

Abstract
Macrophage colony stimulating factor (M-CSF) is known to have profound effects upon vascular pathologies, but potential roles of other colony stimulating factors (CSF) are not well understood. We treated apo E deficient (apo E-/-) mice with granulocyte colony stimulating factor (G-CSF) or vehicle daily for 9 weeks, during which time they were fed a Western-style diet. G-CSF treatment resulted in increased proportions of circulating monocytes (6.9 ± 2.2% vs. 3.8 ± 0.3%; p < 0.05), a trend towards increased neutrophils (33.5 ± 19.1% vs. 22.2 ± 7.8%; p = 0.17), and decreased serum levels of total cholesterol (981 ± 594 vs. 1495 ± 1009 mg/dL; p < 0.005) compared to control mice. There was a trend towards less low density lipoprotein (LDL) in G-CSF treated mice (24.6 ± 2.4% vs. 37.4 ± 12.3%; p = 0.10). A greater proportion of bone marrow cells from G-CSF treated mice expressed membrane type 1 matrix metalloprotease (MT1-MMP) (G-CSF: 14.5 ± 5.5%; Control: 6.2 ± 5.0%; p < 0.05) compared to bone marrow cells from vehicle treated mice. G-CSF treatment was also associated with smaller atheromatous plaque, decreased Oil red O staining, and decreased infiltration of both Monocyte/Macrophage Marker Antibody (MOMA-2) and F4/80 dependent macrophage populations into aortic lesions. However, decreased plaque area appeared to be largely due to lower cholesterol levels in G-CSF-treated mice. Lesions in G-CSF treated mice appeared to be structurally distinct from control mice, containing relatively less lipid and macrophages. Our results suggest important roles for G-CSF in cholesterol metabolism, mobilization of bone marrow stem cells that might alter plaque development, and accumulation of lipids and macrophages into atherosclerotic lesions.
Keywords: Atherosclerosis, G-CSF, apolipoprotein E, lipoproteins, serum cholesterol, macrophage
Introduction
Emerging evidence indicates a prominent role for bone marrow-derived pluripotent cells in vascular pathologies such as atherosclerosis [1]. Bone marrow-derived progenitor cells can differentiate into endothelial cells [2,3] or vascular smooth muscle cells (VSMCs) [4,5] and participate in the formation of microvessels [6,7] and neointima [4,8] and angiogenesis in animal models of graft arteriosclerosis [4,9]. Arteriopathy in cardiac transplant patients has been reported to be associated with a reduction in circulating endothelial progenitors and with increased seeding of recipient-derived endothelial cells at the site of plaque development. Post mortem studies in patients who had undergone bone marrow transplantation years previously indicate the presence of smooth muscle cells (SMCs) that originated from donor marrow cells [10]. In atherosclerosis-prone apolipoprotein E deficient (apo E-/-) mice, bone marrow transplantation with cells from young but not old apo E-/- mice prevented progression of atherosclerosis, suggesting that pluripotent bone marrow cells that may home to sites of vascular lesions might play a preventive or reparative role in atherosclerosis [11]. Despite these intriguing reports, fundamental questions regarding how bone marrow cells are mobilized, home to developing atherosclerotic lesions, develop into terminally differentiated vascular cells, what functional role they perform in the arterial wall and under what set of signals, and how long they may reside and proliferate there remain largely unexplored.
A number of studies suggest that colony stimulating factors (CSFs) may play a crucial role in vascular pathology and homeostasis. For example, mice with genetic deficiency in both macrophage colony stimulating factor (M-CSF) and either apo E [12,13] or low density lipoprotein receptor (LDLR) [14] do not develop significant atherosclerosis. Evidence from a porcine model of neointimal proliferation after balloon injury suggests that M-CSF expression may be involved in the response to injury as well [15]. Several studies suggest that granulocyte macrophage colony stimulating factor (GM-CSF) may have pro-atherogenic properties by regulating macrophage expression of myeloperoxidase or lowering serum cholesterol [16-20]. Administration of M-CSF can also lower serum cholesterol levels both in animal models [21-23] and in humans [23,24]. A number of studies have also reported the significance of granulocyte colony stimulating factor (G-CSF) in vascular pathologies [25,26]. Neointima thickness was reduced by approximately 60% in the G-CSF-treated rats [25]. G-CSF therapy with intracoronary infusion of peripheral blood stem-cells showed improved cardiac function, and promoted angiogenesis in patients with myocardial infarction [26]. Additionally, G-CSF treatment accelerated re-endothelialization and decreased neointima formation following vascular injury in C57BL/6 mice [27]. The G-CSF-induced mobilization of bone marrow derived c-Kit+/Flk-1+ cells contribute to endothelial regeneration in rat [28]. High-dose G-CSF induced neointimal proliferation through excessive inflammation and bone marrow cell mobilization in the early phase [29]. G-CSF accelerated the re-endothelialization of denuded arteries, and the pretreatment with nitric oxide (NO) synthase inhibitor significantly inhibited it in a rabbit model of atherosclerosis [30,31]. These results collectively suggest that G-CSF may play important therapeutic role during the progression of atherosclerosis. Paradoxically, findings also demonstrate that in mice model of atherosclerosis (apo E-/-), administration of G-CSF or GM-CSF failed to show any beneficial therapeutic effect; rather both resulted in a worsening of atherosclerosis [32]. G-CSF may also be involved in other vascular diseases such as Kawasaki Disease [33,34]. Thus, available data suggests that the CSFs may have unique roles in vascular pathologies, but these are not yet well defined. Furthermore, the independent effect of mobilization of pluripotent bone marrow cells on vascular pathologies has not been studied well. Here we explored the potential role of G-CSF in the development of atherosclerosis in apo E-/- mice. We report that chronic injections of G-CSF into apo E-/- mice lowers serum cholesterol, inhibits development of atherosclerotic plaque, alters expression of the pericellular protease MT1-MMP by bone marrow cells, and appears to result in a shift of plaque structure to a less lipid and macrophage-rich phenotype.
Materials and methods
Human recombinant G-CSF was obtained from Amgen, Inc. (Thousand Oaks, CA). Purified mouse monoclonal antibodies to mouse MT1-MMP, and MMP-2 were purchased from Calbiochem, San Diego, CA. Antibodies for MMP-9, c-kit, MOMA-2 and F4/80 were obtained from Abcam, Inc, Cambridge, MA.
Mouse model and treatment
Experimental protocols involving genetically-altered mice were approved by the Institutional Animal Care and Use Committee (IACUC). Mice were sacrificed by isoflurane and cervical dislocation and all efforts were made to minimize suffering. Breeding pairs of Apo E-/- mice on a C57BL/6 background were purchased from Jackson Laboratories (Bar Harbor, ME) and were inbred within our animal colony for at least 7 generations to achieve maximal genetic homogeneity. Mice (10 to 11 weeks of age) were fed a Western style diet containing 0.15% cholesterol (Harlan Teklad) for 9 week. The intraperitoneal injection of G-CSF (300 μg/kg; n = 9) or vehicle was administered daily (excluding weekends) for this duration. At age 19-20 weeks, blood samples were drawn from the periorbital sinus using a capillary pipette, and complete blood counts (CBC), total serum cholesterol and lipoprotein profiles were obtained. Analysis of total cholesterol was performed by spectrophotometric techniques. Lipoprotein analysis was performed using steric exclusion high performance liquid chromatography (HPLC). Blood was obtained, and the heart and aortae were excised and frozen in OCT after perfusion with PBS via left ventricular puncture.
Histochemistry and immunohistochemistry
Serial sections were cut and stained with hematoxylin and counterstained with eosin. Lipid deposits were stained with Oil red O. These stainings were performed using our standard protocol. Computer assisted histomorphometric analysis (ImagePro, Media Cybernetics) was performed to quantitate total cross sectional area, plaque area, and lumen area.
Antibodies against MOMA-2 and F4/80 were used to identify specific macrophage populations. Formalin fixed, paraffin embedded tissue sections were de-paraffinized in xylene and dehydrated in graded ethanol series. Antigen retrieval was performed using high pH antigen unmasking solution (Vector Lab, CA) followed by quenching of endogenous peroxidase activity with 3% hydrogen peroxide. Sections were blocked for non-specific staining for one hour (1 h) at room temperature and then incubated in primary antibody at 4°C for overnight, followed by incubation with biotinylated secondary antibody for 1 h at room temperature. Final visualization was achieved by using the ABC peroxidase kit and 3,3’-diaminobenzidine (DAB; Vector Lab, CA). Sections were counterstained by hematoxylin.
Fluorescence activated cell sorting (FACS)
Blood cells were isolated and FACS analysis was performed. The cells were pelleted by centrifugation and incubated with 20 μg of goat IgG in PBS/0.1% sodium azide on ice for 10 minutes. Primary monoclonal antibody for c-kit and MT1-MMP (0.5 μg per sample to a total volume of 50 μL) was added to cells, which were then incubated on ice for 30 minutes. After 2 washes with PBS containing 1% FCS/0.1% sodium azide, cells were incubated with saturating concentrations of PE-conjugated goat anti-mouse IgG for 30 minutes at 4°C for 10 minutes. After 2 more washes, cells were fixed with 1% paraformaldehyde in PBS. Analysis was performed with FACS (Becton Dickinson). Cell populations were gated according to forward and side scattering. Results were plotted as intensity of fluorescence versus cell number.
Gelatin zymography
MMP-2 and MMP-9 enzymatic activity in tissue extracts was determined by SDS-PAGE gelatin zymography. Briefly, equal amount of serum protein (40 μg) from each sample was denatured in the absence of a reducing agent and was separated on 7.5% SDS-PAGE containing 0.1% (w/v) gelatin. Gels were incubated with 2.5% Triton X-100 at room temperature for 2 hour followed by overnight incubation at 37°C in a buffer containing 10 mM CaCl2, 0.15 M NaCl, and 50 mM Tris (pH 7.5). Gels were then stained with 0.25% coomassie blue, and proteolysis was detected as a white band against a blue background. The activity of MMP-2 and MMP-9 relative to the standards was determined by densitometric scanning of the bands (Gel Imager, Biorad).
Statistical analysis
Results are expressed as mean ± SD. Mean values were compared using Student’s t-tests. The relationships between continuous variables were evaluated using Pearson’s correlation coefficient. A p value of less than 0.05 was considered significant.
Result
G-CSF lowers serum cholesterol and increases circulating monocytes
G-CSF treatment resulted in increased proportions of circulating monocytes (6.9 ± 2.2% vs. 3.8 ± 0.3%; p < 0.05), a trend towards increased neutrophils (33.5 ± 19.1% vs. 22.2 ± 7.8%; p = 0.17). Treatment of apo E-/- mice with G-CSF resulted in significantly lower serum cholesterol levels compared to apo E-/- mice treated with vehicle only (Table 1). In G-CSF treated mice, serum cholesterol values averaged 981 ± 594 mg/dL, compared to 1495 ± 1009 mg/dL in vehicle-treated control mice (p < 0.005, Table 1). We performed HPLC analysis to determine the relative proportions of lipoproteins in the two groups of mice (Figure 1). Results of this analysis revealed a trend towards lower proportions of LDL cholesterol in G-CSF compared to vehicle treated mice (24.6 ± 2.4% vs. 37.4 ± 12.3%). However, this trend did not reach statistical significance (p = 0.10). There were no significant differences in relative proportions of very-low-density lipoprotein (VLDL) and high density lipoprotein (HDL) in the two groups of mice. Thus, treatment with G-CSF appears to lower total serum cholesterol without affecting the relative proportions of lipoproteins significantly.
Table 1.
Effects of G-CSF treatment on circulating monocytes, neutrophils and total cholesterol
| Circulating Monocytes (%) | Neutrophils (%) | Total Cholesterol (mg/dL) | |
|---|---|---|---|
| Control | 3.8 ± 0.3 | 22.2 ± 7.8 | 1495 ± 1009 |
| G-CSF treated | 6.9 ± 2.2* | 33.5 ± 19.1 | 981 ± 594** |
p < 0.05;
p < 0.005.
Figure 1.

Relative proportions of serum lipoproteins in G-CSF-treated and control apo E-/- mice.
Histopathologic effects of G-CSF on atherosclerotic lesions
As shown in Table 2A and Figure 2, the size of intimal plaques was significantly smaller in mice treated with G-CSF compared to vehicle-treated control mice (0.039 ± 0.014; and 0.058 ± 0.005 mm2 respectively; p < 0.01). Accordingly, lumen area was significantly larger in mice treated with G-CSF (G-CSF: 0.174 ± 0.069, control: 0.090 ± 0.032, p < 0.05). When plaque area was expressed as a percentage of the vessel wall cross sectional area (i.e., plaque area ÷ [area within the external elastic lamina (EEL) – area of lumen]), similar findings were noted. In mice treated with G-CSF, there was a trend towards greater total cross sectional area of the aortic root within the external elastic lamina (EEL) compared to control mice, but this difference did not reach statistical significance. (G-CSF: 0.268 ± 0.087, control: 0.181 ± 0.068, p = 0.10).
Table 2A.
Treatment of apo E-/- mice with G-CSF resulted in significantly larger lumen areas, smaller plaque areas, and smaller percent plaque areas compared with control mice treated with vehicle only
| EEL (mm2) | IEL (mm2) | Lumen (mm2) | Plaque (mm2) | Plaque Area (%) | |
|---|---|---|---|---|---|
| Control | 0.181 ± 0.068 | 0.148 ± 0.067 | 0.090 ± 0.032 | 0.058 ± 0.005 | 63.7 ± 18.3 |
| G-CSF | 0.268 ± 0.087 | 0.213 ± 0.075 | 0.174 ± 0.069* | 0.039 ± 0.014* | 41.5 ± 2.9** |
EEL = area within the external elastic lamina; IEL = area within the internal elastic lamina; Lumen = area of the lumen; Plaque = area of plaque (IEL-Lumen); Plaque % Area = (plaque area/[EEL-Lumen Area]).
p < 0.05;
p < 0.01 (G-CSF vs. control).
Figure 2.

Hematoxylin and eosin (A) and Oil red O staining (B) in sections of aortic root from apo E-/- mice treated with G-CSF or vehicle. Magnification × 4.
It is possible that the differences in plaque size observed in mice treated with G-CSF might be explained by the lower serum cholesterol values in these mice relative to vehicle-treated control mice. To evaluate the validity of this notion, we normalized plaque size for total serum cholesterol levels. Plaque size expressed as an area divided by total serum cholesterol was not significantly different between G-CSF-treated and control mice (p = 0.270). When plaque size was expressed as a percentage of arterial cross sectional area (subtracting the lumen area), similar results were obtained (p = 0.382). These results suggest that the effects of G-CSF on plaque size are predominantly mediated by the lipid-lowering actions of G-CSF (Table 2B).
Table 2B.
Comparison of plaque area and percent plaque areas with and without adjustment for total serum cholesterol
| Plaque area mm2 | % Plaque area | total cholesterol mg/dl | Area/Chol | % Area/Chol | |
|---|---|---|---|---|---|
| Control | 0.058 ± 0.005 | 63.7 ± 18.3 | 1495 ± 1009 | 3.88e-5 | 0.043 |
| GCSF | 0.039 ± 0.014* | 41.5 ± 2.9** | 981 ± 594** | 3.98e-5 | 0.042 |
p < 0.05;
p < 0.01.
Effects of G-CSF treatment on plaque lipid content and macrophage infiltration into plaques
We evaluated G-CSF treated animals for differences in plaque lipid content and the accumulation of macrophages. Lesions in G-CSF treated mice appeared to be structurally distinct from control mice, containing relatively less lipid and macrophages. Histochemical Oil red O staining for lipids revealed that G-CSF treatment was associated with decreased lipid staining with smaller atheromatous plaque (Figure 2B). Immunohistochemical analysis using MOMA-2 and F4/80 antibodies to visualize and quantitate macrophage infiltration showed decreased infiltration of both MOMA-2 and F4/80 dependent macrophage populations into aortic lesions (Figure 3A and 3B).
Figure 3.

Immunohistochemical staining for MOMA-2 (A) and F4/80 (B) macrophage antigens in sections of aortic root from apo E-/- mice treated with G-CSF or vehicle. Magnification × 10.
Effects of G-CSF on expression of c-kit and MT1-MMP by bone marrow cells
Pluripotent cells including hematopoietic stem cells are known to express c-kit [35,36]. MT1-MMP is thought to direct a pericellular proteolytic cascade [37], and appears to play an important role in cellular invasion [38] and neovascularization [39]. Sub-endothelial migration of bone marrow-derived cells including mononuclear phagocytic cells is an important feature of all stages of atherogenesis [40]. We reasoned that if bone marrow-derived cells gain entrance to the arterial wall, they might do so by proteolytic degradation of the sub endothelial basement membrane and extracellular matrix components. We therefore hypothesized that G-CSF treatment may enhance expression of both c-kit and MT1-MMP on bone marrow cells. To evaluate this possibility, we performed FACS sorting of bone marrow cells isolated from apo E-/- mice treated with G-CSF or vehicle. As shown in Figure 4, G-CSF treatment resulted in a significantly greater proportion of bone marrow cells that expressed MT1-MMP (G-CSF: 14.5 ± 5.5%; Control: 6.2 ± 5.0%; p < 0.05). There was no significant effect of G-CSF on expression of c-kit by bone marrow cells.
Figure 4.
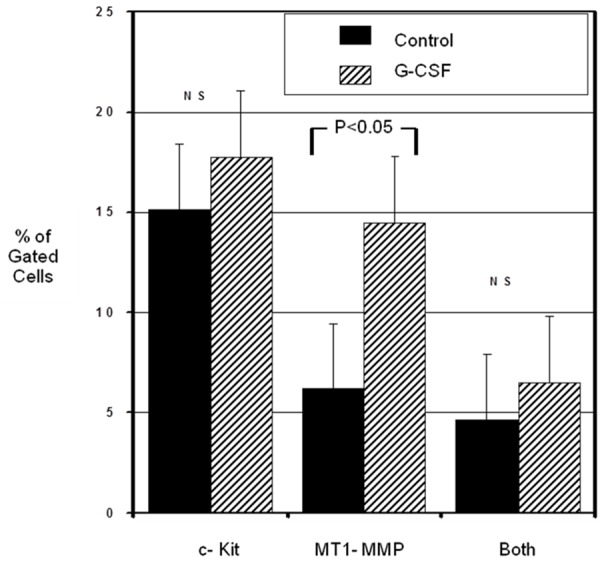
FACS analysis of bone marrow cell expression of c-kit and MT1-MMP.
Gelatin zymography
Among the various MMPs that MT1-MMP can activate are MMP-2 and MMP-9 [37]. To further evaluate whether circulating bone marrow derived cells might assume a more invasive phenotype in response to G-CSF treatment, we performed a gelatinolytic assay to differentiate zymogen and active forms of both MMP-2 and MMP-9 in serum isolated from G-CSF-treated and control apo E-/- mice. These results demonstrated no significant effect of G-CSF treatment on gelatinolytic activity in media from bone marrow cells. Treatment with G-CSF did not appear to alter levels of serum MMP-2 or MMP-9 (Figure 5). Also, the relative proportions of the zymogen and activated forms of these proteases were not affected by treatment with G-CSF.
Figure 5.
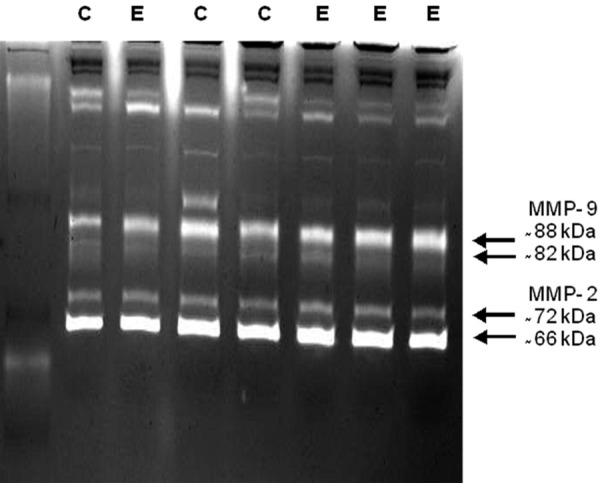
Gelatin zymography on serum obtained from G-CSF (E) and control (C) apo E-/- mice.
Discussion
A number of studies suggest that the CSFs may play important roles in vascular homeostasis and pathologies, though not necessarily independently of their well-documented roles in survival, development, and mobilization of hematopoietic lineages. For example, genetic deficiency of M-CSF in murine models of atherosclerosis results in an increase in serum cholesterol, but little or no atherosclerotic plaque formation [12-14]. However, in these animals, lack of M-CSF causes a profound reduction in mononuclear phagocytic cells, and hence also in tissue macrophages [41]. Hematopoietic cells of the mononuclear phagocytic lineage are well known to be important participants in atherogenesis [40,42]. Therefore, it is not clear whether the effects of M-CSF on atherogenesis are direct, indirect, or both. In addition, two CSFs (M-CSF and GM-CSF) appear to play a role in cholesterol homeostasis [19,20,23,24,43]. Animal models have also suggested a potential function of GM-CSF in development of atheroma [17,18] and have established as a key regulator of intimal cell proliferation in lesions demonstrating that both proliferation and monocyte recruitment contribute to the initiation of atherosclerosis [44]. The function of the CSFs in vascular homeostatic and pathologic processes is further complicated by the fact that CSFs mobilize bone marrow lineages [35,36,45-47], but emerging evidence indicates that bone marrow pluripotent cells are mobilized from bone marrow and migrate to the arterial wall, where they may undergo terminal differentiation into vascular cells, or may possibly play a reparative function [1,48,49]. Thus a comprehensive understanding of how CSFs might impact vascular biology and pathology has not been achieved.
Here we implicate a role for G-CSF in both atherosclerosis and cholesterol metabolism. Our results show that G-CSF inhibits plaque development, decreases lipid and macrophages in plaque that does form, and lowers serum cholesterol. We also found that G-CSF treatment increased bone marrow cell expression of MT1-MMP, but did not change the relative serum levels of the precursor and active forms of MMP-2 and MMP-9. The effects of G-CSF on plaque size may be a function of decreased serum cholesterol levels, but a direct role of G-CSF on atherogenesis cannot be excluded, and is suggested by the decreased macrophage infiltration into plaques that we observed, which together with decreased lipid content, suggest the possibility that G-CSF alters plaque composition. Our results suggest important roles for G-CSF in cholesterol metabolism, mobilization of bone marrow cells that might alter plaque development, and accumulation of lipids and macrophages into atherosclerotic lesions. Recent findings also suggest that appropriate doses of G-CSF reduced atherosclerotic plaque formation and increased plaque stability in cholesterol-fed rabbits [30]. G-CSF significantly prevented an increase in neointima/media ratio at 4 weeks after the treatment in a rabbit model of vascular injury. In this study, G-CSF accelerated the re-endothelialization of denuded arteries, and the pretreatment with nitric oxide synthase inhibitor significantly inhibited it, suggesting a therapeutic potential of G-CSF for the progression of atherosclerosis [31]. Recent studies suggest that regulatory T cells play an important role in the atheroprotective effects of G-CSF [50]. The findings presented in one study, however, demonstrate that in murine model of atherosclerosis (Apo E-/-) mice, administration of G-CSF or GM-CSF, both, resulted in a worsening of atherosclerosis [32].
Conclusions
G-CSF lowers serum cholesterol, increases circulating monocytes, increases bone marrow cell expression of MT1-MMP, inhibits plaque development, and decreases lipid and macrophage infiltration into developing plaque. Effects of G-CSF on plaque size may be a function of decreased serum cholesterol levels. Our results suggest a prominent role for G-CSF in vascular pathology and cholesterol metabolic pathways.
Acknowledgements
This study was supported by grants from The National Institutes of Health HL51980 and HL58555 (to TBR). SKS was supported by NIH grants (U54 MD 007598 from NIMHD and S21 MD 000103) at Charles R. Drew University. VM was supported by a fellowship from the Council of Scientific and Industrial Research (CSIR) India. A research associate funding from the Indian Council of Medical Research (ICMR) supported SN. TBR was supported by a Ramalingaswami Fellowship from the Department of Biotechnology (DBT), Government of India.
Disclosure of conflict of interest
None.
References
- 1.Doherty TM, Shah PK, Rajavashisth TB. Cellular origins of atherosclerosis: towards ontogenetic endgame? FASEB J. 2003;17:592–597. doi: 10.1096/fj.02-0913hyp. [DOI] [PubMed] [Google Scholar]
- 2.Asahara T, Murohara T, Sullivan A, Silver M, van der Zee R, Li T, Witzenbichler B, Schatteman G, Isner JM. Isolation of putative progenitor endothelial cells for angiogenesis. Science. 1997;275:964–967. doi: 10.1126/science.275.5302.964. [DOI] [PubMed] [Google Scholar]
- 3.Shi Q, Rafii S, Wu MH, Wijelath ES, Yu C, Ishida A, Fujita Y, Kothari S, Mohle R, Sauvage LR, Moore MA, Storb RF, Hammond WP. Evidence for circulating bone marrow-derived endothelial cells. Blood. 1998;92:362–367. [PubMed] [Google Scholar]
- 4.Sata M, Saiura A, Kunisato A, Tojo A, Okada S, Tokuhisa T, Hirai H, Makuuchi M, Hirata Y, Nagai R. Hematopoietic stem cells differentiate into vascular cells that participate in the pathogenesis of atherosclerosis. Nat Med. 2002;8:403–409. doi: 10.1038/nm0402-403. [DOI] [PubMed] [Google Scholar]
- 5.Simper D, Stalboerger PG, Panetta CJ, Wang S, Caplice NM. Smooth muscle progenitor cells in human blood. Circulation. 2002;106:1199–1204. doi: 10.1161/01.cir.0000031525.61826.a8. [DOI] [PubMed] [Google Scholar]
- 6.Asahara T, Masuda H, Takahashi T, Kalka C, Pastore C, Silver M, Kearne M, Magner M, Isner JM. Bone marrow origin of endothelial progenitor cells responsible for postnatal vasculogenesis in physiological and pathological neovascularization. Circ Res. 1999;85:221–228. doi: 10.1161/01.res.85.3.221. [DOI] [PubMed] [Google Scholar]
- 7.Takahashi T, Kalka C, Masuda H, Chen D, Silver M, Kearney M, Magner M, Isner JM, Asahara T. Ischemia- and cytokine-induced mobilization of bone marrow-derived endothelial progenitor cells for neovascularization. Nat Med. 1999;5:434–438. doi: 10.1038/7434. [DOI] [PubMed] [Google Scholar]
- 8.Walter DH, Rittig K, Bahlmann FH, Kirchmair R, Silver M, Murayama T, Nishimura H, Losordo DW, Asahara T, Isner JM. Statin therapy accelerates reendothelialization: a novel effect involving mobilization and incorporation of bone marrow-derived endothelial progenitor cells. Circulation. 2002;105:3017–3024. doi: 10.1161/01.cir.0000018166.84319.55. [DOI] [PubMed] [Google Scholar]
- 9.Hu Y, Davison F, Zhang Z, Xu Q. Endothelial replacement and angiogenesis in arteriosclerotic lesions of allografts are contributed by circulating progenitor cells. Circulation. 2003;108:3122–3127. doi: 10.1161/01.CIR.0000105722.96112.67. [DOI] [PubMed] [Google Scholar]
- 10.Caplice NM, Bunch TJ, Stalboerger PG, Wang S, Simper D, Miller DV, Russell SJ, Litzow MR, Edwards WD. Smooth muscle cells in human coronary atherosclerosis can originate from cells administered at marrow transplantation. Proc Natl Acad Sci U S A. 2003;100:4754–4759. doi: 10.1073/pnas.0730743100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rauscher FM, Goldschmidt-Clermont PJ, Davis BH, Wang T, Gregg D, Ramaswami P, Pippen AM, Annex BH, Dong C, Taylor DA. Aging, progenitor cell exhaustion and atherosclerosis. Circulation. 2003;108:457–463. doi: 10.1161/01.CIR.0000082924.75945.48. [DOI] [PubMed] [Google Scholar]
- 12.Lapidot T, Petit I. Current understanding of stem cell mobilization: the roles of chemokines, proteolytic enzymes, adhesion molecules, cytokines and stromal cells. Exp Hematol. 2002;30:973–981. doi: 10.1016/s0301-472x(02)00883-4. [DOI] [PubMed] [Google Scholar]
- 13.Petit I, Szyper-Kravitz M, Nagler A, Lahav M, Peled A, Habler L, Ponomaryov T, Taichman RS, Arenzana-Seisdedos F, Fujii N, Sandbank J, Zipori D, Lapidot T. G-CSF induces stem cell mobilization by decreasing bone marrow SDF-1 and up-regulating CXCR4. Nat Immunol. 2002;3:687–694. doi: 10.1038/ni813. [DOI] [PubMed] [Google Scholar]
- 14.Cottler-Fox MH, Lapidot T, Petit I, Kollet O, DiPersio JF, Link D, Devine S. Stem cell mobilization. Hematology Am Soc Hematol Educ Program. 2003:419–437. doi: 10.1182/asheducation-2003.1.419. [DOI] [PubMed] [Google Scholar]
- 15.Gazitt Y. Comparison between granulocyte colony-stimulating factor and granulocyte-macrophage colony-stimulating factor in the mobilization of peripheral blood stem cells. Curr Opin Hematol. 2002;9:190–198. doi: 10.1097/00062752-200205000-00003. [DOI] [PubMed] [Google Scholar]
- 16.Fischmeister G, Gadner H. Granulocyte colony-stimulating factor versus granulocyte-macrophage colony-stimulating factor for collection of peripheral blood progenitor cells from healthy donors. Curr Opin Hematol. 2000;7:150–155. doi: 10.1097/00062752-200005000-00004. [DOI] [PubMed] [Google Scholar]
- 17.Rajavashisth T, Qiao JH, Tripathi S, Tripathi J, Mishra N, Hua M, Wang XP, Loussararian A, Clinton S, Libby P, Lusis A. Heterozygous osteopetrotic (op) mutation reduces atherosclerosis in LDL receptor- deficient mice. J Clin Invest. 1998;101:2702–2710. doi: 10.1172/JCI119891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Smith JD, Trogan E, Ginsberg M, Grigaux C, Tian J, Miyata M. Decreased atherosclerosis in mice deficient in both macrophage colony-stimulating factor (op) and apolipoprotein E. Proc Natl Acad Sci U S A. 1995;92:8264–8268. doi: 10.1073/pnas.92.18.8264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stanley ER, Guilbert LJ, Tushinski RJ, Bartelmez SH. CSF-1--a mononuclear phagocyte lineage-specific hemopoietic growth factor. J Cell Biochem. 1983;21:151–159. doi: 10.1002/jcb.240210206. [DOI] [PubMed] [Google Scholar]
- 20.Tushinski RJ, Oliver IT, Guilbert LJ, Tynan PW, Warner JR, Stanley ER. Survival of mononuclear phagocytes depends on a lineage-specific growth factor that the differentiated cells selectively destroy. Cell. 1982;28:71–81. doi: 10.1016/0092-8674(82)90376-2. [DOI] [PubMed] [Google Scholar]
- 21.Finkelstein A, Makkar R, Doherty TM, Vegesna VR, Tripathi P, Liu M, Bergman J, Fishbein M, Hausleiter J, Takizawa K, Rukshin V, Shah PK, Rajavashisth TB. Increased expression of macrophage colony-stimulating factor after coronary artery balloon injury is inhibited by intracoronary brachytherapy. Circulation. 2002;105:2411–2415. doi: 10.1161/01.cir.0000016048.03020.6c. [DOI] [PubMed] [Google Scholar]
- 22.Qiao JH, Tripathi J, Mishra NK, Cai Y, Tripathi S, Wang XP, Imes S, Fishbein MC, Clinton SK, Libby P, Lusis AJ, Rajavashisth TB. Role of macrophage colony-stimulating factor in atherosclerosis: studies of osteopetrotic mice. Am J Pathol. 1997;150:1687–1699. [PMC free article] [PubMed] [Google Scholar]
- 23.Sugiyama S, Okada Y, Sukhova GK, Virmani R, Heinecke JW, Libby P. Macrophage myeloperoxidase regulation by granulocyte macrophage colony-stimulating factor in human atherosclerosis and implications in acute coronary syndromes. Am J Pathol. 2001;158:879–891. doi: 10.1016/S0002-9440(10)64036-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang J, Wang S, Lu Y, Weng Y, Gown AM. GM-CSF and M-CSF expression is associated with macrophage proliferation in progressing and regressing rabbit atheromatous lesions. Exp Mol Pathol. 1994;61:109–118. doi: 10.1006/exmp.1994.1030. [DOI] [PubMed] [Google Scholar]
- 25.Ishibashi T, Yokoyama K, Shindo J, Hamazaki Y, Endo Y, Sato T, Takahashi S, Kawarabayasi Y, Shiomi M, Yamamoto T, et al. Potent cholesterol-lowering effect by human granulocyte-macrophage colony-stimulating factor in rabbits. Possible implications of enhancement of macrophage functions and an increase in mRNA for VLDL receptor. Arterioscler Thromb. 1994;14:1534–1541. doi: 10.1161/01.atv.14.10.1534. [DOI] [PubMed] [Google Scholar]
- 26.Shindo J, Ishibashi T, Yokoyama K, Nakazato K, Ohwada T, Shiomi M, Maruyama Y. Granulocyte-macrophage colony-stimulating factor prevents the progression of atherosclerosis via changes in the cellular and extracellular composition of atherosclerotic lesions in watanabe heritable hyperlipidemic rabbits. Circulation. 1999;99:2150–2156. doi: 10.1161/01.cir.99.16.2150. [DOI] [PubMed] [Google Scholar]
- 27.Ishibashi T, Yokoyama K, Shindo J, Hamazaki Y, Endo Y, Sato T, Takahashi S, Kawarabayasi Y, Shiomi M, Yamamoto T, et al. Human granulocyte-macrophage colony-stimulating factor lowers the levels of plasma cholesterol with an increase in mRNA for very low density lipoprotein receptor in rabbits. Ann N Y Acad Sci. 1995;748:630–633. doi: 10.1111/j.1749-6632.1994.tb17377.x. [DOI] [PubMed] [Google Scholar]
- 28.Irie H, Koshiba H, Koyama M, Asakura E, Shibata H, Kimura K, Naito K, Yamauchi T, Yada K, Hanamura T, Hanada S, Abe S, Nakamura N. Effects of recombinant human macrophage colony-stimulating factor on atherosclerotic lesions established in the aorta of high cholesterol-fed rabbits. J Biochem. 2001;129:717–724. doi: 10.1093/oxfordjournals.jbchem.a002911. [DOI] [PubMed] [Google Scholar]
- 29.Stoudemire JB, Garnick MB. Effects of recombinant human macrophage colony-stimulating factor on plasma cholesterol levels. Blood. 1991;77:750–755. [PubMed] [Google Scholar]
- 30.Munn DH, Cheung NK. Preclinical and clinical studies of macrophage colony-stimulating factor. Semin Oncol. 1992;19:395–407. [PubMed] [Google Scholar]
- 31.Shimano H, Yamada N, Motoyoshi K, Matsumoto A, Ishibashi S, Mori N, Takaku F. Plasma cholesterol-lowering activity of monocyte colony-stimulating factor (M-CSF) Ann N Y Acad Sci. 1990;587:362–370. doi: 10.1111/j.1749-6632.1990.tb00177.x. [DOI] [PubMed] [Google Scholar]
- 32.Kong D, Melo LG, Gnecchi M, Zhang L, Mostoslavsky G, Liew CC, Pratt RE, Dzau VJ. Cytokine-induced mobilization of circulating endothelial progenitor cells enhances repair of injured arteries. Circulation. 2004;110:2039–2046. doi: 10.1161/01.CIR.0000143161.01901.BD. [DOI] [PubMed] [Google Scholar]
- 33.Kang HJ, Kim HS, Zhang SY, Park KW, Cho HJ, Koo BK, Kim YJ, Soo Lee D, Sohn DW, Han KS, Oh BH, Lee MM, Park YB. Effects of intracoronary infusion of peripheral blood stem-cells mobilised with granulocyte-colony stimulating factor on left ventricular systolic function and restenosis after coronary stenting in myocardial infarction: the MAGIC cell randomised clinical trial. Lancet. 2004;363:751–756. doi: 10.1016/S0140-6736(04)15689-4. [DOI] [PubMed] [Google Scholar]
- 34.Yoshioka T, Takahashi M, Shiba Y, Suzuki C, Morimoto H, Izawa A, Ise H, Ikeda U. Granulocyte colony-stimulating factor (G-CSF) accelerates reendothelialization and reduces neointimal formation after vascular injury in mice. Cardiovasc Res. 2006;70:61–69. doi: 10.1016/j.cardiores.2005.12.013. [DOI] [PubMed] [Google Scholar]
- 35.Shoji M, Iso Y, Kusuyama T, Omori Y, Soda T, Tsunoda F, Sato T, Koba S, Geshi E, Kobayashi Y, Katagiri T, Suzuki H. High-dose granulocyte-colony stimulating factor promotes neointimal hyperplasia in the early phase and inhibits neointimal hyperplasia in the late phase after vascular injury. Circ J. 2008;72:1885–1893. doi: 10.1253/circj.cj-07-1037. [DOI] [PubMed] [Google Scholar]
- 36.Takamiya M, Okigaki M, Jin D, Takai S, Nozawa Y, Adachi Y, Urao N, Tateishi K, Nomura T, Zen K, Ashihara E, Miyazaki M, Tatsumi T, Takahashi T, Matsubara H. Granulocyte colony-stimulating factor-mobilized circulating c-Kit+/Flk-1+ progenitor cells regenerate endothelium and inhibit neointimal hyperplasia after vascular injury. Arterioscler Thromb Vasc Biol. 2006;26:751–757. doi: 10.1161/01.ATV.0000205607.98538.9a. [DOI] [PubMed] [Google Scholar]
- 37.Matsumoto T, Watanabe H, Ueno T, Tsunemi A, Hatano B, Kusumi Y, Mitsumata M, Fukuda N, Matsumoto K, Saito S, Mugishima H. Appropriate doses of granulocyte-colony stimulating factor reduced atherosclerotic plaque formation and increased plaque stability in cholesterol-fed rabbits. J Atheroscler Thromb. 2010;17:84–96. doi: 10.5551/jat.2279. [DOI] [PubMed] [Google Scholar]
- 38.Hasegawa H, Takano H, Ohtsuka M, Ueda K, Niitsuma Y, Qin Y, Tadokoro H, Shiomi M, Komuro I. G-CSF prevents the progression of atherosclerosis and neointimal formation in rabbits. Biochem Biophys Res Commun. 2006;344:370–376. doi: 10.1016/j.bbrc.2006.03.081. [DOI] [PubMed] [Google Scholar]
- 39.Haghighat A, Weiss D, Whalin MK, Cowan DP, Taylor WR. Granulocyte colony-stimulating factor and granulocyte macrophage colony-stimulating factor exacerbate atherosclerosis in apolipoprotein E-deficient mice. Circulation. 2007;115:2049–2054. doi: 10.1161/CIRCULATIONAHA.106.665570. [DOI] [PubMed] [Google Scholar]
- 40.Samada K, Igarashi H, Shiraishi H, Hatake K, Momoi MY. Increased serum granulocyte colony-stimulating factor correlates with coronary artery dilatation in Kawasaki disease. Eur J Pediatr. 2002;161:538–541. doi: 10.1007/s00431-002-1018-5. [DOI] [PubMed] [Google Scholar]
- 41.Igarashi H, Hatake K, Shiraishi H, Samada K, Tomizuka H, Momoi MY. Elevated serum levels of macrophage colony-stimulating factor in patients with Kawasaki disease complicated by cardiac lesions. Clin Exp Rheumatol. 2001;19:751–756. [PubMed] [Google Scholar]
- 42.Hotary KB, Yana I, Sabeh F, Li XY, Holmbeck K, Birkedal-Hansen H, Allen ED, Hiraoka N, Weiss SJ. Matrix metalloproteinases (MMPs) regulate fibrin-invasive activity via MT1-MMP-dependent and -independent processes. J Exp Med. 2002;195:295–308. doi: 10.1084/jem.20010815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hiraoka N, Allen E, Apel IJ, Gyetko MR, Weiss SJ. Matrix metalloproteinases regulate neovascularization by acting as pericellular fibrinolysins. Cell. 1998;95:365–377. doi: 10.1016/s0092-8674(00)81768-7. [DOI] [PubMed] [Google Scholar]
- 44.Libby P. Vascular biology of atherosclerosis: overview and state of the art. Am J Cardiol. 2003;91:3A–6A. doi: 10.1016/s0002-9149(02)03143-0. [DOI] [PubMed] [Google Scholar]
- 45.Jakubowski AA, Bajorin DF, Templeton MA, Chapman PB, Cody BV, Thaler H, Tao Y, Filippa DA, Williams L, Sherman ML, Garnick MB, Houghton AN. Phase I study of continuous-infusion recombinant macrophage colony-stimulating factor in patients with metastatic melanoma. Clin Cancer Res. 1996;2:295–302. [PubMed] [Google Scholar]
- 46.Libby P, Geng YJ, Aikawa M, Schoenbeck U, Mach F, Clinton SK, Sukhova GK, Lee RT. Macrophages and atherosclerotic plaque stability. Curr Opin Lipidol. 1996;7:330–335. doi: 10.1097/00041433-199610000-00012. [DOI] [PubMed] [Google Scholar]
- 47.Zhu SN, Chen M, Jongstra-Bilen J, Cybulsky MI. GM-CSF regulates intimal cell proliferation in nascent atherosclerotic lesions. J Exp Med. 2009;206:2141–2149. doi: 10.1084/jem.20090866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sata M. Circulating vascular progenitor cells contribute to vascular repair, remodeling, and lesion formation. Trends Cardiovasc Med. 2003;13:249–253. doi: 10.1016/s1050-1738(03)00106-3. [DOI] [PubMed] [Google Scholar]
- 49.Sata M. Molecular strategies to treat vascular diseases: circulating vascular progenitor cell as a potential target for prophylactic treatment of atherosclerosis. Circ J. 2003;67:983–991. doi: 10.1253/circj.67.983. [DOI] [PubMed] [Google Scholar]
- 50.Uchiyama R, Hasegawa H, Kameda Y, Ueda K, Kobayashi Y, Komuro I, Takano H. Role of regulatory T cells in atheroprotective effects of granulocyte colony-stimulating factor. J Mol Cell Cardiol. 2012;52:1038–1047. doi: 10.1016/j.yjmcc.2011.12.016. [DOI] [PubMed] [Google Scholar]