

Abstract
Hexanucleotide repeat expansions of C9orf72 are the most common genetic cause of amyotrophic lateral sclerosis and frontotemporal degeneration. The mutation is associated with reduced C9orf72 expression and the accumulation of potentially toxic RNA and protein aggregates. CpG methylation is known to protect the genome against unstable DNA elements and to stably silence inappropriate gene expression. Using bisulfite cloning and restriction enzyme-based methylation assays on DNA from human brain and peripheral blood, we observed CpG hyper-methylation involving the C9orf72 promoter in cis to the repeat expansion mutation in approximately one-third of C9orf72 repeat expansion mutation carriers. Promoter hypermethylation of mutant C9orf72 was associated with transcriptional silencing of C9orf72 in patient-derived lymphoblast cell lines, resulting in reduced accumulation of intronic C9orf72 RNA and reduced numbers of RNA foci. Furthermore, demethylation of mutant C9orf72 with 5-aza-deoxycytidine resulted in increased vulnerability of mutant cells to oxidative and autophagic stress. Promoter hypermethylation of repeat expansion carriers was also associated with reduced accumulation of RNA foci and dipeptide repeat protein aggregates in human brains. These results indicate that C9orf72 promoter hypermethylation prevents downstream molecular aberrations associated with the hexanucleotide repeat expansion, suggesting that epigenetic silencing of the mutant C9orf72 allele may represent a protective counter-regulatory response to hexanucleotide repeat expansion.
Keywords: Neurodegeneration, Amyotrophic lateral sclerosis, Dementia, Motor neuron disease, Frontotemporal lobar degeneration, Epigenetics
Introduction
Amyotrophic lateral sclerosis (ALS) and frontotemporal degeneration (FTD) exhibit overlapping clinical, pathologic and genetic features [33, 40]. ALS is characterized by weakness and spasticity due to the loss of motor neurons, while FTD is characterized by behavioral and language dysfunction due to the degeneration of the frontal and temporal lobes. However, many individuals exhibit clinical features of both ALS and FTD, and affected CNS regions exhibit similar neuropathologic changes [33, 40]. An intronic GGGGCC hexanucleotide repeat expansion in C9orf72 is the most common genetic cause of ALS and FTD [16, 53]. The C9orf72 mutation is associated with highly variable clinical phenotypes [5, 7, 13, 28, 30, 31, 41, 47, 56, 58]. While the basis for this heterogeneity is largely unknown, this suggests that there are endogenous mechanisms which modulate C9orf72-dependent disease pathways.
Mounting evidence suggests that repeat expansion of C9orf72 results in a toxic gain of function associated with the formation of RNA foci containing repeat RNA [16] or the accumulation of repeat-associated non-ATG mediated translation (RANT) dipeptide aggregates [2, 46]. The repeat expansion is associated with increased vulnerability to cellular stressors [1, 20, 55] and exogenous expression of the hexanucleotide repeat leads to overt toxicity [35]. However, the endogenous mechanisms which modulate the pathogenesis of C9orf72-associated disease are not well understood. Antisense oligonucleotides that target mutant RNA for post-transcriptional degradation have recently been shown to mitigate the molecular signatures of disease in experimental models [20, 32, 55], suggesting that mechanisms which regulate mutant RNA expression may modulate disease pathogenesis.
Repeat expansions have been associated with epigenetic silencing, most notably in the case of trinucleotide repeat mutations of FMR1 in Fragile X syndrome where promoter hypermethylation is linked to a pathogenic loss of function [18]. DNA methylation is an epigenetic modification which protects the genome against deleterious repeat DNA elements and regulates imprinted or developmentally timed gene expression [60]. Recent studies have demonstrated that mutation carriers exhibit C9orf72 promoter hypermethylation [65] in association with repressive histone marks [3] that silence gene expression. However, the downstream effects of epigenetic silencing of C9orf72 have not yet been demonstrated. We hypothesized that endogenous transcriptional gene silencing alters downstream disease pathways by virtue of modulating mutant RNA expression and that understanding the relationship between epigenetic silencing of C9orf72 and repeat expansion-associated toxicity can distinguish whether the C9orf72 mutation causes disease via a loss or gain of function mechanism.
Here, we show that C9orf72 promoter hypermethylation is found in a subset of repeat expansion cases and appears to protect against mutant C9orf72-mediated toxicity and pathology. Using human tissue, we determined that C9orf72 hypermethylation is monoallelic involving a region upstream of the hexanucleotide repeat only in repeat expansion mutation carriers. C9orf72 promoter methylation resulted in transcriptional silencing, inhibiting the accumulation of mutant intronic RNA, reducing vulnerability to oxidative and autophagic stress, decreasing the number of RNA foci, and inhibiting the accumulation of RANT pathology. Collectively, these results support the hypothesis that C9orf72 mutations cause disease through a toxic gain of function, and indicate that C9orf72 promoter hypermethylation may represent an endogenous response that modulates disease via inhibition of mutant C9orf72.
Materials and methods
Patient-derived materials
Human autopsy tissue and peripheral DNA were obtained from the University of Pennsylvania Center for Neurodegenerative Disease Research biorepository [62]. A summary of cases is available in supplemental materials. Lymphoblast cell lines ND16183, ND11836, ND14442 and ND10966 were obtained from the Coriell NINDS Repository (Camden, NJ, USA).
Nucleic acid extraction
Total RNA was extracted using Trizol (Life Technologies, Carlsbad, CA, USA). RNA was digested with 1 unit of RQ1 DNase (Promega, Madison, WI, USA) per μg followed by ethanol precipitation. RNA was quantified using the Qubit RNA HS Assay kit (Life Technologies). RNA was reverse transcribed to cDNA using the High Capacity RNA to cDNA kit (Life Technologies). qPCR was done with 2× FastStart SYBR Green Master (Roche Applied Science, Indianapolis, IN, USA) to quantify C9orf72 RNA and control housekeeping RNAs using primers as listed in the supplemental materials on the StepOne Plus Real-Time PCR Machine (Life Technologies) using the ΔΔ Ct method. For qPCR measuring C9orf72 intronic RNA, reactions containing RNA without reverse transcriptase did not amplify. DNA from tissue or cells was extracted using the DNeasy Blood and Tissue kit (Qiagen, Valencia, CA, USA).
C9orf72 promoter methylation assay
For quantitative assessment of methylation levels, 100 ng of DNA was digested for 16 h with 2 units of HhaI (New England Biolabs, Ipswich, MA, USA) followed by heat inactivation. qPCR was done with 2× FastStart SYBR Green Master (Roche) using primers amplifying the differentially methylated C9orf72 promoter region (see supplemental materials for primer sequences). The difference in the number of cycles to threshold amplification between digested versus mock-digested DNA was used as a measure of CpG methylation. Mock-digested DNA consisted of either undigested DNA or HaeIII-digested DNA as a negative control restriction enzyme which does not cut within the qPCR amplicon. To determine the linearity of this assay, a methylated DNA standard was generated by in vitro methylating DNA from a non-expanded lymphoblast cell line using M.SssI (New England Biolabs) for 4 h at 37 °C. Methylated DNA was purified by phenol:chloroform:isoamyl alcohol extraction, and different ratios of methylated to unmethylated DNA were subject to the C9orf72 promoter methylation assay.
To determine if methylation occurs in cis or trans, 100 ng of DNA from three C9orf72 promoter hypermethylated repeat-expanded patient cases who were heterozygous for the deletion polymorphism (rs200034037) was digested for 16 h with 2 units of HhaI and HpaII (New England Biolabs) vs. a no enzyme mock digestion followed by heat inactivation. DNA was amplified by PCR using primers flanking rs200034037 and the HhaI and HpaII cut sites within the C9orf72 promoter (see supplemental materials for primer sequences). The PCR product was run on a poly-acrylamide gel and imaged with ethidium bromide or used for Sanger sequencing.
Bisulfite cloning
Cerebellar DNA from four C9orf72 expansion carriers and four control cases was bisulfite converted using the EpiTect Bisulfite Kit (Qiagen). Bisulfite-converted DNA was subjected to PCR to amplify both CpG islands. The entire first CpG island was amplified by standard PCR. The second CpG island was divided into two separate reactions. The first half of the 2nd CpG island was amplified by nested PCR, while the second half of the second CpG island was amplified using standard PCR. Primer sequences are available in the supplemental materials. PCR products were run on an agarose gel and gel purified using QIAquick Gel Extraction kit (Qiagen). Amplified DNA was sub-cloned by ligation into the pGEM-T Easy vector (Promega), transformation into competent E. coli and isolation of plasmid DNA from individual bacterial colonies. Five or six individual clones from each case were Sanger sequenced using a T7 promoter sequencing primer.
Hexanucleotide repeat methylation assay
DNA (1 μg) from post-mortem cerebellar tissue was restriction enzyme digested for 4 h with HpaII (10 U), MspI (10 U), or MspJI (4 U) (New England Biolabs) at 37°C followed by phenol:chloroform:isoamyl alcohol extraction. 100 ng of digested DNA was used for repeat primed PCR as described previously [53]. Fragment length analysis was done using the Genetic Analyzer 3130x (Life Technologies) and Peak Scanner software (Life Technologies). To ensure that methylated DNA could be digested by MspJI under these conditions, 1 μg of DNA from repeat expanded or control cerebellum was in vitro methylated using M.SssI (New England Biolabs) for 4 h at 37 °C followed by phenol:chloroform:isoamyl alcohol extraction. 1 μg of purified DNA was digested with 2 units of MspJI at 37 °C for 4 h and purified using phenol:chloroform:isoamyl alcohol prior to repeat primed PCR.
Southern blotting
Southern blot hybridization was performed as previously described [16]. Briefly, 5–10 μg of genomic DNA was digested with EcoRI and HindIII, denatured at 95 °C for 5 min, and run on a 0.8 % agarose gel at 100 V for 4 h. DNA was transferred to a positively charged nylon membrane (GE Life Sciences, Pittsburg, PA, USA) and crosslinked. A 576-bp digoxigenin (DIG)-labeled probe was amplified using PCR DIG Probe Synthesis Kit. Primers are listed in supplemental materials. The blot was hybridized for 16 h at 49 °C, washed in 2× SSC with 0.1 % SDS at room temperature, and then in 0.1× SSC, 0.1 % SDS for 45 min at 71 °C twice. Anti-digoxigenin antibody (1:10,000, Roche) was used to detect the probe, which was visualized with CSPD (Roche). Blots were visualized with an LAS-3000 Luminescent Image Analyzer (Fujifilm).
5-Aza-deoxycytidine treatment
Lymphoblast cell lines derived from either a repeat expansion carrier (ND14442) or a non-expanded ALS patient (ND16183) were grown in RPMI 1640 with 15 % fetal bovine serum and 2 mM L-glutamine in 37 °C with 5 % CO2. Cells were seeded at a concentration of 300,000 cells/ml in 10 ml in a T25 flask, and 0.5 μM 5-aza-dC (Sigma-Aldrich, St. Louis, MO, USA) was spiked daily into lymphoblast cells for 3 days followed by a 3-day recovery period without 5-aza-dC. RNA and DNA were extracted as described above. To assess the toxicity, untreated or 5-aza-dC treated cells were seeded at 300,000 cells/ml and treated 10 μM sodium arsenite or 100 μM chloroquine for 24 h in RPMI 1640 with 5 % FBS and 2 mM L-glutamine. After 24 h, cells and media were collected and measured in triplicate using a lactate dehydrogenase assay (Clontech, Mountain View, CA, USA) to calculate the percent LDH release into the media relative to total LDH from untreated cell pellets.
RNA fluorescent in situ hybridization (FISH)
Lymphoblast cell lines were fixed in 4 % paraformalde-hyde (PFA) in RNase-free PBS for 10 min on ice followed by permeabilization with 0.2 % Triton X-100 in PBS for 10 min on ice. Cells were prehybridized with 40 % formamide (Fisher Scientific)/1× SSC at 37 °C for 10 min. A locked nucleic acid (LNA) probe complementary to the hexanucleotide repeat (FAM-CCCCGGCCCCGGCCCC, batch #613510, Exiqon, Woburn, MA, USA) was denatured at 85 °C for 75 s prior to incubation with cells in hybridization buffer [40 % formamide, 1× SSC, 50 mM sodium phosphate, pH = 7, 10 % RNase-free dextran sulfate (Sigma-Aldrich) with 40 nM LNA probe] at 66 °C for 2 h. Cells were washed once with 0.1 % Tween-20 in 2× SSC at room temperature for 5 min, and three times in 0.1× SSC at 65 °C for 10 min each. Cells were then resuspended in Prolong Gold Antifade Reagent with DAPI (Life Technologies), coverslipped onto glass slides, and imaged on a Leica SPE confocal microscope (Leica Microsystems, Buffalo Grove, IL, USA). RNA foci numbers were scored in 200 cells for each cell line blinded to cell type using a 60× objective.
Frozen cerebellar cortex (100–200 mg) was dounce homogenized in 2 ml of 0.25 M sucrose with TKM (50 mM Tris–HCl, pH 7.5, 25 mM potassium chloride and 5 mM magnesium chloride). Two volumes of 2.3 M sucrose with TKM were mixed with the lysate. Nuclei were pelleted at 10,000×g for 10 min at 4 °C. Nuclei were fixed in 2 % PFA in RNase-free PBS for 10 min at room temperature and quenched with 0.83 M glycine (pH = 7.6). Nuclei were pelleted and resuspended in RNase-free PBS. Nuclei were pre-hybridized with hybridization buffer [40 % formamide, 10 mM ribonucleoside vanadyl complex (New England Biolabs), 1× SSC, 50 mM sodium phosphate, pH = 7, 10 % RNase-free dextran sulfate] for 10 min at 37 °C. The LNA probe complementary to the hexanucleotide repeat was denatured at 100 °C for 10 min in 95 % formamide and then hybridized to nuclei for 12–16 h at 37 °C at 40 nM. Nuclei were washed and imaged as described above for LCLs. RNA foci numbers were scored in at least 100 nuclei for each case using a 60× objective.
RANT immunohistochemistry
Polyclonal anti-(GA)15 and polyclonal anti-(GP)15 antibodies were generated by immunizing rabbits with a purified fusion protein containing his-tagged maltose-binding protein (MBP) with (GA)15 or (GP)15 at the C-terminus. The GA or GP repeats’ fragment was engineered using the primers as described in the supplemental materials (Integrated DNA Technologies). The annealed fragment was ligated into the pDB.His.MBP vector (DNASU Plasmid Repository, AZ, USA) at the NdeI–EcoRI cloning sites. The his-tagged MBP-GA15 and MBP-GP15 fusion proteins were expressed in BL21 bacterial cells upon IPTG induction and purified using Ni–NTA Superflow (Qiagen, CA, USA). (GR)15 antibody was generated by immunizing rabbits with an aminohexanoic acid linked synthetic peptide (C-Ahx-(GR15)) conjugated to KLH and affinity purified using SulfoLinkR-immobilized immunogen peptide (Thermo Scientific). Cerebellar sections were stained with RANT antibodies using standard ABC methods with microwave antigen retrieval [34], and at least 300 granular neurons per case were assessed using a 100× objective blinded to methylation status to determine the percentage of neurons with RANT inclusions.
RANT biochemistry
Frozen cerebellar cortex was homogenized in a series of extraction buffers intermixed with pelleting of insoluble material by ultracentrifugation at 135,000×g for 30 min. The extraction buffers included RIPA buffer (5 ml buffer per gram tissue; 50 mM Tris pH 7.3, 150 mM NaCl, 0.1 % SDS, 0.5 % sodium deoxycholate, 1 % NP-40 with protease inhibitors), myelin floatation buffer (5 ml buffer per gram tissue; 10 mM Tris pH 7.5, 500 mM NaCl, 2 mM EDTA, 1 mM DTT, 30 % sucrose with protease inhibitors), DNAse buffer containing (Promega, 1 unit of RQ DNAse per 10 mg tissue in 1× reaction buffer, incubated at 37° for 30 min) and sarkosyl buffer (5 ml buffer per gram tissue with sonication; 1 % sarkosyl, 10 mM Tris pH 7.5, 500 mM NaCl, 2 mM EDTA, 1 mM DTT, 10 % sucrose with protease inhibitors). The remaining insoluble material was pelleted and sonicated in SDS buffer (1 ml per gram tissue; 2 % SDS, 50 mM Tris, pH 7.6). Five μl of SDS lysates was dot blotted onto nitrocellulose using a Biorad Bio-Dot apparatus and washed with 300 μl of SDS buffer containing β-mercaptoethanol. Dot blots were blocked with 5 % milk, blotted with RANT-specific antibodies and visualized by film using enhanced chemiluminescence.
Statistical analyses
Two-way ANOVA, one-way ANOVA, t tests, linear regression and Fisher’s exact test were used as described using GraphPad Prism software (GraphPad, San Diego, CA, USA). Post-hoc analyses were Bonferroni corrected. All statistical tests were two-sided.
Results
Methylation of the C9orf72 promoter
Methylation of repeat expansions can reduce gene transcription as exemplified by Fragile X syndrome [49]. To determine whether the C9orf72 hexanucleotide repeat expansion is methylated, cerebellar genomic DNA from eight repeat expansion carriers and eight non-expanded controls was digested using three restriction enzymes: (1) MspI that cuts the hexanucleotide repeat irrespective of methylation status, (2) HpaII that cuts the hexanucleotide repeat only if it is not methylated, and (3) MspJI that cuts the hexanucleotide repeat only if it contains methylated CpGs. Digested DNA was then amplified using repeat primed PCR as a qualitative measure of whether the repeat region is methylated. Repeat-primed PCR allows for partial amplification of the expanded hexanucleotide repeat resulting in a characteristic tapering sawtooth pattern, and is used because conventional PCR is unable to amplify repeat expansions due to their size and high GC content [16, 53]. Non-expanded controls demonstrated the pattern expected of unmethylated DNA in which DNA was amplified after mock and MspJI digestion, and was not amplified after HpaII and MspI digestion (Fig. 1a). The same pattern was observed for repeat expansion carriers with amplification of the repeat expansion after MspJI digestion but no amplification after HpaII digestion, indicating that the repeat expansion is not methylated (Fig. 1a). To confirm that MspJI is able to cut the hexanucleotide repeat if it is methylated, DNA from a repeat expansion carrier was in vitro methylated using CpG methyltransferase (M. SssI) and digested with MspJI. Repeat primed PCR analysis showed poor amplification of in vitro methylated DNA relative to unmethylated DNA (Supplemental Figure 1), demonstrating the ability of MspJI to cut methylated GGGGCC repeats.
Fig. 1.

Hypermethylation of the C9orf72 promoter. a Cerebellar DNA from control (n = 8, left) and repeat-expanded cases (n = 8, right) were mock digested (no enzyme) or digested with MspI, HpaII or MspJI. DNA was subject to repeat primed PCR and representative electropherograms are shown. b Top panel shows a schematic of the bisulfite sequenced regions where filled boxes are exons, open boxes are CpG islands, and the star is the GGGGCC repeat expansion. Amplicon A covers the first CpG island, amplicon B covers the first half of the second CpG island and amplicon C covers the second half of the second CpG island. The bottom panels are summaries of bisulfite cloning results in which cerebellar DNA from four C9orf72 repeat expansion carriers and four control cases (n = 20–21 clones per genotype) was sequenced. Each oval represents a single CpG dinucleotide where unfilled oval represents an unmethylated CpG dinucleotide (0–10 % of clones) and a filled oval represents a methylated CpG dinucleotide (10–25 % of clones). Methylation over 25 % was not observed. c Top panel shows a schematic of the 5′ end of C9orf72 including the differentially methylated region (DMR, shaded) upstream of the 1st coding exon (E1) of C9orf72. The dinucleotide deletion polymorphism (rs200034037) and HhaI/HpaII cut sites are shown as arrows and the star is the hexanucleotide repeat expansion upstream of the 2nd coding exon (E2). DNA from C9orf72 promoter hypermethylated repeat-expanded cases (n = 3) that contain the polymorphism was mock digested (no enzyme) or digested with HpaII and HhaI. DNA from case 1 is from a lymphoblastoid cell line (ND14442) while DNA from cases 2 and 3 is from peripheral blood. The region flanking the deletion and restriction enzyme cut sites were amplified and run on a polyacrylamide gel to separate the major vs. minor rs200034037 alleles. Representative sequencing chromatograms of mock digested or HhaI/HpaII-digested DNA are shown where the gray area denotes the sequences demonstrating monoallelic vs. biallelic sequences downstream of rs200034037
We then extended our methylation analysis to include the two CpG islands adjacent to the repeat expansion (open boxes in Fig. 1b). A bisulfite cloning screen was performed on cerebellar DNA from four C9orf72 expansion carriers and four non-expanded controls to identify methylated CpG dinucleotides. PCR was used to amplify three regions covering both CpG islands from bisulfite-converted DNA (amplicons A–C, Fig. 1b). Amplified DNA was cloned, and individual clones (≥5 per case) were sequenced. Increased methylation (10–25 % of clones) was observed in the vicinity of the 5′ CpG island upstream of the hexanucleotide repeat region (amplicon A, Fig. 1b). The second CpG island downstream of the repeat expansion was not methylated in both repeat expanded and control cases (amplicons B and C, Fig. 1b). The sequencing results for all clones are provided in Supplemental Figure 1. These results confirm recently published studies demonstrating promoter hypermethylation in some repeat expansion mutation cases [65].
Since methylation was observed only in C9orf72 mutation cases, we hypothesized that promoter methylation occurs in cis to the repeat expansion. To determine whether methylation was monoallelic vs. biallelic, we identified three hypermethylated C9orf72 mutation cases heterozygous for the rs200034037 polymorphism, a dinucleotide deletion with an allele frequency of ~25.6 %. This polymorphism lies upstream of HhaI and HpaII restriction enzyme cut sites within the differentially methylated region (DMR). Both HhaI and HpaII only cut unmethylated DNA. The DMR was amplified after HhaI/HpaII double digestion and subject to PAGE electrophoresis (Fig. 1c). In all three cases, only the major rs200034037 allele was amplified after digestion of unmethylated DNA, in contrast with mock-digested DNA in which both major and minor alleles were observed. These results were confirmed by Sanger sequencing which showed biallelic sequences after the rs200034037 polymorphism in mock-digested DNA. This is in contrast with HhaI/HpaII-digested DNA which showed only the major allele sequence (color traces, bottom of Fig. 1c). Thus, C9orf72 promoter methylation is monoallelic, consistent with promoter hypermethylation in cis relative to the repeat expansion.
Methylation of repeat expansion carrier brains
To confirm the presence of promoter methylation in a larger cohort of subjects, we developed an alternative method based on the HhaI restriction enzyme recognition site within the DMR (Fig. 2a). HhaI digestion of unmethylated DNA was coupled with quantitative PCR (qPCR) which demonstrated a shift in DNA amplification curves relative to mock-digested DNA (Fig. 2b). This shift was used to calculate the percent DNA that is resistant to HhaI digestion as a measure of methylation status. This method proved to be robust, as standard curves using in vitro methylated DNA showed high linearity from 0 to 100 % methylation (Fig. 2c).
Fig. 2.
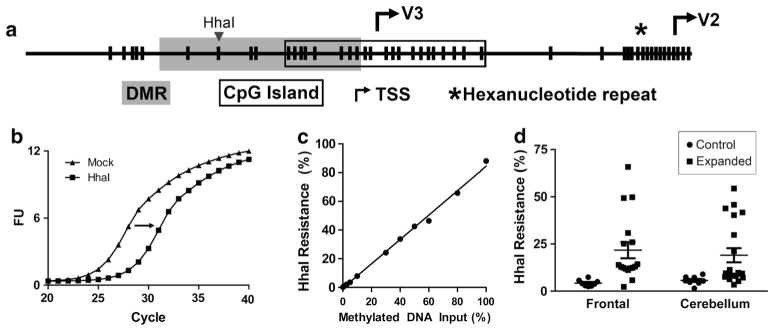
C9orf72 promoter hypermethylation in repeat expanded and control brain. a Schematic representation of the 5′ end of the C9orf72 gene in which individual CpG dinucleotides are designated by vertical bars, the upstream CpG island is designated with an open box, the TSS for V2 and V3 transcripts are designated by arrows, and the hexanucleotide repeat region is designated by a star. The differentially methylated region (DMR) is shaded. The HhaI restriction enzyme recognition site in the differentially methylated region is shown. b Representative qPCR amplification curves for mock (no enzyme) versus HhaI-digested DNA demonstrating a shift in the amplification curve upon DNA digestion. The magnitude of this shift is used to calculate the % DNA resistant to HhaI digestion as a measure of DNA methylation. c DNA from control LCLs was in vitro methylated with MSssl, and various ratios of methylated and mock methylated DNA were tested. Digest qPCR quantification for HhaI resistance was plotted for increasing amounts of in vitro methylated DNA input. d HhaI digest resistance as assessed by digest qPCR of frontal cortex (n = 8–17) or cerebellum (n = 8–20) DNA from control (circles) or repeat expansion cases (squares) is shown. Individual values are plotted in addition to the mean and standard error. Two-way ANOVA: genotype p = 0.0009, region p = 0.8851, interaction p = 0.6445
This assay was used to confirm the presence of C9orf72 promoter methylation using DNA from the frontal cortex and cerebellum of repeat-expanded and non-expanded cases. Both the frontal cortex and cerebellum of C9orf72 mutation carriers exhibited higher methylation than controls (p = 0.0009, Fig. 2d). Despite this highly significant result, C9orf72 promoter methylation for many C9orf72 mutation carriers was low and within the normal range, suggesting that C9orf72 promoter methylation is not driving disease pathogenesis.
C9orf72 RNA expression in lymphoblast cell lines
We analyzed ENCODE CAGE-seq data to determine whether there are differences in C9orf72 transcription across diverse cell lineages [54]. C9orf72 expression is highly variable across different cell lines, with both hematopoietic and neuronal cell lines expressing similar levels of C9orf72 in contrast with epithelial, fibroblastic or other embryonic or mesenchymal cell types (Fig. 3a). CAGE-seq data also indicated that of the three annotated C9orf72 mRNAs, variant 2 (V2) represented the major RNA transcript, representing 92.6 ± 1.2 % of all C9orf72 transcripts (Fig. 3b). Based on these analyses, we turned to lymphoblast cell lines (LCLs) to demonstrate that methylation is associated with reduced C9orf72 expression. LCLs from non-expanded (ND16183) versus expanded ALS cases (ND11836, ND10966 and ND14442) were analyzed by Southern blotting using a probe specific to C9orf72 which confirmed the presence of large repeat expansions in the three mutant LCLs (Fig. 3c). Methylation was very low (<1 %) in non-expanded ND16183 cells and was high in two expanded cell lines (ND10966 and ND14442, Fig. 3d). Despite harboring the hexanucleotide repeat expansion, ND11836 cells were not methylated, akin to our results above which showed many carriers exhibit minimal C9orf72 promoter methylation.
Fig. 3.

C9orf72 methylation inhibits expression of mutant RNA. a ENCODE CAGE-seq quantification of C9orf72 mRNA. The total number of C9orf72 sequence tags was normalized for number of total sequence reads, shown as mean log2 transformed tags per million (TPM) ± SE. Cell lines were divided into different cell lineages as labeled, with other representing various mesenchymal and embryonic stem cell lineages. b ENCODE CAGE-seq quantification of C9orf72 V2 mRNA relative to total C9orf72 mRNA expression. The number of V2 tag sequences was normalized to total C9orf72 tag sequences, shown as mean % of total ± SE. c Southern blot of LCL DNA from non-expanded (ND16183) and expanded (ND11836, ND10966 and ND14442) cultures using a probe specific for C9orf72 that recognizes the normal allele (bottom) and expanded alleles (top). Molecular weight markers are shown as indicated. d HhaI resistance from non-expanded (ND16183) and expanded (ND11836, ND10966 and ND14442) LCLs, shown as mean + SE. One-way ANOVA: p < 0.0001. ****p < 0.0001 relative to ND16183. Each cell line was measured in triplicate. e–h RT-qPCR quantification shown as mean + SE (n = 3–5) for total mRNA (e), V2 mRNA (f), V3 mRNA (g) and intronic RNA (h) from control (left) or expanded (right) lymphoblast cells. *p < 0.05, **p < 0.01, ****p < 0.0001
C9orf72 mRNA was measured using RT-qPCR which demonstrated that the unmethylated but repeat-expanded cell line, ND11836, showed essentially normal total C9orf72 mRNA levels (Fig. 3e). Despite expressing normal levels of total C9orf72 mRNA, ND11836 cells demonstrated increased usage of the alternative upstream TSS, as shown by the increase in V3 C9orf72 mRNA relative to control cells (Fig. 3g). Since transcription from the upstream TSS results in transcription through intron 1 which contains the hexanucleotide repeat, we speculated that the repeat expansion mutation is associated with accumulation of intron 1-containing RNA. Indeed, using primers 5′ to the hexanucleotide repeat within intron 1, unmethylated expanded cells exhibited over fourfold more intronic C9orf72 RNA relative to control cells (Fig. 3h).
In contrast, expanded cells with C9orf72 promoter hypermethylation exhibited reduced total (Fig. 3e) and V2 (Fig. 3f) mRNA relative to control cells. Promoter methylation was also associated with reduced expression from the alternative TSS, as determined by reduced V3 mRNA expression relative to unmethylated expanded cells (Fig. 3g). The reduction of V3 mRNA expression was coupled to reduced intronic RNA accumulation in methylated expanded cells relative to unmethylated expanded cells (Fig. 3h).
To verify that the C9orf72 mutation causes a shift in TSS usage and to confirm that methylation inhibits C9orf72 RNA levels, the effect of DNA demethylation was tested in hypermethylated expanded LCLs using the DNA methyltransferase inhibitor 5-aza-2′-deoxycytidine (5-aza-dC). Repeat-expanded lymphoblast cells (ND14442) and non-expanded control cells (ND16183) were treated with 5-aza-dC for 3 days followed by a 3-day recovery without 5-aza-dC. 5-aza-dC treatment resulted in a 41.8 % reduction in C9orf72 promoter methylation (p < 0.0001, Fig. 4a). 5-aza-dC treatment increased total (p < 0.05, Fig. 4b) and V3 (p < 0.001, Fig. 4d) C9orf72 mRNA expression in expanded but not control cells, demonstrating that C9orf72 promoter methylation is linked to transcriptional silencing of mutant C9orf72. However, V2 mRNA was not significantly changed in expanded cells upon demethylation (Fig. 4c). Thus, promoter demethylation reverses transcriptional silencing of C9orf72 but not of the major V2 transcript. Rather, demethylation rescued only total and V3 mRNA indicating that the expanded allele is preferentially transcribed from the upstream TSS. This alternative TSS usage translated into a marked increase in intronic C9orf72 accumulation (p < 0.01, Fig. 4e). Thus, methylation inhibits the accumulation of potentially toxic intronic RNA by inhibiting transcription through the repeat expansion.
Fig. 4.

C9orf72 promoter demethylation promotes toxic RNA accumulation. a HhaI resistance shown as mean + SE (n = 5) from control (left) or expanded (right) lymphoblast cells treated with 5-aza-dC (open) or untreated (filled). Two-way ANOVA: genotype p < 0.0001, treatment p < 0.0001, interaction p < 0.0001. ****p < 0.0001. b–e RT-qPCR quantification shown as mean + SE (n = 5) for total mRNA (b), V2 mRNA (c), V3 mRNA (d) and intronic RNA (e) from control (left) or expanded (right) lymphoblast cells treated with 5-aza-dC (open) or untreated (filled). Two-way ANOVA for total (genotype p = 0.0087, treatment p = 0.0264, interaction p = 0.0681), V2 (genotype p < 0.0001, treatment p = 0.5197, interaction p = 0.3579), V3 (genotype p = 0.026, treatment p = 0.0006, interaction p = 0.0076) and intronic RNA (genotype p = 0.0001, treatment p = 0.0259, interaction p = 0.0397). *p < 0.05, **p < 0.01. f–g Control (f) and expanded cells (g) that were untreated (filled) or 5-aza-dC treated (open) were assessed for toxicity (%LDH release relative to untreated, mean + SE, n = 4–8) after 24 h of no additional treatment (left), arsenite (10 μM, middle) or chloroquine (100 μM, right). Two-way ANOVA for control cells (5-aza-dC p = 0.0358, stressor p < 0.0001, interaction p = 0.0773) and mutant cells (5-aza-dC p < 0.0001, stressor p < 0.0001, interaction p = 0.1471). *p < 0.05, **p < 0.01, ***p < 0.001
Selective vulnerability of hypomethylated cells
The accumulation of repeat-containing intronic RNA has been postulated to promote neurodegeneration. Since promoter hypermethylation inhibited intronic RNA levels, we hypothesized that hypermethylation may protect cells from cellular insults, such as oxidative stressors which lead to stress granule formation or altered proteostasis. Both processes have been implicated in ALS and FTD [36, 37]. To test this hypothesis, expanded and control cells were dem-ethylated with 5-aza-dC and exposed to sodium arsenite or chloroquine, known to induce stress granules or inhibit autophagy, respectively. Toxicity was assessed by measuring lactate dehydrogenase (LDH) release into media. In both control and expanded cell lines, 5-aza-dC resulted in mild toxicity compared to untreated cells consistent with the known toxicity profile of 5-aza-dC (control p < 0.01, mutant p < 0.05, Fig. 4f, g). Importantly, demethylation of control non-expanded cells with 5-aza-dC had no effect on arsenite or chloroquine toxicity, indicating that global hypo-methylation does not result in an altered response to these cellular stressors (Fig. 4f). In contrast, hypomethylation of mutant cells with 5-aza-dC resulted in enhanced sensitivity to both arsenite and chloroquine (arsenite p < 0.05, chloroquine p < 0.001, Fig. 4g). Thus, reactivation of mutant C9orf72 transcription which results in the accumulation of mutant intronic C9orf72 RNA appears to be associated with increased vulnerability to cellular stressors.
Hypermethylation, RNA foci and RANT pathology
Having demonstrated that C9orf72 promoter hypermethylation was associated with lower levels of intronic RNA using RT-qPCR, we hypothesized that hypermethylation is associated with reduced accumulation of RNA foci. LCLs were studied using in situ hybridization with a fluorescently labeled locked nucleic acid (LNA) probe that recognizes the GGGGCC hexanucleotide repeat. RNA foci were observed in expanded cell lines but not in non-expanded cells (Fig. 5a). LCLs were scored for the presence of RNA foci which demonstrated that 12 % of ND11836 cells (expanded and unmethylated) exhibited RNA foci, with several cells exhibiting multiple RNA foci (Fig. 5c). In contrast, expanded and methylated cell lines, ND10966 and ND14442, exhibited significantly fewer RNA foci compared to ND11836 (1.5 % for ND10966, p < 0.0001; 3 % for ND14442, p = 0.00094, Fisher’s exact test, Fig. 5c).
Fig. 5.

C9orf72 promoter hypermethylation inhibits RNA foci accumulation. a Representative in situ hybridization images of non-expanded (ND16183), unmethylated and expanded (ND11836), and hypermethylated and expanded (ND10966 and ND14442) LCLs. RNA foci in green are highlighted with arrows, and nuclei are counterstained blue with DAPI. Scale bar 10 μm. b Representative in situ hybridization images of non-expanded (left), hypomethylated and expanded (middle), and hypermethylated and expanded (right) cerebellar nuclei. RNA foci in green are highlighted with arrows, and nuclei are counterstained blue with DAPI. Arrowhead points to non-specific autofluorescence. Scale bar 5 μm. c The number of RNA foci in LCLs was scored (n = 200 cells per cell line), shown as a stacked bar graph to demonstrate the proportion of LCLs with zero, one or multiple RNA foci. d The percentage of cerebellar nuclei with RNA foci from 12 repeat-expanded cases is shown as a function of C9orf72 methylation. A linear regression was performed (R2 = 0.4633, p = 0.0148)
Human brain tissue was also used to test whether C9orf72 hypermethylation is associated with reduced accumulation of RNA foci. Intact nuclei were isolated from post-mortem cerebellum and stained for RNA foci. RNA foci were only observed in repeat-expanded cases and not in non-expanded control cases (Fig. 5b). The percentage of nuclei containing RNA foci was quantified from 12 repeat-expanded cases which revealed an inverse relationship between C9orf72 methylation and the percentage of nuclei with RNA foci (p = 0.0148, Fig. 5d).
RANT aggregates accumulate as a consequence of non-ATG translation of mutant C9orf72 RNA which may contribute to disease pathogenesis [2, 46]. Given that hypermethylation reduced mutant C9orf72 expression, it seemed likely that hypermethylation would be associated with reduced RANT pathology. To understand the relationship between methylation and RANT pathology, cerebellar tissue sections from hypomethylated (<12 % HhaI resistance) and hypermethylated (>12 % HhaI resistance) repeat expansion carriers were stained with antibodies specific for GA (glycine–alanine), GP (glycine–proline), or GR (glycine–arginine) repeats corresponding to the three different sense reading frames of the repeat expansion. The 12 % threshold corresponds to three standard deviations above the mean HhaI resistance values for control, non-expanded cases. Immunohistochemistry showed abundant numbers of inclusions in hypomethylated C9orf72 expansion cases and markedly fewer RANT inclusions in hypermethylated cases (Fig. 6a). Quantification of the percent of cerebellar granule neurons with RANT inclusions demonstrated a significant reduction in RANT pathology burden associated with C9orf72 promoter hypermethylation (Fig. 6b). To confirm this finding, insoluble proteins were extracted from hypermethylated and hypomethylated cerebellum and assayed by dot blot analysis which again demonstrated that hypermethylation was associated with reduced accumulation of GA, GP, and GR proteins (Fig. 6c). These results show a tight relationship between C9orf72 promoter methylation and RANT pathology, and indicate that endogenous transcriptional silencing reduces the downstream pathologic features associated with the hexanucleotide repeat expansion.
Fig. 6.

C9orf72 promoter hypermethylation and RANT accumulation in C9orf72 repeat expansion carriers. a Representative immunohisto-chemistry of cerebellar granular neurons using antibodies recognizing glycine–alanine (GA), glycine–proline (GP) and glycine–arginine (GR) dipeptide repeat proteins. Aggregates appear brown while nuclei are counterstained with hematoxylin. Scale bar 10 μm. b Quantification of RANT pathology in 11 hypomethylated and 7 hypermethylated C9orf72 repeat expansion carriers shown as mean + standard error of the percentage of cerebellar granular neurons containing GA (left), GP (middle) or GR (right) aggregates. Two-way ANOVA: p < 0.0001 (pathology type), p = 0.0080 (methylation), 0.0002 (interaction). c Sarkosyl-insoluble material was biochemically extracted and subject to dot blot analysis using antibodies that recognize GA, GP or GR dipeptide repeat proteins in five non-expanded controls (top row), five hypermethylated C9orf72 repeat expansion carriers (middle row), and five hypomethylated C9orf72 repeat expansion carriers (bottom row)
Discussion
We examined the downstream consequences of C9orf72 promoter hypermethylation and provide evidence that C9orf72 promoter methylation found in a subset of C9orf72 expansion carriers may be protective against toxicity associated with the C9orf72 hexanucleotide repeat expansion. Hypermethylation [65] and repressive histone marks [3] have been previously observed in the promoter region of C9orf72 in repeat expansion carriers, but the downstream consequences of these changes were not known. We found that (1) C9orf72 promoter but not the hexanucleotide repeat is hypermethylated in a subset of mutation carriers in cis with the repeat expansion mutation, (2) the repeat expansion is associated with alternative TSS usage, (3) promoter methylation leads to transcriptional gene silencing of both V2 and V3 C9orf72 transcripts, (4) methylation inhibits the accumulation of potentially toxic intronic RNA in LCLs and brain, (5) demethylation of C9orf72 promoter appears to be associated with increased vulnerability to cellular stressors, and (6) hypermethylation is associated with reduced accumulation of RNA foci and RANT aggregates.
Repeat expansion mutations can cause disease through a variety of means including gain and loss of function mechanisms. Mounting evidence suggests that the C9orf72 mutation results in a toxic gain of function linked to the accumulation of either mutant intronic RNA within RNA foci, or the accumulation of RANT aggregates. In this regard, the C9orf72 mutation may be similar to the trinucleotide repeat expansion mutation affecting DM1 associated with myotonic dystrophy where repeat-expanded RNA accumulates as RNA which sequesters muscleblind protein [44]. In fact, Pur α[66], hnRNP A3 [45], hnRNP-H [35] and ADARB2 [20] have been shown to bind to GGGGCC repeats and may be sequestered within RNA foci, resulting in altered RNA metabolism. Furthermore, toxicity through increased vulnerability to cellular stressors caused by autophagy inhibitors and glutamate in induced pluripotent stem cell-derived neurons are linked to expression of the hexanucleotide repeat [1, 20]. Hexanucleotide repeat length also correlates with toxicity in neuroblastoma cells [35]. Collectively, these data support the hypothesis that the accumulation of mutant intronic RNA through transcription of the repeat expansion contributes to vulnerability to cellular stress.
Alternatively, the repeat expansion mutation has also been associated with a reduction in C9orf72 mRNA and protein together with various epigenetic marks indicative of transcriptional silencing [3, 64, 65]. This would be analogous to Fragile X syndrome where repeat expansion mutations in FMR1 cause dense promoter hypermethylation and transcriptional silencing, resulting in a loss of function [49, 61]. Notably, both nonsense and missense FMR1 mutations are also associated with Fragile X syndrome, supporting the loss of function hypothesis [11, 12, 25, 48]. In contrast, nonsense and missense C9orf72 mutations were not found in a large ALS cohort [29]. Moreover, individuals homozygous for the C9orf72 mutation are phenotypically similar to heterozygous carriers, which argues against a loss of function mechanism [14, 24]. Experimental data are conflicting regarding C9orf72 haploinsufficiency, as reduction of C9orf72 in mice with antisense oligonucleotides does not result in a neurodegenerative phenotype in mice while knockdown of the C9orf72 homolog in zebrafish results in motor neuron axonal degeneration [8, 32].
We demonstrate here that these two mechanisms may not be independent of each other, but rather that transcriptional silencing of C9orf72 appears to inhibit the formation of RNA foci and RANT aggregates (Fig. 7). Given that promoter methylation can suppress hexanucleotide repeat expansion-associated pathologies, we propose that epigenetic silencing of C9orf72 may be a protective counter-regulatory response to the presence of the hexanucleotide repeat expansion. This is in contrast with epigenetic silencing seen in Fragile X syndrome, where silencing of FMR1 causes disease. While additional studies are clearly needed to determine the functional consequences of reduced C9orf72 expression, our observation that demethylation of the C9orf72 promoter appears to be associated with reduced cellular vulnerability supports the hypothesis that the C9orf72 mutation causes disease due to a deleterious gain of function.
Fig. 7.
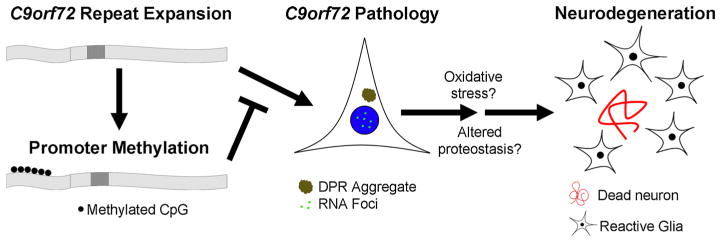
C9orf72 promoter methylation model. C9orf72 repeat expansions are associated with promoter hypermethylation in subset of mutation carriers. Expression of the repeat expansion leads to the accumulation of mutation-specific pathologies, namely RNA foci and DPR aggregates. The accumulation of mutant RNA is associated with increased vulnerability to cellular stressors, including oxidative and autophagic stress. Promoter hypermethylation inhibits these downstream effects by reducing the accumulation of RNA foci and/or DPR aggregates
Our study identifies two molecular mechanisms which contribute to the accumulation of repeat-expanded intronic RNA. First, the presence of the repeat expansion mutation is associated with increased usage of the upstream TSS. Expression from this alternative TSS results in expression of the hexanucleotide repeat. We observed alternative TSS usage in unmethylated mutant LCLs, and confirmed this finding using 5-aza-dC treated LCLs which showed that demethylation of the C9orf72 promoter reactivates V3 but not V2 expression. These results are consistent with a recent report showing alternative TSS usage in repeat-expanded neurons differentiated from induced pluripotent stem cells [55]. Alternative usage of the upstream TSS results in transcription through the hexanucleotide repeat expansion within intron 1, resulting in increased intronic RNA accumulation and the formation of RNA foci. The reason for alternative TSS usage is not clear, although the hexa-nucleotide repeat is known to form very stable configurations including G quadraplexes that may alter local chromatin structure and affect transcription [23, 26, 51]. The second mechanism that regulates intronic RNA accumulation is promoter methylation. Indeed, promoter methylation was associated with reduced levels of C9orf72 transcripts regardless of which TSS was used. Reduced transcription led to reduced intronic RNA accumulation and reduced numbers of RNA foci. This supports a model in which the repeat expansion mutation leads to downstream molecular pathologies which are inhibited by promoter methylation in a subset of cells and individuals (Fig. 7).
RANT aggregates have been proposed to be toxic [2, 46], perhaps through disruption of cellular pathways like autophagy or altered proteostasis [9, 50]. However, RANT pathology does not associate with severity of neurodegeneration and it is unclear whether RANT aggregates are pathogenic [39]. Regardless, we observed a strong correlation between hypermethylation and reduced C9orf72 RANT pathology within hypermethylated C9orf72 mutation carriers, again indicating that promoter hypermethylation is an endogenous pathway which inhibits hexanucleotide repeat expansion-associated aberrations. Indeed, C9orf72 promoter hypermethylation is the first known epigenetic modification that appears to inhibit the pathology in a neurodegenerative disease.
If methylation indeed modulates C9orf72 mutation pathways, C9orf72 methylation status may be used as a diagnostic biomarker to separate individuals into epigenetic subtypes. C9orf72 mutation carriers are clinically heterogeneous, ranging from rapidly fatal motor neuron disease to the so-called “slowly progressive” form of frontotemporal dementia [5, 13, 30, 31, 47, 56]. The molecular basis for this heterogeneity is largely unknown. Hexanucleotide repeat length measured from peripheral blood does not appear to correlate with disease phenotypes [4, 19, 63]. Due to the relatively low numbers of cases studied here, we did not observe any clinical parameters associated with C9orf72 hypermethylation.
In other polynucleotide expansion diseases, hypermethylation in the vicinity of repeat expansions has been linked to clinical outcomes. In Fragile X Syndrome, FMR1 methylation inversely correlates with intelligence quotient scores [15, 27, 38, 42, 43, 52, 57]. Similarly, FXN methylation in Friedreich’s ataxia also inversely correlates with disease severity [6, 22]. In both Fragile X syndrome and Friedreich’s ataxia, repeat expansion mutations result in a loss of function mutation that cause disease, consistent with hypermethylation leading to a more severe disease phenotype. In the case of FMR1, the repeat expansion is thought to result in epigenetic silencing of the mutant allele in cis, linked to the formation of RNA–DNA hybrids in early development [10, 17, 21]. Whether this represents a general mechanism responsible for hypermethylation adjacent to repeat expansion loci remains to be determined. In contrast with Fragile X syndrome and Friedreich’s ataxia, our study supports the hypothesis that the C9orf72 mutation causes disease through a gain of function mutation. Therefore, we predict that C9orf72 hypermethylation may be associated with milder clinical disease. It will be important to determine whether C9orf72 methylation correlates with clinical parameters relevant to disease in a larger cohort of C9orf72 mutation carriers.
Experimental models have also demonstrated heterogeneity in terms of C9orf72 RNA expression and molecular phenotypes. Given that methylation affects many of the downstream effects of the hexanucleotide repeat mutation, current efforts to develop and characterize cellular or other experimental models should include promoter methylation analysis [1, 20, 55]. Methylation status may be a confounding factor when trying to compare cell lines derived from different patients. Indeed, we observed significant differences between LCLs based on methylation status. Reported discrepancies regarding C9orf72 expression between various cell lines may similarly be linked to differences in methylation [55].
Finally, determining whether C9orf72 hypermethylation indeed modifies disease progression in C9orf72 mutation carriers will help guide the development of novel molecular therapies. Given that the C9orf72 mutation reduces gene expression, one approach may be to increase C9orf72 expression to reverse any deleterious effects of low C9orf72 expression [8]. While it remains possible that reduced expression of C9orf72 is deleterious, our results suggest that efforts to increase C9orf72 expression as a potential therapy for hexanucleotide repeat expansion carriers should proceed with caution as such approaches may lead to increased accumulation of potentially toxic RNA. In contrast, our results bolster recent efforts to develop molecular therapies for C9orf72 mutation carriers based on antisense oligonucleotides that target hexanucleotide repeat-expanded RNA for post-transcriptional degradation [20, 32, 55]. Another therapeutic possibility includes development of therapies that promote transcriptional silencing of mutant C9orf72, thereby reducing toxic RNA and downstream RANT pathology.
In summary, a subset of C9orf72 mutation carriers demonstrates C9orf72 promoter hypermethylation which may represent an endogenous protective response to the hexanucleotide repeat expansion. Promoter hypermethylation results in stable silencing of the mutant gene and reduction in the downstream pathologies associated with the C9orf72 mutation. Since transcriptional silencing is associated with a protective phenotype, this study supports the hypothesis that the C9orf72 hexanucleotide repeat expansion causes disease by a gain of toxic function as opposed to haploinsufficiency, and highlights an endogenous molecular pathway which may be amenable to future therapy development.
Supplementary Material
Acknowledgments
Cell lines (ND16183, ND11836, ND10966, and ND14442) and clinical data from the NINDS Repository (ccr.coriell.org/ninds) were used. We thank Dr. Linda Kwong, Yan Xu and the Center for Neurodegenerative Disease Research for providing RANT antibodies and autopsy materials. The authors would like to thank the patients and patients’ families who made this research possible. This study was supported in part by a grant from the Judith & Jean Pape Adams Foundation and by the National Institutes of Health (K08AG039510, T32AG00255, P30AG10125, P01AG017586, P01AG032953).
Footnotes
Electronic supplementary material: The online version of this article (doi:10.1007/s00401-014-1286-y) contains supplementary material, which is available to authorized users.
Contributor Information
Elaine Y. Liu, Translational Neuropathology Research Laboratory, Perelman School of Medicine at the University of Pennsylvania, 605B Stellar Chance Laboratories, 422 Curie Blvd, Philadelphia, PA 19104, USA. Department of Pathology and Laboratory Medicine, Perelman School of Medicine at the University of Pennsylvania, Philadelphia, USA
Jenny Russ, Translational Neuropathology Research Laboratory, Perelman School of Medicine at the University of Pennsylvania, 605B Stellar Chance Laboratories, 422 Curie Blvd, Philadelphia, PA 19104, USA. Department of Pathology and Laboratory Medicine, Perelman School of Medicine at the University of Pennsylvania, Philadelphia, USA.
Kathryn Wu, Translational Neuropathology Research Laboratory, Perelman School of Medicine at the University of Pennsylvania, 605B Stellar Chance Laboratories, 422 Curie Blvd, Philadelphia, PA 19104, USA. Department of Pathology and Laboratory Medicine, Perelman School of Medicine at the University of Pennsylvania, Philadelphia, USA.
Donald Neal, Department of Pathology and Laboratory Medicine, Perelman School of Medicine at the University of Pennsylvania, Philadelphia, USA.
Eunran Suh, Department of Pathology and Laboratory Medicine, Perelman School of Medicine at the University of Pennsylvania, Philadelphia, USA.
Anna G. McNally, Translational Neuropathology Research Laboratory, Perelman School of Medicine at the University of Pennsylvania, 605B Stellar Chance Laboratories, 422 Curie Blvd, Philadelphia, PA 19104, USA. Department of Pathology and Laboratory Medicine, Perelman School of Medicine at the University of Pennsylvania, Philadelphia, USA
David J. Irwin, Department of Pathology and Laboratory Medicine, Perelman School of Medicine at the University of Pennsylvania, Philadelphia, USA. Department of Neurology, Perelman School of Medicine at the University of Pennsylvania, Philadelphia, USA
Vivianna M. Van Deerlin, Department of Pathology and Laboratory Medicine, Perelman School of Medicine at the University of Pennsylvania, Philadelphia, USA
Edward B. Lee, Email: edward.lee@uphs.upenn.edu, Translational Neuropathology Research Laboratory, Perelman School of Medicine at the University of Pennsylvania, 605B Stellar Chance Laboratories, 422 Curie Blvd, Philadelphia, PA 19104, USA
References
- 1.Almeida S, Gascon E, Tran H, Chou HJ, Gendron TF, Degroot S, Tapper AR, Sellier C, Charlet-Berguerand N, Karydas A, Seeley WW, Boxer AL, Petrucelli L, Miller BL, Gao FB. Modeling key pathological features of frontotemporal dementia with C9ORF72 repeat expansion in iPSC-derived human neurons. Acta Neuropathol. 2013;126(3):385–399. doi: 10.1007/s00401-013-1149-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ash PE, Bieniek KF, Gendron TF, Caulfield T, Lin WL, Dejesus-Hernandez M, van Blitterswijk MM, Jansen-West K, Paul JW, 3rd, Rademakers R, Boylan KB, Dickson DW, Petrucelli L. Unconventional translation of C9ORF72 GGGGCC expansion generates insoluble polypeptides specific to c9FTD/ALS. Neuron. 2013;77(4):639–646. doi: 10.1016/j.neuron.2013.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Belzil VV, Bauer PO, Prudencio M, Gendron TF, Stetler CT, Yan IK, Pregent L, Daughrity L, Baker MC, Rademakers R, Boylan K, Patel TC, Dickson DW, Petrucelli L. Reduced C9orf72 gene expression in c9FTD/ALS is caused by histone trimethyla-tion, an epigenetic event detectable in blood. Acta Neuropathol. 2013;126(6):895–905. doi: 10.1007/s00401-013-1199-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Benussi L, Rossi G, Glionna M, Tonoli E, Piccoli E, Fostinelli S, Paterlini A, Flocco R, Albani D, Pantieri R, Cereda C, Forloni G, Tagliavini F, Binetti G, Ghidoni R. C9ORF72 hexanucleotide repeat number in frontotemporal lobar degeneration: a genotype–phenotype correlation study. J Alzheimers Dis. 2013 doi: 10.3233/JAD-131028. [DOI] [Google Scholar]
- 5.Boeve BF, Boylan KB, Graff-Radford NR, DeJesus-Hernandez M, Knopman DS, Pedraza O, Vemuri P, Jones D, Lowe V, Murray ME, Dickson DW, Josephs KA, Rush BK, Machulda MM, Fields JA, Ferman TJ, Baker M, Rutherford NJ, Adamson J, Wszolek ZK, Adeli A, Savica R, Boot B, Kuntz KM, Gavrilova R, Reeves A, Whitwell J, Kantarci K, Jack CR, Jr, Parisi JE, Lucas JA, Petersen RC, Rademakers R. Characterization of fron-totemporal dementia and/or amyotrophic lateral sclerosis associated with the GGGGCC repeat expansion in C9ORF72. Brain. 2012;135(Pt 3):765–783. doi: 10.1093/brain/aws004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Castaldo I, Pinelli M, Monticelli A, Acquaviva F, Giacchetti M, Filla A, Sacchetti S, Keller S, Avvedimento VE, Chiariotti L, Cocozza S. DNA methylation in intron 1 of the frataxin gene is related to GAA repeat length and age of onset in Friedreich ataxia patients. J Med Genet. 2008;45(12):808–812. doi: 10.1136/jmg.2008.058594. [DOI] [PubMed] [Google Scholar]
- 7.Chio A, Borghero G, Restagno G, Mora G, Drepper C, Traynor BJ, Sendtner M, Brunetti M, Ossola I, Calvo A, Pugliatti M, Sotgiu MA, Murru MR, Marrosu MG, Marrosu F, Marinou K, Mandrioli J, Sola P, Caponnetto C, Mancardi G, Mandich P, La Bella V, Spataro R, Conte A, Monsurro MR, Tedeschi G, Pisano F, Bartolomei I, Salvi F, Lauria Pinter G, Simone I, Logroscino G, Gambardella A, Quattrone A, Lunetta C, Volanti P, Zollino M, Penco S, Battistini S, Renton AE, Majounie E, Abramzon Y, Conforti FL, Giannini F, Corbo M, Sabatelli M. Clinical characteristics of patients with familial amyotrophic lateral sclerosis carrying the pathogenic GGGGCC hexanucleotide repeat expansion of C9ORF72. Brain. 2012;135(Pt 3):784–793. doi: 10.1093/brain/awr366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ciura S, Lattante S, Le Ber I, Latouche M, Tostivint H, Brice A, Kabashi E. Loss of function of C9orf72 causes motor deficits in a zebrafish model of amyotrophic lateral sclerosis. Ann Neurol. 2013 doi: 10.1002/ana.23946. [DOI] [PubMed] [Google Scholar]
- 9.Cleary JD, Ranum LP. Repeat-associated non-ATG (RAN) translation in neurological disease. Hum Mol Genet. 2013;22(R1):R45–R51. doi: 10.1093/hmg/ddt371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Colak D, Zaninovic N, Cohen MS, Rosenwaks Z, Yang WY, Gerhardt J, Disney MD, Jaffrey SR. Promoter-bound tri-nucleotide repeat mRNA drives epigenetic silencing in fragile X syndrome. Science. 2014;343(6174):1002–1005. doi: 10.1126/science.1245831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Collins SC, Bray SM, Suhl JA, Cutler DJ, Coffee B, Zwick ME, Warren ST. Identification of novel FMR1 variants by massively parallel sequencing in developmentally delayed males. Am J Med Genet A. 2010;152A(10):2512–2520. doi: 10.1002/ajmg.a.33626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Collins SC, Coffee B, Benke PJ, Berry-Kravis E, Gilbert F, Oostra B, Halley D, Zwick ME, Cutler DJ, Warren ST. Array-based FMR1 sequencing and deletion analysis in patients with a fragile X syndrome-like phenotype. PLoS One. 2010;5(3):e9476. doi: 10.1371/journal.pone.0009476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cooper-Knock J, Hewitt C, Highley JR, Brockington A, Milano A, Man S, Martindale J, Hartley J, Walsh T, Gelsthorpe C, Baxter L, Forster G, Fox M, Bury J, Mok K, McDermott CJ, Traynor BJ, Kirby J, Wharton SB, Ince PG, Hardy J, Shaw PJ. Clinico-pathological features in amyotrophic lateral sclerosis with expansions in C9ORF72. Brain. 2012;135(Pt 3):751–764. doi: 10.1093/brain/awr365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cooper-Knock J, Higginbottom A, Connor-Robson N, Bayatti N, Bury JJ, Kirby J, Ninkina N, Buchman VL, Shaw PJ. C9ORF72 transcription in a frontotemporal dementia case with two expanded alleles. Neurology. 2013;81(19):1719–1721. doi: 10.1212/01.wnl.0000435295.41974.2e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.de Vries BB, Jansen CC, Duits AA, Verheij C, Willemsen R, van Hemel JO, van den Ouweland AM, Niermeijer MF, Oostra BA, Halley DJ. Variable FMR1 gene methylation of large expansions leads to variable phenotype in three males from one fragile X family. J Med Genet. 1996;33(12):1007–1010. doi: 10.1136/jmg.33.12.1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.DeJesus-Hernandez M, Mackenzie IR, Boeve BF, Boxer AL, Baker M, Rutherford NJ, Nicholson AM, Finch NA, Flynn H, Adamson J, Kouri N, Wojtas A, Sengdy P, Hsiung GY, Karydas A, Seeley WW, Josephs KA, Coppola G, Geschwind DH, Wszolek ZK, Feldman H, Knopman DS, Petersen RC, Miller BL, Dickson DW, Boylan KB, Graff-Radford NR, Rademakers R. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron. 2011;72(2):245–256. doi: 10.1016/j.neuron.2011.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Devys D, Biancalana V, Rousseau F, Boue J, Mandel JL, Oberle I. Analysis of full fragile X mutations in fetal tissues and monozygotic twins indicate that abnormal methylation and somatic heterogeneity are established early in development. Am J Med Genet. 1992;43(1–2):208–216. doi: 10.1002/ajmg.1320430134. [DOI] [PubMed] [Google Scholar]
- 18.Dion V, Wilson JH. Instability and chromatin structure of expanded trinucleotide repeats. Trends Genet. 2009;25(7):288–297. doi: 10.1016/j.tig.2009.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dols-Icardo O, Garcia-Redondo A, Rojas-Garcia R, Sanchez-Valle R, Noguera A, Gomez-Tortosa E, Pastor P, Hernandez I, Esteban-Perez J, Suarez-Calvet M, Anton-Aguirre S, Amer G, Ortega-Cubero S, Blesa R, Fortea J, Alcolea D, Capdevila A, Antonell A, Llado A, Munoz-Blanco JL, Mora JS, Galan-Davila L, Rodriguez De Rivera FJ, Lleo A, Clarimon J. Characterization of the repeat expansion size in C9orf72 in amyotrophic lateral sclerosis and frontotemporal dementia. Hum Mol Genet. 2013 doi: 10.1093/hmg/ddt460. [DOI] [PubMed] [Google Scholar]
- 20.Donnelly CJ, Zhang PW, Pham JT, Heusler AR, Mistry NA, Vidensky S, Daley EL, Poth EM, Hoover B, Fines DM, Maragakis N, Tienari PJ, Petrucelli L, Traynor BJ, Wang J, Rigo F, Bennett CF, Blackshaw S, Sattler R, Rothstein JD. RNA toxicity from the ALS/FTD C9ORF72 expansion is mitigated by antisense intervention. Neuron. 2013;80(2):415–428. doi: 10.1016/j.neuron.2013.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Eiges R, Urbach A, Malcov M, Frumkin T, Schwartz T, Amit A, Yaron Y, Eden A, Yanuka O, Benvenisty N, Ben-Yosef D. Developmental study of fragile X syndrome using human embryonic stem cells derived from preimplantation genetically diagnosed embryos. Cell Stem Cell. 2007;1(5):568–577. doi: 10.1016/j.stem.2007.09.001. [DOI] [PubMed] [Google Scholar]
- 22.Evans-Galea MV, Carrodus N, Rowley SM, Corben LA, Tai G, Saffery R, Galati JC, Wong NC, Craig JM, Lynch DR, Regner SR, Brocht AF, Perlman SL, Bushara KO, Gomez CM, Wilmot GR, Li L, Varley E, Delatycki MB, Sarsero JP. FXN methylation predicts expression and clinical outcome in Friedreich ataxia. Ann Neurol. 2012;71(4):487–497. doi: 10.1002/ana.22671. [DOI] [PubMed] [Google Scholar]
- 23.Fratta P, Mizielinska S, Nicoll AJ, Zloh M, Fisher EMC, Parkinson G, Isaacs AM. C9orf72 hexanucleotide repeat associated with amyotrophic lateral sclerosis and fronto-temporal dementia forms RNA G-quadruplexes. Sci Rep. 2012 doi: 10.1038/srep01016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fratta P, Poulter M, Lashley T, Rohrer JD, Polke JM, Beck J, Ryan N, Hensman D, Mizielinska S, Waite AJ, Lai MC, Gendron TF, Petrucelli L, Fisher EM, Revesz T, Warren JD, Collinge J, Isaacs AM, Mead S. Homozygosity for the C9orf72 GGGGCC repeat expansion in frontotemporal dementia. Acta Neuropathol. 2013;126(3):401–409. doi: 10.1007/s00401-013-1147-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gronskov K, Brondum-Nielsen K, Dedic A, Hjalgrim H. A nonsense mutation in FMR1 causing fragile X syndrome. Eur J Hum Genet. 2011;19(4):489–491. doi: 10.1038/ejhg.2010.223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Haeusler AR, Donnelly CJ, Periz G, Simko EA, Shaw PG, Kim MS, Maragakis NJ, Troncoso JC, Pandey A, Sattler R, Rothstein JD, Wang J. C9orf72 nucleotide repeat structures initiate molecular cascades of disease. Nature. 2014;507(7491):195–200. doi: 10.1038/nature13124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hagerman RJ, Hull CE, Safanda JF, Carpenter I, Staley LW, O’Connor RA, Seydel C, Mazzocco MM, Snow K, Thibodeau SN, et al. High functioning fragile X males: demonstration of an unmethylated fully expanded FMR-1 mutation associated with protein expression. Am J Med Genet. 1994;51(4):298–308. doi: 10.1002/ajmg.1320510404. [DOI] [PubMed] [Google Scholar]
- 28.Harms M, Benitez BA, Cairns N, Cooper B, Cooper P, Mayo K, Carrell D, Faber K, Williamson J, Bird T, Diaz-Arrastia R, Foroud TM, Boeve BF, Graff-Radford NR, Mayeux R, Chakraverty S, Goate AM, Cruchaga C. C9orf72 hexanucleotide repeat expansions in clinical Alzheimer disease. JAMA Neurol. 2013;70(6):736–741. doi: 10.1001/2013.jamaneurol.537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Harms MB, Cady J, Zaidman C, Cooper P, Bali T, Allred P, Cruchaga C, Baughn M, Libby RT, Pestronk A, Goate A, Ravits J, Baloh RH. Lack of C9ORF72 coding mutations supports a gain of function for repeat expansions in amyotrophic lateral sclerosis. Neurobiol Aging. 2013;34(9):2234. e2213–e2239. doi: 10.1016/j.neurobiolaging.2013.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hsiung GY, DeJesus-Hernandez M, Feldman HH, Sengdy P, Bouchard-Kerr P, Dwosh E, Butler R, Leung B, Fok A, Rutherford NJ, Baker M, Rademakers R, Mackenzie IR. Clinical and pathological features of familial frontotemporal dementia caused by C9ORF72 mutation on chromosome 9p. Brain. 2012;135(Pt 3):709–722. doi: 10.1093/brain/awr354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Khan BK, Yokoyama JS, Takada LT, Sha SJ, Rutherford NJ, Fong JC, Karydas AM, Wu T, Ketelle RS, Baker MC, Hernandez MD, Coppola G, Geschwind DH, Rademakers R, Lee SE, Rosen HJ, Rabinovici GD, Seeley WW, Rankin KP, Boxer AL, Miller BL. Atypical, slowly progressive behavioural variant fronto-temporal dementia associated with C9ORF72 hexanucleotide expansion. J Neurol Neurosurg Psychiatry. 2012;83(4):358–364. doi: 10.1136/jnnp-2011-301883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lagier-Tourenne C, Baughn M, Rigo F, Sun S, Liu P, Li HR, Jiang J, Watt AT, Chun S, Katz M, Qiu J, Sun Y, Ling SC, Zhu Q, Polymenidou M, Drenner K, Artates JW, McAlonis-Downes M, Markmiller S, Hutt KR, Pizzo DP, Cady J, Harms MB, Baloh RH, Vandenberg SR, Yeo GW, Fu XD, Bennett CF, Cleveland DW, Ravits J. Targeted degradation of sense and antisense C9orf72 RNA foci as therapy for ALS and frontotemporal degeneration. Proc Natl Acad Sci USA. 2013;110(47):E4530–E4539. doi: 10.1073/pnas.1318835110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lee EB, Lee VM, Trojanowski JQ. Gains or losses: molecular mechanisms of TDP43-mediated neurodegeneration. Nat Rev Neurosci. 2012;13(1):38–50. doi: 10.1038/nrn3121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lee EB, Russ J, Jung H, Elman LB, Chahine LM, Kremens D, Miller BL, Branch Coslett H, Trojanowski JQ, Van Deerlin VM, McCluskey LF. Topography of FUS pathology distinguishes late-onset BIBD from a FTLD-U. Acta Neuropathol Commun. 2013;1(9):1–11. doi: 10.1186/2051-5960-1-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lee Y-B, Chen H-J, Peres João N, Gomez-Deza J, Attig J, talekar M, Troakes C, Nishimura Agnes L, Scotter Emma L, Vance C, Adachi Y, Sardone V, Miller Jack W, Smith Bradley N, Gallo J-M, Ule J, Hirth F, Rogelj B, Houart C, Shaw Christopher E. Hexanucleotide repeats in ALS/FTD form length-dependent RNA foci, sequester RNA binding proteins, and are neuro-toxic. Cell reports. 2013 doi: 10.1016/j.celrep.2013.10.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li YR, King OD, Shorter J, Gitler AD. Stress granules as crucibles of ALS pathogenesis. J Cell Biol. 2013;201(3):361–372. doi: 10.1083/jcb.201302044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ling SC, Polymenidou M, Cleveland DW. Converging mechanisms in ALS and FTD: disrupted RNA and protein homeostasis. Neuron. 2013;79(3):416–438. doi: 10.1016/j.neuron.2013.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Loesch DZ, Huggins R, Hay DA, Gedeon AK, Mulley JC, Sutherland GR. Genotype–phenotype relationships in fragile X syndrome: a family study. Am J Hum Genet. 1993;53(5):1064–1073. [PMC free article] [PubMed] [Google Scholar]
- 39.Mackenzie IR, Arzberger T, Kremmer E, Troost D, Lorenzl S, Mori K, Weng SM, Haass C, Kretzschmar HA, Edbauer D, Neumann M. Dipeptide repeat protein pathology in C9ORF72 mutation cases: clinico-pathological correlations. Acta Neuropathol. 2013;126(6):859–879. doi: 10.1007/s00401-013-1181-y. [DOI] [PubMed] [Google Scholar]
- 40.Mackenzie IR, Frick P, Neumann M. The neuropathology associated with repeat expansions in the C9ORF72 gene. Acta Neuropathol. 2014;127(3):347–357. doi: 10.1007/s00401-013-1232-4. [DOI] [PubMed] [Google Scholar]
- 41.Mahoney CJ, Beck J, Rohrer JD, Lashley T, Mok K, Shakespeare T, Yeatman T, Warrington EK, Schott JM, Fox NC, Rossor MN, Hardy J, Collinge J, Revesz T, Mead S, Warren JD. Fron-totemporal dementia with the C9ORF72 hexanucleotide repeat expansion: clinical, neuroanatomical and neuropathological features. Brain. 2012;135(Pt 3):736–750. doi: 10.1093/brain/awr361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.McConkie-Rosell A, Lachiewicz AM, Spiridigliozzi GA, Tarleton J, Schoenwald S, Phelan MC, Goonewardena P, Ding X, Brown WT. Evidence that methylation of the FMR-I locus is responsible for variable phenotypic expression of the fragile X syndrome. Am J Hum Genet. 1993;53(4):800–809. [PMC free article] [PubMed] [Google Scholar]
- 43.Merenstein SA, Sobesky WE, Taylor AK, Riddle JE, Tran HX, Hagerman RJ. Molecular–clinical correlations in males with an expanded FMR1 mutation. Am J Med Genet. 1996;64(2):388–394. doi: 10.1002/(SICI)1096-8628(19960809)64:2<388:AID-AJMG31>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 44.Miller JW, Urbinati CR, Teng-Umnuay P, Stenberg MG, Byrne BJ, Thornton CA, Swanson MS. Recruitment of human muscleblind proteins to (CUG)(n) expansions associated with myotonic dystrophy. EMBO J. 2000;19(17):4439–4448. doi: 10.1093/emboj/19.17.4439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mori K, Lammich S, Mackenzie IR, Forné I, Zilow S, Kretzschmar H, Edbauer D, Janssens J, Kleinberger G, Cruts M, Herms J, Neumann M, Van Broeckhoven C, Arzberger T, Haass C. hnRNP A3 binds to GGGGCC repeats and is a constituent of p62-positive/TDP43-negative inclusions in the hippocampus of patients with C9orf72 mutations. Acta Neuropathol. 2013;125(3):413–423. doi: 10.1007/s00401-013-1088-7. [DOI] [PubMed] [Google Scholar]
- 46.Mori K, Weng SM, Arzberger T, May S, Rentzsch K, Kremmer E, Schmid B, Kretzschmar HA, Cruts M, Van Broeckhoven C, Haass C, Edbauer D. The C9orf72 GGGGCC repeat is translated into aggregating dipeptide-repeat proteins in FTLD/ALS. Science. 2013;339(6125):1335–1338. doi: 10.1126/science.1232927. [DOI] [PubMed] [Google Scholar]
- 47.Murray ME, Bieniek KF, Banks Greenberg M, DeJesus-Hernandez M, Rutherford NJ, van Blitterswijk M, Niemantsverdriet E, Ash PE, Gendron TF, Kouri N, Baker M, Goodman IJ, Petrucelli L, Rademakers R, Dickson DW. Progressive amnestic dementia, hippocampal sclerosis, and mutation in C9ORF72. Acta Neuropathol. 2013;126(4):545–554. doi: 10.1007/s00401-013-1161-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Myrick L, Nakamoto-Kinoshita M, Lindor N, Kirmani S, Cheng X, Warren S. Fragile X syndrome due to a missense mutation. Eur J Hum Genet. 2014 doi: 10.1038/ejhg.2013.311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.O’Donnell WT, Warren ST. A decade of molecular studies of fragile X syndrome. Annu Rev Neurosci. 2002;25:315–338. doi: 10.1146/annurev.neuro.25.112701.142909. [DOI] [PubMed] [Google Scholar]
- 50.Pearson CE, Nichol Edamura K, Cleary JD. Repeat instability: mechanisms of dynamic mutations. Nat Rev Genet. 2005;6(10):729–742. doi: 10.1038/nrg1689. [DOI] [PubMed] [Google Scholar]
- 51.Reddy K, Zamiri B, Stanley SY, Macgregor RB, Jr, Pearson CE. The disease-associated r(GGGGCC)n repeat from the C9orf72 gene forms tract length-dependent uni- and multi-molecular RNA G-quadruplex structures. J Biol Chem. 2013;288(14):9860–9866. doi: 10.1074/jbc.C113.452532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Reiss AL, Freund LS, Baumgardner TL, Abrams MT, Denckla MB. Contribution of the FMR1 gene mutation to human intellectual dysfunction. Nat Genet. 1995;11(3):331–334. doi: 10.1038/ng1195-331. [DOI] [PubMed] [Google Scholar]
- 53.Renton AE, Majounie E, Waite A, Simon-Sanchez J, Rollinson S, Gibbs JR, Schymick JC, Laaksovirta H, van Swieten JC, Myllykangas L, Kalimo H, Abramzon Y, Remes AM, Kaganovich A, Scholz SW, Duckworth J, Ding J, Harmer DW, Hernandez DG, Johnson JO, Mok K, Ryten M, Trabzuni D, Guerreiro RJ, Orrell RW, Neal J, Murray A, Pearson J, Jansen IE, Sondervan D, Seelaar H, Blake D, Young K, Halliwell N, Callister JB, Toulson G, Richardson A, Gerhard A, Snowden J, Mann D, Neary D, Nalls MA, Peuralinna T, Jansson L, Isoviita VM, Kaivorinne AL, Holtta-Vuori M, Ikonen E, Sulkava R, Benatar M, Wuu J, Chio A, Restagno G, Borghero G, Borghero G, Sabatelli M, Consortium I, Heckerman D, Rogaeva E, Zinman L, Rothstein JD, Sendtner M, Drepper C, Eichler EE, Alkan C, Abdullaev Z, Pack SD, Dutra A, Pak E, Hardy J, Singleton A, Williams NM, Heutink P, Pickering-Brown S, Morris HR, Tienari PJ, Traynor BJ. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron. 2011;72(2):257–268. doi: 10.1016/j.neuron.2011.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rosenbloom KR, Sloan CA, Malladi VS, Dreszer TR, Learned K, Kirkup VM, Wong MC, Maddren M, Fang R, Heitner SG, Lee BT, Barber GP, Harte RA, Diekhans M, Long JC, Wilder SP, Zweig AS, Karolchik D, Kuhn RM, Haussler D, Kent WJ. ENCODE data in the UCSC genome browser: year 5 update. Nucleic Acids Res. 2013;41(Database issue):D56–D63. doi: 10.1093/nar/gks1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sareen D, O’Rourke JG, Meera P, Muhammad AK, Grant S, Simpkinson M, Bell S, Carmona S, Ornelas L, Sahabian A, Gendron T, Petrucelli L, Baughn M, Ravits J, Harms MB, Rigo F, Bennett CF, Otis TS, Svendsen CN, Baloh RH. Targeting RNA Foci in iPSC-derived motor neurons from ALS patients with a C9ORF72 repeat expansion. Sci Transl Med. 2013;5(208):208ra149. doi: 10.1126/scitranslmed.3007529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Simon-Sanchez J, Dopper EG, Cohn-Hokke PE, Hukema RK, Nicolaou N, Seelaar H, de Graaf JR, de Koning I, van Schoor NM, Deeg DJ, Smits M, Raaphorst J, van den Berg LH, Schelhaas HJ, De Die-Smulders CE, Majoor-Krakauer D, Rozemuller AJ, Willemsen R, Pijnenburg YA, Heutink P, van Swieten JC. The clinical and pathological phenotype of C9ORF72 hexanucleotide repeat expansions. Brain. 2012;135(Pt 3):723–735. doi: 10.1093/brain/awr353. [DOI] [PubMed] [Google Scholar]
- 57.Smeets HJ, Smits AP, Verheij CE, Theelen JP, Willemsen R, van de Burgt I, Hoogeveen AT, Oosterwijk JC, Oostra BA. Normal phenotype in two brothers with a full FMR1 mutation. Hum Mol Genet. 1995;4(11):2103–2108. doi: 10.1093/hmg/4.11.2103. [DOI] [PubMed] [Google Scholar]
- 58.Snowden JS, Rollinson S, Thompson JC, Harris JM, Stopford CL, Richardson AM, Jones M, Gerhard A, Davidson YS, Robinson A, Gibbons L, Hu Q, DuPlessis D, Neary D, Mann DM, Pickering-Brown SM. Distinct clinical and pathological characteristics of frontotemporal dementia associated with C9ORF72 mutations. Brain. 2012;135(Pt 3):693–708. doi: 10.1093/brain/awr355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Stepto A, Gallo JM, Shaw CE, Hirth F. Modelling C9ORF72 hexanucleotide repeat expansion in amyotrophic lateral sclerosis and frontotemporal dementia. Acta Neuropathol. 2014;127(3):377–389. doi: 10.1007/s00401-013-1235-1. [DOI] [PubMed] [Google Scholar]
- 60.Straussman R, Nejman D, Roberts D, Steinfeld I, Blum B, Benvenisty N, Simon I, Yakhini Z, Cedar H. Developmental programming of CpG island methylation profiles in the human genome. Nature Struct Mol Biol. 2009;16(5):564–571. doi: 10.1038/nsmb.1594. [DOI] [PubMed] [Google Scholar]
- 61.Sutcliffe JS, Nelson DL, Zhang F, Pieretti M, Caskey CT, Saxe D, Warren ST. DNA methylation represses FMR-1 transcription in fragile X syndrome. Hum Mol Genet. 1992;1(6):397–400. doi: 10.1093/hmg/1.6.397. [DOI] [PubMed] [Google Scholar]
- 62.Toledo JB, Van Deerlin VM, Lee EB, Suh E, Baek Y, Robinson JL, Xie SX, McBride J, Wood EM, Schuck T, Irwin DJ, Gross RG, Hurtig H, McCluskey L, Elman L, Karlawish J, Schellenberg G, Chen-Plotkin A, Wolk D, Grossman M, Arnold SE, Shaw LM, Lee VM, Trojanowski JQ. A platform for discovery: the University of Pennsylvania integrated neurodegenerative disease biobank. Alzheimers Dement. 2013 doi: 10.1016/j.jalz.2013.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.van Blitterswijk M, DeJesus-Hernandez M, Niemantsverdriet E, Murray ME, Heckman MG, Diehl NN, Brown PH, Baker MC, Finch NA, Bauer PO, Serrano G, Beach TG, Josephs KA, Knopman DS, Petersen RC, Boeve BF, Graff-Radford NR, Boylan KB, Petrucelli L, Dickson DW, Rademakers R. Association between repeat sizes and clinical and pathological characteristics in carriers of C9ORF72 repeat expansions (Xpansize-72): a cross-sectional cohort study. Lancet Neurol. 2013;12(10):978–988. doi: 10.1016/S1474-4422(13)70210-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Waite AJ, Baumer D, East S, Neal J, Morris HR, Ansorge O, Blake DJ. Reduced C9orf72 protein levels in frontal cortex of amyotrophic lateral sclerosis and frontotemporal degeneration brain with the C9ORF72 hexanucleotide repeat expansion. Neurobiol Aging. 2014 doi: 10.1016/j.neurobiolaging.2014.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Xi Z, Zinman L, Moreno D, Schymick J, Liang Y, Sato C, Zheng Y, Ghani M, Dib S, Keith J, Robertson J, Rogaeva E. Hypermethylation of the CpG island near the G4C2 repeat in ALS with a C9orf72 expansion. Am J Hum Genet. 2013;92(6):981–989. doi: 10.1016/j.ajhg.2013.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Xu Z, Poidevin M, Li X, Li Y, Shu L, Nelson DL, Li H, Hales CM, Gearing M, Wingo TS, Jin P. Expanded GGGGCC repeat RNA associated with amyotrophic lateral sclerosis and fronto-temporal dementia causes neurodegeneration. Proc Natl Acad Sci USA. 2013;110(19):7778–7783. doi: 10.1073/pnas.1219643110. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.