

Abstract
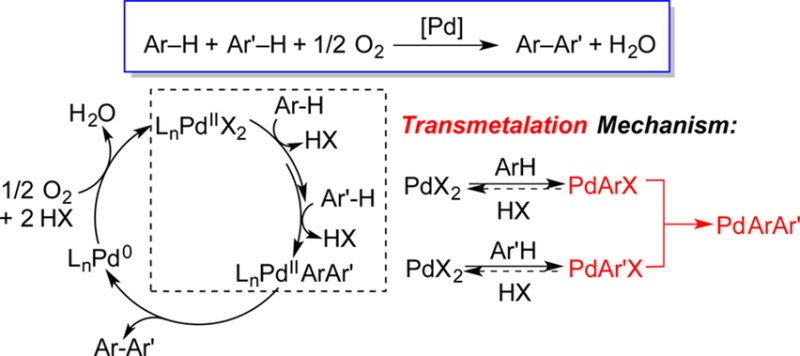
Pd-catalyzed aerobic oxidative coupling of arenes provides efficient access to biaryl compounds. The biaryl product forms via C–H activation of two arenes to afford a PdIIArAr′ intermediate, which then undergoes C–C reductive elimination. The key PdIIArAr′ intermediate could form via a “monometallic” pathway involving sequential C–H activation at a single PdII center, or via a “bimetallic” pathway involving parallel C–H activation at separate PdII centers, followed by a transmetalation step between two PdII–aryl intermediates. Here, we investigate the oxidative coupling of o-xylene catalyzed by a PdX2/2-fluoropyridine catalyst (X = trifluoroacetate, acetate). Kinetic studies, H/D exchange experiments, and kinetic isotope effects provide clear support for a bimetallic/transmetalation mechanism.
The oxidative coupling of hydrocarbons (eq 1) is an important contemporary topic with significant commercial implications. Prominent targets include the conversion of methane to higher hydrocarbons and the coupling of arenes to biaryls. The latter application is relevant to the industrial production of heat transfer fluids (e.g., biphenyl1) and monomers for high-performance polymers (e.g., Upilex2). Moreover, oxidative cross-coupling of arenes represents an efficient route to unsymmetrical biaryls relevant to the pharmaceutical, agrochemical, and fine chemical industries.3 PdII catalysts show significant promise in oxidative coupling reactions as they are eminently compatible with O2 as the stoichiometric oxidant and therefore could be accomplished with near-ideal atom economy. Mechanistic understanding of key steps could facilitate the development of new and/or improved reactions of this type.4 Here, we address unresolved mechanistic questions related to C–H activation and C–C bond formation in the Pd-catalyzed aerobic oxidative homocoupling of o-xylene, a reaction relevant to Upilex production.5
| 1 |
The first example of PdII-mediated oxidative biaryl coupling, reported by van Helden and Verberg in 1965,6 featured the oxidative coupling of benzene with stoichiometric PdCl2 and NaOAc. Within a few years, several groups reported PdII-catalyzed biaryl coupling methods that used O2 as the oxidant.7 Related catalytic methods were later developed for the commercial synthesis of 3,4,3′,4′-tetramethyl biphenyltetracarboxylate, a monomeric precursor to Upilex.8 In subsequent decades, numerous additional examples of Pd-catalyzed methods for oxidative homocoupling of arenes have been reported.3,9 The vast majority of work in this area has focused on empirical development of new catalyst systems, but important mechanistic questions remain unanswered.
A reasonable mechanism for Pd-catalyzed oxidative coupling of arenes consists of a PdII/Pd0 catalytic cycle with three general steps (Scheme 1): (i) PdII-mediated activation of two aryl C–H bonds to produce a PdIIArAr′ intermediate, (ii) reductive elimination of Ar–Ar′ from PdIIArAr′,10 (iii) and aerobic oxidation of Pd0 to PdII.11 While the mechanism of arene C–H activation by PdII has been the focus of considerable investigation,12 the pathway for formation of PdIIArAr′ is not clear. At least two different mechanisms are possible. A “monometallic” mechanism (Scheme 1A) involves sequential C–H activation at a single PdII center, while a “bimetallic” mechanism (Scheme 1B) involves parallel C–H activation at two separate PdII centers followed by transmetalation between the two PdII–aryl species.
Scheme 1. Pd-Catalyzed Aerobic Oxidative Biaryl Coupling and Two Mechanistic Possibilities for PdArAr′ Formation.

The “monometallic” mechanism has been commonly invoked in recent reports, especially in the context of Pd-catalyzed oxidative cross-coupling reactions,13 and C–H activation of arenes by PdIIArX species has been studied experimentally14 and computationally.15 The bimetallic pathway was suggested in 1968 by Davidson and Triggs;16 however, no experimental data were provided to support the proposal and few subsequent reports consider this pathway.17 Transmetalation of an aryl group between well-defined PdII–aryl species has been investigated,18 but its direct relevance to oxidative biaryl coupling has not been established.
Our mechanistic studies focus on the recently reported aerobic homocoupling of o-xylene with a PdX2/2Fpy catalyst system [X = TFA (trifluoroacetate) or OAc (acetate), 2Fpy = 2-fluoropyridine].5b This reaction proceeds with high selectivity to the symmetrical biaryl product shown in Figure 1. Kinetic studies were carried out to establish the dependence of the reaction rate on [Pd] (2–50 mM, 0.04–1.0 mol % with respect to o-xylene) (Figure 1). Initial rates of the reactions, monitored by gas chromatography, reveal a second-order dependence at low [Pd] (≤5 mM; inset, Figure 1) and a first-order dependence at high [Pd] (≥15 mM; Figure 1). To our knowledge, this is the first observation of a second-order [Pd] dependence in oxidative biaryl coupling, and it provides preliminary support for the bimetallic mechanism in Scheme 1.
Figure 1.

Kinetic dependence of the rate of Pd-catalyzed oxidative coupling of o-xylene on [Pd]. Reaction conditions: [o-xylene] = 4.5 M (4 mmol), [Pd] = 2–50 mM (0.0016–0.04 mmol), HOAc (0.4 mL), O2 (1 atm), 80 °C, 0–4 h.
Deuterium kinetic isotope effects (KIEs) were evaluated to gain insight into the contribution of C–H activation to the overall turnover rate. The oxidative coupling of o-xylene and o-xylene-d10, monitored independently with 0.5 mol % [Pd] in HOAc (Figure 2), reveal a very large KIE: kH/kD = 24 ± 2. This value is significantly larger than KIEs observed previously for Pd-based oxidative biaryl coupling reactions (kH/kD ≈ 2–5).19 The reaction was also performed in DOAc to probe solvent isotope effects, and a significant inverse KIE was observed: kHOAc/kDOAc = 0.31 ± 0.02 (Figure 2).
Figure 2.
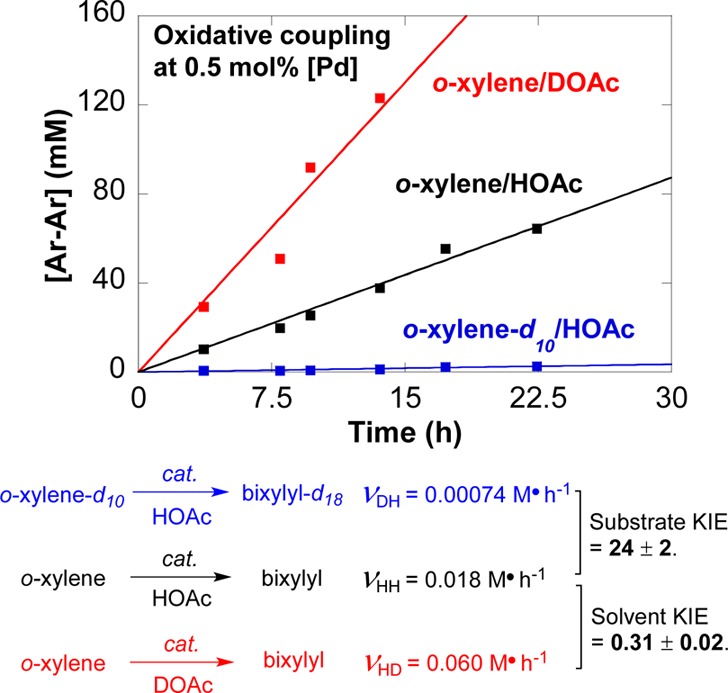
Kinetic isotope effects derived from initial rate measurements (ν) for oxidative coupling of o-xylene. Reaction conditions: [arene] = 4.5 M (4 mmol), [Pd] = 22.5 mM (0.02 mmol), solvent (0.4 mL), O2 (1 atm), 80 °C, 0–4 h.
Further insights into PdII-mediated C–H activation and the origin of the substrate and solvent isotope effects were obtained from H/D exchange experiments. The reaction of o-xylene-d10 in HOAc (0.5 mol % [Pd]) shows that H/D exchange occurs ∼30-fold faster than biaryl product formation (Figure 3A); however, this ratio changes at different [Pd]. For example, H/D exchange is favored over biaryl formation by a ratio of ∼120:1 at 0.1 mol % [Pd]. Plots of H/D exchange and biaryl product yields show that H/D exchange exhibits a first-order dependence on [Pd] while biaryl product formation is second-order in [Pd] (Figure 3B). The latter observation complements the second-order [Pd]-dependence data at low [Pd], shown in Figure 1. H/D exchange occurs exclusively at aromatic positions and favors exchange at meta positions.20 The 2:1 meta/ortho regioselectivity for H/D exchange is substantially lower than the regioselectivity of biaryl product formation, which exhibits ∼10:1 selectivity for meta–meta over ortho–meta coupling (ortho–ortho coupling is not observed).
Figure 3.
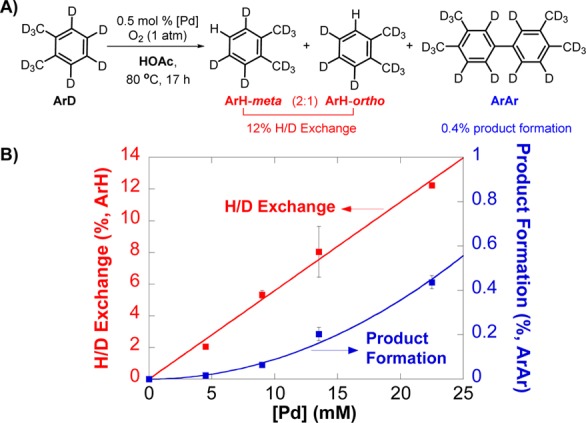
(A) H/D exchange experiments at 0.5 mol % [Pd] (B) H/D exchange and product yields at different [Pd]. Reaction conditions: [o-xylene-d10] = 4.5 M (4 mmol), [Pd] = 5–25 mM (0.0045–0.0225 mmol), HOAc (0.4 mL), O2 (1 atm), 80 °C, 17 h.
The above data led us to undertake a more systematic assessment of kinetic isotope effects as a function of [Pd]. Substrate and solvent KIEs were determined at four different [Pd] (2.5–67.5 mM, 0.06–1.5 mol % of substrate) (Figure 4; see Supporting Information for individual kinetic plots), and the data reveal that the substrate KIE can vary from 7.9 to 25, depending on the [Pd] and solvent (HOAc vs DOAc) (Figure 4A). The reciprocal of the solvent KIE (i.e., kDOAc/kHOAc) varies similarly from 3.1 to 23 (Figure 4B). At least three general trends are worth noting. The substrate KIEs are significantly larger in HOAc (KIE = 18–25) than in DOAc (KIE = 7.9–12). The reciprocal solvent KIEs increase significantly as the [Pd] is lowered and also are substantially larger with o-xylene-d10 than with o-xylene.
Figure 4.

Substrate and solvent KIE values at different [Pd]. Reaction conditions: [arene] = 4.5 M (4 mmol), [Pd] = 2.7–67.5 mM (0.0024–0.06 mmol), solvent (0.4 mL), O2 (1 atm), 80 °C.
Collectively, the kinetic data, KIEs, and H/D-exchange results clearly distinguish between the two possible mechanisms in Scheme 1. Rate laws derived for both mechanisms (eqs 2–4; see SI for derivations) show that the monometallic mechanism always exhibits a first-order dependence on [Pd], while the bimetallic mechanism could have a first-order or second-order dependence on [Pd], depending on the identity of the rate-limiting step. The switch from second-order to first-order kinetic behavior as [Pd] is increased (cf. Figure 1) provides support for the bimetallic mechanism. At low [Pd], a lower steady-state concentration of the [LnPdIIArX] intermediate leads to rate-limiting transmetalation (cf. eq 4). At higher [Pd], the bimetallic transmetalation rate will increase faster than the unimolecular C–H activation step, and C–H activation can become rate-limiting.
|
|
| 2 |
|
|
| 3 |
|
|
| 4 |
The bimetallic mechanism in Scheme 1 also rationalizes the H/D exchange results. Under the conditions of the experiments depicted in Figure 3, H/D exchange is more prominent at lower [Pd] because this process (i.e., protonolysis of a LnPdArX intermediate) requires only a single Pd species, while the competing transmetalation process requires two LnPdArX intermediates. The higher regioselectivity observed for biaryl product formation relative to H/D exchange suggests that the transmetalation step exhibits a strong preference for meta- rather than ortho-substituted PdII-xylyl intermediates. Further, this transmetalation selectivity is higher than the selectivity of PdII-mediated C–H activation. These observations have important implications for controlling the regioselectivity of oxidative C–H/C–H coupling reactions.21
Finally, the bimetallic mechanism explains the unusually large KIE values observed for this reaction. The largest substrate KIEs will be obtained when transmetalation is rate-limiting because the isotope effect associated with C–H activation (k1′) is squared (eq 4). The reversibility of the C–H cleavage does not reduce the KIE because the acidic solvent means that k–1′ is the same for both substrate isotopologs (Scheme 1B and eq 4). Previously reported KIEs for PdII-mediated arene C–H activation vary from 3 to 5,22 which corresponds to a net KIE of 9–25 when this value is squared. Therefore, the KIEs of 18–25 in Figure 4A are not unusual and fit in the range expected for a bimetallic mechanism when transmetalation is rate-limiting.23 Under conditions that lead to rate-limiting C–H activation, the KIE will approach the intrinsic isotope effect associated with C–H activation (i.e., 3–5). We never fully reach this limit under the conditions of our experiments, and the lowest KIE that we observe is 7.9 (Figure 4A).
Inverse solvent isotope effects are evident in all of the reactions described here, and they arise from faster protonolysis of the LnPdArX intermediate by HOAc relative to DOAc. The slower protonolysis with DOAc will lead to a higher steady-state concentration of the LnPdArX intermediate and result in a higher rate of product formation via transmetalation from this species. The inverse solvent KIE will be most substantial when transmetalation is rate-limiting. Under these conditions, C–H activation is reversible, and the isotope effect associated with protonolysis of LnPdArX (k-1′) is squared in the denominator of eq 4 (because two LnPdArX species are needed for the transmetalation step). This scenario is favored when [Pd] is low and o-xylene-d10 is the substrate, as evident in Figure 4B. Both conditions reduce the steady-state concentration of LnPdArX.24
This mechanistic study of aerobic oxidative coupling of o-xylene with the PdX2 (X = OAc, TFA)/2Fpy catalyst system provides clear evidence for a bimetallic/transmetalation mechanism. Evidence for this pathway includes a bimolecular kinetic dependence on [Pd] (Figure 1) and unusually large substrate and solvent KIEs (Figures 2 and 4). The bimetallic mechanism, but not the monometallic mechanism, readily accommodates these observations. The insights obtained from the relatively simple catalytic reaction described here provide an important foundation for future studies of more complex Pd-catalyzed oxidative biaryl coupling reactions, such as those that employ redox-active cocatalysts (e.g., CuII and polyoxometalates), as well as cross-coupling reactions between two different arenes.
Acknowledgments
We thank Dr. Paul White for helpful discussion of the reaction mechanism. Financial support of this work was provided by the NIH (R01 GM67173) and Mitsubishi Chemical Corporation (YI). NMR spectroscopy facilities were provided using a generous gift from Paul J. Bender.
Supporting Information Available
Experimental procedures, reaction time courses, additional mechanistic experiments and discussion. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Thompson Q. E.Biphenyl and Terphenyls. In Kirk-Othmer Encyclopedia of Chemical Technology; Wiley: Hoboken, NJ, 2000; pp 1–13. [Google Scholar]
- a Mohri Y. Seni Gakkaishi 1994, 50, 96. [Google Scholar]; b Kreuz J. A.; Edman J. R. Adv. Mater. 1998, 10, 1229. [Google Scholar]; c Takeoshi T.Polyimides. In Advances in Polymer Science; Springer-Verlag: Berlin, Germany, 1990; Vol. 94, pp 2–22. [Google Scholar]
- For reviews on oxidative biaryl coupling, see:; a Yeung C. S.; Dong V. M. Chem. Rev. 2011, 111, 1215. [DOI] [PubMed] [Google Scholar]; b Liu C.; Zhang H.; Shi W.; Lei A. Chem. Rev. 2011, 111, 1780. [DOI] [PubMed] [Google Scholar]; c Lyons T. W.; Sanford M. S. Chem. Rev. 2010, 110, 1147. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Ashenhurst J. A. Chem. Soc. Rev. 2010, 39, 540. [DOI] [PubMed] [Google Scholar]
- Stahl S. S. Angew. Chem., Int. Ed. 2004, 43, 3400. [DOI] [PubMed] [Google Scholar]
- a Itatani H.; Yoshimoto H.; Shiotani A.; Yokota A. Yoshikiyo M. U.S. Patent 4,294,976, 1981.; b Izawa Y.; Stahl S. S. Adv. Synth. Catal. 2010, 352, 3223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Helden R.; Verberg G. Recl. Trav. Chim. Pays-Bas 1965, 84, 1263. [Google Scholar]
- For leading references, see:; a Davidson J. M.; Triggs C. J. Chem. Soc. A 1968, 1331. [Google Scholar]; b Yoshimoto H.; Itatani H. J. Catal. 1973, 31, 8. [Google Scholar]; c Iataaki H.; Yoshimoto H. J. Org. Chem. 1973, 38, 76. [Google Scholar]; d Stock L. M.; Tse K.-t.; Vorvick L. J.; Walstrum S. A. J. Org. Chem. 1981, 46, 1757. [Google Scholar]
- a Itatani H.; Shiotani A.; Yokota A. U.S. Patent 4,338,456, 1982.; b Shiotani A.; Itatani H.; Inagaki T. J. Mol. Catal. 1986, 34, 57. [Google Scholar]
- For leading references, see:; a Mukhopadhyay S.; Rothenberg G.; Lando G.; Agbaria K.; Kazanci M.; Sasson Y. Adv. Synth. Catal. 2001, 343, 455. [Google Scholar]; b Yokota T.; Sakaguchi S.; Ishii Y. Adv. Synth. Catal. 2002, 344, 849. [Google Scholar]; c Lee S. H.; Lee K. H.; Lee J. S.; Jung J. D.; Shim J. S. J. Mol. Catal. A: Chem. 1997, 115, 241. [Google Scholar]; d Rong Y.; Li R.; Lu W. Organometallics 2007, 26, 4376. [Google Scholar]; e Hull K. L.; Lanni E. L.; Sanford M. S. J. Am. Chem. Soc. 2006, 128, 14047. [DOI] [PubMed] [Google Scholar]; f Masui K.; Ikegami H.; Mori A. J. Am. Chem. Soc. 2004, 126, 5074. [DOI] [PubMed] [Google Scholar]
- a Hartwig J. F. Inorg. Chem. 2007, 46, 1936. [DOI] [PubMed] [Google Scholar]; b Brown J. M.; Cooley N. A. Chem. Rev. 1988, 88, 1031. [Google Scholar]; c Dedieu A. Chem. Rev. 2000, 100, 543. [DOI] [PubMed] [Google Scholar]; d Xue L.; Lin Z. Chem. Soc. Rev. 2010, 39, 1692. [DOI] [PubMed] [Google Scholar]
- a Konnick M. M.; Guzei I. A.; Stahl S. S. J. Am. Chem. Soc. 2004, 126, 10212. [DOI] [PubMed] [Google Scholar]; b Popp B. V.; Stahl S. S. Chem.—Eur. J. 2009, 15, 2915. [DOI] [PubMed] [Google Scholar]; c Stahl S. S.; Thorman J. L.; Nelson R. C.; Kozee M. A. J. Am. Chem. Soc. 2001, 123, 7188. [DOI] [PubMed] [Google Scholar]; d Muzart J. Chem.—Asian J. 2006, 1, 508. [DOI] [PubMed] [Google Scholar]
- a Ryabov A. D.; Sakodinskaya I. K.; Yatsimirsky A. K. J. Chem. Soc., Dalton Trans. 1985, 14, 2629. [Google Scholar]; b Karig G.; Moon M.-T.; Thasana N.; Gallagher T. Org. Lett. 2002, 4, 3115. [DOI] [PubMed] [Google Scholar]; c García-Cuadrado D.; Braga A. A. C.; Maseras F.; Echavarren A. M. J. Am. Chem. Soc. 2006, 128, 1066. [DOI] [PubMed] [Google Scholar]; d Ackermann L. Chem. Rev. 2011, 111, 1315. [DOI] [PubMed] [Google Scholar]; e Balcells D.; Clot E.; Eisenstein O. Chem. Rev. 2010, 110, 749. [DOI] [PubMed] [Google Scholar]
- a Stuart D. R.; Fagnou K. Science 2007, 316, 1172. [DOI] [PubMed] [Google Scholar]; b Lyons T. W.; Hull K. L.; Sanford M. S. J. Am. Chem. Soc. 2011, 133, 4455. [DOI] [PubMed] [Google Scholar]; c Li B.-J.; Tian S.-L.; Fang Z.; Shi Z.-J. Angew. Chem., Int. Ed. 2008, 47, 1115. [DOI] [PubMed] [Google Scholar]; d Potavathri S.; Dumas A. S.; Dwight T. A.; Naumiec G. R.; Hammann J. M.; DeBoef B. Tetrahedron Lett. 2008, 49, 4050. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Zhao X.; Yeung C. S.; Dong V. M. J. Am. Chem. Soc. 2010, 132, 5837. [DOI] [PubMed] [Google Scholar]; f Liu B.; Huang Y.; Lan J.; Song F.; You J. Chem. Sci. 2013, 4, 2163. [Google Scholar]
- See, for example:; a Hull K. L.; Sanford M. S. J. Am. Chem. Soc. 2009, 131, 9651. [DOI] [PubMed] [Google Scholar]; b Tan Y.; Hartwig J. F. J. Am. Chem. Soc. 2011, 133, 3308. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Sun H.-Y.; Gorelsky S. I.; Stuart D. R.; Campeau L.-C.; Fagnou K. J. Org. Chem. 2010, 75, 8180. [DOI] [PubMed] [Google Scholar]
- a Gorelsky S. I.; Lapointe D.; Fagnou K. J. Am. Chem. Soc. 2008, 130, 10848. [DOI] [PubMed] [Google Scholar]; b Gorelsky S. I.; Lapointe D.; Fagnou K. J. Org. Chem. 2012, 77, 658. [DOI] [PubMed] [Google Scholar]
- Davidson J. M.; Triggs C. J. Chem. Soc. A 1968, 1324. [Google Scholar]
- a Yatsimirsky A. K.; Deiko S. A.; Ryabov A. D. Tetrahedron 1983, 39, 2381. [Google Scholar]; b Ackerman L. J.; Sadighi J. P.; Kurtz D. M.; Labinger J. A.; Bercaw J. E. Organometallics 2003, 22, 3884. [Google Scholar]; c Lotz M. D.; Remy M. S.; Lao D. B.; Ariafard A.; Yates B. F.; Canty A. J.; Mayer J. M.; Sanford M. S. J. Am. Chem. Soc. 2014, 136, 8237. [DOI] [PubMed] [Google Scholar]
- See, for example:; a Ozawa F.; Fujimori M.; Yamamoto T.; Yamamoto A. Organometallics 1986, 5, 2144. [Google Scholar]; b Casado A. L.; Caseras J. A.; Espinet P. Organometallics 1997, 16, 5730. [Google Scholar]; c Tan Y.; Barrios-Landeros F.; Hartwig J. F. J. Am. Chem. Soc. 2012, 134, 3683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- See, for example, ref (16) and the following:Kashima M.; Yoshimoto H.; Itatani H. J. Catal. 1973, 29, 92. [Google Scholar]
- See Supporting Information for details.
- For other studies that probe factors controlling the regioselectivity of oxidative coupling reactions, see refs (13)a, (13b), (13d), and the following:; a Stuart D. R.; Villemure E.; Fagnou K. J. Am. Chem. Soc. 2007, 129, 12072. [DOI] [PubMed] [Google Scholar]; b Campbell A. N.; Meyer E. B.; Stahl S. S. Chem. Commun. 2011, 47, 10257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For intrinsic KIE in Pd-mediated arene C–H activation, see:; a Hennessy E. J.; Buchwald S. L. J. Am. Chem. Soc. 2003, 125, 12084. [DOI] [PubMed] [Google Scholar]; b Campeau L. C.; Parisien M.; Jean A.; Fagnou K. J. Am. Chem. Soc. 2006, 128, 581. [DOI] [PubMed] [Google Scholar]; c Chiong H. A.; Pham Q.-N.; Daugulis O. J. Am. Chem. Soc. 2007, 129, 9879. [DOI] [PubMed] [Google Scholar]; d Geary L. M.; Hultin P. G. Eur. J. Org. Chem. 2010, 5563. [DOI] [PubMed] [Google Scholar]
- Transmetalation will be comparatively slow when [Pd] is low and/or protonolysis of the LnPdArX intermediate is fast (e.g., in HOAc vs DOAc; cf. Figure 4A).
- See Supporting Information for additional discussion on the KIE result and its consistency with the bimetallic mechanism.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.