

Abstract
The long-term use of proton pump inhibitors (PPIs) exacerbates corpus atrophic gastritis in patients with Helicobacter pylori (H. pylori) infection. To identify a potential mechanism for this change, we discuss interactions between pH, bile acids, and H. pylori. Duodenogastric reflux, which includes bile, occurs in healthy individuals, and bile reflux is increased in patients with gastroesophageal reflux disease (GERD). Diluted human plasma and bile acids have been found to be significant chemoattractants and chemorepellents, respectively, for the bacillus H. pylori. Although only taurine conjugates, with a pKa of 1.8-1.9, are soluble in an acidic environment, glycine conjugates, with a pKa of 4.3-5.2, as well as taurine-conjugated bile acids are soluble in the presence of PPI therapy. Thus, the soluble bile acid concentrations in the gastric contents of patients with GERD after continuous PPI therapy are considerably higher than that in those with intact acid production. In the distal stomach, the high concentration of soluble bile acids is likely to act as a bactericide or chemorepellent for H. pylori. In contrast, the mucous layer in the proximal stomach has an optimal bile concentration that forms chemotactic gradients with plasma components required to direct H. pylori to the epithelial surface. H. pylori may then colonize in the stomach body rather than in the pyloric antrum, which may explain the occurrence of corpus-predominant gastritis after PPI therapy in H. pylori-positive patients with GERD.
Keywords: Helicobacter pylori, Proton pump inhibitor, Corpus-predominant gastritis, Bile acids, Gastroesophageal reflux disease, Chemotactic gradient
Core tip: It has been widely accepted that the long-term use of proton pump inhibitors (PPIs) exacerbates corpus atrophic gastritis in patients with Helicobacter pylori (H. pylori) infection. Recently, we successfully demonstrated that long-term PPI administration promotes corpus atrophic gastritis in Mongolian gerbils, which are excellent models of H. pylori-related gastritis and adenocarcinoma. Here, we suggest a potential mechanism for corpus-predominant gastritis after PPI therapy in H. pylori-positive patients with gastroesophageal reflux disease that includes interactions between bile acids, pH, and H. pylori.
INTRODUCTION
It has been widely accepted that the long-term use of proton pump inhibitors (PPIs) exacerbates corpus atrophic gastritis in patients with Helicobacter pylori (H. pylori) infection[1-3]. Furthermore, we successfully demonstrated that long-term PPI administration promotes corpus atrophic gastritis in Mongolian gerbils, which are excellent models of H. pylori-related gastritis and adenocarcinoma[4-6]. Kuipers hypothesized that corpus atrophic gastritis is caused by a change from antral-predominant to corpus-predominant gastritis related to PPI therapy in H. pylori-positive patients[7]. However, the mechanism for this change has not been determined. In this article, we suggest a potential mechanism for corpus-predominant gastritis related to PPI therapy in H. pylori-positive patients with gastroesophageal reflux disease (GERD) that includes interactions between pH, bile acids, and H. pylori.
BILE REFLUX AND GERD
The use of PPIs for GERD treatment has increased substantially over the past 2 decades[8]. Duodenogastric reflux, which includes bile, occurs in healthy individuals[9,10], but increases in patients with GERD[11], and bile acids have been detected in esophageal aspiration samples in up to 86% of patients with GERD[12]. These findings suggest that patients with GERD are likely to have increased concentration of bile acids in their gastric contents.
BILE ACIDS AND pKa
The concentration of bile acids in the duodenum ranges from 2-3 mmol/L at fasting to 6-10 mmol/L after a meal[13,14]. The chemical characteristics of bile in humans are governed by the pKa of the individual bile acids. Free bile acids have a pKa of approximately 7 and comprise approximately 2% of bile. Glycine-conjugated bile acids have a pKa of 4.3-5.2 and comprise more than 60% of bile, and taurine-conjugated bile acids have a pKa of 1.8-1.9 and comprise 20% of bile, resulting in a ratio of glycine to taurine conjugates of about 3:1[15,16]. Although both glycine and taurine conjugates are soluble in neutral solutions, only taurine conjugates are soluble in an acidic environment. However, both glycine- and taurine-conjugated bile acids are soluble in the presence of PPI therapy. Thus, the concentrations of soluble bile acids markedly increase (approximately 4 times) with PPI administration.
BILE ACIDS AND H. PYLORI
Experimentally, highly diluted human plasma and gallbladder bile (2% diluted in buffer) were found to be significant chemoattractants and chemorepellents, respectively, for the bacillus H. pylori[17]. Of the bile acids, which were all chemorepellents, the greatest effects were demonstrated by taurocholic and taurodeoxycholic acids[17]. In contrast, another study reported that glycine-conjugated bile acids markedly inhibit H. pylori growth and more so than the taurine conjugates[18]. It is strongly suspected that important chemotactic gradients are formed from chemoattractant plasma components that transude from capillaries into the mucosa and chemorepellent duodenal bile contents that reflux into the stomach[17].
It has also been reported that bile is bactericidal for H. pylori at higher concentrations; H. pylori is reduced by 0.5%-1% bile and inhibited by 5% bile[19]. Several studies have reported an inverse relationship between bile reflux and the presence of H. pylori[20-22], which may account for the absence of H. pylori in the stomach with persistent biliary reflux and in the lower gastrointestinal tract[19]. These results imply that an optimal bile concentration that forms chemotactic gradients with plasma is required to direct H. pylori to the epithelial surface from the gastric lumen and this concentration is likely to be < 5%.
EVIDENCE OF DUODENAL ULCER TREATMENT BY ANTISECRETORY THERAPY
Since the 1940s, it has been recognized that the duodenal bulb pH is lower in patients with ulcer disease than in those without[23] and that antacids and antisecretory therapy, which reduce the duodenal acid load, accelerate ulcer healing. Graham et al[18] reported that the combination of a high duodenal acid load and H. pylori infection is possibly a critical event in the pathogenesis of H. pylori-related duodenal ulcer disease. They suggested that, because glycine-conjugated bile acids are precipitated at an acidic pH, H. pylori can survive in gastric metaplasia, and any event that leads to an increase in the duodenal acid load predisposes patients with H. pylori infection to duodenal ulcer diseases[24,25]. Furthermore, any condition that reduces the duodenal acid load (e.g., antisecretory therapy) allows the bile acids to remain in the solution, inhibits the growth of H. pylori, and promotes ulcer healing[18]. Therefore, inhibition of acid by antisecretory therapy increases the duodenal bulb pH, allowing soluble glycine-conjugated bile acids to inhibit H. pylori growth[24].
POTENTIAL MECHANISM OF CORPUS-PREDOMINANT GASTRITIS AFTER PPI THERAPY
In patients with intact gastric acid production, the pyloric antrum mucous layer may have the optimal bile concentration to direct H. pylori to the epithelial surface and thus enable H. pylori colonization, while the stomach body may be less optimal for H. pylori colonization[17] (Figure 1A). The relative bile concentrations, particularly those of both soluble glycine- and taurine-conjugated bile acids, in the gastric contents in patients with GERD, in whom the gastric acid production is inhibited by maintenance PPI therapy, are considerably higher than in those with intact acid production. Therefore, in the distal stomach, the high concentration of soluble bile acids likely acts as a bactericide or chemorepellent for H. pylori, and chemotactic gradients with plasma components are not formed to direct the H. pylori toward the epithelial surface. In contrast, the mucous layer in the proximal stomach has the optimal bile acid concentration to form chemotactic gradients with plasma components for H. pylori colonization in the presence of PPI therapy. H. pylori may then colonize in the stomach body and not in the pyloric antrum (Figure 1B). This may explain the mechanism for corpus-predominant gastritis after PPI therapy in H. pylori-positive patients with GERD.
Figure 1.
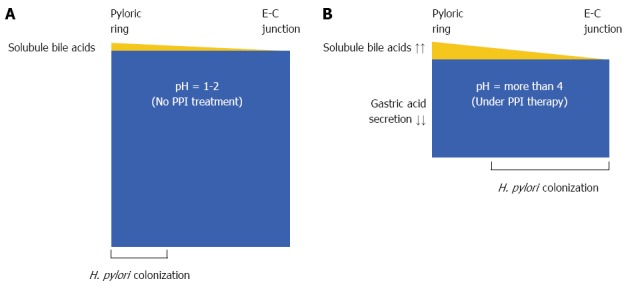
Potential mechanism for the changes in Helicobactor pylori colonization with proton pump inhibitor therapy. A: Normal subject with intact acid production; B: Patient with gastroesophageal reflux disease (GERD) and proton pump inhibitor (PPI) therapy. These figures show the intragastric conditions. The yellow area represents the reflux of soluble bile acids, and the blue area represents gastric juice. In normal subjects, the concentration of soluble bile acids may be < 5% in the gastric contents of the pyloric antrum (A). In contrast, increased soluble bile acid reflux with decreased gastric acid secretion acts as a bactericide for Helicobacter pylori (H. pylori) in the distal stomach; the concentration of soluble bile acids in patients with GERD under PPI maintenance therapy is considerably higher than that in normal subjects with intact acid production, especially in the distal stomach (B). The colonization of H. pylori therefore changes the pattern from antral-predominant to corpus-predominant. E-C junction: Esophago-gastric junction.
In conclusion, interactions between bile acids, pH, and H. pylori seem to be associated with the occurrence of corpus-predominant gastritis after PPI therapy in H. pylori-positive patients with GERD. It has been reported that the corpus-predominant gastritis might promote adenocarcinoma development[7]. In the clinical setting, it is recommended that patients being considered for long-term PPI therapy should be tested for H. pylori infection, and if present, this pathogen should be eradicated[6].
Footnotes
P- Reviewer: Kim BW, Reyes VE S- Editor: Ma YJ L- Editor: A E- Editor: Ma S
References
- 1.Kuipers EJ, Uyterlinde AM, Peña AS, Hazenberg HJ, Bloemena E, Lindeman J, Klinkenberg-Knol EC, Meuwissen SG. Increase of Helicobacter pylori-associated corpus gastritis during acid suppressive therapy: implications for long-term safety. Am J Gastroenterol. 1995;90:1401–1406. [PubMed] [Google Scholar]
- 2.Kuipers EJ, Lundell L, Klinkenberg-Knol EC, Havu N, Festen HP, Liedman B, Lamers CB, Jansen JB, Dalenback J, Snel P, et al. Atrophic gastritis and Helicobacter pylori infection in patients with reflux esophagitis treated with omeprazole or fundoplication. N Engl J Med. 1996;334:1018–1022. doi: 10.1056/NEJM199604183341603. [DOI] [PubMed] [Google Scholar]
- 3.García Rodríguez LA, Lagergren J, Lindblad M. Gastric acid suppression and risk of oesophageal and gastric adenocarcinoma: a nested case control study in the UK. Gut. 2006;55:1538–1544. doi: 10.1136/gut.2005.086579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Watanabe T, Tada M, Nagai H, Sasaki S, Nakao M. Helicobacter pylori infection induces gastric cancer in mongolian gerbils. Gastroenterology. 1998;115:642–648. doi: 10.1016/s0016-5085(98)70143-x. [DOI] [PubMed] [Google Scholar]
- 5.Kudo T, Lu H, Wu JY, Ohno T, Wu MJ, Genta RM, Graham DY, Yamaoka Y. Pattern of transcription factor activation in Helicobacter pylori-infected Mongolian gerbils. Gastroenterology. 2007;132:1024–1038. doi: 10.1053/j.gastro.2007.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hagiwara T, Mukaisho K, Nakayama T, Sugihara H, Hattori T. Long-term proton pump inhibitor administration worsens atrophic corpus gastritis and promotes adenocarcinoma development in Mongolian gerbils infected with Helicobacter pylori. Gut. 2011;60:624–630. doi: 10.1136/gut.2010.207662. [DOI] [PubMed] [Google Scholar]
- 7.Kuipers EJ. Proton pump inhibitors and gastric neoplasia. Gut. 2006;55:1217–1221. doi: 10.1136/gut.2005.090514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Targownik LE, Metge CJ, Leung S, Chateau DG. The relative efficacies of gastroprotective strategies in chronic users of nonsteroidal anti-inflammatory drugs. Gastroenterology. 2008;134:937–944. doi: 10.1053/j.gastro.2008.01.010. [DOI] [PubMed] [Google Scholar]
- 9.Müller-Lissner SA, Fimmel CJ, Sonnenberg A, Will N, Müller-Duysing W, Heinzel F, Müller R, Blum AL. Novel approach to quantify duodenogastric reflux in healthy volunteers and in patients with type I gastric ulcer. Gut. 1983;24:510–518. doi: 10.1136/gut.24.6.510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.King PM, Adam RD, Pryde A, McDicken WN, Heading RC. Relationships of human antroduodenal motility and transpyloric fluid movement: non-invasive observations with real-time ultrasound. Gut. 1984;25:1384–1391. doi: 10.1136/gut.25.12.1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dixon MF, Neville PM, Mapstone NP, Moayyedi P, Axon AT. Bile reflux gastritis and Barrett’s oesophagus: further evidence of a role for duodenogastro-oesophageal reflux. Gut. 2001;49:359–363. doi: 10.1136/gut.49.3.359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kauer WK, Peters JH, DeMeester TR, Feussner H, Ireland AP, Stein HJ, Siewert RJ. Composition and concentration of bile acid reflux into the esophagus of patients with gastroesophageal reflux disease. Surgery. 1997;122:874–881. doi: 10.1016/s0039-6060(97)90327-5. [DOI] [PubMed] [Google Scholar]
- 13.Northfield TC, McColl I. Postprandial concentrations of free and conjugated bile acids down the length of the normal human small intestine. Gut. 1973;14:513–518. doi: 10.1136/gut.14.7.513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dowling RH. The enterohepatic circulation. Gastroenterology. 1972;62:122–140. [PubMed] [Google Scholar]
- 15.Nair PP, Kritchenvski D. The Bile Acids: Chemistry, Physiology and Metabolism Vol. 1: Chemistry. New York: Plenum Press; 1971. [Google Scholar]
- 16.Stamp DH. Three hypotheses linking bile to carcinogenesis in the gastrointestinal tract: certain bile salts have properties that may be used to complement chemotherapy. Med Hypotheses. 2002;59:398–405. doi: 10.1016/s0306-9877(02)00125-1. [DOI] [PubMed] [Google Scholar]
- 17.Worku ML, Karim QN, Spencer J, Sidebotham RL. Chemotactic response of Helicobacter pylori to human plasma and bile. J Med Microbiol. 2004;53:807–811. doi: 10.1099/jmm.0.45636-0. [DOI] [PubMed] [Google Scholar]
- 18.Graham DY, Osato MS. H. pylori in the pathogenesis of duodenal ulcer: interaction between duodenal acid load, bile, and H. pylori. Am J Gastroenterol. 2000;95:87–91. doi: 10.1111/j.1572-0241.2000.01704.x. [DOI] [PubMed] [Google Scholar]
- 19.Tompkins DS, West AP. Campylobacter pylori, acid, and bile. J Clin Pathol. 1987;40:1387. doi: 10.1136/jcp.40.11.1387-a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.O'Connor HJ, Newbold KM, Alexander-Williams J, Thompson H, Drumm J, Donovan IA. Effect of Roux-en-Y biliary diversion on Campylobacter pylori. Gastroenterology. 1989;97:958–964. doi: 10.1016/0016-5085(89)91504-7. [DOI] [PubMed] [Google Scholar]
- 21.Sobala GM, O’Connor HJ, Dewar EP, King RF, Axon AT, Dixon MF. Bile reflux and intestinal metaplasia in gastric mucosa. J Clin Pathol. 1993;46:235–240. doi: 10.1136/jcp.46.3.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.O'Connor HJ, Dixon MF, Wyatt JI, Axon AT, Ward DC, Dewar EP, Johnston D. Effect of duodenal ulcer surgery and enterogastric reflux on Campylobacter pyloridis. Lancet. 1986;2:1178–1181. doi: 10.1016/s0140-6736(86)92193-8. [DOI] [PubMed] [Google Scholar]
- 23.Berk JE, Rehfuss ME, Thomas JE. Duodenal bulb acidity under fasting conditions in patients with duodenal ulcer. Arch Surg. 1942;45:406–415. [Google Scholar]
- 24.Han SW, Evans DG, el-Zaatari FA, Go MF, Graham DY. The interaction of pH, bile, and Helicobacter pylori may explain duodenal ulcer. Am J Gastroenterol. 1996;91:1135–1137. [PubMed] [Google Scholar]
- 25.Graham DY, Genta RM, Go MF. Which is the most important factor in duodenal ulcer pathogenesis: the strain of Helicobacter pylori or the host. In: Hunt RH, Tytgat GNJ, eds , editors. Helicobacter pylori: Basic mechanisms to clinical cure. Lancaster: Kluwer Academic Publishers; 1996. pp. 85–91. [Google Scholar]