

Abstract
Excessive accumulation of β-amyloid peptide (Aβ) is one of the major mechanisms responsible for neuronal death in Alzheimer's disease. Flavonoids, primarily antioxidants, are a group of polyphenolic compounds synthesized in plant cells. The present study aimed to identify flavonoid compounds that could inhibit Aβ-induced neuronal death by examining the effects of various flavonoids on the neurotoxicity of Aβ fragment 25-35 (Aβ25-35) in mouse cortical cultures. Aβ25-35 induced concentration- and exposure-time-dependent neuronal death. Neuronal death induced by 20 µM Aβ25-35 was significantly inhibited by treatment with either Trolox or ascorbic acid. Among 10 flavonoid compounds tested [apigenin, baicalein, catechin, epicatechin, epigallocatechin gallate (EGCG), kaempferol, luteolin, myricetin, quercetin, and rutin], all except apigenin showed strong 1,1-diphenyl-2-pycrylhydrazyl (DPPH) scavenging activity under cell-free conditions. The flavonoid compounds except apigenin at a concentration of 30 µM also significantly inhibited neuronal death induced by 20 µM Aβ25-35 at the end of 24 hours of exposure. Epicatechin, EGCG, luteolin, and myricetin showed more potent and persistent neuroprotective action than did the other compounds. These results demonstrated that oxidative stress was involved in Aβ-induced neuronal death, and antioxidative flavonoid compounds, especially epicatechin, EGCG, luteolin, and myricetin, could inhibit neuronal death. These findings suggest that these four compounds may be developed as neuroprotective agents against Alzheimer's disease.
Keywords: β-Amyloid peptide, Alzheimer's disease, Flavonoids
INTRODUCTION
Flavonoids make up the most common group of polyphenolic compounds in the human diet that are synthesized in plant cells. Flavonoids have a 15-carbon skeleton consisting of two fused six-membered rings (an aromatic ring and a heterocyclic ring) connected through a carbon-carbon bridge to an aromatic ring.1 The flavonoids can be divided into six different major classes (flavonols, flavanones, flavones, isoflavones, catechins, and anthocyanidins) on the basis of differences in the structure of their molecular backbone.2,3 The major sources of flavonoids include fruits, vegetables, tea, wine, and cocoa.4 The flavonol quercetin and the flavone apigenin are found in onions, apples, broccoli, and berries. The flavanone naringenin is found in citrus foods. Catechins are abundant in green tea. Cyanidin and other anthocyanidins are largely responsible for the deep colors of berries, grapes, and red wine. Genistein is an isoflavone found predominantly in legumes. The flavonoid consumed most, in general, is quercetin, and the richest sources of flavonoids consumed in general are tea, onions, and apples.5 Flavonoids contain a number of phenolic hydroxyl groups attached to ring structures, which may act as antioxidants,6,7,8 as radical-scavenging agents,9,10 or as agents of anti-inflammation.11,12 Recently, significant evidence has emerged to indicate that the consumption of flavonoid-rich foods is associated with lower rates of dementia13 and beneficial effects on memory and learning.13,14
Alzheimer's disease (AD) is the major subtype of dementia in all age groups.15 AD is a progressive neurodegenerative disorder associated with a disruption of neuronal function and a clinically gradual deterioration in remembering recent events, thinking abilities, and behavior. AD is pathologically characterized by the presence of neuritic plaques, neurofibrillary tangles, and the loss of cortical neurons and synapses,16 which is a hallmark feature of the AD brain. The β-amyloid peptide (Aβ) is the major component of extracellular neuritic plaques. Aβ is 39 to 43 amino acid fragments in length and is generated by proteolytic cleavage of the larger amyloid precursor protein (APP), a ubiquitously expressed transmembrane glycoprotein.17,18 An excessive accumulation of Aβ is one of the significant factors responsible for neuronal death in AD.19,20
Among the Aβ fragments (Aβ25-35, Aβ1-40, and Aβ1-42) that have toxic effects on neurons in vitro,21,22,23,24 Aβ25-35 is one of the short peptide sequences that retains biological activity comparable with that of full-length Aβ.25 Although the precise mechanisms mediating Aβ-induced neuronal death have yet to be fully established, it is well known that Aβ-induced neuronal death is associated with the generation of reactive oxygen species in the brain.26 Also, Aβ initiates a cascade of intracellular events that culminates in neuronal death via calcium-permeable cation channels as well as free-radical-like action.23,24,27,28
To determine the antioxidant potencies of 10 types of flavonoids in vitro, and to identify some flavonoid compounds that could inhibit Aβ-induced neuronal death, we investigated the effects of various flavonoids on neuronal death induced by Aβ25-35 in mouse cortical cultures.
MATERIALS AND METHODS
1. Materials
Eagle's minimal essential medium (MEM) and Hanks' balanced solution were purchased from Gibco (Gaithersburg, MD, USA). Fetal bovine serum (FBS) and horse serum were obtained from Hyclone (Logan, UT, USA). HEPES (acid), glucose, NaHCO3, NaCl, KCl, MgCl2, CaCl2, NaOH, phenol red, trypsin, cytosine arabinoside, epidermal growth factor, sucrose, ascorbic acid, cycloheximide, apigenin, baicalein, catechin, epicatechin, epigallocatechin gallate (EGCG), kaempferol, luteolin, myricetin, quercetin, rutin, nifedipine, flunarizine, diltiazem, NMDA, MK-801, Trolox, and Aβ25-35 were all purchased from Sigma-Aldrich (St. Louis, MO, USA).
2. Methods
1) Cortical cell cultures
Mixed cortical cell cultures, including both neurons and glia, were prepared with minor modification of the methods described by Choi et al.29 Pregnant mice (Institute of Cancer Research, London, UK) at 15 to 17 days of gestation were euthanized by cervical dislocation under halothane anesthesia. Fetal mice were rapidly removed and decapitated. Mouse brains were excised and then rinsed in cold Ca2+/Mg2+-free Hanks' balanced salt solution supplemented with 5 mg/mL glucose, 7 mg/mL sucrose, and 0.35 mg/mL sodium bicarbonate (DM). By use of fine-tipped forceps and a microsurgical knife, the meninges were carefully removed from the brain tissue under a stereomicroscope. The cerebral cortex was dissected free and minced into 1-2 mm3 sized pieces with a sterile scalpel. The cortex pieces were incubated in DM with 0.25% trypsin added at 37℃ for 15 minutes and then centrifuged at 1000×g for 5 minutes. After removal of supernatant, the tissue pellet was suspended in 1-2 mL plating medium with Eagle's MEM (Gibco, Gaithersburg, MD) containing 2 mM glutamine, 5% FBS, and 5% horse serum. Cells were separated by 8 or 10 trituration passages by using a flame-narrowed pipette. Dissociated cortical cells were plated onto the previously established glial layer in 24-well multi-well plates at a density of 3 hemispheres/plate (approximately 2.5×105 cells per well). The plates were plated in an incubator (Forma Company, USA) at 37℃, 5% CO2, with humidified air. Cytosine arabinoside was added to produce a final concentration of 10 µM at 5 days in vitro and the plates were maintained for 2 days to halt non-neuronal cell division. The culture medium was changed twice a week after 7 days in vitro. Cultures were used at 13 or 14 days in vitro for the experiments.
Cortical glial cultures were prepared from postnatal mice (Institute of Cancer Research, London, UK) aged 1 to 2 days. Dissection and dissociation were as described above for mixed cortical cell cultures, and cells were plated in 24-well multi-well plates at a density of 0.5 hemisphere/plate. The plating medium was supplemented with Eagle's MEM containing 2 mM glutamine, 10% FBS, 10% horse serum, and 10 ng/mL of epidermal growth factor. The plates were maintained in the same incubator. The culture medium was changed once a week after 14 days in vitro. Glial cultures were used for the plating of mixed cortical cell cultures between 18 and 24 days in vitro.
The procedures involving experimental animals complied with the regulations for the care and use of laboratory animals of the animal ethical committee of Chonnam National University.
2) Neurotoxicity and flavonoid experiments
Cultured cells in 24-well multi-well plates were used for experimentation at 13 to 14 days in vitro. After washing with MEM, cultures were exposed to Aβ25-35, Trolox, ascorbic acid, and various flavonoids for 24 or 48 hours.
Each row of the 24-well plates had 4 wells that were part of the same experiment. The four wells in the first row were treated with sham wash, NMDA (500 µM) treatment was given for full killing of neurons in the second row, and the third to the sixth rows were treated with drugs. The amount of each drug was 4 to 8 µL in each well with culture media. To evaluate the protective effects of these drugs on Aβ25-35-induced injury, the authors treated the cultures with two drugs simultaneously.
3) Analysis of neuronal survival
Cell viability of the cultures was assessed according to morphological characteristics under phase-contrast microscopy.30 The extent of neuronal injury was evaluated at the termination of Aβ exposure for 24 hours and was based on the amount of lactate dehydrogenase (LDH) released into the medium at the end of the exposure period. Neuronal injury was quantified by measurement of LDH in the culture medium. LDH activity in the medium was measured as described by Koh and Choi.31 For the measurement of LDH, 25 µL of culture medium was collected from each well of the 24-well plate and mixed with 125 µL of a buffer solution and 100 µL of 0.3 mg/mL NADH and then added to 30 µL of 22.7 mM pyruvate. We measured the density at 340 nm for 4 minutes by using a microplate reader (Molecular Device Co., USA). We used Sigma's control enzyme as the standard enzyme. Each condition was represented in three or four wells per experiment and was repeated 3 or 4 times in independent experiments. The measured values were calculated from 0 for the sham wash group to 100 for the full kill group and were reported as means±SEMs.
4) DPPH assay
An amount of 190 µL of 200 µM DPPH (1,1-diphenyl-2-picrylhydrazyl) with ethanol solution was added to 10 µL of DMSO and absorbance was measured immediately at 517 nm for the control reading.
For each of the flavonoid compounds, the sample volume was made uniformly to 10 µL by using ethanol and then 190 µL of 200 µM DPPH was added. Absorbance was measured after 30 minutes at 517 nm by using ethanol as the blank on a UV-visible spectrometer (Shimadzu, UV-1601, Japan). The concentration of each flavonoid compound was checked 3 times. The IC50 values for each compound and for the standard preparation were calculated. The DPPH free radical scavenging activity was calculated by using the following formula:
| %Scavenging = [(Absorbance of control - Absorbance of test sample)/Absorbance of control]×100 |
5) Data analysis
Experiments were set up with 8 to 12 replicate samples. Results are expressed as mean±SEM values, and the agents' effects were statistically evaluated with analysis of variance (one-way ANOVA) followed by the Student-Newman-Keuls post hoc test. Statistical significance was considered at value of p<0.05.
RESULTS
1. Aβ25-35-induced neurotoxicity of cultured neurons
To evaluate the neurotoxicity of Aβ25-35, primary cultures of mouse cortical cells containing neuronal and non-neuronal cells maintained for 13 to 14 days in vitro were exposed to Aβ25-35 at each concentration (5, 10, 20, 40, and 80 µM) for 24 or 48 hours. Aβ25-35 within a concentration range of 5 to 20 µM produced concentration-dependent neurotoxicity in cultured cortical neurons. Therefore, we used 20 µM Aβ25-35 to induce neuronal cell damage in the following experiments.
Neuronal cell damage (Fig. 1A, B) was observed under a phase-contrast microscope after exposure to 20 µM Aβ25-35 for 24 hours. On the basis of LDH measurements in the culture media, at a concentration of 20 µM Aβ25-35, approximately 20% or 50% of cells were damaged after exposure for 24 or 48 hours, respectively (Fig. 1C). Even when the concentration of Aβ25-35 was increased to 40 and 80 µM, the LDH measurement was similar with that induced by 20 µM of Aβ25-35 (Fig. 1C). Neurotoxicity was not induced by the reverse sequence of the Aβ35-25 peptide.
FIG. 1.

Aβ25-35-induced neuronal death in mouse cortical cultures. Phase-contrast photomicrographs from typical representative fields (200×field) of cells were taken after a 24-hour exposure to (A) sham wash or (B) 20 µM Aβ25-35 showing neurotoxic action of 20 µM Aβ25-35 in mouse cortical cultures (200×field). Treatment with Aβ25-35 induced concentration- and exposure-time-dependent neuronal death in mixed cortical cultures (C). Each point and bars are the mean±SEM from 8-20 cultured wells (C).
2. Effects of antioxidants on Aβ25-35-induced neuronal death
Free radical injury is one of the most common and important ways of inducing neuronal death. Aβ-induced neuronal death is also related to oxidative cell damage. To evaluate whether antioxidants such as Trolox and ascorbic acid could prevent Aβ25-35-induced neuronal death in cultured neurons, the amount of LDH release in the culture media was measured after exposure of the cultured neurons to 20 µM Aβ25-35 for 24 or 48 hours. On the basis of LDH measurements in the culture media, approximately 16±2.6% or 52±3.1% of cells were damaged by 20 µM Aβ25-35 treatment for 24 hours (n=12) or 48 hours (n=12), respectively (Fig. 2A). Pretreatment with 100 µM Trolox reduced Aβ25-35-induced LDH release to 4±3% (n=8) for 24 hours and 8±2.7% (n=8) for 48 hours, and 100 µM ascorbic acid reduced LDL release to 0±1.4% (n=8) for 24 hours and 5±2.5% (n=8) for 48 hours (Fig. 2).
FIG. 2.

Effect of co-treatment with Trolox (100 µM) or ascorbic acid (AA, 100 µM) on 20 µM Aβ25-35-induced neuronal death at the end of 24 and 48 hours of exposure. Each bar is the mean±SEM from 8-16 cultured wells. *Significantly different from 24-hour-treated control group (p<0.05). #Significantly different from 48-hour-treated control group (p<0.05).
3. Antioxidant activities of flavonoid compounds
According to the above data, the antioxidants Trolox and ascorbic acid strongly inhibited Aβ25-35-induced neuronal death. To test the antioxidant effects of flavonoid compounds in neuronal death, we used apigenin, baicalein, catechin, epicatechin, EGCG, kaempferol, luteolin, myricetin, quercetin, and luteolin as the flavonoid compounds. Scavenging of free radical DPPH is the basis of a common antioxidant assay. We therefore used the stable free radical DPPH to estimate the antioxidant activity of the flavonoid compounds. This method examines the use of the parameter IC50 (the concentration of substrate that causes 50% loss of the DPPH activity). All of the flavonoid compounds except for apigenin had strong antioxidant effects. Compared with the antioxidant effects of Trolox, kaempferol had similar effects, whereas the others had stronger antioxidant effects (Table 1).
TABLE 1.
Antioxidant ability of various flavonoids against the free radical DPPH
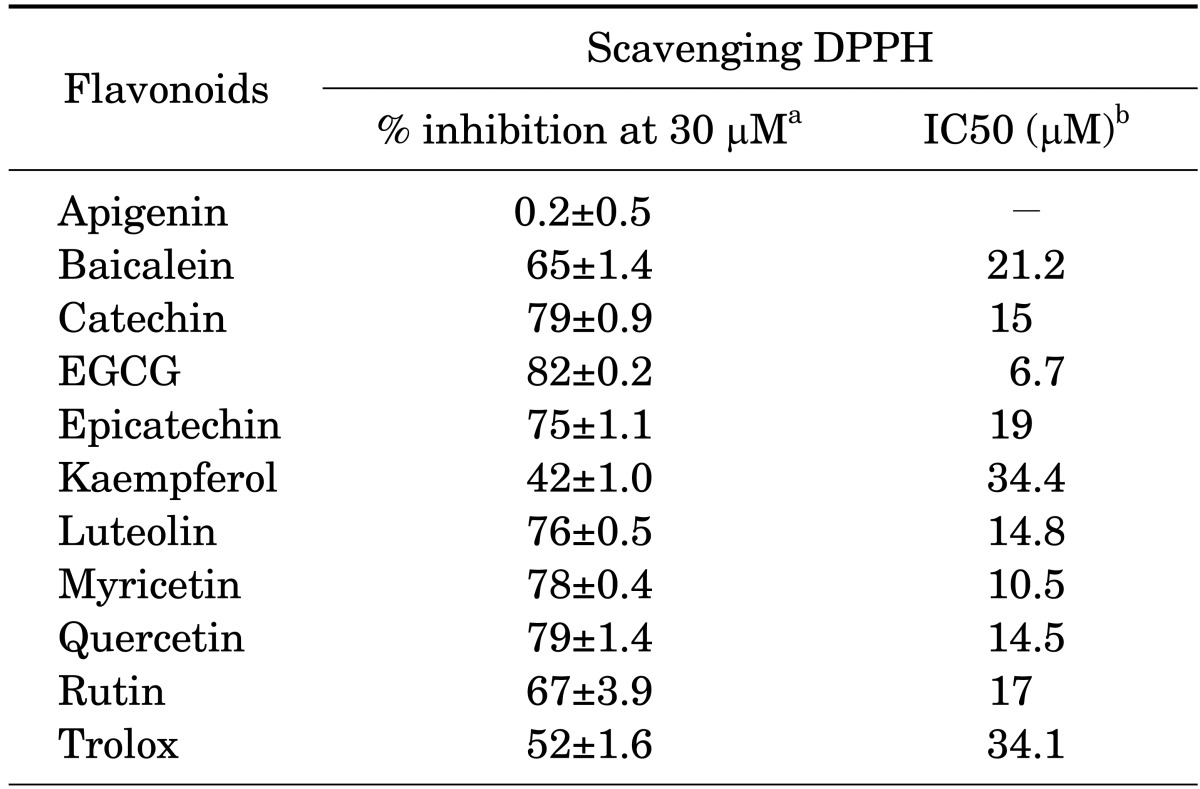
DPPH: 1,1-diphenyl-2-pycrylhydrazyl, EGCG: epigallocatechin gallate. aRepresents the inhibition (%) of DPPH activity at the 30 µM concentration of each flavonoid. bRepresents the concentration of substrate that causes 50% loss of the DPPH activity.
4. Effects of flavonoids on Aβ25-35-induced neuronal death
Cultured neurons were treated with 20 µM Aβ25-35 for 24 or 48 hours and 30 µM of each flavonoid, and the amount of LDH release in the culture media was measured. Except for apigenin, all flavonoids inhibited Aβ25-35-induced neuronal death for 24 hours, but these protective effects were not maintained for 48 hours for baicalein, catechin, kaempferol, quercetin, and rutin (Table 2).
TABLE 2.
Effects of some flavonoids on 20 µM Aβ25-35-induced neuronal death
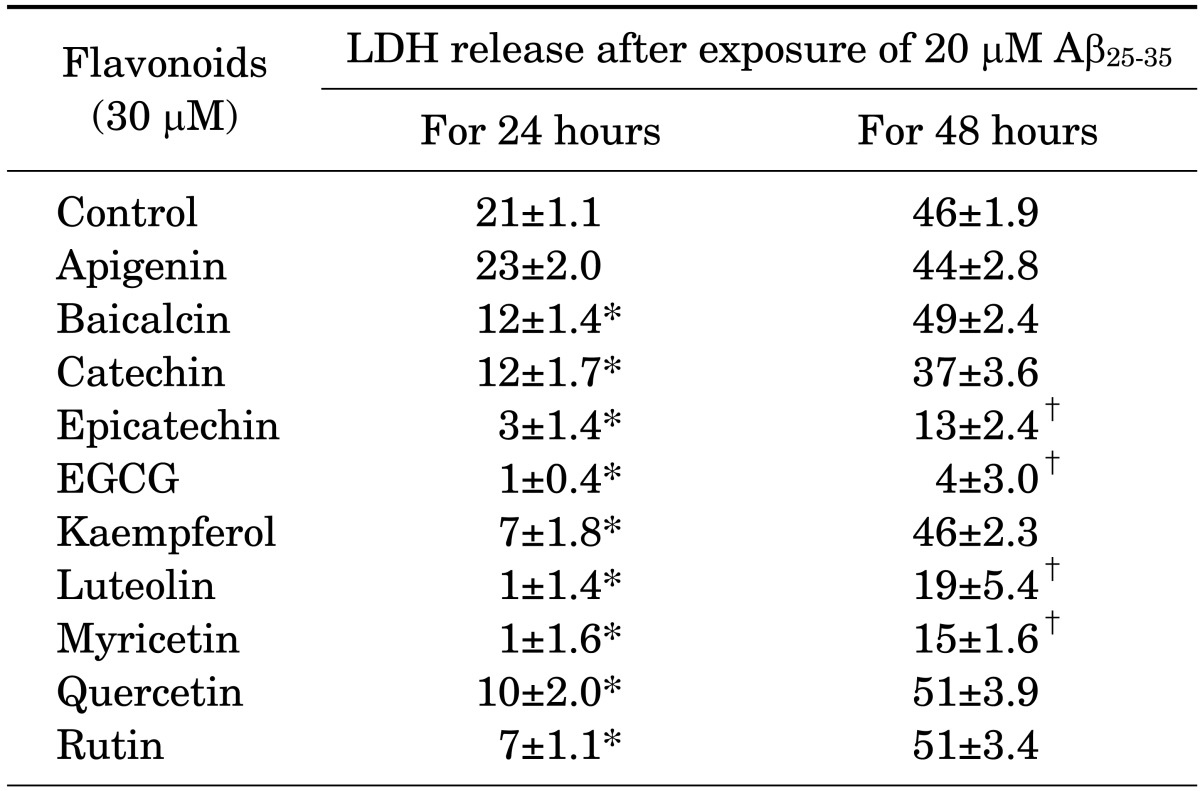
Aβ25-35: β-amyloid peptide fragment 25-35, LDH: lactate dehydrogenase, EGCG: epigallocatechin gallate. Numerals are the calculated percentage (mean±SE) of LDH release from 4×52 wells compared with complete neuronal death. * and †represent significant differences for 24 hours and 48 hours compared with the control group, respectively (p<0.05).
To determine whether the 3 flavonoids EGCG, luteolin, and myricetin could prevent Aβ25-35-induced neuronal death in cultured neurons, LDH release was measured after exposure of the neurons to 20 µM Aβ25-35 for 24 and 48 hours. To determine the flavonoid concentration for neuroprotective effects, we selected 3 flavonoids (EGCG, luteolin, myricetin) that inhibited Aβ25-35-induced neuronal death for 24 and 48 hours. Treatment with EGCG (3, 10, or 30 µM) significantly attenuated LDH release by Aβ25-35-induced neuronal death in a dose-dependent manner compared with the control (Fig. 3A). Compared with LDH release after treatment with 20 µM Aβ25-35 (21±2.3% at 24 hours, n=12, and 46±3.2% at 48 hours, n=12), pretreatment with 3, 10, or 30 µM EGCG reduced LDL release to 9±1.6%, 4±1.6%, and 1±04% at 24 hours (n=12) and to 20±2.5%, 8±1.6%, and 4±3.0% at 48 hours (n=12), respectively (Fig. 3A). Compared with LDH release after treatment with 20 µM Aβ25-35 (20±2.7% at 24 hours, n=12, and 42±1.8% at 48 hours, n=12), pretreatment with 3, 10, or 30 µM luteolin reduced LDH release to 2±1.6%, 0±2.2%, and 1±1.4% at 24 hours (n=12) and to 36±3.4%, 38±2.6%, and 19±4.3% at 48 hours (n=12), respectively (Fig. 3B). At 24 hours, all concentrations of luteolin from 3 µM to 30 µM almost completely inhibited Aβ25-35-induced neuronal death, but at 48 hours, only 30 µM luteolin significantly inhibited Aβ25-35-induced neuronal death (Fig. 3B). In the experiment with myricetin, when cortical neurons were exposed to 20 µM Aβ25-35 for 24 or 48 hours, the amounts of LDH release in the culture media were 23±1.7% (n=12) and 50±1.9% (n=12), respectively. Pretreatment with myricetin (3, 10, and 30 µM) reduced Aβ25-35-induced LDH release to 3±1.8%, 1±2.1%, and 1±1.6% at 24 hours (n=12) and to 34±4.9%, 31±2.8%, and 15±1.6% at 48 hours (n=12), respectively (Fig. 3C).
FIG. 3.

(A) Effects of epigallocatechin gallate (EGCG, 3, 10, 30 µM) on 20 µM Aβ25-35-induced neuronal death at the end of 24 or 48 hours of exposure. (B) Effects of luteolin (LTL, 3, 10, 30 µM) on 20 µM Aβ25-35-induced neuronal death at the end of 24 or 48 hours of exposure. (C) Effects of myricetin (MYR, 3, 10, 30 µM) on 20 µM Aβ25-35-induced neuronal death at the end of 24 or 48 hours of exposure. Each bar is the mean±SEM from 8-12 cultured wells. Other legends are the same as in Fig. 2.
DISCUSSION
The results of the present study showed that Aβ25-35 treatment induced neuronal death that was dependent on dosage and exposure time, whereas the inverted amino acid sequence Aβ35-25 failed to induce neuronal death. These results suggest that Aβ25-35-induced neuronal death is specific. In addition, although increasing the concentration of Aβ25-35 to at least 20 µM made a difference between a 24-hour time frame and a 48-hour time frame, there was no significant difference in the maximum effect within the same time frame. Subsequently, these results imply that Aβ25-35 acted selectively on only a fraction of the cultured neurons, which suggests that further investigation is needed.
In this study, treatment with the antioxidants Trolox and ascorbic acid noticeably blocked Aβ-induced neuronal death. In addition, among the 10 different flavonoid compounds tested in this study, the free radical scavenging inactive compound apigenin failed to block Aβ-induced neuronal death in DPPH scavenging-activity tests, whereas all other compounds significantly blocked Aβ-induced neuronal death after a 24-hour time frame. These results indicate that Aβ-induced neuronal death is associated with oxidative damage due to free radical formation. In fact, many studies suggest that oxidative damage is associated with Aβ-induced neuronal death.23,24,32,33
A search on free radical DPPH scavenging activity yielded a sequence of highest to lowest activity as follows: EGCG > myricetin > quercetin = luteolin = catechin ≥ rutin ≥ epicatechin ≥ baicalein > kaempferol = Trolox (Table 1). Such results indicate that the majority of the flavonoid compounds used in this study had a higher level of antioxidant activity than that of Trolox. Additionally, the inhibitory action of flavonoid compounds at a concentration of 30 µM on Aβ-induced neuronal death was (highest to lowest) as follows: EGCG = luteolin = myricetin = epicatechin > kaempferol = rutin ≥ quercetin ≥ baicalein = catechin (Table 2). Although there are some discrepancies between the rankings for antioxidant activity and those for the inhibitory activity of Aβ-induced neuronal death for some of these flavonoids, the finding that EGCG, the compound with the most antioxidant activity, also had the highest inhibitory activity on Aβ-induced neuronal death implies a possible correlation. In a previous study,34 free radical DPPH scavenging activity was related with total phenolic content, which was related to cell viability in Aβ25-35-induced hippocampal neuronal death. Rice-Evans et al.6 reported that upon measuring an alternate antioxidant activity marker such as Trolox-equivalent antioxidant activity (TEAC) in various flavonoid compounds, apigenin had greater antioxidant activity than kaempferol, and all other flavonoid compounds still had higher antioxidant activity than Trolox. The present study found that apigenin had negligible free radical DPPH scavenging activity as well as no influence on Aβ-induced neuronal death, which is not compatible with the findings of previous studies.6,35 This result may be due to the use of free radical DPPH scavenging activity, which better reflects antioxidant activity on Aβ compared with TEAC. However, an explanation for why apigenin exhibited differing results for free radical DPPH scavenging activity versus TEAC remains to be investigated.
Among the flavonoid compounds used in this study, epicatechin, EGCG, luteolin, and myricetin showed the strongest inhibitory effects, which were maintained after a 48-hour time frame. The dose response tests of EGCG, luteolin, and myricetin showed that a low concentration of 3 to 10 µM significantly blocked Aβ-induced neuronal death. In particular, luteolin and myricetin had stronger inhibitory effects than did EGCG. On the basis of these results and considering that EGCG exhibited stronger inhibitory effects than luteolin or myricetin for free radical DPPH scavenging activity, the inhibitory effects of these compounds on Aβ-induced neuronal death may not always be proportional to antioxidant activity. Nevertheless, the inhibitory effect of EGCG on Aβ-induced neuronal death was substantially stronger after a 48-hour time frame. The difference between the levels of inhibition at 24 hours compared with 48 hours may be dependent on the chemical properties of the compounds, but this will require further inquiry.
Ginkgo biloba extract EGb 761 has been gaining attention for its effects in memory enhancement in the treatment of Alzheimer's disease.36 The structure of EGb 761 as a polymer made up of basic flavonoids37 and reports that this drug blocks Aβ-induced neuronal death in cultured neurons23 suggest the potential of the various flavonoid compounds analyzed in this study to be developed as neuroprotectant agents. Currently, active studies are being done to develop compounds, including both flavonoids and antioxidants, for use as possible drugs to treat neurodegenerative diseases.38,39,40,41
In conclusion, this study was undertaken to analyze the effects of 10 different flavonoid compounds (apigenin, baicalein, catechin, epicatechin, EGCG, kaempferol, luteolin, myricetin, quercetin, and rutin) to search for a flavonoid compound that can be developed as a new drug to treat Aβ-induced neuronal death. Aβ25-35-induced neuronal death was dependent on dosage and exposure time. The results of this study indicated the presence of oxidative damage during Aβ-induced neuronal death and that flavonoid compounds with free radical scavenging activity can block Aβ-induced neuronal death. Finally, the results suggest that compounds that have strong inhibitory effects against Aβ25-35-induced neuronal death, such as epicatechin, EGCG, luteolin, and myricetin, have the potential to be developed into new drugs for the treatment of Alzheimer's disease.
ACKNOWLEDGEMENTS
This study was supported by a grant of the Korean Health Technology R&D Project A121751 (to KH Choi) funded by the Ministry of Health & Welfare, Republic of Korea, and a grant from the Brain Research Program through the National Research Foundation of Korea funded by the Ministry of Science, ICT & Future Planning NRF-2013M3C7A1072997 (to BC Kim).
Footnotes
None declared.
References
- 1.Robards K, Antolovich M. Analytical chemistry of fruit bioflavonoids. A review. Analyst. 1997;122:11R–34R. [Google Scholar]
- 2.Lu MF, Xiao ZT, Zhang HY. Where do health benefits of flavonoids come from? Insights from flavonoid targets and their evolutionary history. Biochem Biophys Res Commun. 2013;434:701–704. doi: 10.1016/j.bbrc.2013.04.035. [DOI] [PubMed] [Google Scholar]
- 3.Beecher GR. Overview of dietary flavonoids: nomenclature, occurrence and intake. J Nutr. 2003;133:3248S–3254S. doi: 10.1093/jn/133.10.3248S. [DOI] [PubMed] [Google Scholar]
- 4.Harnly JM, Doherty RF, Beecher GR, Holden JM, Haytowitz DB, Bhagwat S, et al. Flavonoid content of U.S. fruits, vegetables, and nuts. J Agric Food Chem. 2006;54:9966–9977. doi: 10.1021/jf061478a. [DOI] [PubMed] [Google Scholar]
- 5.Hertog MG, Feskens EJ, Hollman PC, Katan MB, Kromhout D. Dietary antioxidant flavonoids and risk of coronary heart disease: the Zutphen Elderly Study. Lancet. 1993;342:1007–1011. doi: 10.1016/0140-6736(93)92876-u. [DOI] [PubMed] [Google Scholar]
- 6.Rice-Evans CA, Miller NJ, Paganga G. Structure-antioxidant activity relationships of flavonoids and phenolic acids. Free Radic Biol Med. 1996;20:933–956. doi: 10.1016/0891-5849(95)02227-9. [DOI] [PubMed] [Google Scholar]
- 7.Pietta PG. Flavonoids as antioxidants. J Nat Prod. 2000;63:1035–1042. doi: 10.1021/np9904509. [DOI] [PubMed] [Google Scholar]
- 8.Zeng LH, Zhang HD, Xu CJ, Bian YJ, Xu XJ, Xie QM, et al. Neuroprotective effects of flavonoids extracted from licorice on kainate-induced seizure in mice through their antioxidant properties. J Zhejiang Univ Sci B. 2013;14:1004–1012. doi: 10.1631/jzus.B1300138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dugas AJ, Jr, Castañeda-Acosta J, Bonin GC, Price KL, Fischer NH, Winston GW. Evaluation of the total peroxyl radical-scavenging capacity of flavonoids: structure-activity relationships. J Nat Prod. 2000;63:327–331. doi: 10.1021/np990352n. [DOI] [PubMed] [Google Scholar]
- 10.Cho JG, Song NY, Nam TG, Shrestha S, Park HJ, Lyu HN, et al. Flavonoids from the grains of C1/R-S transgenic rice, the transgenic Oryza sativa spp. japonica, and their radical scavenging activities. J Agric Food Chem. 2013;61:10354–10359. doi: 10.1021/jf403072c. [DOI] [PubMed] [Google Scholar]
- 11.Nijveldt RJ, van Nood E, van Hoorn DE, Boelens PG, van Norren K, van Leeuwen PA. Flavonoids: a review of probable mechanisms of action and potential applications. Am J Clin Nutr. 2001;74:418–425. doi: 10.1093/ajcn/74.4.418. [DOI] [PubMed] [Google Scholar]
- 12.Thilakarathna SH, Rupasinghe HP. Flavonoid bioavailability and attempts for bioavailability enhancement. Nutrients. 2013;5:3367–3387. doi: 10.3390/nu5093367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Beking K, Vieira A. Flavonoid intake and disability-adjusted life years due to Alzheimer's and related dementias: a population-based study involving twenty-three developed countries. Public Health Nutr. 2010;13:1403–1409. doi: 10.1017/S1368980009992990. [DOI] [PubMed] [Google Scholar]
- 14.Letenneur L, Proust-Lima C, Le Gouge A, Dartigues JF, Barberger-Gateau P. Flavonoid intake and cognitive decline over a 10-year period. Am J Epidemiol. 2007;165:1364–1371. doi: 10.1093/aje/kwm036. [DOI] [PubMed] [Google Scholar]
- 15.Fratiglioni L, Launer LJ, Andersen K, Breteler MM, Copeland JR, Dartigues JF, et al. Neurologic Diseases in the Elderly Research Group. Incidence of dementia and major subtypes in Europe: A collaborative study of population-based cohorts. Neurology. 2000;54(11 Suppl 5):S10–S15. [PubMed] [Google Scholar]
- 16.Terry RD, Masliah E, Salmon DP, Butters N, DeTeresa R, Hill R, et al. Physical basis of cognitive alterations in Alzheimer's disease: synapse loss is the major correlate of cognitive impairment. Ann Neurol. 1991;30:572–580. doi: 10.1002/ana.410300410. [DOI] [PubMed] [Google Scholar]
- 17.Masters CL, Simms G, Weinman NA, Multhaup G, McDonald BL, Beyreuther K. Amyloid plaque core protein in Alzheimer disease and Down syndrome. Proc Natl Acad Sci U S A. 1985;82:4245–4249. doi: 10.1073/pnas.82.12.4245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kang J, Lemaire HG, Unterbeck A, Salbaum JM, Masters CL, Grzeschik KH, et al. The precursor of Alzheimer's disease amyloid A4 protein resembles a cell-surface receptor. Nature. 1987;325:733–736. doi: 10.1038/325733a0. [DOI] [PubMed] [Google Scholar]
- 19.Small DH, McLean CA. Alzheimer's disease and the amyloid beta protein: What is the role of amyloid? J Neurochem. 1999;73:443–449. doi: 10.1046/j.1471-4159.1999.0730443.x. [DOI] [PubMed] [Google Scholar]
- 20.Small DH, Mok SS, Bornstein JC. Alzheimer's disease and Abeta toxicity: from top to bottom. Nat Rev Neurosci. 2001;2:595–598. doi: 10.1038/35086072. [DOI] [PubMed] [Google Scholar]
- 21.Pike CJ, Burdick D, Walencewicz AJ, Glabe CG, Cotman CW. Neurodegeneration induced by beta-amyloid peptides in vitro: the role of peptide assembly state. J Neurosci. 1993;13:1676–1687. doi: 10.1523/JNEUROSCI.13-04-01676.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jordán J, Galindo MF, Miller RJ. Role of calpain- and interleukin-1 beta converting enzyme-like proteases in the beta-amyloid-induced death of rat hippocampal neurons in culture. J Neurochem. 1997;68:1612–1621. doi: 10.1046/j.1471-4159.1997.68041612.x. [DOI] [PubMed] [Google Scholar]
- 23.Bastianetto S, Ramassamy C, Doré S, Christen Y, Poirier J, Quirion R. The Ginkgo biloba extract (EGb 761) protects hippocampal neurons against cell death induced by beta-amyloid. Eur J Neurosci. 2000;12:1882–1890. doi: 10.1046/j.1460-9568.2000.00069.x. [DOI] [PubMed] [Google Scholar]
- 24.Bisaglia M, Venezia V, Piccioli P, Stanzione S, Porcile C, Russo C, et al. Acetaminophen protects hippocampal neurons and PC12 cultures from amyloid beta-peptides induced oxidative stress and reduces NF-kappaB activation. Neurochem Int. 2002;41:43–54. doi: 10.1016/s0197-0186(01)00136-x. [DOI] [PubMed] [Google Scholar]
- 25.Pike CJ, Walencewicz-Wasserman AJ, Kosmoski J, Cribbs DH, Glabe CG, Cotman CW. Structure-activity analyses of beta-amyloid peptides: contributions of the beta 25-35 region to aggregation and neurotoxicity. J Neurochem. 1995;64:253–265. doi: 10.1046/j.1471-4159.1995.64010253.x. [DOI] [PubMed] [Google Scholar]
- 26.Smid SD, Maag JL, Musgrave IF. Dietary polyphenol-derived protection against neurotoxic β-amyloid protein: from molecular to clinical. Food Funct. 2012;3:1242–1250. doi: 10.1039/c2fo30075c. [DOI] [PubMed] [Google Scholar]
- 27.Hensley K, Carney JM, Mattson MP, Aksenova M, Harris M, Wu JF, et al. A model for beta-amyloid aggregation and neurotoxicity based on free radical generation by the peptide: relevance to Alzheimer disease. Proc Natl Acad Sci U S A. 1994;91:3270–3274. doi: 10.1073/pnas.91.8.3270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mattson MP, Cheng B, Davis D, Bryant K, Lieberburg I, Rydel RE. Beta-Amyloid peptides destabilize calcium homeostasis and render human cortical neurons vulnerable to excitotoxicity. J Neurosci. 1992;12:376–389. doi: 10.1523/JNEUROSCI.12-02-00376.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Choi DW, Maulucci-Gedde M, Kriegstein AR. Glutamate neurotoxicity in cortical cell culture. J Neurosci. 1987;7:357–368. doi: 10.1523/JNEUROSCI.07-02-00357.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pike CJ, Walencewicz AJ, Glabe CG, Cotman CW. In vitro aging of beta-amyloid protein causes peptide aggregation and neurotoxicity. Brain Res. 1991;563:311–314. doi: 10.1016/0006-8993(91)91553-d. [DOI] [PubMed] [Google Scholar]
- 31.Koh JY, Choi DW. Quantitative determination of glutamate mediated cortical neuronal injury in cell culture by lactate dehydrogenase efflux assay. J Neurosci Methods. 1987;20:83–90. doi: 10.1016/0165-0270(87)90041-0. [DOI] [PubMed] [Google Scholar]
- 32.Goodman Y, Steiner MR, Steiner SM, Mattson MP. Nordihydroguaiaretic acid protects hippocampal neurons against amyloid beta-peptide toxicity, and attenuates free radical and calcium accumulation. Brain Res. 1994;654:171–176. doi: 10.1016/0006-8993(94)91586-5. [DOI] [PubMed] [Google Scholar]
- 33.Bruce AJ, Malfroy B, Baudry M. beta-Amyloid toxicity in organotypic hippocampal cultures: protection by EUK-8, a synthetic catalytic free radical scavenger. Proc Natl Acad Sci U S A. 1996;93:2312–2316. doi: 10.1073/pnas.93.6.2312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Okada Y, Okada M. Protective effects of plant seed extracts against amyloid β-induced neurotoxicity in cultured hippocampal neurons. J Pharm Bioallied Sci. 2013;5:141–147. doi: 10.4103/0975-7406.111819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu R, Zhang T, Yang H, Lan X, Ying J, Du G. The flavonoid apigenin protects brain neurovascular coupling against amyloid-β25-35-induced toxicity in mice. J Alzheimers Dis. 2011;24:85–100. doi: 10.3233/JAD-2010-101593. [DOI] [PubMed] [Google Scholar]
- 36.Oken BS, Storzbach DM, Kaye JA. The efficacy of Ginkgo biloba on cognitive function in Alzheimer disease. Arch Neurol. 1998;55:1409–1415. doi: 10.1001/archneur.55.11.1409. [DOI] [PubMed] [Google Scholar]
- 37.Drieu K. Preparation and definition of Ginkgo biloba extract. Presse Med. 1986;15:1455–1457. [PubMed] [Google Scholar]
- 38.Moosmann B, Behl C. Antioxidants as treatment for neurodegenerative disorders. Expert Opin Investig Drugs. 2002;11:1407–1435. doi: 10.1517/13543784.11.10.1407. [DOI] [PubMed] [Google Scholar]
- 39.Youdim KA, Spencer JP, Schroeter H, Rice-Evans C. Dietary flavonoids as potential neuroprotectants. Biol Chem. 2002;383:503–519. doi: 10.1515/BC.2002.052. [DOI] [PubMed] [Google Scholar]
- 40.Gao J, Inagaki Y, Li X, Kokudo N, Tang W. Research progress on natural products from traditional Chinese medicine in treatment of Alzheimer's disease. Drug Discov Ther. 2013;7:46–57. [PubMed] [Google Scholar]
- 41.Jones QR, Warford J, Rupasinghe HP, Robertson GS. Target-based selection of flavonoids for neurodegenerative disorders. Trends Pharmacol Sci. 2012;33:602–610. doi: 10.1016/j.tips.2012.08.002. [DOI] [PubMed] [Google Scholar]