile plaques and neurofibrillary tangles, combined with massive neuronal loss, mainly in the hippocampus and association regions of the neocortex. AD has complex etiology and pathogenesis. Its cause is not clear yet but known to encompass many genetic and environmental risk factors, inflammation, oxidative stress, energy metabolism, changes in the expression of many genes, and up regulation of multiple pathogenic pathways such as amyloid β peptide (Aβ) deposition, tau hyperphosphorylation, and aberrant cell cycle control/apoptosis [1].
The majority of cases of AD are sporadic, and likely several genetic and environmental factors contribute to their development. Reversible epigenetic alteration is expected to be a potential mechanism for explaining unsolved phenomena beyond genetic association with sporadic AD. Studies have established a link between epigenetic changes and AD pathogenesis. One of the most studied epigenetic modifications is the change of methylation patterns of CpG rich regions in the promoters of specific genes, which modifies gene expression by interfering the binding between DNA and transcription factors, and then results in gene silencing (hypermethylation) or overexpression (hypomethylation) [2].
Folate metabolism, also known as one-carbon metabolism, is required for the production of S-adenosylmethionine (SAM), which is the major DNA methylating agent. Studies have indicated that folate values were reduced in the plasma of AD individuals [3,4]. Impaired folate metabolism and subsequent reduction of SAM levels might result in epigenetic modifications of the promoters of AD-related genes leading to increased Aβ peptide production. Studies performed in mice and in neuronal cell cultures indicated that the depletion of folate, respectively from the diet or from the media, resulted in epigenetic modifications of AD-related genes, with a subsequent increased production of Presenilin 1 (PS1), beta-site APP-cleaving enzyme 1 (BACE1), and Aβ fragments [5-8].
DNA methyltransferase (Dnmt) 1, the enzyme maintenancing DNA methylation, and Dnmt 3a, who initiates DNA methylation de novo, coregulated the methylation status of genes [9]. DNMT1 and components of the methyl-CpG binding protein 2/methyl-CpG binding domain protein (MECP2/MBD2) methylation complex were significantly reduced in the entorhinal cortex of AD subjects than in controls [10]. DNMT1, 3a and 3b were differently modulated, in response to hypomethylating (folate and B vitamin deficiency) and hypermethylating (S-adenosyl-L-methionine supplementation) alterations of the one-carbon metabolism in AD models, in line with the changes of PS1 methylation pattern [11].
Efforts were made to determine aging- and AD-related genome-wide methylation pattern or module. Actually, some researchers have chosen peripheral blood as their research material, and the gene methylation pattern of peripheral blood has been successfully addressed [12,13]. It should be noted that lymphocytes may be an important neural and genetic probe in AD-related studies.
Death receptors 4 (DR4), also known as tumor necrosis factor superfamily, member 10 (TNFSF10A), is activated by tumor necrosis factor-related apoptosis inducing ligand, (TRAIL) and thus transduces cell death signal and induces cell apoptosis [14]. DR4 has been shown to mediate oligomeric Aβ-induced cerebral microvascular endothelial cell apoptosis [15]. Aberrant promoter methylation and resultant silencing of DR4 were reported in cancers, strongly attenuating TRAIL- or DR4-mediated apoptosis in cancers [16]. An inverse association between cancer and AD has been found. One possible explanation for this association is that both diseases arise via malfunction of an underlying common mechanism, which could regulate the capability of the cells of switching the cell machinery from a prone-to-death state (AD phenotype) to a prone-to-survive/grow state (cancer phenotype). Genetic polymorphisms or variance in their DNA methylation could explain such opposing effects [17,18].
To date little is known about the relevance of this epigenetic modification in AD. This work was to evaluate the patterns of gene expression and promoter methylation of DR4 from peripheral circulating blood lymphocytes of AD patients and folate-deficiency medium cultured neuroblast cells, and also expression levels of DNMT1, DNMT3a, and MECP2.
Materials and methods
Cell culture
Human SH-SY5Y neuroblastoma cells were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) without folate (Cat# D2429, Sigma, Yorba Linda, California, USA), supplemented with folate with different final folate concentrations (normal DMEM contains folate with concentrations as 4 ug/ml), complemented with 1xNon Essential amino acids, 1 mM Sodium Pyruvate, 1.5 g/L Sodium bicarbonate, and 10% fetal bovine serum and incubated in 5% CO2 at 37°C. Cultures were re-fed every second day. All assays were repeated at least three times.
Cell viability assay
Cell viability of SH-SY5Y cells was evaluated by the 3-(4,5-di-methylthiazol-2-yl)-2,5-diphenyl tetrazolium (MTT) assay. MTT was purchased from Sigma. Briefly, cells after incubation for specified times were plated in 96-well plates containing different mediums (supplemented with folate concentration 0 ug/ml, 1 ug/ml, 4 ug/ml, 8 ug/ml, and 16 ug/ml, respectively) and incubated for 24 h, and 10 μl of the MTT solution (5 mg/mL) was added to each well and went on with incubation for 4 hour at 37°C in incubator. After removing the supernatant, the cells were added with 100 uL DMSO each well for shaking for 15 min on a microtiter plate shaker. The absorbance was measured at 570 nm using a microplate reader (Thermo, MULTISKAN MK3, USA). All experiments were repeated 3 times. The viability ratio was calculated as follow: viability% = (ODnegative-ODsample) / (ODcontrol-ODblank) × 100%.
Patients and controls
All procedures were done in accordance with the Declaration of Helsinki and approved by the institutional reviewing board of the Second Hospital of Shandong University. A written consent form was obtained from every participant signed by self or caregiver. Sporadic AD patients were diagnosed and recruited based on the American Psychiatric Association “Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition, the diagnostic criteria of the DSM-IV-R” standards for dementia and National Institute of Neurological and Communicative Diseases and Stroke; Alzheimer Disease and Related Disorders Association, NINCDS-ADRDA diagnostic criteria for likely AD.
Inclusion criteria: 1) more than 60-year-old men or postmenopausal women; 2) meet the DSM-IV-R diagnosis standards of dementia; 3) meet the NINCDS-ADRDA diagnostic criteria for possible or likely AD; 4) no family history of AD or other genetic disease; 5) the subjects or their caregivers voluntarily agreed to participate in the trial and signed the informed consent form.
Exclusion criteria: 1) not greater than 60 years of age; 2) the family history of dementia or other genetic diseases; 3) patients with Hamilton Depression Scale (HAMD) ≥ 15 with obvious symptoms of depression or Hachinski Ischemic score ≥ 7 with vascular dementia; 4) patients suffering from other diseases or their control siblings suffering from other diseases; 5) the patients or their families do not agree to participate. The controls were same age, same sex, and non-dementia healthy subjects (see Table 1 for detailed information). Mini-Mental State Examination (MMSE) was calculated and fasting blood samples were intravenously drawn.
Table 1.
Characteristics of objects
| ID (AD) | Gender | Age (Years) | Time since Diagnosis | MMSE | ID (Control) | Gender | Age (Years) | |
|---|---|---|---|---|---|---|---|---|
| A1 | Male | 86 | 3 Years | 18 | C1 | Male | 86 | 28 |
| A2 | Male | 84 | 2 Years | 11 | C2 | Male | 84 | 28 |
| A3 | Male | 70 | 1 Year | 13 | C3 | Male | 70 | 30 |
| A4 | Female | 79 | 3 Years | 14 | C4 | Female | 79 | 29 |
| A5 | Female | 76 | 4 Years | 3 | C5 | Female | 76 | 30 |
| A6 | Female | 82 | 1 Year | 3 | C6 | Female | 82 | 28 |
| A7 | Female | 75 | 2 Years | 9 | C7 | Female | 75 | 30 |
| A8 | Female | 69 | 4 Years | 7 | C8 | Female | 69 | 30 |
| A9 | Male | 72 | 4 Years | 13 | C9 | Male | 72 | 30 |
| A10 | Female | 66 | 8 Years | 7 | C10 | Female | 66 | 30 |
| A11 | Male | 64 | 10 Months | 2 | C11 | Male | 64 | 30 |
| A12 | Male | 72 | 3 Years | 6 | C12 | Male | 72 | 30 |
| A13 | Female | 80 | 3 Years | 9 | C13 | Female | 80 | 28 |
| A14 | Female | 77 | 1 Year | 12 | C14 | Female | 77 | 28 |
| A15 | Female | 90 | 1 Year | 15 | C15 | Female | 90 | 27 |
| A16 | Male | 62 | 2 Years | 5 | C16 | Male | 62 | 30 |
| A17 | Female | 63 | 4 Years | 13 | C17 | Female | 63 | 30 |
| A18 | Male | 71 | 7 Years | 8 | C18 | Male | 71 | 30 |
| A19 | Female | 78 | 5 Years | 10 | C19 | Female | 78 | 29 |
| A20 | Female | 86 | 3 Years | 3 | C20 | Female | 86 | 27 |
| A21 | Female | 62 | 7 Months | 2 | C21 | Female | 62 | 30 |
| A22 | Female | 61 | 4 Months | 10 | C22 | Female | 61 | 30 |
| A23 | Female | 85 | 1 Year | 16 | C23 | Female | 85 | 27 |
| A24 | Female | 86 | 3 Years | 5 | C24 | Female | 86 | 27 |
| A25 | Male | 90 | 4 Years | 2 | C25 | Male | 90 | 27 |
Bisulfite sequencing PCR
Cell cultures were stopped after 144 h for methylation assays. Genomic DNA from blood lymphocytes and SH-SY5Y cells were extracted using Qiagen (Hilden, Mettmann, Germany) DNeasy Blood & Tissue Kit (Cat# 69504) according to manufacturer’s instruction. About 2 μg DNA was transferred into an Eppendorf tube and diluted to 50 μL with ddH2O, then mixed with 5.5 μL of freshly prepared 3 M NaOH and incubated at 42°C for 30 min before mixed with 30 μL of 10 mM hydroquinone and 520 μL 0f 3.6 M sodium bisulfate and incubated in dark at 50°C for 16 hours. Treated DNA was purified with kit from Sangon Biotech (Shanghai, China, Cat# SK1261). Briefly, 300 μL of binding solution (6 M guanidine hydrochloride) was mixed with reaction mixture and transferred into UNIQ column. Stand at room temperature for 2 min before centrifugation at 8000 rpm for 1 min. Wash twice with 650 μL of washing buffer with an additional centrifugation at 12000 rpm for 1 min. Then DNA was eluted with 50 μL ddH2O.
We got the gene sequence in the Ensembl database online (http://www.ensembl.org). The CpG islands of the promoter of DR4, 600 bp upstream of transcription start site, were predicted with the MethPrimer software online (http://www.urogene.org/cgi-bin/methprimer), with the indexes set as, criteria used: island size > 100, GC Percent > 50.0, Obs/Exp > 0.6. Two CpG islands were found in the promoter sequence. Island 1 is from 155 bp to 334 bp with the size of 180 bp, including 9 CpG sites. And Island 2 is from 360 bp to 541 bp with the size of 182 bp with 13 CpG sites (Figure 1A, 1B). Target sequences including these two islands were sequenced.
Figure 1.
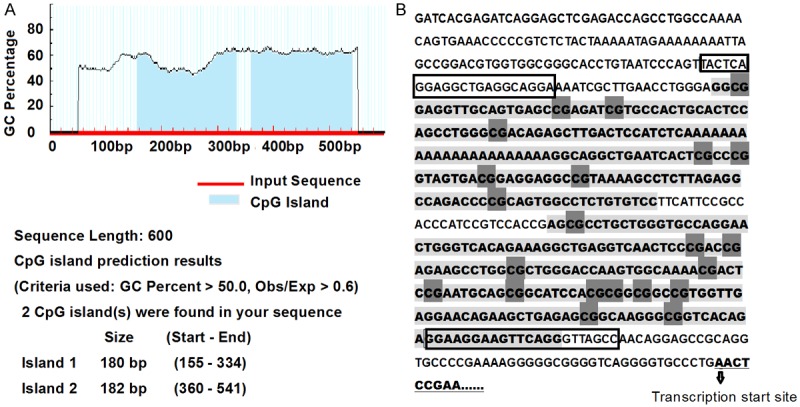
The CpG islands prediction and sequence of the promoter of DR4. A. Two CpG islands were predicted with the MethPrimer software in the promoter of DR4; B. Sequence of the promoter of DR4. Light gray shaded sequence: CpG islands; Dark gray shaded: CpG sites; Framed: primers binding sites.
The sequences were PCR amplified with gene-specific primer pairs (F: TATTTAGGAGGTTGAGGTAGGA; R: AACTAACCCTAAACTTCCTTCC, Product 435 bp) using bisulfite-treated genomic DNA as templates.
PCR products were cleared and cloned into PUCm-T vector and sequenced with M13 primers. Five clones of each sample were sequenced.
Transcription factor binding sites searching
The TFSEARCH: Searching Transcription Factor Binding Sites (ver 1.3) software online (http://www.cbrc.jp/research/db/TFSEARCH.html) was used to search transcription factor binding sites in the target fragments including the CpG sites, searching highly correlated sequence fragments versus TFMATRIX transcription factor binding site profile database. The threshold was set as 80.0 point.
Reverse transcription and quantitative real-time PCR (qRT-PCR)
Cell cultures were stopped after 168 h for gene expression analyses. Total RNA from blood samples and cells were purified with RNeasy Mini Kit (Qiagen, Cat# 74104). Reverse transcription (RT) was performed in a total volume of 20 μL using PrimeScriptTM RT reagent Kit With gDNA Eraser (TaKaRa Bio.Inc., Dalian, China). 2.0 μl of 5 × gDNA Eraser Buffer, 1.0 μL of gDNA Eraser, total RNA (1.0 μg) were combined in a PCR reaction tube, and RNase-free water was added to make up the final volume to 10 μL. The RNA was denatured at 42°C for 2 min and then at 4°C for 30 sec. And then 4.0 μL of 5 × PrimeScript Buffer 2 (for real time), 1.0 μl of PrimeScript RT Enzyme Mix I and 1.0 μl of RT Primer Mix were added, and RNase-free water was added to make up the final volume to 20 μL. The mixture was incubated at 37°C for 15 min, 85°C for 5 sec and then at 4°C for 30 sec.
The expression levels of DR4, DNMT1, DNMT3a and MECP2 were analyzed by quantitative real-time PCR (see Table 2 for primer sequences) on an ABI Prism 7500 HT sequence detection system (Applied Biosystems, Foster, California, USA). The primers were synthesized by TaKaRa Bio.Inc. Each reaction contained 5 μL of the 2 × SYBR® Premix Ex Taq™ II.
Table 2.
Oligonucleotide sequences for qRT-PCR
| Gene | Primer sequence |
|---|---|
| β-Actin | F: TGCGCAGAAAACAAGATGAG |
| R: CACCTTCACCGTTCCAGTTT | |
| DR4 | F: CATGTCAGTGCAAACCAGGAA |
| R: CGATGTCACTCCAGGGCGTA | |
| DNMT1 | F: GAGCTACCACGCAGACATCA |
| R: CGAGGAAGTAGAAGCGGTTG | |
| DNMT3a | F: CCGGAACATTGAGGACATCT |
| R: CAGCAGATGGTGCAGTAGGA | |
| MeCP2 | F: CCCCACCCTGCCTGAA |
| R: GATGTGTCGCCTACCTTTTCG |
0.4 μL of 10 μmol/L each primer pair, 2 μL of template cDNA from the RT reaction and RNase-free water was added to make up the final volume to 10 μL. PCR conditions included a denaturing cycle at 95°C for 30 s, then followed by 45 cycles of 95°C for 5 s, 60°C for 30 s. Melting analysis was performed as 95°C for 15 s, 60°C for 30 s, and then 95°C for 15 s. After the reaction, PCR products were visualized on a 1.5% agarose-ethidium bromide gel under UV light.
Folate concentration of blood samples
Serum was separated from each blood sample immediately after drawn, and serum folate concentration was measured by photochemical method on automated chemiluminescence immunoassay system (Beckman Access, Fullerton, California, USA).
Statistical analysis
The data were analyzed using the SPSS statistical package (SPSS, version 13.0). Paired t-test was used to compare the clinical data to identify sites with statistically significant difference between AD patients and paired normal controls. For the data of cultured cells, t-test was used. Values presented are means±se. P-value was required to be ≤ 0.05 to be considered significant. Asterisks in figures evidence the statistically significant differences; differences lacking of remarks are to be considered nonsignificant
Results
Folate deficiency inhibits the growth of SH-SY5Y cells
We examined the effect of folate deficiency on the viability of SH-SY5Y cells by MTT assay, and we observed both folate deficiency and folate overdose inhibited the growth of SH-SY5Y cells. As shown in Figure 2A, 2B, cell viability was significantly decreased cultured in medium added with folate 0 μg/ml, 1 μg/ml, and 16 μg/ml for 144 h than that in medium with folate 4 μg/ml, and cell growth was remarkably inhibited in medium added with folate 0 μg/ml after treatment for 144 h. It is suggested that both folate deficiency and overdose inhibit cell growth as compared with the control, and folate deficiency induced inhibition of cell growth is time-dependently.
Figure 2.
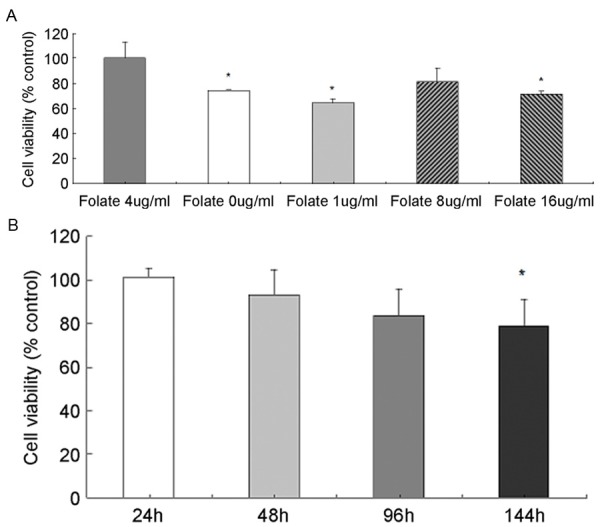
Cell viability examination by MTT assay in SH-SY5Y cells, which was administrated various concentration of folate for 144 h (A) and folate 0 ug/ml for various times (B). *P < 0.05, N-3.
So we set cells cultured in medium with folate concentration 0 ug/ml as folate-deficiency group (folate-), and 4 μg/ml as normal group for next methylation assays (cultured after 144 h) and gene expression analyses (cultured after 168 h).
DNA hypomethylation of the promoter of DR4 in cells cultured with folate deficient medium
Two CpG islands located between -445 to -266 bp (numbered from the first base of exon 1) and -240 to -59 bp upstream of transcription start site of the promoter of DR4 were analyzed, and there were 9 CpG sites in CpG island 1, and 13 sites in CpG island 2. (Figure 1).
Figure 3 shows the DNA methylation status of DR4 promoter in cultured cells. These data evidence that, cells cultured with folate deficient medium showed a trend of hypomethylation than that in control medium (Figure 3A and 3B).
Figure 3.
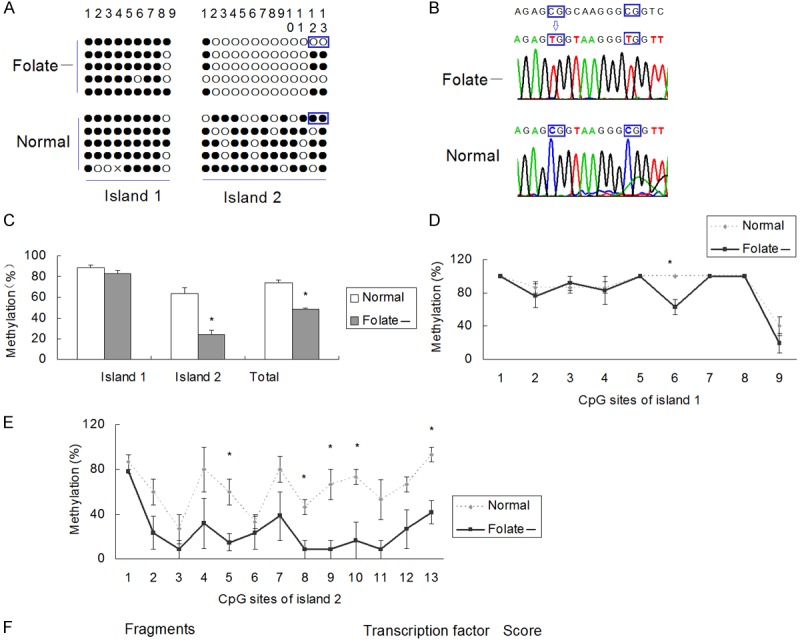
DR4 promoter methylation in SH-SY5Y cell lines cultured in different mediums. A. DNA methylation assay results of DR4 promoter. The sequences including the framed CpG sites were shown in Figure 2B. Solid circles indicate methylated CpG sites, and hollow circle nonmethylated. “Folate-” labels indicate cells cultured in medium supplemented with folate concentration 0 μg/ml, and “normal” labels folate concentration 4 μg/ml; B. Sequences of the 12th and 13th CpG sites of CpG island 2. They are both unmethylated in folate deficient cells and methylated in normal cells; C. Overall methylation % of CpG sites in CpG island 1, CpG island 2, and both islands in the promoter of DR4. *P < 0.05; **P < 0.01; N=3; D. CpG methylation pattern of each CpG site of DR4 promoter in CpG island 1. The methylation rate of the 6th CpG site is lower in folate deficient cells than that in normal cells (P < 0.05); E. CpG methylation pattern of each CpG site of DR4 promoter in CpG island 2. The methylation rates of the 5th, 8th, 9th,10th, and 13th CpG site are lower in folate deficient cells than those in normal cells (P < 0.05); F. Results of search transcription factor binding sites using TFSEARCH software in fragments including target CpG sites.
Total methylation rate was 48.33±1.97% in folate deficient cells and 73.94±3.49% in normal controls (P < 0.05). The first CpG island was methylated in samples with a methylation rate of 88.89±3.39% in folate deficient cells and 82.96±5.19% in normal controls (P > 0.05), while the second CpG island with a methylation rate of 24.10±6.55% in folate deficient cells and 63.59±5.19% in normal controls (P < 0.05) (Figure 3C).
Analysis of methylation status of each CpG site evidences some important differences in spatial methylation pattern in DR4 promoter. The methylation rates of the 6th CpG site of the first island (Figure 3D), and also the 8th, 9th,10th and 13th CpG sites of the second island (Figure 3E) were lower in the folate deficient cells than those in normal cells (P < 0.05).
And then we used the TFSEARCH software online to search transcription factor binding sites in those fragments including those CpG sites respectively. We found that the fragment including the 8th, 9th and 10th CpG site of the second island was the binding sites of transcription factor GATA1, GATA2, v-yes-1 Yamaguchi sarcoma viral oncogene homolog pseudogene (SYR), upstream transcription factor (USF), v-myb avian myeloblastosis viral oncogene homolog (v-Myb) and v-myc avian myelocytomatosis viral oncogene neuroblastoma derived homolog (N-myc), and the fragment including the 13th CpG site of the second island was the binding sites of cellular E26 transformation-specific (c-Ets), and Ether-a-go-go-like potassium 1 (Elk1).
DNA hypomethylation of the promoter of DR4 in AD patients
Blood plasma samples from 25 AD patients and 25 age- and sex-matched elderly normal controls were collected. The mean age was 75.44±9.10 years, and the male-to-female ratio was 1:1.8 in both groups. Time since diagnosis of AD patients 3.11±2.36 years. MMSE was 8.64±4.71 in AD patients and 28.9±1.26 in normal controls (P < 0.05).
Figure 4 shows the DNA methylation status of DR4 promoter in AD patients and controls. Figure 4A showed the DNA methylation status of A1 and C1. Figure 4B shows the sequence including the 1st and 2nd CpG sites of island 1.
Figure 4.
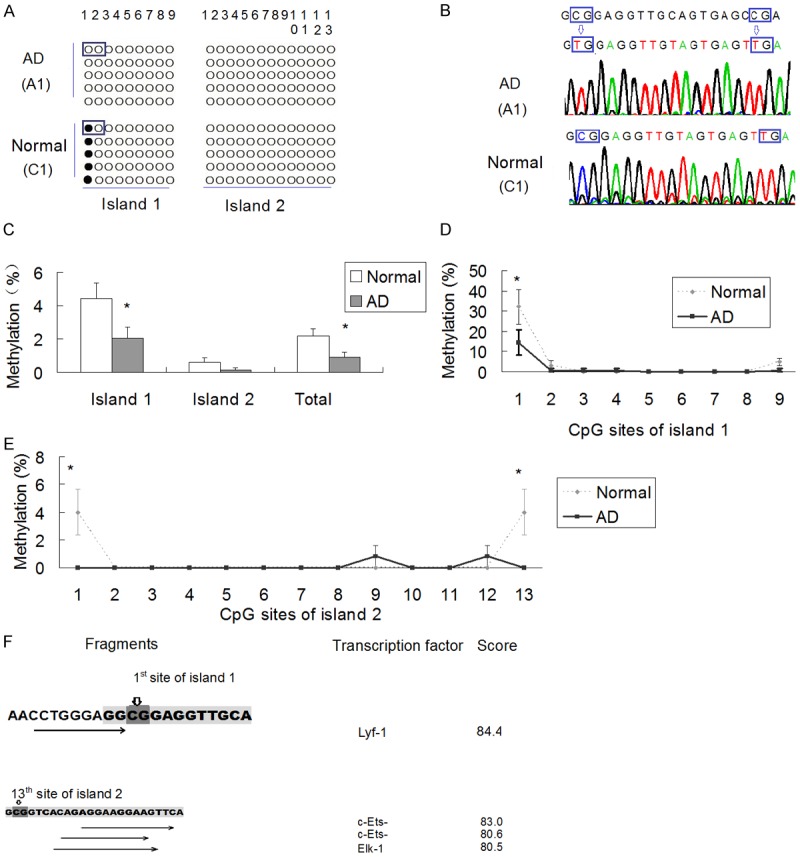
DR4 promoter methylation in AD patients and normal controls. A. DNA methylation assay results of DR4 promoter of AD patient (A1) and control (C1). The sequences of framed CpG sites are shown in Figure 3B; B. Sequences of the 1st and 2nd CpG sites of CpG island 1. The 1st site is unmethylated in A1 and methylated in C1, and the 2nd site is unmethylated in both subjects; C. Overall methylation % of CpG sites in CpG island 1, CpG island 2, and both islands in the promoter of DR4. *P < 0.05; N=25; D. CpG methylation pattern of each CpG site of DR4 promoter in CpG island 1. The methylation rates of the 1st CpG site is lower in AD patients than that in controls (P < 0.05); E. CpG methylation pattern of each CpG site of DR4 promoter in CpG island 2. The methylation rates of the 1st and 13th CpG site of the second island are lower in AD patients than those in controls (P < 0.05); F. TFSEARCH results of the fragment including the 1st and 13th CpG site of the second island.
The methylation rate was lower in AD patients than that in normal controls (0.87±0.30% vs. 2.18±0.41%, P < 0.05) in total CpG sites, and it was also lower in the first CpG island (methylation rate 2.04±0.71% in AD patients and 4.44±0.93% in normal controls, P < 0.05). However, no significant difference of methylation rate was found in the second CpG island (0.13±0.12% vs. 0.62±0.25%, P > 0.05) (Figure 4C).
Analysis of methylation status of each CpG site evidences that he methylation rates of the 1st CpG site of the first island, (Figure 4D) and also the 1st and 13th CpG site of the second island (Figure 4E) were lower in AD patients than those in normal controls (P < 0.05).
And TFSEARCH software showed that the fragment including the 1st CpG site of the second island was nearby the binding sites of transcription factor Lymphoid transcription factor 1 (Lyf1), and 13th CpG site was nearby the binding sites of c-Ets, and Elk1 (Figure 4F).
Overexpression of DR4 in cells cutured with folate deficient medium and AD patients and by qRT-PCR
Gene expression analyses by qRT-PCR evidenced the overexpression of DR4 in AD patients and folate-deficiency cells. Figure 5A shows DR4 expression in human SH-SY5Y cells cultured in different conditions, DR4 expression level significantly increased in folate deficient cells comparing to that of normal controls (P < 0.01). The expression level of DR4 in AD patients was about 1.5 folds higher than that of controls (P < 0.05) (Figure 5B). Folate concentration in AD patients was 8.92±1.12 ng/ml, and 14.09±1.11 ng/ml in controls (P < 0.01) (Figure 5C).
Figure 5.
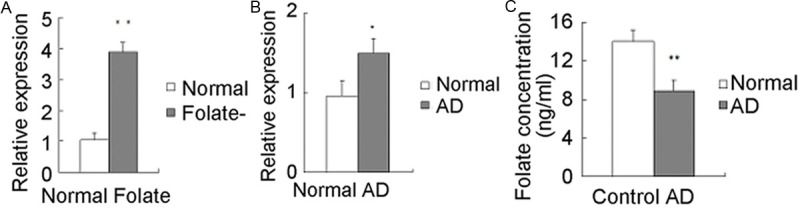
DR4 expression. qRT-PCR analyses of DR4 mRNA expression in clinical subjects (A) and cell cultures (B), and folate concentrations in clinical subjects (C). *P < 0.05; cells, N=3; clinical subjects, N=25.
DNMT1 and DNMT3a were upregulated in cells cultured with folate deficient medium and AD patients
We used qRT-PCR analysis to quantify the expression levels of genes that encode DNMT1, DNMT3a, and MECP2. Expression of DNMT1 and DNMT3a both increased in cells cultured with folate deficient medium (Figure 6A) and also in AD patients (Figure 6B) (P < 0.05); while expression of MECP2 showed no differences neither in cells cultured with folate deficient medium (Figure 6A), nor in AD patients (Figure 6B) (P > 0.05).
Figure 6.
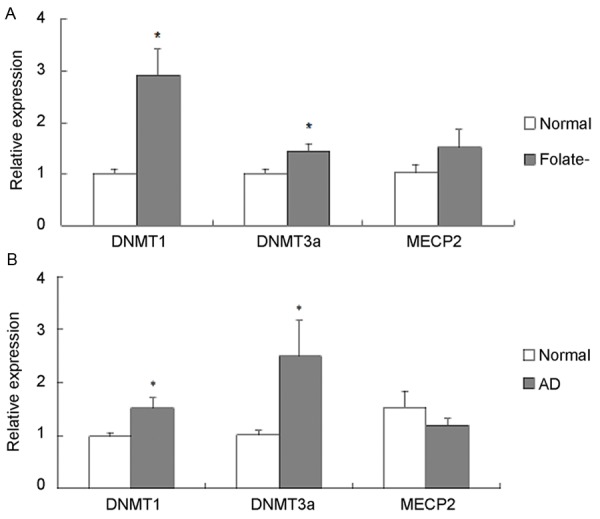
DNMT1, DNMT3a, and MECP2 expressions by qRT-PCR analyses. DNMT1, DNMT3a, and MECP2 mRNA expression in clinical subjects (A) and cell cultures (B). *P < 0.05; cells, N=3; clinical subjects, N=25.
Discussion
AD is an age-related progressive neurodegenerative disease resulting in dementia and death. The vast majority of AD is late-onset forms (LOAD) likely due to the interplay of environmental influences and individual genetic susceptibility. Epigenetic mechanisms, including DNA methylation, histone modifications and non-coding RNAs, constitute dynamic intracellular processes for translating environmental stimuli into modifications in gene expression [19]. The change of DNA methylation status of major AD risk genes has been detected in AD patients and aged individuals. There are reports on the relationship between AD and the expression and promoter methylation status of AD-risk genes [20].
Increasing evidence supports interplay between one-carbon metabolism and epigenetic modifications in the onset of AD. Studies in rodents suggested that dietary restriction of folate and other B vitamins resulted in epigenetic modifications of AD related genes in the animal brains [21]. Similarly, studies performed on human neuronal cell cultures revealed that folate and other B vitamins deprivation from the media resulted in epigenetic modification of the AD-risk genes [6]. There have been efforts to develop a blood AD biomarker panel based on DNA methylation [10,22] and transcriptional change [23,24].
DR4 is a member of the TNF-receptor superfamily, transducing cell death signal and induces cell apoptosis [14]. Evidence from literature indicates that the DNA methylation-mediated down-regulation of DR4 could be a significant mechanism through which tumor cells avoid apoptosis [9].
AD is a degenerative disease characterized by massive neuronal loss in pathology, caused by abnormal expression of apoptosis-related genes. The apoptosis of neurons was thought to be nonreversible, and this made AD difficult to be cured [1]. As known to us, apoptosis can be induced by folate deficiency [25]. Methylation modification of some apoptosis-related genes in AD may present some clues for the treatment. DR4 has been shown to mediate oligomeric Aβ-induced cerebral cell apoptosis [15]. In present study, methylation patterns and gene expression level of DR4 in leucocytes of AD patients and cells cultured in folate deficient medium were investigated. We found that, in leucocytes of AD patients and folate deficient cells, DR4 was hypomethylated, and qRT-PCR analysis detected significant increase of DR4 expression in AD blood samples and also folate deficient cells, accordance with the excessive apoptosis in AD. To our knowledge data presented here about the role of methylation and expression of DR4 in AD is the first who have dealt with this topic.
DNA methylation is initiated by de novo Dnmt3a and 3b, which is propagated in the newly replicated DNA strand by the maintenance Dnmt1 [26,27]. Altered levels of Dnmts can cause hyper- and hypo-methylation, which are associated with downregulating and reactivating gene expression, respectively [28]. The increased Dnmt-1 activity is associated with genome-wide hypomethylation and regional hypermethylation in cytosine guanine dinucleotide islands of some disease risk genes [29]. DNMT1 and DNMT3a have been shown to be required for the methylation-induced silencing of genes of members of the TNF-receptor superfamily, death decoy receptors (DcR) 1 and 2 [9].
DNMTs have been shown to be modulated by folate. However, the association between DNMTs and folate status is controversial. Some studies demonstrated that folate was associated with DNMT activation. Mandatory fortification with folic acid was associated with increased expression of Dnmt1 in the cervix [29]. Folate could decrease DNMTs activity and induce proliferation of neural stem cells [30]. Folate supplementation could increase DNMT1 expression in jejunum of newborn intrauterine growth retarded piglets [31]. And folate deficiency downregulated DNMT3a and DNMT3b in cultured SK-N-BE cells [11]. However, other studies observed that Dnmt1 and Dnmt3a were upregulated in rats fed with folate- and methyl-deficient diet [32]. And the levels of DNMT1 and DNMT3a were upregulated by dietary folate deficiency in mice during early pregnancy [33].
Unexpectedly perhaps, more recent observations strongly suggest that DNMTs, particular DNMT1, are directly involved in DNA damage repair systems via what is likely to be a DNA-methylation-independent mechanism [34-38]. Dnmt-1 has been shown to recognize and binds with high affinity to DNA lesions such as base mispairs, uracil, and other unusual DNA conformations [39,40]. And those DNA lesions are thought to be present in AD. There have been many studies on DNA damage and its role in AD. DNA damage provokes apoptotic cell death in terminally differentiated cells such as neurons [41]. Disturbance in DNA damage and repair leads to the accumulation of DNA damage leading to the deleterious effects like neurodegeneration in AD [42]. The DNA damage can be caused by folate deficiency [43,44]. Some studies showed that folate deficiency exacerbated DNA damage induced by Aβ [45], and impaired DNA repair in neurons, which sensitized them to oxidative damage induced by Aβ [46]. Biomarkers of DNA damage were also found to be increased in the leukocytes of AD patients [47,48].
Present study demonstrated that DNMT1 and DNMT3a were upregulated in AD patients, with lower folate concentration. And in cells cultured with folate-deficient medium, we found that DNMT1 and DNMT3a expression levels also increased. We suppose that, elevated DNMTs may be associated with DNA lesion induced by folate deficiency.
In conclusion, reversible epigenetic alteration is expected to be a potential mechanism for explaining unsolved phenomena beyond genetic association with sporadic AD. Our presented results showed that DNA methylation levels of DR4 in AD blood samples and also cells cultured with folate-deficient medium changed in a site-specific manner. The hypomethylation and upregulated expression of DR4, together with decreased folate concentration and elevated DNMTs expression levels, exacerbate the process of neurodegeneration in AD.
Acknowledgements
This study is supported by Youth Fund of the 2nd Hospital of Shandong University (Y2013010022), National Natural Science Fund (81171214 and 81371420), Independent Innovation Foundation of Shandong University, IIFSDU (2010JC016), Science and Technology Development Plan Project of Shandong Province (2011GSF11811), and Shandong Provincial Natural Science Foundation (ZR2010HL039).
Disclosure of conflict of interest
None.
References
- 1.Elias-Sonnenschein LS, Bertram L, Visser PJ. Relationship between genetic risk factors and markers for Alzheimer’s disease pathology. Biomark Med. 2012;6:477–495. doi: 10.2217/bmm.12.56. [DOI] [PubMed] [Google Scholar]
- 2.Zawia NH, Lahiri DK, Cardozo-Pelaez F. Epigenetics, oxidative stress, and Alzheimer disease. Free Radic Biol Med. 2009;46:1241–1249. doi: 10.1016/j.freeradbiomed.2009.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Koseoglu E, Karaman Y. Relations between homocysteine, folate and vitamin B12 in vascular dementia and in Alzheimer disease. Clin Biochem. 2007;40:859–863. doi: 10.1016/j.clinbiochem.2007.04.007. [DOI] [PubMed] [Google Scholar]
- 4.Balk EM, Raman G, Tatsioni A, Chung M, Lau J, Rosenberg IH. Vitamin B6, B12, and folic acid supplementation and cognitive function: a systematic review of randomized trials. Arch Intern Med. 2007;167:21–30. doi: 10.1001/archinte.167.1.21. [DOI] [PubMed] [Google Scholar]
- 5.Scarpa S, Fuso A, D’Anselmi F, Cavallaro RA. Presenilin 1 gene silencing by S-adenosylmethionine: a treatment for Alzheimer disease? FEBS Lett. 2003;541:145–148. doi: 10.1016/s0014-5793(03)00277-1. [DOI] [PubMed] [Google Scholar]
- 6.Fuso A, Seminara L, Cavallaro RA, D’Anselmi F, Scarpa S. S-adenosylmethionine/homocysteine cycle alterations modify DNA methylation status with consequent deregulation of PS1 and BACE and beta-amyloid production. Mol Cell Neurosci. 2005;28:195–204. doi: 10.1016/j.mcn.2004.09.007. [DOI] [PubMed] [Google Scholar]
- 7.Lin HC, Hsieh HM, Chen YH, Hu ML. S-Adenosylhomocysteine increases beta-amyloid formation in BV-2 microglial cells by increased expressions of beta-amyloid precursor protein and presenilin 1 and by hypomethylation of these gene promoters. Neurotoxicology. 2009;30:622–627. doi: 10.1016/j.neuro.2009.03.011. [DOI] [PubMed] [Google Scholar]
- 8.Chen KL, Wang SS, Yang YY, Yuan RY, Chen RM, Hu CJ. The epigenetic effects of amyloid-beta (1-40) on global DNA and neprilysin genes in murine cerebral endothelial cells. Biochem Biophys Res Commun. 2009;378:57–61. doi: 10.1016/j.bbrc.2008.10.173. [DOI] [PubMed] [Google Scholar]
- 9.Venza M, Visalli M, Catalano T, Fortunato C, Oteri R, Teti D, Venza I. Impact of DNA methyltransferases on the epigenetic regulation of tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) receptor expression in malignant melanoma. Biochem Biophys Res Commun. 2013;441:743–750. doi: 10.1016/j.bbrc.2013.10.114. [DOI] [PubMed] [Google Scholar]
- 10.Mastroeni D, Grover A, Delvaux E, Whiteside C, Coleman PD, Rogers J. Epigenetic changes in Alzheimer’s disease: decrements in DNA methylation. Neurobiol Aging. 2010;31:2025–2037. doi: 10.1016/j.neurobiolaging.2008.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fuso A, Nicolia V, Cavallaro RA, Scarpa S. DNA methylase and demethylase activities are modulated by one-carbon metabolism in Alzheimer’s disease models. J Nutr Biochem. 2011;22:242–251. doi: 10.1016/j.jnutbio.2010.01.010. [DOI] [PubMed] [Google Scholar]
- 12.Bakulski KM, Dolinoy DC, Sartor MA, Paulson HL, Konen JR, Lieberman AP, Albin RL, Hu H, Rozek LS. Genome-wide DNA methylation differences between late-onset Alzheimer’s disease and cognitively normal controls in human frontal cortex. J Alzheimers Dis. 2012;29:571–588. doi: 10.3233/JAD-2012-111223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang SC, Oelze B, Schumacher A. Age-specific epigenetic drift in late-onset Alzheimer’s disease. PLoS One. 2008;3:e2698. doi: 10.1371/journal.pone.0002698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Andera L. Signaling activated by the death receptors of the TNFR family. Biomed Pap Med Fac Univ Palacky Olomouc Czech Repub. 2009;153:173–180. doi: 10.5507/bp.2009.029. [DOI] [PubMed] [Google Scholar]
- 15.Fossati S, Ghiso J, Rostagno A. TRAIL death receptors DR4 and DR5 mediate cerebral microvascular endothelial cell apoptosis induced by oligomeric Alzheimer’s Abeta. Cell Death Dis. 2012;3:e321. doi: 10.1038/cddis.2012.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Carvalho JR, Filipe L, Costa VL, Ribeiro FR, Martins AT, Teixeira MR, Jeronimo C, Henrique R. Detailed analysis of expression and promoter methylation status of apoptosis-related genes in prostate cancer. Apoptosis. 2010;15:956–965. doi: 10.1007/s10495-010-0508-6. [DOI] [PubMed] [Google Scholar]
- 17.Behrens MI, Lendon C, Roe CM. A common biological mechanism in cancer and Alzheimer’s disease? Curr Alzheimer Res. 2009;6:196–204. doi: 10.2174/156720509788486608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tremolizzo L, Rodriguez-Menendez V, Brighina L, Ferrarese C. Is the inverse association between Alzheimer’s disease and cancer the result of a different propensity to methylate DNA? Med Hypotheses. 2006;66:1251–1252. doi: 10.1016/j.mehy.2005.12.022. [DOI] [PubMed] [Google Scholar]
- 19.Mastroeni D, Grover A, Delvaux E, Whiteside C, Coleman PD, Rogers J. Epigenetic mechanisms in Alzheimer’s disease. Neurobiol Aging. 2011;32:1161–1180. doi: 10.1016/j.neurobiolaging.2010.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.van Eijk KR, de Jong S, Boks MP, Langeveld T, Colas F, Veldink JH, de Kovel CG, Janson E, Strengman E, Langfelder P, Kahn RS, van den Berg LH, Horvath S, Ophoff RA. Genetic analysis of DNA methylation and gene expression levels in whole blood of healthy human subjects. BMC Genomics. 2012;13:636. doi: 10.1186/1471-2164-13-636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fuso A, Nicolia V, Cavallaro RA, Ricceri L, D’Anselmi F, Coluccia P, Calamandrei G, Scarpa S. B-vitamin deprivation induces hyperhomocysteinemia and brain S-adenosylhomocysteine, depletes brain S-adenosylmethionine, and enhances PS1 and BACE expression and amyloid-beta deposition in mice. Mol Cell Neurosci. 2008;37:731–746. doi: 10.1016/j.mcn.2007.12.018. [DOI] [PubMed] [Google Scholar]
- 22.Horvath S, Zhang Y, Langfelder P, Kahn RS, Boks MP, van Eijk K, van den Berg LH, Ophoff RA. Aging effects on DNA methylation modules in human brain and blood tissue. Genome Biol. 2012;13:R97. doi: 10.1186/gb-2012-13-10-r97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Booij BB, Lindahl T, Wetterberg P, Skaane NV, Saebo S, Feten G, Rye PD, Kristiansen LI, Hagen N, Jensen M, Bardsen K, Winblad B, Sharma P, Lonneborg A. A gene expression pattern in blood for the early detection of Alzheimer’s disease. J Alzheimers Dis. 2011;23:109–119. doi: 10.3233/JAD-2010-101518. [DOI] [PubMed] [Google Scholar]
- 24.Rye PD, Booij BB, Grave G, Lindahl T, Kristiansen L, Andersen HM, Horndalsveen PO, Nygaard HA, Naik M, Hoprekstad D, Wetterberg P, Nilsson C, Aarsland D, Sharma P, Lonneborg A. A novel blood test for the early detection of Alzheimer’s disease. J Alzheimers Dis. 2011;23:121–129. doi: 10.3233/JAD-2010-101521. [DOI] [PubMed] [Google Scholar]
- 25.Mattson MP, Shea TB. Folate and homocysteine metabolism in neural plasticity and neurodegenerative disorders. Trends Neurosci. 2003;26:137–146. doi: 10.1016/S0166-2236(03)00032-8. [DOI] [PubMed] [Google Scholar]
- 26.Chen T, Ueda Y, Dodge JE, Wang Z, Li E. Establishment and maintenance of genomic methylation patterns in mouse embryonic stem cells by Dnmt3a and Dnmt3b. Mol Cell Biol. 2003;23:5594–5605. doi: 10.1128/MCB.23.16.5594-5605.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hsieh CL. The de novo methylation activity of Dnmt3a is distinctly different than that of Dnmt1. BMC Biochem. 2005;6:6. doi: 10.1186/1471-2091-6-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Morey Kinney SR, Smiraglia DJ, James SR, Moser MT, Foster BA, Karpf AR. Stage-specific alterations of DNA methyltransferase expression, DNA hypermethylation, and DNA hypomethylation during prostate cancer progression in the transgenic adenocarcinoma of mouse prostate model. Mol Cancer Res. 2008;6:1365–1374. doi: 10.1158/1541-7786.MCR-08-0040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Piyathilake CJ, Celedonio JE, Macaluso M, Bell WC, Azrad M, Grizzle WE. Mandatory fortification with folic acid in the United States is associated with increased expression of DNA methyltransferase-1 in the cervix. Nutrition. 2008;24:94–99. doi: 10.1016/j.nut.2007.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li W, Yu M, Luo S, Liu H, Gao Y, Wilson JX, Huang G. DNA methyltransferase mediates dose-dependent stimulation of neural stem cell proliferation by folate. J Nutr Biochem. 2013;24:1295–1301. doi: 10.1016/j.jnutbio.2012.11.001. [DOI] [PubMed] [Google Scholar]
- 31.Liu J, Chen D, Mao X, Yu B. Effects of maternal folic acid supplementation on morphology and apoptosis-related gene expression in jejunum of newborn intrauterine growth retarded piglets. Arch Anim Nutr. 2011;65:376–385. doi: 10.1080/1745039x.2011.594352. [DOI] [PubMed] [Google Scholar]
- 32.Ghoshal K, Li X, Datta J, Bai S, Pogribny I, Pogribny M, Huang Y, Young D, Jacob ST. A folate- and methyl-deficient diet alters the expression of DNA methyltransferases and methyl CpG binding proteins involved in epigenetic gene silencing in livers of F344 rats. J Nutr. 2006;136:1522–1527. doi: 10.1093/jn/136.6.1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ding YB, He JL, Liu XQ, Chen XM, Long CL, Wang YX. Expression of DNA methyltransferases in the mouse uterus during early pregnancy and susceptibility to dietary folate deficiency. Reproduction. 2012;144:91–100. doi: 10.1530/REP-12-0006. [DOI] [PubMed] [Google Scholar]
- 34.Milutinovic S, Zhuang Q, Niveleau A, Szyf M. Epigenomic stress response. Knockdown of DNA methyltransferase 1 triggers an intra-S-phase arrest of DNA replication and induction of stress response genes. J Biol Chem. 2003;278:14985–14995. doi: 10.1074/jbc.M213219200. [DOI] [PubMed] [Google Scholar]
- 35.Unterberger A, Andrews SD, Weaver IC, Szyf M. DNA methyltransferase 1 knockdown activates a replication stress checkpoint. Mol Cell Biol. 2006;26:7575–7586. doi: 10.1128/MCB.01887-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen T, Hevi S, Gay F, Tsujimoto N, He T, Zhang B, Ueda Y, Li E. Complete inactivation of DNMT1 leads to mitotic catastrophe in human cancer cells. Nat Genet. 2007;39:391–396. doi: 10.1038/ng1982. [DOI] [PubMed] [Google Scholar]
- 37.Loughery JE, Dunne PD, O’Neill KM, Meehan RR, McDaid JR, Walsh CP. DNMT1 deficiency triggers mismatch repair defects in human cells through depletion of repair protein levels in a process involving the DNA damage response. Hum Mol Genet. 2011;20:3241–3255. doi: 10.1093/hmg/ddr236. [DOI] [PubMed] [Google Scholar]
- 38.Ha K, Lee GE, Palii SS, Brown KD, Takeda Y, Liu K, Bhalla KN, Robertson KD. Rapid and transient recruitment of DNMT1 to DNA double-strand breaks is mediated by its interaction with multiple components of the DNA damage response machinery. Hum Mol Genet. 2011;20:126–140. doi: 10.1093/hmg/ddq451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Klimasauskas S, Roberts RJ. M. HhaI binds tightly to substrates containing mismatches at the target base. Nucleic Acids Res. 1995;23:1388–1395. doi: 10.1093/nar/23.8.1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yang AS, Shen JC, Zingg JM, Mi S, Jones PA. HhaI and HpaII DNA methyltransferases bind DNA mismatches, methylate uracil and block DNA repair. Nucleic Acids Res. 1995;23:1380–1387. doi: 10.1093/nar/23.8.1380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yang JL, Weissman L, Bohr VA, Mattson MP. Mitochondrial DNA damage and repair in neurodegenerative disorders. DNA Repair (Amst) 2008;7:1110–1120. doi: 10.1016/j.dnarep.2008.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Obulesu M, Rao DM. DNA damage and impairment of DNA repair in Alzheimer’s disease. Int J Neurosci. 2010;120:397–403. doi: 10.3109/00207450903411133. [DOI] [PubMed] [Google Scholar]
- 43.Fenech M. Folate (vitamin B9) and vitamin B12 and their function in the maintenance of nuclear and mitochondrial genome integrity. Mutat Res. 2012;733:21–33. doi: 10.1016/j.mrfmmm.2011.11.003. [DOI] [PubMed] [Google Scholar]
- 44.Courtemanche C, Huang AC, Elson-Schwab I, Kerry N, Ng BY, Ames BN. Folate deficiency and ionizing radiation cause DNA breaks in primary human lymphocytes: a comparison. FASEB J. 2004;18:209–211. doi: 10.1096/fj.03-0382fje. [DOI] [PubMed] [Google Scholar]
- 45.Lee SL, Thomas P, Fenech M. Extracellular amyloid beta 42 causes necrosis, inhibition of nuclear division, and mitotic disruption under both folate deficient and folate replete conditions as measured by the cytokinesis-block micronucleus cytome assay. Environ Mol Mutagen. 2014;55:1–14. doi: 10.1002/em.21811. [DOI] [PubMed] [Google Scholar]
- 46.Kruman II, Kumaravel TS, Lohani A, Pedersen WA, Cutler RG, Kruman Y, Haughey N, Lee J, Evans M, Mattson MP. Folic acid deficiency and homocysteine impair DNA repair in hippocampal neurons and sensitize them to amyloid toxicity in experimental models of Alzheimer’s disease. J Neurosci. 2002;22:1752–1762. doi: 10.1523/JNEUROSCI.22-05-01752.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gackowski D, Rozalski R, Siomek A, Dziaman T, Nicpon K, Klimarczyk M, Araszkiewicz A, Olinski R. Oxidative stress and oxidative DNA damage is characteristic for mixed Alzheimer disease/vascular dementia. J Neurol Sci. 2008;266:57–62. doi: 10.1016/j.jns.2007.08.041. [DOI] [PubMed] [Google Scholar]
- 48.Mecocci P, Polidori MC, Cherubini A, Ingegni T, Mattioli P, Catani M, Rinaldi P, Cecchetti R, Stahl W, Senin U, Beal MF. Lymphocyte oxidative DNA damage and plasma antioxidants in Alzheimer disease. Arch Neurol. 2002;59:794–798. doi: 10.1001/archneur.59.5.794. [DOI] [PubMed] [Google Scholar]