

Abstract
Pancreatic ductal adenocarcinoma (PDAC) is a devastating disease with a median overall survival time of 5 mo and the five years survival less than 5%, a rate essentially unchanged over the course of the years. A well defined progression model of accumulation of genetic alterations ranging from single point mutations to gross chromosomal abnormalities has been introduced to describe the origin of this disease. However, due to the its subtle nature and concurring events PDAC cure remains elusive. Nuclear receptors (NR) are members of a large superfamily of evolutionarily conserved ligand-regulated DNA-binding transcription factors functionally involved in important cellular functions ranging from regulation of metabolism, to growth and development. Given the nature of their ligands, NR are very tempting drug targets and their pharmacological modulation has been widely exploited for the treatment of metabolic and inflammatory diseases. There are now clear evidences that both classical ligand-activated and orphan NR are involved in the pathogenesis of PDAC from its very early stages; nonetheless many aspects of their role are not fully understood. The purpose of this review is to highlight the striking connections that link peroxisome proliferator activated receptors, retinoic acid receptors, retinoid X receptor, androgen receptor, estrogen receptors and the orphan NR Nur, chicken ovalbumin upstream promoter transcription factor II and the liver receptor homologue-1 receptor to PDAC development, connections that could lead to the identification of novel therapies for this disease.
Keywords: Peroxisome proliferator activated receptor, Pancreatic intraepithelial neoplasia, COUP-TFII, Nuclear receptors, Orphan nuclear receptor, Nuclear receptors 4A2, Nuclear receptors 2F2, Pancreatic cancer, Retinoid X receptor, Testicular receptor 3
Core tip: Pancreatic cancer is a devastating disease with well defined genetic alterations made deadly by its subtle nature and the lack of effective drugs. Nuclear receptors (NR) are ligand-regulated transcription factors involved in important cellular functions and tempting targets for drug development. There are now evidences that classical ligand-activated peroxisome proliferator activated receptors, retinoic acid receptors, retinoid X receptors, androgen receptor, estrogen receptors and orphan Nur, chicken ovalbumin upstream promoter transcription factor II and liver receptor homologue-1 NR are involved in the pathogenesis of pancreatic cancer. No clinical application of these NR in pancreatic cancer cure is reported but a more comprehensive analysis of NR action could lead to the identification of new treatments for this disease.
INTRODUCTION
The most frequent form of pancreatic cancer is pancreatic ductal adenocarcinoma (PDAC) one of the most lethal cancer and the fifth cause of cancer death in the Developed Countries[1]. Treatment of PDAC is primarily a combination of curative surgery and adjuvant chemotherapy with modestly effective drugs[2]. Unfortunately, due to absence of specific symptoms, a high percentage of patients at the time of diagnosis present a incurable locally advanced or metastatic disease that precludes a successful surgical resection, the only possible curative methods for PDAC, at least for early stages diseases.
Approved in 1996[2], Gemcitabine is the frontline standard chemotherapy used essentially in monotherapy for the treatment of pancreatic cancer with modest results; the innate or acquired resistance to chemotherapy drugs of PDAC remains the major obstacle to its successful control[3]. Early detection of PDAC is difficult if not impossible: benign and malignant lesions share similar clinical presentations and imaging features, making imaging detection of early disease difficult[4], and the majority of available molecular markers possess low specificity[5]. Therefore the median overall survival time is 5 mo and the five years survival is less than 5%, a rate essentially unchanged over the course of the years[6]. Consequently, a deeper understanding the pathobiology of this disease is essential to lead to new targeting strategies.
The route to PDAC
Despite the effort of the scientific community, the etiology of PDAC is still poorly understood undermining the efforts for its prevention and cure. A large number of epidemiological studies suggested that tobacco smoking, alcohol consumptions, obesity, chronic pancreatitis, genetic risk factors and diabetes are risk factors of PDAC[7-9].
Pancreatic adenocarcinoma arises from three precursor lesions: the microscopic pancreatic intraepithelial neoplasia (PanIN) and the macroscopic intraductal papillary mucinous neoplasm and mucinous cystic neoplasm (Figure 1)[10,11].
Figure 1.
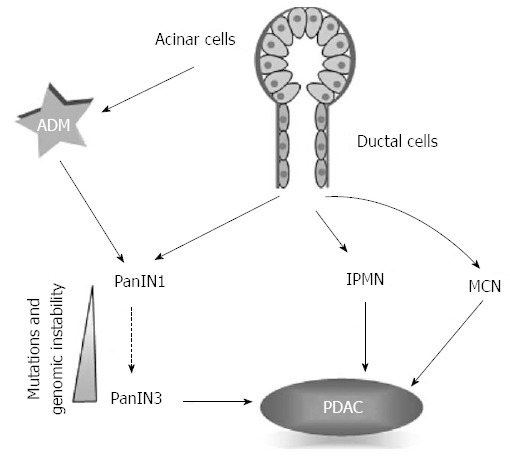
Origin of pancreatic ductal adenocarcinoma. It is widely accepted that pancreatic ductal carcinoma (PDAC) arises from precursor lesions derived from ductal cells; however, recently another model has been proposed where PDAC arises from acinar cells through a process called “acinar to ductal metaplasia” (ADM). PanIN: Intraepithelial neoplasm; IPMN: Intraductal papillary mucinous neoplasm; MCN: Mucinous cystic neoplasm.
PanINs are the more frequent preneoplastic precursors of PDAC[12]; they are classified according to the accumulation of architectural, cytologic, and genetic alterations: from PanIN 1 with the appearance of columnar cells with mucin, to PanIN3 (also called carcinoma in situ) characterized by a severe cyto-architectural athypia[10].
PDAC is a disease with a well defined progression model of accumulation of genetic alterations ranging from single point mutations to gross chromosomal abnormalities[13-17]. The most frequent and studied alterations determine the activation of epidermal growth factor receptor-KRAS pathway[15]. Although almost all patients posses at least one of these mutations, the late stage disease is characterized by increased genome instability and heterogeneity with an average of 63 genetic alterations, the majority of which are point mutations, grouped in a core set of 12 cellular signaling pathways[18]. Morphological progression from PanIN to PDAC is paralleled by the accumulation of these genetic alterations in a progression model resembling the colon cancer model. Ductal origin of PanIN and PDAC is however questioned and a new model has been proposed where the cancer ductal cells in PanIN lesions originate from metaplastic acinar cells in a process called “acinar to ductal metaplasia” (ADM)[19].
Nuclear receptors: Classification, structural features, and ligands
Nuclear receptors (NRs) are members of a large superfamily of evolutionarily related ligand-regulated DNA-binding transcription factors present in most metazoan[20].
The first NR was only cloned in the ‘80s by Professor Evans R[21] long after the presence of NR was detected biochemically[22,23]; so far 48 NR has been identified in human by genome sequencing. All NR share characteristic structural features or domains named A to F from the N-terminal to the C-terminal; however, defining features of NR are only the presence of two highly conserved regions: the DNA binding domain (DBD) and the ligand binding domain (LBD), that can function independently[24] and are located in the region C and in the region E of the protein, respectively (Figure 2).
Figure 2.
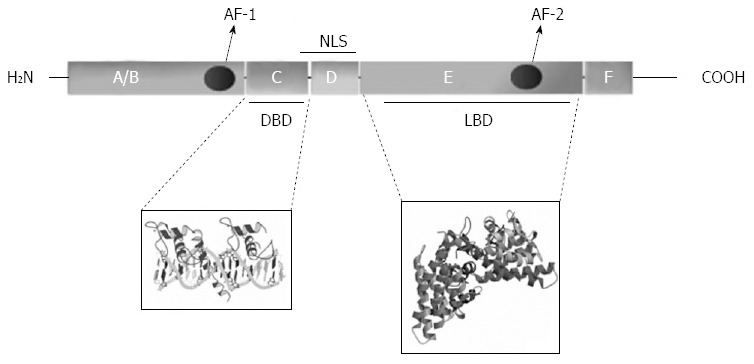
Domains and structural features of a classical nuclear receptor. A typical nuclear receptor (NR) consists of 6 region (A to F); region F may or may not be present. Region D (hinge) contains the nuclear localization signal (NLS); other NLSs may be present in region E. AF-1: Activator function 1; AF-2: Activator function 2.
NR are classified into six different subfamilies on homology basis: NR1 (thyroid hormone like), NR2 (HNF4-like), NR3 (estrogen like), NR4 (nerve growth factor IB-like), NR5 (fushi tarazu-F1 like), and NR6 (germ cell nuclear factor like), all originally named from the first member identified. A seventh subclass, NR0, has been introduced to classify two receptors, DAX-1 and SHP, that do not possess the DBD[25] (Table 1, information on ligands obtained from the “nuclear receptor signaling atlas”, NURSA, www.nursa.org).
Table 1.
Nomenclature of nuclear receptors
| Subfamily official name (class) | NRNC group | Member trivial name | Official name | Abbreviation | Ligand |
| NR0 (domain-depleted receptors) | B [DAX-like receptors (DAX, SHP)] | Dosage-sensitive sex reversal-adrenal hypoplasia congenita critical region on the X chromosome, gene 1 | NR0B1 | DAX-1 | Orphan |
| NR1 (TR/RAR/PPAR/VDR-like receptors) | A (thyroid hormone receptors) | Short heterodimer partner | NR0B2 | SHP | Orphan |
| Thyroid hormone receptor α | NR1A1 | TRα | GC-1, thyroid hormone | ||
| B (retinoic acid receptors) | Thyroid hormone receptor β | NR1A2 | TRβ | GC-1, thyroid hormone | |
| Retinoic acid receptor α | NR1B1 | RARα | Am 580, all-trans-Retinoic acid, Arotinoid acid | ||
| Retinoic acid receptor β | NR1B2 | RARβ | Am 580, all-trans-Retinoic acid, Arotinoid acid | ||
| C (peroxisome proliferator activated receptors) | Retinoic acid receptor γ | NR1B3 | RARγ | Am 580, all-trans-Retinoic acid, Arotinoid acid | |
| Peroxisome proliferator-activated receptor α | NR1C1 | PPARα | GW409544, GW7647, GW6471, Pirinixic acid, Palmitic acid, Leukotriene B4 | ||
| Peroxisome proliferator-activated receptor β | NR1C2 | PPARβ/δ | Eicosapentaenoic acid, GW0742 | ||
| Peroxisome proliferator-activated Receptor γ | NR1C3 | PPARγ | Rosiglitazone, GW1929, GW9662, GW409544, GW7647, 15-Deoxy-∆-12,14-prostaglandin, 15-Deoxy-∆-12,14-PGJ2 | ||
| D [Rev-Erb (NRD, E75)]F (RAR-related orphan receptors (ROR, HR3) | Rev-erbα | NR1D1 | Rev-erbα | Orphan | |
| NR1D2 | Rev-erbβ | Orphan | |||
| Retinoic acid receptor-related orphan receptor α | NR1F1 | RORα | Melatonin, CGP 52608 | ||
| Retinoic acid receptor-related orphan receptor β | NR1F2 | RORβ | Melatonin, CGP 52608 | ||
| Retinoic acid receptor-related orphan receptor γ | NR1F3 | RORγ | Melatonin,CGP52608 | ||
| Liver X receptor β | NR1H2 | LXRβ | GW3969, T0901317, oxysterols | ||
| H (ecdysone-like receptors) | Liver X receptor α | NR1H3 | LXRα | GW3969, T0901317, oxysterols | |
| Farnesoid X receptor α | NR1H4 | FXRα | GW4064, bile acid chenodeoxycholic acid | ||
| I (vitamin D3-like receptors) | Vitamin D3 receptor | NR1I1 | VDR | 1,25-dihydroxyvitamin D3 | |
| Pregnane X receptor | NR1I2 | PXR | Hyperforin, SR12813, rifampicin, pregnenolone carbonitrile, T0901317, 24(S),25-epoxycholesterol, butamben | ||
| Constitutive androstane receptor | NR1I3 | CAR | Androstanol, CITCO, phenobarbital, ATE | ||
| NR2 (HNF4/RXR/TLL/COUP-like receptors) | A (hepatocyte nuclear factor 4) | Hepatocyte nuclear factor 4 α | NR2A1 | HNF4α | Palmitoyl coenzyme A |
| Hepatocyte nuclear factor 4 γ | NR2A2 | HNF4γ | Orphan | ||
| B (retinoid X receptors) | Retinoid x receptor α | NR2B1 | RXRα | LGD 100268, GW0791, 9-retinoic acid | |
| Retinoid x receptor β | NR2B2 | RXRβ | LGD 100268 | ||
| Retinoid x receptor γ | NR2B3 | RXRγ | LGD 100268 | ||
| Testis receptor | NR2C1 | TR2 | Orphan | ||
| E (tailless-like receptors) | NR2C2 | TR4 | Orphan | ||
| Tailless | NR2E2 | TLL | Orphan | ||
| Photoreceptor-specific nuclear receptor | NR2E3 | PNR | Orphan | ||
| F [COUP-TF-like receptors (COUP-TF, SVP, EAR2)] | Chicken ovalbumin upstream promoter transcription factor IChicken ovalbumin upstream promoter transcription factor II | NR2F1 | COUP-TFI | Orphan | |
| NR2F2 | COUP-TFII | Orphan | |||
| ErbA2-related gene-2 | NR2F6 | EAR2 | Orphan | ||
| NR3 (ER/ERR/GR/MR/PR/AR) | A | Estrogen receptor | NR3A1 | Erα | Fulvestrant, 17β-estradiol, 4-hydroxytamoxifen, Raloxifene |
| NR3A2 | Erβ | Fulvestrant, 17β-estradiol, 4-hydroxytamoxifen, Raloxifene | |||
| B | Estrogen receptor-related receptor | NR3B1 | ERRα | Orphan | |
| NR3B2 | ERRβ | GSK4716, Diethylstilbestrol | |||
| NR3B3 | ERRγ | GSK4716, 4-hydroxytamoxifen | |||
| C | Glucocorticoid receptor | NR3C1 | GR | Dexamethasone, hydrocortisone, RU486 | |
| Mineralocorticoid receptor | NR3C2 | MR | Spironolactone, aldosterone, RU486 | ||
| Progesteron receptor | NR3C3 | PR | R5020, progesterone, RU486 | ||
| Androgen receptor | NR3C4 | AR | Dihydrotestosterone, RU486, Bicalutamide, R1881 | ||
| NR4 (NGFIB-like receptors ) | A [nerve growth factor IB-like receptors (NGFIB, NURR, NOR, HR38, CNR-8)] | Growth factor-inducible immediate early gene Nur77 | NR4A1 | Nur77 | Orphan |
| Nur-related protein 1 | NR4A2 | NURR1 | Orphan | ||
| Neuron-derived orphan receptor 1 | NR4A3 | NOR1 | Orphan | ||
| NR5 (FTZ-F1/SF1-like receptors) | A [fushi tarazu F1-like receptors (SF1, FTF, FTZ-F1)] | Steroidogenic factor-1/ELP | NR5A1 | SF1 | Orphan |
| Liver receptor homolog 1 | NR5A2 | LRH-1 | Orphan | ||
| NR6 (GCNF) | A (germ cell nuclear factor) | Germ cell nuclear factor 1 | NR6A1 | GCNF1 | Orphan |
PPAR: Peroxisome proliferator-activated receptor; PDAC: Pancreatic ductal carcinoma; TZD: Thiazolidinediones; IFN: Interferon; RAR: Retinoic acid receptor; RXR: Retinoid X receptor; AR: Androgen receptor; ER: Estrogen receptor; LHR-1: Liver receptor homologue-1 receptor; COUP-TFII: Chicken ovalbumin upstream promoter transcription factor II.
NR ligands are small hydrophobic molecules that bind to the LBD; retinoids, fatty acids, cholesterol, lipophilic hormones and vitamins, as well as some antibiotics, xenobiotics and synthetic drugs are all NR ligands. The ligand binding induces a conformational change that modify the DBD ability to bind specific DNA sequences called response elements. NR act as monomeres, homodimers or heterodimers with the retinoid-X-receptor (RXR) (Figure 3). Upon DNA binding, the final transcriptional activity depends on the presence of co-activator or co-repressor molecules[26].
Figure 3.
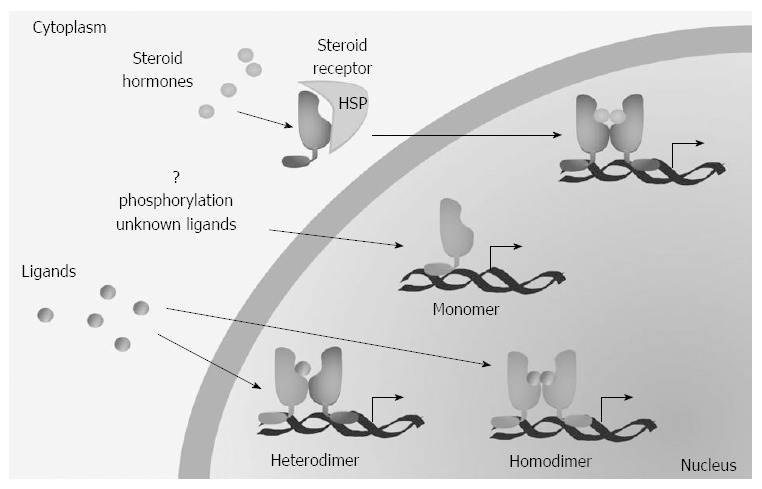
Mechanisms of action of nuclear receptors. Type I nuclear receptors (NR) (steroid receptors) are complexed with heat shock proteins (HSP) and maintained in the cytoplasm in the absence of ligands. The other receptors are instead mainly nuclear and the ligands induce hetero- (for type II receptors) or homo-dimerization (for type III receptors). Furthermore, a group of receptors (type IV) whose regulation is poorly known act as monomers.
NR physiologic functions vary from the regulation of metabolism, to growth and development; NR are also implicated with a number of diseases such as cancer. The association of NR with major diseases has transformed these proteins in the most popular and promising drug targets thanks also the properties of their ligands that can easily cross the cell membrane[27].
NR IN PANCREATIC CANCER
NRs are important in the development and homeostasis of the pancreas and their role in PDAC development is the subject of intense study by the scientific community. Here, we will describe the role of a group of these receptors, specifically the peroxisome proliferator activated receptors, the retinoid receptors, the androgen and estrogen receptors, and the orphan NRs.
PEROXISOME PROLIFERATOR ACTIVATED RECEPTORS
The peroxisome proliferator activated receptors (PPARs) belong to the NR1 (thyroid-like) subfamily of NR. Three PPARs are known: PPARα (NR1C1), PPARβ/δ (NR1C2) and PPARγ (NR1C3). PPARγ is the only PPAR with three isoforms with different spatial distribution[28]. They were identified as NR that responded to peroxisome proliferators, heterogenous chemicals that increase the number of peroxisomes (making them “proliferate”) in hepatocytes[29]. Natural ligands for PPARs are free fatty acids and PPARγ is also activated by 15-Deoxy-delta (12,14)-prostaglandin J(02)(15d-PGJ2)[30]. Some other PPAR ligands are the PPARα agonists hypolipidemic drugs fibrates and leukotriene B4 (LB4), the PPARγ specific agonist and antidiabetic drugs thiazolidinediones (TZD), and the PPARβ/δ specific agonist GW501516[31].
PPARs show a different expression pattern, and PPARβ is the most widely expressed[32]; they act as heterodimers with RXR and regulate complex gene networks especially in energy homeostasis and inflammation[28,33,34]. Furthermore they are involved in a spectrum of disease such as alcoholic liver disease and may mediates oxidative stress response[28,30,32,34].
PPARα
PPARα sustained activation leads to the development of cancers in the liver, testis and pancreas in rodents[35,36]. Moreover, PDX-1, an oncogene for pancreatic cancer that is overexpressed in PDAC[37], is a PPARα -dependent gene and its expression is downregulated by MK886, a specific PPARα antagonist[38]. However PPARα -dependent carcinogenesis has been recently questioned[39].
PPARβ/δ
Recently it has been reported that PPAR signaling, especially PPARβ/δ, is reduced in pancreatic cancer relapse, compared to primitive cancer[40], but PPARβ/δ has been suggested to be a critical component of the angiogenetic switch in pancreatic cancer[41,42]. Abdollahi[42] showed that the expression of PPARβ/δ detected by immunohistochemistry in human pancreatic specimens high-density tissue microarrays correlated with tumor staging; indeed PPARβ/δ staining intensity increased from normal pancreas to chronic pancreatitis, pancreatic cancer and metastasis and the up-regulation of PPARβ/δ is actually more enhanced in the tumor vasculature and in the tumor stroma[42]. High expression of PPARβ/δ in tumor is also confirmed by a recent paper[31]. Elevated PPARβ/δ expression levels are also highly correlated with advanced stages of tumor progression and with increased risk for tumor recurrence or distant metastasis[42] and PPARβ/δ has been proposed as a “central hub” in tumor angiogenesis given that tumor growth and angiogenesis were greatly reduced when tumor cells were implanted in PPARβ/δ null mice[42].
PPARβ/δ is also expressed in human pancreatic cancer cells and its activation regulates the metallo-protease matrix metalloproteinases (MMP)-9, decreasing cancer cells ability to transverse the basement membrane[31]. PPARβ/δ activation reduces the tumor necrosis factor (TNF)α-induced expression of various genes implicated in metastasis increasing the availability of the transcriptional repressor B-cell lymphoma (BCL)-6. BCL-6 is bound to PPARβ/δ and is released after GW501516 treatment resulting in decreased MMP-9 expression. These results suggest that increased expression of PPARβ/δ regulates pancreatic cancer cell invasion sequestering BCL-6 and hence inducing MMP-9 mediated invasion[31].
PPARγ
PPARγ is not only implicated in adipocyte differentiation, lipid accumulation, and glucose homeostasis but it is also an important regulator of inflammation via the inhibition of, or the interference with, proinflammatory signalings such as signal transducers and activators of transcription (STATs), nuclear factor-κB (NF-κB), and activator protein-1 (AP-1)[28,30].
PPARγ is expressed in primary PDAC[43-46] and strongly correlates with a more advanced clinical stage; PPARγ staining is also associated with shorter overall survival and its has proved to be an independent prognostic factor in uni- and multi-variates analysis[43,45]. However, whereas Kristiansen et al[45] found a strong overexpression of PPARγ by expression profiling in 19 microdissected carcinoma compared to 14 ductal epithelia, Pazienza et al[44] did not find significant alterations in the expression levels of PPARγ in pancreatic cancer. A possible explanation of this discrepancy may be that is the expression “per se” of PPARγ, and not its levels, important in pancreatic cancer progression. Interestingly, a large number of studies have demonstrated that TZD reduce the risk of PDAC[9].
A genetic association PPARγ/PDAC has been tested by two different groups[47,48] analyzing the expression of the single nucleotide polymorphisms (SNP) Pro12Ala that has been associated with reduced risk of diabetes and some cancers[47]. Fesinmeyer et al[48] demonstrated that this SNP is associated with increased risk of PDAC in a high-risk sample of smokers randomized to high-dose vitamin A; however, two years later, a similar study in obese and diabetic patients demonstrated a protective role of the SNP, prompting the need of further studies[47].
In vitro, the role of PPARγ is controversial but it is generally accepted that the receptor acts as a tumor inhibitor at multiple levels (Figure 4). PPARγ is expressed in human pancreatic cancer cell lines[46,49-55] and its expression follows a circadian rhythmicity with a period of 24 h that could potentially influence the cell phenotype and the human disease behavior[55]. Agonists of PPARγ such as TZD, its natural ligand 15d-PGJ2, or 1,1-Bis(3-indolyl)-1-(p-substituted phenyl)methanes (C-DIMs), induce cell cycle arrest in G1, apoptosis and ductal differentiation in pancreatic cancer cells[46,53,54,56-58]. However some of the effects associated with PPARγ activation are instead receptor independent: this is especially true for TZD agonists whose receptor independent effects have been widely described[51,52,59-69].
Figure 4.
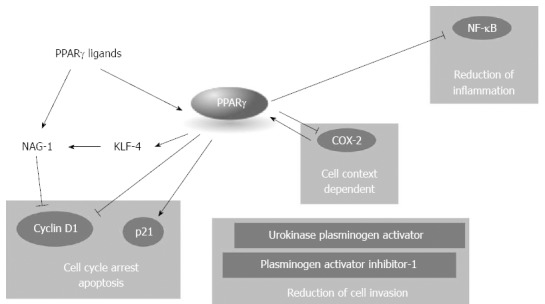
Peroxisome proliferator activated receptors and pancreatic ductal carcinoma. Peroxisome proliferator activated receptor (PPAR)γ acts as a tumor inhibitor at multiple levels, blocking cell cycle progression, inflammation, and cell invasion. NAG-1: Non steroidal anti-inflammatory drug-activated gene-1; Cox-2: Ciclooxigenase-2; NF-κB: Nuclear factor-κB.
Cell cycle arrest is associated to decreased PPARγ-dependent expression of cyclin D1[53,54,57,58] whereas the reported induction of p21 might be PPARγ-dependent or PPARγ-independent[52,54]. Deletion analysis of the p21 promoter indicates that PPARγ-dependent activation of p21 requires GC-rich sites in the proximal region of the promoter[54]. It is worth to note that some agonists of PPARγ[61] may induce a PPARγ independent down-regulation of cyclin D1, due to induction of non steroidal anti-inflammatory drug-activated gene-1 (NAG-1). NAG-1 is a member of the TGF-β superfamily involved in tumor progression acting as a pro-apoptotic gene. Interestingly, it has been reported that NAG-1 expression is positively regulated by PPARγ: MCC-555, a PPARγ agonist, induces the expression of the transcription factor KLF-4 in PPARγ-dependent manner who subsequently enhances the NAG-1 promoter activity[51,52]. PPARγ agonists reduce the invasive capacity of PDAC cells with a PPARγ dependent mechanism[50]. The PPARγ ligands 15d-PGJ2 and ciglitazone attenuate pancreatic cancer cell invasion increasing plasminogen activator inhibitor-1 and decreasing urokinase plasminogen activator levels resulting in the reduction of total urokinase activity in pancreatic cancer cells[50]. Interestingly, the PPARγ antagonist T0070907 suppresses pancreatic cancer cell motility by altering the localization of p120 catenin and by suppressing the activity of the Ras-homologous GTPases Rac1 and Cdc42[49].
The effects of PPARγ activation or inhibition by specific molecules sinergize or interact with other pathways or other NR activations[53,56,62-64]. Combination of recombinant interferon-β (IFN-β) and the PPARγ agonist troglitazone induces a synergistic effect on the growth inhibition of pancreatic cancer cells, through the counteraction of the IFN-β-induced activation of STAT-3, MAPK and AKT and the increase in the binding of both STAT-1 related complexes and PPARγ with specific DNA responsive elements. The combination induces also an increase in autophagy and a decrease in anti-autophagic bcl-2/beclin-1 complex formation, mediated by the inactivation of the AKT/mTOR-dependent pathway[64]. PPARγ form mandatory heterodimers with RXRα and its activity is maximal in the presence of RXRα agonists; it is not surprising then that co-treatment with PPARγ and RXRα agonists exacerbates the effects of PPARγ increasing the inhibition of cell growth[53,62,63]. Synergistic effects on growth inhibition are also visible when inhibitors of ciclooxigenase-2 (Cox-2) and PPARγ agonists are used in combination[56,65]. Cox-2 is an inducible ciclooxygenase that contributes to the metabolism of arachidonic acid forming prostaglandin H2, a precursor of 15d-PGJ2[66]. Cox-2 is a downtarget of PPARγ and its expression may be either induced or repressed by the NR, depending on the cell context[30]. However, selective Cox-2 inhibitors have opposite effects in pancreatic cancer depending on Cox-2 expression: in high Cox-2-expressing cells the inhibitors reduced tumor growth; conversely, in Cox-2 negative or low expressing cancer cells the inhibitors, at very high concentrations, enhance tumor progression increasing intratumoral VEGF and tumor angiogenesis in a PPARγ-dependent way[65].
PPARγ is known to reduce tumor growth in mice in vivo[49,62,65,67] and its activation increases the gemcitabine mediated tumor suppression[68]. Tumor growth inhibition mediated by PPARγ may be due to reduced inflammation and increased activation of anti-inflammatory genes[30,67]. Genetic deletion of Ikk2, a component of the canonical NF-κB signaling pathway, in the Kras(G12D)Pdx1-cre mouse model of pancreatic cancer, substantially delays pancreatic oncogenesis and results in downregulation of the classical Notch target genes Hes1 and Hey1[67]; in the same model TNF-α stimulation resulted in increased Hes1 expression and consequent suppression of PPARγ expression facilitating the formation of a inflammatory pro-tumoral enviroment; induction of PPARγ instead may block NF-κB induced processes[30] reducing or delaying tumor formation[67].
Despite all these intriguing discoveries on PPARγ role in PDAC, clinical application of PPARγ modulation has recently suffered yet another failure when a new oral anticancer agent with LB4 antagonist and PPARγ agonist properties[69,70], the LY29311, did not demonstrate any benefit in association with gemcitabine in unpretreated patients with advanced PDAC[69].
RETINOIC AND RETINOID RECEPTORS
Retinoic acid receptors (RARs) and RXRs are NRs transcription factors that bind retinoids, natural and synthetic molecules structurally and/or functionally related to vitamin A, and regulate cell differentiation, proliferation, and survival[25,71,72]. A list of retinoids with biological functions comprises, but is not limited to, all trans retinoic acid (atRA), 9-cis-retinoic acid (9-cis-RA), 11-cis-RA, 13-cis-RA, being the atRA the predominant physiological form; retinoids that specifically bind to RXR are called rexinoids and have been effective in cancer treatment. RARs can be activated by both atRA and 9-cis RA, while RXRs are exclusively activated by 9-cis-RA, initially identified as a bona fide RXR ligand in vitro[71], but never detected in vivo[73]. Other RXR natural ligands have been identified in vivo but they are not RXR specific ligands[74-76].
RARs and RXRs are each encoded by three different genes that give rise to the -α, -β, -γ isoforms of RXR and RAR, each presenting transcription variants, and characterized by different spatial distribution[77,78]. RXRs were identified as cofactor for efficient binding of RAR to its DNA response elements[79], but unique among the NRs, RXR play a modulatory role along multiple pathways forming mandatory dimers with thyroid hormone receptor, PPAR, vitamin D receptor (VDR), RAR, Nur77, etc.[25,71]; in these heterodimers RXR may function as an active partner (such in the case of PPARγ:RXR dimers) meaning that the dimers respond to 9-cis-RA, or as silent partner and the dimers do not respond to RA.
Due to their regulatory potential, these NRs are major drug targets for a number of pathologies, including cancer and metabolic diseases.
RAR and RXR receptors are expressed during pancreatic organogenesis and are essential for ductal differentiation[80,81]. Retinoid receptors are more expressed in the exocrine compartment, usually during late gestation, with a strong lineage specificity. Exogenous 9-cis-RA induces predominantly ducts instead of acini, plus more mature endocrine architecture, whereas exogenous atRA induces predominantly acini instead of ducts, with no apparent endocrine effect[80]. RAR-selective agonists mimicked the acinar suppressive effect of 9-cis-RA, suggesting that RAR-RXR heterodimers are critical to ductal differentiation; however, retinoids do not regulate exocrine lineage selection cell-autonomously but epithelial-mesenchymal interactions are mandatory given that 9-cis -RA does not induce ductal differentiation in the absence of mesenchyme and requires the presence of laminin-1[81]. The ability to restore a more differentiated phenotype and to regulates ductal differentiation may explain the effects of retinoids.
Expression of RXR and RAR has been described in pancreatic cancer cell lines and PDAC[42,53,62,63,82-88] but their biological and clinical significance is not clear: in most cases RXR and RAR receptors apparently act as tumor suppressors in PDAC cancer both in vivo and in vitro[43,53,62,63,84-88], inducing arrest in cell proliferation and differentiation although results suggestive of a pro-oncogenic role are also reported[89-91].
A differential expression of RAR-α, -β, and -γ, and RXRα was detected in histological sections of human PDAC and their adjacent normal tissues. Whereas all four receptors were detected in adjacent normal pancreatic tissue specimens, RARβ mRNA transcripts were detected in only 67% of the malignant tissues and when expressed, the level of expression was significantly lower than that of the corresponding adjacent normal tissues, especially in moderately- and poorly-differentiated cancers[92]; these results are in agreement with previous papers showing that RARβ expression is lost during PDAC malignant transformation[84]. The mechanisms at the basis of RARβ mRNA downexpression are not known but it is worth noting that in pancreatic endocrine neoplams the NR promoter is often hypermethylated[93,94], suggesting that in certain pancreatic carcinomas the reduction or loss of RARβ expression by epigenetic mechanisms might be associated with the development or progression of tumors[92]; interestingly one missense mutation in RARβ has also been identified in PDAC[18]. The anti-tumoral role of RARβ is confirmed by its overexpression in DAN-G pancreatic cancer cells that results in induction of differentiation and inhibition of proliferation in vivo and in vitro[84].
Immunohistochemical evaluation of PPARγ and RXRα protein expression in 65 PDAC patients statistically analyzed in relation to clinicopathological characteristics, tumor proliferative capacity, and patients’ survival showed that 75% of patients tested positive for PPARγ and 85% stained positive for RXRα. Interestingly, RXRα positivity was significantly associated with tumor proliferative capacity and PPARγ positivity but RXRα failed to predict patients’ survival[43].
In vitro, RXR and RAR involvement in PDAC has been usually tested by means of specific agonists or antagonists. Retinoids may be useful agents for the treatment of pancreatic cancer; however, RAR-selective retinoids produce unwanted side effects. In contrast, RXR-selective retinoids produce fewer side effects. The reported results indicate that these receptors often possess antiproliferative and pro-differentiative effects whereas reports on induction of apoptosis are mixed[82,83,88].
In 13 cell lines established from patients who underwent surgery for PDAC Albrechtsson et al[86] detected the expression of the RAR and RXR subtypes and evaluated the effect of atRA and 9-cis-RA on cell proliferation. They demonstrated that RARα, β and RXRβ were expressed in most of the cell line. RXRγ was expressed in about half of them and RARγ in only one whereas the RXRα receptor was expressed in all cell lines. Incubation of the cells with atRA or 9-cis-RA reduced cell proliferation, although only about half of the cell lines responded to the latter[86]. These results partially contradict a previous paper that showed that pancreatic cancer cell lines in vitro responded to 9-cis-RA but not atRA at clinically relevant concentrations[87]. Moreover, as previously reported[53], 9-cis-RA acts additively with the TZD Troglitazone blocking the cells in G1 phase through reduction of cyclin D1 levels.
The RXR-selective retinoid, AGN194204 inhibits the proliferation of pancreatic cancer cells more efficiently than RAR-selective retinoids, but does not increase the apoptosis, whereas other retinoids are also able to induce apoptosis[82]. Block of cell proliferation in these cells is associated with reduced cyclin E and cyclin dependent kinase 6 levels, an effect reversed by the RXR antagonist AGN195393 but not by RAR antagonist AGN193109. Treatment of MIAPaCa-2 cells with AGN194204 and cytotoxic agents such as gemcitabine, 5-fluorouracil, or IFNγ resulted in an additive but not synergistic reduction in cell number[95]. Interestingly, the retinoid-related ligand AGN193198 reduces BxPC-3, MIAPaCa-2 and AsPC-1 cell proliferation (blocking the cell in the S phase) more efficiently than high-affinity RAR- or RXR-selective retinoids and induces apotosis[88]; however the compound does not activate transcription from RAR or RXR response elements and its effects on cell survival are not reversed by treatment with RAR- or RXR receptor-selective antagonists. These results suggest that AGN193198 (but we may not exclude also other retinoids) might act independently of the classical retinoid receptors.
Treatment of pancreatic cancer cells with 9-cis-RA induces apoptosis lowering the ratio Bcl2/Bax2 and requires the presence of RARγ[83]. Interestingly, 9-cis-RA acting on RXRα may induce the nuclear export of Nur77[96,97], facilitating its interaction with Bcl2 and hence increasing the apoptosis (see later for details).
Retinoids possess the ability to induce pancreatic cancer differentiation in vitro[82,89,98]. The differentiation phenotype changes are associated with increase in aerobic metabolism, expression of mucins, synthesis and secretion of TGF-β, and reduction of EGF receptor expression[82]. This differentiation effects are dependent on TGF-β, because co-treatment with atRA and a pan-TGF-β neutralizing antibody abolishes the anti-proliferative and pro-differentiative effect of the retinoid and reduces MUC4 expression[82,89]. As previously described, ADM might play an important role in PDAC development and DSL-6A/C1 cells, who expresses RAR-α and -β and RXRα, represent an in vitro model of this carcinogenic sequence[98]. Treatment of DSL-6A/C1 cells with retinoids results in a time- and dose-dependent inhibition of cell growth, paralleled by a retinoid-mediated transactivation of a pTK:betaRAREx2-luciferase reporter; growth inhibition is reverted by the RARα specific antagonist Ro 41-5253, suggesting that the RARα might influence ADM[98].
Regulation of expression of mucins (MUCs) by retinoids however raises questions regarding the response of pancreatic tumor cell in vivo. RAR and RXR receptors have been reported to influence the expression of MUC4 and MUC17[89-91] and RXR:VDR response elements are present in the promoter region of MUC17 gene[90]. Both mucins are associated to the progression of pancreatic cancer: MUC17 is linked to the presence of lymph node metastasis[99] and MUC4 expression increases during progression of PDAC from PanIN1 to PanIN3, and it is highly expressed in invasive adenocarcinomas[100-102]. The expression of MUC17 gene is regulated by a 1146-bp DNA fragment upstream of MUC17 that contains GATA, NF-κB, Cdx-2 and RXR:VDR response elements, but no data are available on the role of the latter. Instead, retinoids directly regulates the expression of MUC4[91], sinergistically with IFNγ and dependently of TGF-β. Interestingly, IFNγ has been shown to possess antitumor activity and it is well known that TGF-β possess tumor suppressive and oncogenic activities[103]. At early stages TGF-β acts as a tumor suppressor, whereas at later stages tumor cells become resistant to its antiproliferative effects but continue to secrete high quantity of the factor. Indeed, pancreatic tumors overexpress all the three TGF isoforms and this correlates with decreased patient survival[104] and induction of epithelial to mesenchymal transition (EMT). On the other hand, although IFNγ is antiproliferative in vitro against pancreatic cancer cells, the temporal aspect of this process has never been studied. Indeed, the expression of MUC4 does not require the continuing presence of IFNγ or RA which instead are required for the priming of MUC4 expression[91]. From a pathological point of view, aberrant expression of mucins on the surface of PDAC cells may provide protection against the host’s activated immune system while conferring antiadhesive properties upon the cells and hence favoring the EMT-mediated metastatization, casting a shadow on the use of retinoids in vivo. Nonetheless, retinoids have been used in phase II clinical trials in the past[105-107] with mostly disappointing results. In 1998, basing on promising in vitro results, one trial in which patients with advanced PDAC were treated with 13-cis-RA and IFNα resulted in prolonged stable disease in two third of the patients[105]; this however contradict a 1995 phase II trial where the same therapeutic regimen did not improve patients condition[107]. Furthermore, the combination of 13-cis-RA with gemcitabine in a more recent phase II clinical trial, although well tolerated, did not determine an improvement in the response[106]. PDAC resistance to retinoid treatment might be dependent on the relative intracellular expression of the retinoids-binding proteins fatty acid-binding protein 5 (FABP5) and cellular retinoic acid-binding protein 2 (CRABP2)[108] that were shown to be critical for either antisurvival (CRABP2) or prosurvival (FABP5) effects of retinoic acid[109].
ANDROGEN AND ESTROGEN RECEPTORS
Androgen receptor (AR, NR3C4) and estrogen receptors (ER)-α and -β (NR3A1 and NR3A2) belong to the steroid receptor subfamily (NR3). These NR regulate multiple physiological processes including sexual development and are implicated in multiple cancers[110-113].
Their involvement in PDAC has long been suggested by the evidence that pancreatic cancer shows an apparent hormonal imbalance in the incidence with a male to female ratio ranging from 1.25-1.75:1[114,115], that approach the 1:1 ratio with advancing age[116] (for recent reviews see[117,118]).
In the early ‘90s, the presence of AR in PDAC was questioned, but recent papers clearly demonstrated that pancreatic cancer cells express detectable levels of AR[117,119,120]. In vitro, PDAC cancer cells variably respond to the treatment with the agonist testosterone, showing a modest increase in cell proliferation[119,121]. Flutamide, an AR blocker used for the treatment of prostate cancer, induces a reduction in cell proliferation that does not however correlate with AR expression levels[119]. Furthermore, flutamide treatment does not alter the cell response to gemcitabine in vitro and in vivo[119]. AR activity is modulated by IL-6, an inflammatory cytokine overexpressed in pancreatic cancer[120,122]. IL-6 enhances STAT3 and MAPK pathways that in turn increase the AR transcriptional activity; IL-6 also enhances pancreatic cancer cell migration in the presence of AR, an effect blocked by the silencing of the receptor[120] (Figure 5A). In a double-blind placebo-controlled trial, flutamide doubled the survival duration when administered in a dosage of 250 mg three times daily[123]. This excellent result has not been confirmed by other small phase II trials where flutamide was used in monotherapy or in combination with gemcitabine[124,125]. The lack of AR response in PDAC patients suggests that the tumor cells, although expressing the AR, are in a hormone-refractory proliferative status, as observed in prostate cancer[119].
Figure 5.
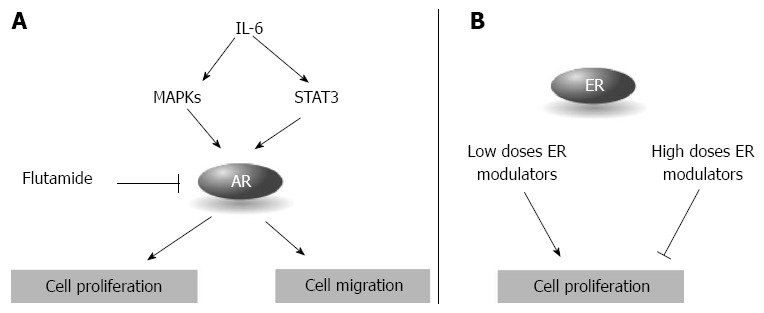
Steroid receptors in pancreatic ductal carcinoma. A: Androgen receptor (AR) regulates cell proliferation and migration and it is activated by the inflammatory cytokine interleukin-6 (IL-6); B: The effect of estrogen receptors (ER) on cell proliferation depends on the concentrations of the ER modulators.
The role of ER in PDAC is controversial: although several papers described the presence of ER (usually ERα) in primary PDAC, other reports did not detect the receptors at all[118,126,127] and the antiestrogen tamoxifen has been used in clinical trials with no benefit[118,128]. Expression of both ERα and -β has been described in pancreatic cancer cells[129] and a recent proteomic validation study in formalin-fixed paraffin-embedded tissues identified several proteins tightly associated through ERα[130]. In vitro, PDAC cells respond to the treatment with estrogens modulating agents: lower concentrations usually induce cell proliferation whereas high concentrations arrest cell proliferation (Figure 5B)[129]. The response seems dependent on the expression of ERα and ERβ, specifically to their ratio. ERα and ERβ share an almost perfect homology in the LBD that allows both of them to bind estrogen; other domains are instead less conserved with the most divergent region being the A/B domain, characterized by the absence of the activator function-1 in the ERβ. Analyzing the expression of the two estrogen receptors Iwao et al[129] found that pancreatic cancers showed significantly lower ERα mRNA levels than ER-negative breast cancers while ERβ mRNA levels (that were higher in ER-negative than ER-positive breast cancers) were significantly higher than ER-negative breast cancers; in seven out of eight pancreatic cancer cell lines ERβ outweighs ERα and cells with lower ERα/ERβ ratio tend to have higher responsiveness[128], suggesting that ERβ may play a more important role in PDAC[118,128]. Interestingly, phytoestrogens such as genistein block pancreatic cancer growth in vitro and show higher affinity for ERβ.
OTHER NRS: ORPHAN NRS IN THE SPOTLIGHT
Roughly half of the 48 human NR are classified as orphan NRs. The orphan NRs form a specific subgroup of the NR proteins characterized by different functional and evolutionary origin; orphan NR are distributed along the all the six NR subfamilies. These proteins have in common only the term “orphan receptor” conied few decades ago to describe, by definition, gene products that appear to belong to the nuclear receptor family on the basis of sequence identity but for whom no ligands is known[131,132]. Orphan receptors diverge also at the structural levels with various examples of members without all the classical features of nuclear receptor, such as the LBD or the DBD[132]. The discovery of the orphan receptors drastically changed endocrinology introducing the “reverse endocrinology” approach where orphan receptors are used to identify new hormones and their associated biology, conversely to traditional endocrinology that identified hormones starting from their physiological or pathological effect[131].
Although the term “receptor” implies the existence of a natural ligand, this assumption is debated and not necessarily true for at least some of the orphan NRs[25]. In time, the number of orphan receptors has diminished due to the discovery of natural ligands for some of them that have then become “adopted”, such in the case of PPARγ or FXR for example[25].
Here we will discuss recent findings on orphan receptors, specifically the NR4As, the liver receptor homologue-1 receptor (LRH-1) and the chicken ovalbumin upstream promoter transcription factor II (COUP-TFII) discussing their role in PDAC.
Nur77: A “two faces” action in pancreatic cancer
The orphan NR subfamily 4 subgroup A (NR4A) is comprised by three members: NR4A1 (also known as Nur77, testicular receptor TR3, or nerve growth factor 1b NGFI-B), NRA42 (Nurr-related factor 1) and NR4A3 (neuron-derived orphan receptor1, Nor-1)[25,133]. The first member of NR4A family was identified by differential hybridization in the rat pheochromocytoma cell line PC-12 cell as encoded by an immediate early gene (i.e., a gene that is rapidly and transiently transcribed in response to stimuli) induced by the nerve growth factor[134]. As in the case of other NRs, the NR4A members show common features of a classic nuclear receptor and an high degree of sequence homology specifically in the DBD and LBD regions, where homology may be as high as 95% (Figure 6).
Figure 6.

Clustal sequence alignment of human NR4As. Shaded sequences correspond to the DNA binding domain where homology among NR4A1-3 is very high.
All three members are localized in the nucleus due to the presence of a nuclear localization signal in the DBD (three signals in the case of NR4A1). The three members of the family show a different and often overlapping expression in adult tissues, being Nur77 more abundantly and broadly expressed[133]. NR4A receptors might act as monomere or as homo- hetero-dimers with different affinity to DNA response elements; dimers show stronger activity over monomers[133]. The crystallographic analysis of NR4A receptors suggests that the members of this subfamily are constitutively activated: their LBD is almost completely occupied by bulky aminoacid side chains conferring a 3D structural conformation similar to that of agonist-bound receptors[132,133]. Consequently, unlike others NR, the activity of NR4As is not regulated by stimuli through a ligand binding but instead by modulation of their expression or via post-translational modifications[132,133]. Expression of these receptors is induced by a range of stimuli, including stimuli associated with metabolic functions, such as: fatty acid, growth factors, prostaglandines, membrane depolarization, cold, glucose, cholesterol, TZD and hormones[135]. Other hormones may regulate NR4A transcriptional activity interacting with the NR4 heterodimers, such 9-cis-RA on NR4A:RXR dimers, and new molecules have been identified acting as agonists or antagonists[96,97,136].
The involvement of NR4A2 in pancreatic cancer in vitro has been reported by two papers[133,136]. NR4A2 is highly expressed in many cancer cell lines including Panc1 and Panc28 pancreatic cancer cells. Structure-dependent activation of NR4A2 by a series of C-DIM analogs, especially a p-bromophenyl analog (DIM-C-pPhBr), alters the expression of NR4A2 target genes; among the altered genes the drug determines a NR4A2-dependent repression of neuropilin (NP)-2 in both cell lines, whereas it induces the expression of NP-1 in PANC28 but not PANC1[136]. NPs are important in the progression of pancreatic cancer and are currently seen as potential target for PDAC treatment[137] and it is conceivable that differential expression of these molecules by NR4A2 might be important for PDAC development. Moreover, in PC3 cells NR4A2 acts as a pro-survival antiapoptotic factor[133]: silencing of the receptor greatly reduced the anchorage independent growth, with minimal effect in anchorage-dependent growth, largely due to increase anoikis; NR4A2 silencing also impaired the formation of tumors in nude mice[133].
More data link NR4A1 to pancreatic cancer, depicting a complex mechanism of action where Nur77 acts as having “two faces”, being pro-survival and anti- and pro-apoptotic at the same time.
NR4A1 is expressed as a nuclear protein in pancreatic cancer cells[136,138-142] and was found to be overexpressed in PDAC tissues primarily in the nucleus, whereas 83% of non-tumor pancreatic tissues did not express it[142].
c-DIMs are a class of molecules that activate PPARγ and might act on NR4A receptors[136,138,139]; a c-DIM, specifically the 1,1-Bis(3’-indolyl)-1-(p-anisyl)methane (DIM-C-pPhOCH3), is the first identified Nur77 agonist[139]. DIM-C-pPhOCH3 activates GAL4-Nur77 chimeras expressing wild-type and the ligand binding domain of Nur77. In Panc-28 pancreatic cancer cells, Nur77 agonists decrease cell survival activating the cell death pathways, including tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) and poly(ADP-ribose) polymerase (PARP) cleavage. Activation of TRAIL and PARP was further confirmed using Nur77 siRNA in Panc-28 cells. Nur77 agonists also inhibit tumor growth in vivo in athymic mice bearing Panc-28 cell xenografts[139]. DIM-C-pPhOCH3 arrests pancreatic cancer cell in G0-G1, by a Nur77-dependent, but KLF-4-independent, expression of cyclin-dependent kinase inhibitor p21. Interestingly, regulation of p21 does not require the presence of Nur77 response elements but involve a GC-rich promoter region and requires the presence of the Sp proteins Sp1 and Sp4, but not Sp3[140]. Microarray analysis of L3.6pL pancreatic cancer cells treated with DIM-C-pPhOCH3 demonstrated a NR4A1-dependent induction of genes associated with metabolism, homeostasis, signal transduction, transcription, stress, transport, immune responses, growth inhibition and apoptosis. Among the most highly induced growth inhibitory and proapoptotic genes were activating transcription factor 3 (ATF3), p21, cystathionase, dual specificity phosphatase 1 and growth differentiation factor 15. Furthermore, DIM-C-pPhOCH3 induced Fas ligand and TRAIL, the latter with a ATF3 dependent mechanism[141].
Although activation of Nur77 by specific c-DIM suggests that Nur77 is a tumor suppressor, experiments with the antagonist 1,1-bis(3’-indolyl)-1-(p-hydroxyphenyl)methane (DIM-C-pPhOH) suggest the contrary[142]. Blocking of endogenous Nur77 results in increased cell death and reduced cell proliferation; moreover expression of antiapoptotic genes Bcl-2 and Survivin is also reduced. When administered in vivo, DIM-C-pPhOH inhibits tumor growth, acting on the same antiapoptotic markers observed in vitro. Survivin is overexpressed in pancreatic cancer[143,144] and its expression increases during PanIN progression to PDAC[144]. Transcriptional regulation of Survivin by Nur77 is Sp1-dependent, paralleling the p21 regulation[140], and it is co-regulated by p300. Thus, activation of nuclear Nur77 by the agonist DIM-C-pPhOCH3 or inactivation by the antagonist DIM-C-pPhOH reduces proliferation and induces apoptosis through two different transcription pathways: the first involves the induction of expression of apoptosis promoter genes such as p21 and TRAIL, whereas the latter is dependent on suppression of pro-survival genes. Consequently, Nur77 acts both as a tumor suppressor and as a tumor promoting gene in pancreatic cancer (Figure 7).
Figure 7.
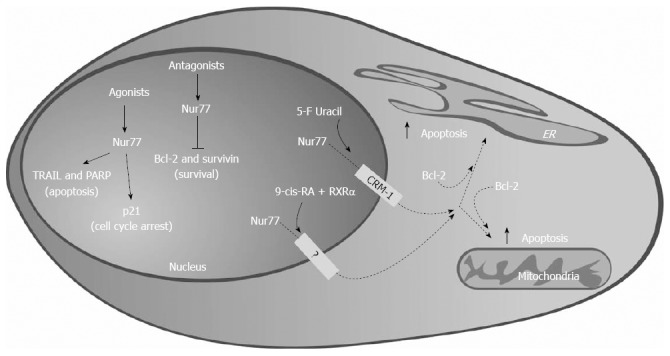
Nur77 acts as a tumor suppressor and as tumor promoting gene in pancreatic ductal carcinoma, inducing pro-apototic and anti-proliferative genes or repressing pro-survival genes. Dashed lines: Effects yet to be demonstrated in pancreatic ductal adenocarcinoma (PDAC). TRAIL: Tumor necrosis factor-related apoptosis-inducing ligand; PARP: Poly(ADP-ribose) polymerase; RXR: Retinoid X receptor; ER: Endoplasmic reticulum.
Interestingly, Nur77 may act as an apoptotic inducer agent in several cancer cells after nuclear export[96,97,145-147]; although not yet described in PDAC it is conceivable that this extra-nuclear action may also be present in pancreatic cancer cells. Inducers of apoptosis, including 5’-fluorouracil which is used in PDAC treatment, stimulate nuclear export of Nur77 mediated by the export receptor CRM1[145,147]. NR4A1 nuclear export may also be induced by 9-cis-RA, requires RXRα as a carrier[97], and targets Nur77 to mitochondria[96]. Despite lacking classical mitochondria targeting sequences, Nur77 might translocate to mitochondria in response to cell death stimuli, through interaction with anti-apoptotic Bcl-2[147]. Bcl-2 acts forming channels in the mitochondria membrane to regulate apoptosis[148]. The interaction Bcl-2/Nur77 is mediated by the N-terminal loop of Bcl-2 and by the NR4A1 LBD: this binding induces a conformational change that exposes the BH3 region of Bcl-2, resulting in its transformation in an inducer of apoptosis[147]. Interestingly, Nur77 may also translocate to other organelles, specifically endoplasmic reticulum (ER). Cell treatment with CD437 induces a nucleus-cytoplasmic translocation of Nur77, followed by ER localization: this requires again the interaction with Bcl-2 and triggers the release of Ca2+ from ER inducing apoptosis. Again, this effect has been demonstrated in human neuroblastoma, esophageal squamous carcinoma and hepatocarcinoma cells[145,146] but not in PDAC cells (Figure 7).
LRH-1 in pancreatic cancer
LHR-1 (NR5A2) is an orphan NR that is essential during development and necessary in the adult for the function of the pancreas, liver, intestine, and ovary[149,150]. LHR-1 recognizes specific DNA sequences to whom it binds as monomere[151].
The status of LHR-1 as “orphan” is debated and some scientists suggest that it may be classified as “adopted”[132,151]. The structure of the mouse LHR-1 LDB shows an active conformation with a large hydrophobic, but empty, ligand binding pocket resulting in a constitutive active receptor[132]; instead, the crystallographic analysis of the human LHR-1 revealed the presence of phosphatidyl inositol in the binding pocket[151]. Phosphatidyl inositol is required for activation[151] but it is not clear if it may enter and leave the pocket acting as a proper ligand[132]. Nonetheless, new molecules acting as antagonists have been recently identified by screening of commercially available compounds[152].
Chromatin immunoprecipitation-seq and RNA-seq analyses revealed that LRH-1 directly induces expression of genes encoding digestive enzymes and secretory and mitochondrial proteins and cooperates with the pancreas transcription factor 1-L complex in regulating exocrine pancreas-specific gene expression[153].
LHR-1 is important in maintaining acinar identity but is not required for acinar development[154] and mice with a selective deletion of LHR-1 in the pancreas did not display histological abnormalities[153-155]. A genome-wide association study conducted in pancreatic cancer patients and unaffected controls identified 5 SNP in the vicinity of NR5A2 associated with the risk of PDAC[156], that were confirmed by later studies[47,157,158].
In normal human pancreas, the LRH-1 protein is expressed at low levels in the nucleus and cytoplasm of both acini and ducts cells; in contrast PDAC show heightened levels of the protein and in some neoplastic cells the receptor appeared to localize predominantly in the cytoplasm[159]. An increased presence of LRH-1 was also detected in the acinar cells affected by pancreatitis and in PanIN lesions[159]. Overexpression of the receptor was also detected in pancreatic cancer cells in vitro[159].
Treatment of pancreatic cancer cells in vitro with LHR-1 antagonists or with LHR-1 siRNA significantly inhibits cell proliferation inducing a G0/G1 block associated with a reduction of of cyclins D1 and E1[152,159], suggesting that in vitro LHR-1 promotes tumor proliferation. In vivo, selective inactivation of one NR5A2 allele in pancreatic epithelial cells is sufficient to cause impaired recovery from pancreatitis[154] and conditional pancreatic deletion of LHR-1 leads to destabilization of the mature acinar differentiation state, increased inflammation, ADM and loss of regenerative capacity following acute caerulein pancreatitis[154,155]; loss of both alleles also dramatically accelerates the development of oncogenic Kras driven ADM and PanIN lesions[154,155].
The in vivo studies clearly show that LHR-1 inhibits the ductal transformation of adult acinar cells by mutant Kras and prevent PDAC progression; however in vitro studies show that NR5A2 promotes, instead of inhibiting, tumorigenesis. It has been hypothesized that NR5A2 exercises an inhibitory action in the early phase of PDAC development, blocking RAS with unknown mechanisms, while it will have an opposite effect later on[160].
COUP-TFII expression predicts survival in PDAC
COUP-TFII is a orphan nuclear receptor encoded by the NR2F2 gene localized in the chromosome region 15q26, a region frequently amplified in pancreatic cancer[14], and it is a down target of multiple pathways altered in pancreatic cancer[46,161-163]. In mouse two different transcription variants are described whereas in human at least four different variant are expressed. They differ in the N-terminal region and only one variant presents the structural features of NR being the others without the DBD. The role of these variants is not fully understood and two recent papers gave contradictory results describing one of the DBD lacking forms acting either as enhancer of COUP-TFII transcriptional activity or as a repressor, increasing the cytoplasmic localization of full length COUP-TFII, suggesting a cell specific function for this truncated NR[164,165]. COUP-TFII exists in a autorepressed conformation that prevents recruitment of coactivators, and might respond to retinoids that promote COUP-TFII to recruit coactivators[166]. Full length COUP-TFII exerts an important role during development and in adulthood[167], and it is implicated in the progression of various type of cancers[168]. COUP-TFII is expressed at low levels in adult normal exocrine pancreas[168,169] and recently we demonstrated its involvement in pancreatic cancer in vitro and in vivo[168] (Figure 8). COUP-TFII was expressed in 69% of tested primary samples correlating with the presence of lymph and distant metastasis as well as clinical stage; PDAC patients stained positive for the NR showed a significant reduction of survival compared to NR-negative patients. In vitro silencing of COUP-TFII reduces the cell growth and invasiveness and it strongly inhibits angiogenesis, an effect mediated by the regulation of VEGF-C. The reduced proliferation is associated with a block in G1 and decreased expression of E2F1, but not apoptosis; moreover, COUP-TFII silencing reduces OCT4 and increases Nanog expression. In vitro effects were confirmed in nude mice where COUP-TFII silencing reduces tumor growth by 40%[168].
Figure 8.
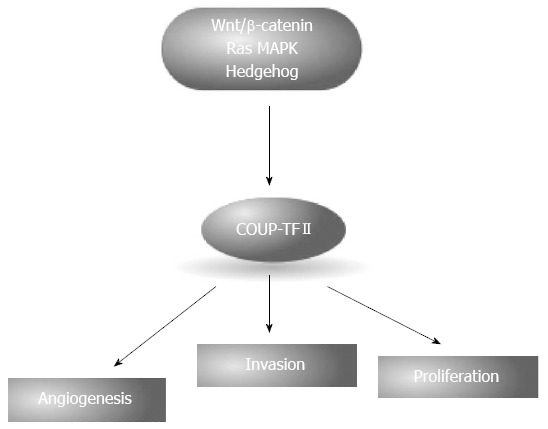
Chicken ovalbumin upstream promoter transcription factor II is involved in the regulation of angiogenesis, invasion and tumor proliferation. Expression of chicken ovalbumin upstream promoter transcription factor II (COUP-TFII) is induced by several pathways altered in pancreatic ductal carcinoma, including Wnt/β-catenin, RAS-MAPK and Hedgehog.
CONCLUSION
PDAC is a devastating disease originating from well defined genetic alterations. However, due to the its subtle nature, the lack of efficient diagnostic methods and of effective drugs it is a deadly disease with a dismal prognosis. NR are ligand-regulated transcription factors functionally involved in important cellular functions ranging from regulation of metabolism, to growth and development. Given the nature of their ligands, NR are very tempting drug targets and their pharmacological modulation has been widely exploited. There are now clear evidences that both classical ligand-activated and orphan NR are involved in the pathogenesis of pancreatic cancer disease from its very early stages. From the review of the literature PPARs, RARs, RXRs, AR, ERα and ERβ and the orphan NR Nur, COUP-TFII and LHR-1 show striking connections with PDAC development, that for certainty will need more experimental confirmations, especially for the orphans. Although clinical application of NR modulators in the PDAC treatment still suffers from failure (Table 2), a more comprehensive analysis of NR action in PDAC could lead to the identification of novel therapies for this disease.
Table 2.
Expression of nuclear receptors and clinical trials
| Nuclear receptor | Expression in primary PDAC | Clinical trials and results |
| PPAR (-α, -β, -γ) | PPARα: Unknown | None for PPARa and β/δ; |
| PPARβ/δ: overexpressed; expression correlates with tumor stage, recurrence, and distant metastasis | PPARγ: TZD apparently reduce the risk of PDAC but PPARγ agonists do not improve survival | |
| PPARg: maybe overexpressed; expression correlates with shorter overall survival | ||
| RAR and RXR (-α, -β, -γ) | Yes (with the exception of RARb that is apparently lost during cancer development) | 13-cis-RA in combination with IFN-γ: prolonged stable disease as well no improvement have been reported |
| AR | Yes | Phase II trials with the antagonist flutamide: increase in survival as well no effect have been reported |
| ER (-α, -β) | Yes | Tamoxifen with no benefit |
| NR4A1, NR4A2, NR4A3 | NR4A1: overexpressed | None |
| NR4A2 and NR4A3: unknown | ||
| LHR-1 | Overexpressed | None |
| COUP-TFII | Overexpressed; expression correlates with shorter overall survival, tumor stage, and presence of metastasis | None |
PPAR: Peroxisome proliferator-activated receptor; PDAC: Pancreatic ductal carcinoma; TZD: Thiazolidinediones; IFN: Interferon; RAR: Retinoic acid receptor; RXR: Retinoid X receptor; AR: Androgen receptor; ER: Estrogen receptor; LHR-1: Liver receptor homologue-1 receptor; COUP-TFII: Chicken ovalbumin upstream promoter transcription factor II.
Footnotes
Supported by Fondo per gli Investimenti della Ricerca di Base (FIRB) (RBAP10MY35_002), by Ente Cassa di Risparmio di Firenze and by FiorGen ONLUS to Galli A
P- Reviewer: Gu DS, Muscarella P S- Editor: Gou SX L- Editor: A E- Editor: Liu XM
References
- 1.Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61:69–90. doi: 10.3322/caac.20107. [DOI] [PubMed] [Google Scholar]
- 2.Burris HA, Moore MJ, Andersen J, Green MR, Rothenberg ML, Modiano MR, Cripps MC, Portenoy RK, Storniolo AM, Tarassoff P, et al. Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: a randomized trial. J Clin Oncol. 1997;15:2403–2413. doi: 10.1200/JCO.1997.15.6.2403. [DOI] [PubMed] [Google Scholar]
- 3.Adesso L, Calabretta S, Barbagallo F, Capurso G, Pilozzi E, Geremia R, Delle Fave G, Sette C. Gemcitabine triggers a pro-survival response in pancreatic cancer cells through activation of the MNK2/eIF4E pathway. Oncogene. 2013;32:2848–2857. doi: 10.1038/onc.2012.306. [DOI] [PubMed] [Google Scholar]
- 4.Chari ST. Detecting early pancreatic cancer: problems and prospects. Semin Oncol. 2007;34:284–294. doi: 10.1053/j.seminoncol.2007.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Goggins M. Markers of pancreatic cancer: working toward early detection. Clin Cancer Res. 2011;17:635–637. doi: 10.1158/1078-0432.CCR-10-3074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Siegel R, Naishadham D, Jemal A. Cancer statistics, 2012. CA Cancer J Clin. 2012;62:10–29. doi: 10.3322/caac.20138. [DOI] [PubMed] [Google Scholar]
- 7.Li D. Molecular epidemiology of pancreatic cancer. Cancer J. 2001;7:259–265. [PubMed] [Google Scholar]
- 8.Gold EB, Goldin SB. Epidemiology of and risk factors for pancreatic cancer. Surg Oncol Clin N Am. 1998;7:67–91. [PubMed] [Google Scholar]
- 9.Bao B, Wang Z, Li Y, Kong D, Ali S, Banerjee S, Ahmad A, Sarkar FH. The complexities of obesity and diabetes with the development and progression of pancreatic cancer. Biochim Biophys Acta. 2011;1815:135–146. doi: 10.1016/j.bbcan.2010.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hruban RH, Adsay NV, Albores-Saavedra J, Compton C, Garrett ES, Goodman SN, Kern SE, Klimstra DS, Klöppel G, Longnecker DS, et al. Pancreatic intraepithelial neoplasia: a new nomenclature and classification system for pancreatic duct lesions. Am J Surg Pathol. 2001;25:579–586. doi: 10.1097/00000478-200105000-00003. [DOI] [PubMed] [Google Scholar]
- 11.Matthaei H, Schulick RD, Hruban RH, Maitra A. Cystic precursors to invasive pancreatic cancer. Nat Rev Gastroenterol Hepatol. 2011;8:141–150. doi: 10.1038/nrgastro.2011.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gaujoux S, Brennan MF, Gonen M, D’Angelica MI, DeMatteo R, Fong Y, Schattner M, DiMaio C, Janakos M, Jarnagin WR, et al. Cystic lesions of the pancreas: changes in the presentation and management of 1,424 patients at a single institution over a 15-year time period. J Am Coll Surg. 2011;212:590–600; discussion 600-603. doi: 10.1016/j.jamcollsurg.2011.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Delpu Y, Hanoun N, Lulka H, Sicard F, Selves J, Buscail L, Torrisani J, Cordelier P. Genetic and epigenetic alterations in pancreatic carcinogenesis. Curr Genomics. 2011;12:15–24. doi: 10.2174/138920211794520132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Birnbaum DJ, Adélaïde J, Mamessier E, Finetti P, Lagarde A, Monges G, Viret F, Gonçalvès A, Turrini O, Delpero JR, et al. Genome profiling of pancreatic adenocarcinoma. Genes Chromosomes Cancer. 2011;50:456–465. doi: 10.1002/gcc.20870. [DOI] [PubMed] [Google Scholar]
- 15.Hidalgo M. Pancreatic cancer. N Engl J Med. 2010;362:1605–1617. doi: 10.1056/NEJMra0901557. [DOI] [PubMed] [Google Scholar]
- 16.Feldmann G, Beaty R, Hruban RH, Maitra A. Molecular genetics of pancreatic intraepithelial neoplasia. J Hepatobiliary Pancreat Surg. 2007;14:224–232. doi: 10.1007/s00534-006-1166-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bardeesy N, Aguirre AJ, Chu GC, Cheng KH, Lopez LV, Hezel AF, Feng B, Brennan C, Weissleder R, Mahmood U, et al. Both p16(Ink4a) and the p19(Arf)-p53 pathway constrain progression of pancreatic adenocarcinoma in the mouse. Proc Natl Acad Sci USA. 2006;103:5947–5952. doi: 10.1073/pnas.0601273103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jones S, Zhang X, Parsons DW, Lin JC, Leary RJ, Angenendt P, Mankoo P, Carter H, Kamiyama H, Jimeno A, et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science. 2008;321:1801–1806. doi: 10.1126/science.1164368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pinho AV, Chantrill L, Rooman I. Chronic pancreatitis: a path to pancreatic cancer. Cancer Lett. 2014;345:203–209. doi: 10.1016/j.canlet.2013.08.015. [DOI] [PubMed] [Google Scholar]
- 20.Sladek FM. What are nuclear receptor ligands? Mol Cell Endocrinol. 2011;334:3–13. doi: 10.1016/j.mce.2010.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hollenberg SM, Weinberger C, Ong ES, Cerelli G, Oro A, Lebo R, Thompson EB, Rosenfeld MG, Evans RM. Primary structure and expression of a functional human glucocorticoid receptor cDNA. Nature 1985- 1986;318:635–641. doi: 10.1038/318635a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jensen EV. On the mechanism of estrogen action. Perspect Biol Med. 1962;6:47–59. doi: 10.1353/pbm.1963.0005. [DOI] [PubMed] [Google Scholar]
- 23.Jensen EV, Khan SA. A two-site model for antiestrogen action. Mech Ageing Dev. 2004;125:679–682. doi: 10.1016/j.mad.2004.08.006. [DOI] [PubMed] [Google Scholar]
- 24.Green S, Walter P, Kumar V, Krust A, Bornert JM, Argos P, Chambon P. Human oestrogen receptor cDNA: sequence, expression and homology to v-erb-A. Nature. 1986;320:134–139. doi: 10.1038/320134a0. [DOI] [PubMed] [Google Scholar]
- 25.Germain P, Staels B, Dacquet C, Spedding M, Laudet V. Overview of nomenclature of nuclear receptors. Pharmacol Rev. 2006;58:685–704. doi: 10.1124/pr.58.4.2. [DOI] [PubMed] [Google Scholar]
- 26.Rosenfeld MG, Lunyak VV, Glass CK. Sensors and signals: a coactivator/corepressor/epigenetic code for integrating signal-dependent programs of transcriptional response. Genes Dev. 2006;20:1405–1428. doi: 10.1101/gad.1424806. [DOI] [PubMed] [Google Scholar]
- 27.Chen T. Nuclear receptor drug discovery. Curr Opin Chem Biol. 2008;12:418–426. doi: 10.1016/j.cbpa.2008.07.001. [DOI] [PubMed] [Google Scholar]
- 28.Varga T, Czimmerer Z, Nagy L. PPARs are a unique set of fatty acid regulated transcription factors controlling both lipid metabolism and inflammation. Biochim Biophys Acta. 2011;1812:1007–1022. doi: 10.1016/j.bbadis.2011.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lock EA, Mitchell AM, Elcombe CR. Biochemical mechanisms of induction of hepatic peroxisome proliferation. Annu Rev Pharmacol Toxicol. 1989;29:145–163. doi: 10.1146/annurev.pa.29.040189.001045. [DOI] [PubMed] [Google Scholar]
- 30.Polvani S, Tarocchi M, Galli A. PPARγ and Oxidative Stress: Con(β) Catenating NRF2 and FOXO. PPAR Res. 2012;2012:641087. doi: 10.1155/2012/641087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Coleman JD, Thompson JT, Smith RW, Prokopczyk B, Vanden Heuvel JP. Role of Peroxisome Proliferator-Activated Receptor β/δ and B-Cell Lymphoma-6 in Regulation of Genes Involved in Metastasis and Migration in Pancreatic Cancer Cells. PPAR Res. 2013;2013:121956. doi: 10.1155/2013/121956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mello T, Polvani S, Galli A. Peroxisome proliferator-activated receptor and retinoic x receptor in alcoholic liver disease. PPAR Res. 2009;2009:748174. doi: 10.1155/2009/748174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Desvergne B, Wahli W. Peroxisome proliferator-activated receptors: nuclear control of metabolism. Endocr Rev. 1999;20:649–688. doi: 10.1210/edrv.20.5.0380. [DOI] [PubMed] [Google Scholar]
- 34.Kwak BR, Mulhaupt F, Mach F. The role of ppargamma ligands as regulators of the immune response. Drug News Perspect. 2002;15:325–332. doi: 10.1358/dnp.2002.15.6.701652. [DOI] [PubMed] [Google Scholar]
- 35.Kennedy GL, Butenhoff JL, Olsen GW, O’Connor JC, Seacat AM, Perkins RG, Biegel LB, Murphy SR, Farrar DG. The toxicology of perfluorooctanoate. Crit Rev Toxicol. 2004;34:351–384. doi: 10.1080/10408440490464705. [DOI] [PubMed] [Google Scholar]
- 36.Klaunig JE, Babich MA, Baetcke KP, Cook JC, Corton JC, David RM, DeLuca JG, Lai DY, McKee RH, Peters JM, et al. PPARalpha agonist-induced rodent tumors: modes of action and human relevance. Crit Rev Toxicol. 2003;33:655–780. doi: 10.1080/713608372. [DOI] [PubMed] [Google Scholar]
- 37.Liu SH, Rao DD, Nemunaitis J, Senzer N, Zhou G, Dawson D, Gingras MC, Wang Z, Gibbs R, Norman M, et al. PDX-1 is a therapeutic target for pancreatic cancer, insulinoma and islet neoplasia using a novel RNA interference platform. PLoS One. 2012;7:e40452. doi: 10.1371/journal.pone.0040452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sun Y, Zhang L, Gu HF, Han W, Ren M, Wang F, Gong B, Wang L, Guo H, Xin W, et al. Peroxisome proliferator-activated receptor-alpha regulates the expression of pancreatic/duodenal homeobox-1 in rat insulinoma (INS-1) cells and ameliorates glucose-induced insulin secretion impaired by palmitate. Endocrinology. 2008;149:662–671. doi: 10.1210/en.2007-1275. [DOI] [PubMed] [Google Scholar]
- 39.Guyton KZ, Chiu WA, Bateson TF, Jinot J, Scott CS, Brown RC, Caldwell JC. A reexamination of the PPAR-alpha activation mode of action as a basis for assessing human cancer risks of environmental contaminants. Environ Health Perspect. 2009;117:1664–1672. doi: 10.1289/ehp.0900758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chang ZY, Sun R, Ma YS, Fu D, Lai XL, Li YS, Wang XH, Zhang XP, Lv ZW, Cong XL, et al. Differential gene expression of the key signalling pathway in para-carcinoma, carcinoma and relapse human pancreatic cancer. Cell Biochem Funct. 2014;32:258–267. doi: 10.1002/cbf.3009. [DOI] [PubMed] [Google Scholar]
- 41.Bishop-Bailey D. PPARs and angiogenesis. Biochem Soc Trans. 2011;39:1601–1605. doi: 10.1042/BST20110643. [DOI] [PubMed] [Google Scholar]
- 42.Abdollahi A, Schwager C, Kleeff J, Esposito I, Domhan S, Peschke P, Hauser K, Hahnfeldt P, Hlatky L, Debus J, et al. Transcriptional network governing the angiogenic switch in human pancreatic cancer. Proc Natl Acad Sci USA. 2007;104:12890–12895. doi: 10.1073/pnas.0705505104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Giaginis C, Katsamangou E, Tsourouflis G, Zizi-Serbetzoglou D, Kouraklis G, Theocharis S. Peroxisome proliferator-activated receptor-gamma and retinoid X receptor-alpha expression in pancreatic ductal adenocarcinoma: association with clinicopathological parameters, tumor proliferative capacity, and patients’ survival. Med Sci Monit. 2009;15:BR148–BR156. [PubMed] [Google Scholar]
- 44.Pazienza V, Tavano F, Benegiamo G, Vinciguerra M, Burbaci FP, Copetti M, di Mola FF, Andriulli A, di Sebastiano P. Correlations among PPARγ, DNMT1, and DNMT3B Expression Levels and Pancreatic Cancer. PPAR Res. 2012;2012:461784. doi: 10.1155/2012/461784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kristiansen G, Jacob J, Buckendahl AC, Grützmann R, Alldinger I, Sipos B, Klöppel G, Bahra M, Langrehr JM, Neuhaus P, et al. Peroxisome proliferator-activated receptor gamma is highly expressed in pancreatic cancer and is associated with shorter overall survival times. Clin Cancer Res. 2006;12:6444–6451. doi: 10.1158/1078-0432.CCR-06-0834. [DOI] [PubMed] [Google Scholar]
- 46.Ceni E, Mello T, Tarocchi M, Crabb DW, Caldini A, Invernizzi P, Surrenti C, Milani S, Galli A. Antidiabetic thiazolidinediones induce ductal differentiation but not apoptosis in pancreatic cancer cells. World J Gastroenterol. 2005;11:1122–1130. doi: 10.3748/wjg.v11.i8.1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tang H, Dong X, Hassan M, Abbruzzese JL, Li D. Body mass index and obesity- and diabetes-associated genotypes and risk for pancreatic cancer. Cancer Epidemiol Biomarkers Prev. 2011;20:779–792. doi: 10.1158/1055-9965.EPI-10-0845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fesinmeyer MD, Stanford JL, Brentnall TA, Mandelson MT, Farin FM, Srinouanprachanh S, Afsharinejad Z, Goodman GE, Barnett MJ, Austin MA. Association between the peroxisome proliferator-activated receptor gamma Pro12Ala variant and haplotype and pancreatic cancer in a high-risk cohort of smokers: a pilot study. Pancreas. 2009;38:631–637. doi: 10.1097/MPA.0b013e3181a53ef9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nakajima A, Tomimoto A, Fujita K, Sugiyama M, Takahashi H, Ikeda I, Hosono K, Endo H, Yoneda K, Iida H, et al. Inhibition of peroxisome proliferator-activated receptor gamma activity suppresses pancreatic cancer cell motility. Cancer Sci. 2008;99:1892–1900. doi: 10.1111/j.1349-7006.2008.00904.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sawai H, Liu J, Reber HA, Hines OJ, Eibl G. Activation of peroxisome proliferator-activated receptor-gamma decreases pancreatic cancer cell invasion through modulation of the plasminogen activator system. Mol Cancer Res. 2006;4:159–167. doi: 10.1158/1541-7786.MCR-05-0257. [DOI] [PubMed] [Google Scholar]
- 51.Cekanova M, Lee SH, McEntee MF, Baek SJ. MCC-555-induced NAG-1 expression is mediated in part by KLF4. Eur J Pharmacol. 2010;637:30–37. doi: 10.1016/j.ejphar.2010.03.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Min KW, Zhang X, Imchen T, Baek SJ. A peroxisome proliferator-activated receptor ligand MCC-555 imparts anti-proliferative response in pancreatic cancer cells by PPARgamma-independent up-regulation of KLF4. Toxicol Appl Pharmacol. 2012;263:225–232. doi: 10.1016/j.taap.2012.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Toyota M, Miyazaki Y, Kitamura S, Nagasawa Y, Kiyohara T, Shinomura Y, Matsuzawa Y. Peroxisome proliferator-activated receptor gamma reduces the growth rate of pancreatic cancer cells through the reduction of cyclin D1. Life Sci. 2002;70:1565–1575. doi: 10.1016/s0024-3205(01)01524-7. [DOI] [PubMed] [Google Scholar]
- 54.Hong J, Samudio I, Liu S, Abdelrahim M, Safe S. Peroxisome proliferator-activated receptor gamma-dependent activation of p21 in Panc-28 pancreatic cancer cells involves Sp1 and Sp4 proteins. Endocrinology. 2004;145:5774–5785. doi: 10.1210/en.2004-0686. [DOI] [PubMed] [Google Scholar]
- 55.Pazienza V, Tavano F, Francavilla M, Fontana A, Pellegrini F, Benegiamo G, Corbo V, di Mola FF, Di Sebastiano P, Andriulli A, et al. Time-Qualified Patterns of Variation of PPARγ, DNMT1, and DNMT3B Expression in Pancreatic Cancer Cell Lines. PPAR Res. 2012;2012:890875. doi: 10.1155/2012/890875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sun WH, Chen GS, Ou XL, Yang Y, Luo C, Zhang Y, Shao Y, Xu HC, Xiao B, Xue YP, et al. Inhibition of COX-2 and activation of peroxisome proliferator-activated receptor gamma synergistically inhibits proliferation and induces apoptosis of human pancreatic carcinoma cells. Cancer Lett. 2009;275:247–255. doi: 10.1016/j.canlet.2008.10.023. [DOI] [PubMed] [Google Scholar]
- 57.Hashimoto K, Ethridge RT, Evers BM. Peroxisome proliferator-activated receptor gamma ligand inhibits cell growth and invasion of human pancreatic cancer cells. Int J Gastrointest Cancer. 2002;32:7–22. doi: 10.1385/IJGC:32:1:7. [DOI] [PubMed] [Google Scholar]
- 58.Chintharlapalli S, Papineni S, Liu S, Jutooru I, Chadalapaka G, Cho SD, Murthy RS, You Y, Safe S. 2-cyano-lup-1-en-3-oxo-20-oic acid, a cyano derivative of betulinic acid, activates peroxisome proliferator-activated receptor gamma in colon and pancreatic cancer cells. Carcinogenesis. 2007;28:2337–2346. doi: 10.1093/carcin/bgm189. [DOI] [PubMed] [Google Scholar]
- 59.Galli A, Ceni E, Mello T, Polvani S, Tarocchi M, Buccoliero F, Lisi F, Cioni L, Ottanelli B, Foresta V, et al. Thiazolidinediones inhibit hepatocarcinogenesis in hepatitis B virus-transgenic mice by peroxisome proliferator-activated receptor gamma-independent regulation of nucleophosmin. Hepatology. 2010;52:493–505. doi: 10.1002/hep.23669. [DOI] [PubMed] [Google Scholar]
- 60.Galli A, Ceni E, Crabb DW, Mello T, Salzano R, Grappone C, Milani S, Surrenti E, Surrenti C, Casini A. Antidiabetic thiazolidinediones inhibit invasiveness of pancreatic cancer cells via PPARgamma independent mechanisms. Gut. 2004;53:1688–1697. doi: 10.1136/gut.2003.031997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Jutooru I, Chadalapaka G, Chintharlapalli S, Papineni S, Safe S. Induction of apoptosis and nonsteroidal anti-inflammatory drug-activated gene 1 in pancreatic cancer cells by a glycyrrhetinic acid derivative. Mol Carcinog. 2009;48:692–702. doi: 10.1002/mc.20518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Dong YW, Wang XP, Wu K. Suppression of pancreatic carcinoma growth by activating peroxisome proliferator-activated receptor gamma involves angiogenesis inhibition. World J Gastroenterol. 2009;15:441–448. doi: 10.3748/wjg.15.441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tsujie M, Nakamori S, Okami J, Takahashi Y, Hayashi N, Nagano H, Dono K, Umeshita K, Sakon M, Monden M. Growth inhibition of pancreatic cancer cells through activation of peroxisome proliferator-activated receptor gamma/retinoid X receptor alpha pathway. Int J Oncol. 2003;23:325–331. [PubMed] [Google Scholar]
- 64.Vitale G, Zappavigna S, Marra M, Dicitore A, Meschini S, Condello M, Arancia G, Castiglioni S, Maroni P, Bendinelli P, et al. The PPAR-γ agonist troglitazone antagonizes survival pathways induced by STAT-3 in recombinant interferon-β treated pancreatic cancer cells. Biotechnol Adv. 2012;30:169–184. doi: 10.1016/j.biotechadv.2011.08.001. [DOI] [PubMed] [Google Scholar]
- 65.Eibl G, Takata Y, Boros LG, Liu J, Okada Y, Reber HA, Hines OJ. Growth stimulation of COX-2-negative pancreatic cancer by a selective COX-2 inhibitor. Cancer Res. 2005;65:982–990. [PubMed] [Google Scholar]
- 66.Ghosh N, Chaki R, Mandal V, Mandal SC. COX-2 as a target for cancer chemotherapy. Pharmacol Rep. 2010;62:233–244. doi: 10.1016/s1734-1140(10)70262-0. [DOI] [PubMed] [Google Scholar]
- 67.Maniati E, Bossard M, Cook N, Candido JB, Emami-Shahri N, Nedospasov SA, Balkwill FR, Tuveson DA, Hagemann T. Crosstalk between the canonical NF-κB and Notch signaling pathways inhibits Pparγ expression and promotes pancreatic cancer progression in mice. J Clin Invest. 2011;121:4685–4699. doi: 10.1172/JCI45797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Koga H, Selvendiran K, Sivakumar R, Yoshida T, Torimura T, Ueno T, Sata M. PPARγ potentiates anticancer effects of gemcitabine on human pancreatic cancer cells. Int J Oncol. 2012;40:679–685. doi: 10.3892/ijo.2011.1237. [DOI] [PubMed] [Google Scholar]
- 69.Saif MW, Oettle H, Vervenne WL, Thomas JP, Spitzer G, Visseren-Grul C, Enas N, Richards DA. Randomized double-blind phase II trial comparing gemcitabine plus LY293111 versus gemcitabine plus placebo in advanced adenocarcinoma of the pancreas. Cancer J. 2009;15:339–343. doi: 10.1097/PPO.0b013e3181b36264. [DOI] [PubMed] [Google Scholar]
- 70.Adrian TE, Hennig R, Friess H, Ding X. The Role of PPARgamma Receptors and Leukotriene B(4) Receptors in Mediating the Effects of LY293111 in Pancreatic Cancer. PPAR Res. 2008;2008:827096. doi: 10.1155/2008/827096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Germain P, Chambon P, Eichele G, Evans RM, Lazar MA, Leid M, De Lera AR, Lotan R, Mangelsdorf DJ, Gronemeyer H. International Union of Pharmacology. LXIII. Retinoid X receptors. Pharmacol Rev. 2006;58:760–772. doi: 10.1124/pr.58.4.7. [DOI] [PubMed] [Google Scholar]
- 72.Germain P, Chambon P, Eichele G, Evans RM, Lazar MA, Leid M, De Lera AR, Lotan R, Mangelsdorf DJ, Gronemeyer H. International Union of Pharmacology. LX. Retinoic acid receptors. Pharmacol Rev. 2006;58:712–725. doi: 10.1124/pr.58.4.4. [DOI] [PubMed] [Google Scholar]
- 73.Kane MA, Chen N, Sparks S, Napoli JL. Quantification of endogenous retinoic acid in limited biological samples by LC/MS/MS. Biochem J. 2005;388:363–369. doi: 10.1042/BJ20041867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ziouzenkova O, Orasanu G, Sharlach M, Akiyama TE, Berger JP, Viereck J, Hamilton JA, Tang G, Dolnikowski GG, Vogel S, et al. Retinaldehyde represses adipogenesis and diet-induced obesity. Nat Med. 2007;13:695–702. doi: 10.1038/nm1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ziouzenkova O, Orasanu G, Sukhova G, Lau E, Berger JP, Tang G, Krinsky NI, Dolnikowski GG, Plutzky J. Asymmetric cleavage of beta-carotene yields a transcriptional repressor of retinoid X receptor and peroxisome proliferator-activated receptor responses. Mol Endocrinol. 2007;21:77–88. doi: 10.1210/me.2006-0225. [DOI] [PubMed] [Google Scholar]
- 76.de Urquiza AM, Liu S, Sjöberg M, Zetterström RH, Griffiths W, Sjövall J, Perlmann T. Docosahexaenoic acid, a ligand for the retinoid X receptor in mouse brain. Science. 2000;290:2140–2144. doi: 10.1126/science.290.5499.2140. [DOI] [PubMed] [Google Scholar]
- 77.Mangelsdorf DJ, Borgmeyer U, Heyman RA, Zhou JY, Ong ES, Oro AE, Kakizuka A, Evans RM. Characterization of three RXR genes that mediate the action of 9-cis retinoic acid. Genes Dev. 1992;6:329–344. doi: 10.1101/gad.6.3.329. [DOI] [PubMed] [Google Scholar]
- 78.Dollé P. Developmental expression of retinoic acid receptors (RARs) Nucl Recept Signal. 2009;7:e006. doi: 10.1621/nrs.07006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Thomas M, Sukhai MA, Kamel-Reid S. An emerging role for retinoid X receptor α in malignant hematopoiesis. Leuk Res. 2012;36:1075–1081. doi: 10.1016/j.leukres.2012.05.022. [DOI] [PubMed] [Google Scholar]
- 80.Kadison A, Kim J, Maldonado T, Crisera C, Prasadan K, Manna P, Preuett B, Hembree M, Longaker M, Gittes G. Retinoid signaling directs secondary lineage selection in pancreatic organogenesis. J Pediatr Surg. 2001;36:1150–1156. doi: 10.1053/jpsu.2001.25734. [DOI] [PubMed] [Google Scholar]
- 81.Kobayashi H, Spilde TL, Bhatia AM, Buckingham RB, Hembree MJ, Prasadan K, Preuett BL, Imamura M, Gittes GK. Retinoid signaling controls mouse pancreatic exocrine lineage selection through epithelial-mesenchymal interactions. Gastroenterology. 2002;123:1331–1340. doi: 10.1053/gast.2002.35949. [DOI] [PubMed] [Google Scholar]
- 82.El-Metwally TH, Hussein MR, Pour PM, Kuszynski CA, Adrian TE. High concentrations of retinoids induce differentiation and late apoptosis in pancreatic cancer cells in vitro. Cancer Biol Ther. 2005;4:602–611. doi: 10.4161/cbt.4.5.1762. [DOI] [PubMed] [Google Scholar]
- 83.Pettersson F, Dalgleish AG, Bissonnette RP, Colston KW. Retinoids cause apoptosis in pancreatic cancer cells via activation of RAR-gamma and altered expression of Bcl-2/Bax. Br J Cancer. 2002;87:555–561. doi: 10.1038/sj.bjc.6600496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kaiser A, Herbst H, Fisher G, Koenigsmann M, Berdel WE, Riecken EO, Rosewicz S. Retinoic acid receptor beta regulates growth and differentiation in human pancreatic carcinoma cells. Gastroenterology. 1997;113:920–929. doi: 10.1016/s0016-5085(97)70188-4. [DOI] [PubMed] [Google Scholar]
- 85.Jasinski P, Zwolak P, Terai K, Vogel RI, Borja-Cacho D, Dudek AZ. MT477 acts in tumor cells as an AURKA inhibitor and strongly induces NRF-2 signaling. Anticancer Res. 2011;31:1181–1187. [PubMed] [Google Scholar]
- 86.Albrechtsson E, Ohlsson B, Axelson J. The expression of retinoic acid receptors and the effects in vitro by retinoids in human pancreatic cancer cell lines. Pancreas. 2002;25:49–56. doi: 10.1097/00006676-200207000-00013. [DOI] [PubMed] [Google Scholar]
- 87.Vickers SM, Sampson LK, Ying W, Phillips JO. Receptor-dependent growth inhibition of human pancreatic cancer by 9-cis retinoic acid. J Gastrointest Surg. 1997;1:174–181; discussion 181. doi: 10.1016/s1091-255x(97)80106-0. [DOI] [PubMed] [Google Scholar]
- 88.Balasubramanian S, Chandraratna RA, Eckert RL. A novel retinoid-related molecule inhibits pancreatic cancer cell proliferation by a retinoid receptor independent mechanism via suppression of cell cycle regulatory protein function and induction of caspase-associated apoptosis. Oncogene. 2005;24:4257–4270. doi: 10.1038/sj.onc.1208586. [DOI] [PubMed] [Google Scholar]
- 89.Choudhury A, Singh RK, Moniaux N, El-Metwally TH, Aubert JP, Batra SK. Retinoic acid-dependent transforming growth factor-beta 2-mediated induction of MUC4 mucin expression in human pancreatic tumor cells follows retinoic acid receptor-alpha signaling pathway. J Biol Chem. 2000;275:33929–33936. doi: 10.1074/jbc.M005115200. [DOI] [PubMed] [Google Scholar]
- 90.Moniaux N, Junker WM, Singh AP, Jones AM, Batra SK. Characterization of human mucin MUC17. Complete coding sequence and organization. J Biol Chem. 2006;281:23676–23685. doi: 10.1074/jbc.M600302200. [DOI] [PubMed] [Google Scholar]
- 91.Andrianifahanana M, Agrawal A, Singh AP, Moniaux N, van Seuningen I, Aubert JP, Meza J, Batra SK. Synergistic induction of the MUC4 mucin gene by interferon-gamma and retinoic acid in human pancreatic tumour cells involves a reprogramming of signalling pathways. Oncogene. 2005;24:6143–6154. doi: 10.1038/sj.onc.1208756. [DOI] [PubMed] [Google Scholar]
- 92.Xu X, Stier U, Rosewicz S, Elnaggar A, Lotan R. Selective suppression of nuclear retinoic acid receptor beta gene expression in human pancreatic carcinomas. Int J Oncol. 1996;8:445–451. doi: 10.3892/ijo.8.3.445. [DOI] [PubMed] [Google Scholar]
- 93.House MG, Guo M, Iacobuzio-Donahue C, Herman JG. Molecular progression of promoter methylation in intraductal papillary mucinous neoplasms (IPMN) of the pancreas. Carcinogenesis. 2003;24:193–198. doi: 10.1093/carcin/24.2.193. [DOI] [PubMed] [Google Scholar]
- 94.House MG, Herman JG, Guo MZ, Hooker CM, Schulick RD, Lillemoe KD, Cameron JL, Hruban RH, Maitra A, Yeo CJ. Aberrant hypermethylation of tumor suppressor genes in pancreatic endocrine neoplasms. Ann Surg. 2003;238:423–431; discussion 431-432. doi: 10.1097/01.sla.0000086659.49569.9e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Balasubramanian S, Chandraratna RA, Eckert RL. Suppression of human pancreatic cancer cell proliferation by AGN194204, an RXR-selective retinoid. Carcinogenesis. 2004;25:1377–1385. doi: 10.1093/carcin/bgh122. [DOI] [PubMed] [Google Scholar]
- 96.Cao X, Liu W, Lin F, Li H, Kolluri SK, Lin B, Han YH, Dawson MI, Zhang XK. Retinoid X receptor regulates Nur77/TR3-dependent apoptosis [corrected] by modulating its nuclear export and mitochondrial targeting. Mol Cell Biol. 2004;24:9705–9725. doi: 10.1128/MCB.24.22.9705-9725.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Lin XF, Zhao BX, Chen HZ, Ye XF, Yang CY, Zhou HY, Zhang MQ, Lin SC, Wu Q. RXRalpha acts as a carrier for TR3 nuclear export in a 9-cis retinoic acid-dependent manner in gastric cancer cells. J Cell Sci. 2004;117:5609–5621. doi: 10.1242/jcs.01474. [DOI] [PubMed] [Google Scholar]
- 98.Brembeck FH, Kaiser A, Detjen K, Hotz H, Foitzik T, Buhr HJ, Riecken EO, Rosewicz S. Retinoic acid receptor alpha mediates growth inhibition by retinoids in rat pancreatic carcinoma DSL-6A/C1 cells. Br J Cancer. 1998;78:1288–1295. doi: 10.1038/bjc.1998.673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Hirono S, Yamaue H, Hoshikawa Y, Ina S, Tani M, Kawai M, Ushijima M, Matsuura M, Saiki Y, Saiura A, et al. Molecular markers associated with lymph node metastasis in pancreatic ductal adenocarcinoma by genome-wide expression profiling. Cancer Sci. 2010;101:259–266. doi: 10.1111/j.1349-7006.2009.01359.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Swartz MJ, Batra SK, Varshney GC, Hollingsworth MA, Yeo CJ, Cameron JL, Wilentz RE, Hruban RH, Argani P. MUC4 expression increases progressively in pancreatic intraepithelial neoplasia. Am J Clin Pathol. 2002;117:791–796. doi: 10.1309/7Y7N-M1WM-R0YK-M2VA. [DOI] [PubMed] [Google Scholar]
- 101.Rachagani S, Macha MA, Ponnusamy MP, Haridas D, Kaur S, Jain M, Batra SK. MUC4 potentiates invasion and metastasis of pancreatic cancer cells through stabilization of fibroblast growth factor receptor 1. Carcinogenesis. 2012;33:1953–1964. doi: 10.1093/carcin/bgs225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Rachagani S, Torres MP, Kumar S, Haridas D, Baine M, Macha MA, Kaur S, Ponnusamy MP, Dey P, Seshacharyulu P, et al. Mucin (Muc) expression during pancreatic cancer progression in spontaneous mouse model: potential implications for diagnosis and therapy. J Hematol Oncol. 2012;5:68. doi: 10.1186/1756-8722-5-68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Akhurst RJ, Derynck R. TGF-beta signaling in cancer--a double-edged sword. Trends Cell Biol. 2001;11:S44–S51. doi: 10.1016/s0962-8924(01)02130-4. [DOI] [PubMed] [Google Scholar]
- 104.Friess H, Yamanaka Y, Büchler M, Ebert M, Beger HG, Gold LI, Korc M. Enhanced expression of transforming growth factor beta isoforms in pancreatic cancer correlates with decreased survival. Gastroenterology. 1993;105:1846–1856. doi: 10.1016/0016-5085(93)91084-u. [DOI] [PubMed] [Google Scholar]
- 105.Riecken EO, Rosewicz S. Retinoids in pancreatic cancer. Ann Oncol. 1999;10 Suppl 4:197–200. [PubMed] [Google Scholar]
- 106.Michael A, Hill M, Maraveyas A, Dalgleish A, Lofts F. 13-cis-retinoic acid in combination with gemcitabine in the treatment of locally advanced and metastatic pancreatic cancer--report of a pilot phase ii study. Clin Oncol (R Coll Radiol) 2007;19:150–153. doi: 10.1016/j.clon.2006.11.008. [DOI] [PubMed] [Google Scholar]
- 107.Moore DFJ, Pazdur R, Sugarman S, Jones D3, Lippman SM, Bready B, Abbruzzese JL. Pilot phase ii trial of 13-cis-retinoic acid and interferon-alpha combination therapy for advanced pancreatic adenocarcinoma. Am J Clin Oncol. 1995;18:525–527. doi: 10.1097/00000421-199512000-00013. [DOI] [PubMed] [Google Scholar]
- 108.Gupta S, Pramanik D, Mukherjee R, Campbell NR, Elumalai S, de Wilde RF, Hong S, Goggins MG, De Jesus-Acosta A, Laheru D, et al. Molecular determinants of retinoic acid sensitivity in pancreatic cancer. Clin Cancer Res. 2012;18:280–289. doi: 10.1158/1078-0432.CCR-11-2165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Schug TT, Berry DC, Shaw NS, Travis SN, Noy N. Opposing effects of retinoic acid on cell growth result from alternate activation of two different nuclear receptors. Cell. 2007;129:723–733. doi: 10.1016/j.cell.2007.02.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Roy AK, Tyagi RK, Song CS, Lavrovsky Y, Ahn SC, Oh TS, Chatterjee B. Androgen receptor: structural domains and functional dynamics after ligand-receptor interaction. Ann N Y Acad Sci. 2001;949:44–57. doi: 10.1111/j.1749-6632.2001.tb04001.x. [DOI] [PubMed] [Google Scholar]
- 111.Culig Z, Klocker H, Bartsch G, Hobisch A. Androgen receptors in prostate cancer. Endocr Relat Cancer. 2002;9:155–170. doi: 10.1677/erc.0.0090155. [DOI] [PubMed] [Google Scholar]
- 112.Culig Z, Bartsch G, Hobisch A. Interleukin-6 regulates androgen receptor activity and prostate cancer cell growth. Mol Cell Endocrinol. 2002;197:231–238. doi: 10.1016/s0303-7207(02)00263-0. [DOI] [PubMed] [Google Scholar]
- 113.Higa GM, Fell RG. Sex hormone receptor repertoire in breast cancer. Int J Breast Cancer. 2013;2013:284036. doi: 10.1155/2013/284036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Andrén-Sandberg A, Hoem D, Bäckman PL. Other risk factors for pancreatic cancer: hormonal aspects. Ann Oncol. 1999;10 Suppl 4:131–135. [PubMed] [Google Scholar]
- 115.Imamura Y, Mizuno S. Comparison of pancreatic cancer mortality in five countries: france, italy, japan, uk and usa from who mortality database (1960-2000) Jpn J Clin Oncol. 2005;35:283–286. doi: 10.1093/jjco/hyi082. [DOI] [PubMed] [Google Scholar]
- 116.Kreiger N, Lacroix J, Sloan M. Hormonal factors and pancreatic cancer in women. Ann Epidemiol. 2001;11:563–567. doi: 10.1016/s1047-2797(01)00219-8. [DOI] [PubMed] [Google Scholar]
- 117.Nacusi LP, Debes JD. Primers on molecular pathways: nuclear receptors in pancreatic cancer. The ligand-independent way. Pancreatology. 2008;8:422–424. doi: 10.1159/000151479. [DOI] [PubMed] [Google Scholar]
- 118.Satake M, Sawai H, Go VL, Satake K, Reber HA, Hines OJ, Eibl G. Estrogen receptors in pancreatic tumors. Pancreas. 2006;33:119–127. doi: 10.1097/01.mpa.0000226893.09194.ec. [DOI] [PubMed] [Google Scholar]
- 119.Konduri S, Schwarz MA, Cafasso D, Schwarz RE. Androgen receptor blockade in experimental combination therapy of pancreatic cancer. J Surg Res. 2007;142:378–386. doi: 10.1016/j.jss.2006.09.034. [DOI] [PubMed] [Google Scholar]
- 120.Okitsu K, Kanda T, Imazeki F, Yonemitsu Y, Ray RB, Chang C, Yokosuka O. Involvement of interleukin-6 and androgen receptor signaling in pancreatic cancer. Genes Cancer. 2010;1:859–867. doi: 10.1177/1947601910383417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Selvan RS, Metzgar RS, Petrow V. Growth modulatory effects of some 6-methylenic steroids on human and hamster pancreatic adenocarcinoma cells in vitro. Drug Des Discov. 1992;9:119–133. [PubMed] [Google Scholar]
- 122.Noh KW, Pungpapong S, Wallace MB, Woodward TA, Raimondo M. Do cytokine concentrations in pancreatic juice predict the presence of pancreatic diseases? Clin Gastroenterol Hepatol. 2006;4:782–789. doi: 10.1016/j.cgh.2006.03.026. [DOI] [PubMed] [Google Scholar]
- 123.Greenway BA. Androgen receptor-blocking agents: potential role in pancreatic cancer. Drugs Aging. 2000;17:161–163. doi: 10.2165/00002512-200017030-00001. [DOI] [PubMed] [Google Scholar]
- 124.Corrie P, Mayer A, Shaw J, D’Ath S, Blagden S, Blesing C, Price P, Warner N. Phase II study to evaluate combining gemcitabine with flutamide in advanced pancreatic cancer patients. Br J Cancer. 2002;87:716–719. doi: 10.1038/sj.bjc.6600523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Negi SS, Agarwal A, Chaudhary A. Flutamide in unresectable pancreatic adenocarcinoma: a randomized, double-blind, placebo-controlled trial. Invest New Drugs. 2006;24:189–194. doi: 10.1007/s10637-005-3536-2. [DOI] [PubMed] [Google Scholar]
- 126.Glass JP, Parasher G, Arias-Pulido H, Donohue R, Prossnitz ER, Cerilli LA. Mesothelin and GPR30 staining among a spectrum of pancreatic epithelial neoplasms. Int J Surg Pathol. 2011;19:588–596. doi: 10.1177/1066896911409575. [DOI] [PubMed] [Google Scholar]
- 127.Wei S, Said-Al-Naief N, Hameed O. Estrogen and progesterone receptor expression is not always specific for mammary and gynecologic carcinomas: a tissue microarray and pooled literature review study. Appl Immunohistochem Mol Morphol. 2009;17:393–402. doi: 10.1097/PAI.0b013e31819faa07. [DOI] [PubMed] [Google Scholar]
- 128.Konduri S, Schwarz RE. Estrogen receptor beta/alpha ratio predicts response of pancreatic cancer cells to estrogens and phytoestrogens. J Surg Res. 2007;140:55–66. doi: 10.1016/j.jss.2006.10.015. [DOI] [PubMed] [Google Scholar]
- 129.Iwao K, Miyoshi Y, Ooka M, Ishikawa O, Ohigashi H, Kasugai T, Egawa C, Noguchi S. Quantitative analysis of estrogen receptor-alpha and -beta messenger RNA expression in human pancreatic cancers by real-time polymerase chain reaction. Cancer Lett. 2001;170:91–97. doi: 10.1016/s0304-3835(01)00563-8. [DOI] [PubMed] [Google Scholar]
- 130.Kojima K, Bowersock GJ, Kojima C, Klug CA, Grizzle WE, Mobley JA. Validation of a robust proteomic analysis carried out on formalin-fixed paraffin-embedded tissues of the pancreas obtained from mouse and human. Proteomics. 2012;12:3393–3402. doi: 10.1002/pmic.201100663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Kliewer SA, Lehmann JM, Willson TM. Orphan nuclear receptors: shifting endocrinology into reverse. Science. 1999;284:757–760. doi: 10.1126/science.284.5415.757. [DOI] [PubMed] [Google Scholar]
- 132.Benoit G, Cooney A, Giguere V, Ingraham H, Lazar M, Muscat G, Perlmann T, Renaud JP, Schwabe J, Sladek F, et al. International Union of Pharmacology. LXVI. Orphan nuclear receptors. Pharmacol Rev. 2006;58:798–836. doi: 10.1124/pr.58.4.10. [DOI] [PubMed] [Google Scholar]
- 133.Li QX, Ke N, Sundaram R, Wong-Staal F. NR4A1, 2, 3--an orphan nuclear hormone receptor family involved in cell apoptosis and carcinogenesis. Histol Histopathol. 2006;21:533–540. doi: 10.14670/HH-21.533. [DOI] [PubMed] [Google Scholar]
- 134.Milbrandt J. Nerve growth factor induces a gene homologous to the glucocorticoid receptor gene. Neuron. 1988;1:183–188. doi: 10.1016/0896-6273(88)90138-9. [DOI] [PubMed] [Google Scholar]
- 135.Pearen MA, Muscat GE. Minireview: Nuclear hormone receptor 4A signaling: implications for metabolic disease. Mol Endocrinol. 2010;24:1891–1903. doi: 10.1210/me.2010-0015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Li X, Lee SO, Safe S. Structure-dependent activation of NR4A2 (Nurr1) by 1,1-bis(3’-indolyl)-1-(aromatic)methane analogs in pancreatic cancer cells. Biochem Pharmacol. 2012;83:1445–1455. doi: 10.1016/j.bcp.2012.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Muders MH. Neuropilin and neuropilin associated molecules as new molecular targets in pancreatic adenocarcinoma. Anticancer Agents Med Chem. 2011;11:442–447. doi: 10.2174/187152011795677481. [DOI] [PubMed] [Google Scholar]
- 138.Guo J, Chintharlapalli S, Lee SO, Cho SD, Lei P, Papineni S, Safe S. Peroxisome proliferator-activated receptor gamma-dependent activity of indole ring-substituted 1,1-bis(3’-indolyl)-1-(p-biphenyl)methanes in cancer cells. Cancer Chemother Pharmacol. 2010;66:141–150. doi: 10.1007/s00280-009-1144-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Chintharlapalli S, Burghardt R, Papineni S, Ramaiah S, Yoon K, Safe S. Activation of Nur77 by selected 1,1-Bis(3’-indolyl)-1-(p-substituted phenyl)methanes induces apoptosis through nuclear pathways. J Biol Chem. 2005;280:24903–24914. doi: 10.1074/jbc.M500107200. [DOI] [PubMed] [Google Scholar]
- 140.Lee SO, Chintharlapalli S, Liu S, Papineni S, Cho SD, Yoon K, Safe S. p21 expression is induced by activation of nuclear nerve growth factor-induced Balpha (Nur77) in pancreatic cancer cells. Mol Cancer Res. 2009;7:1169–1178. doi: 10.1158/1541-7786.MCR-08-0473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Yoon K, Lee SO, Cho SD, Kim K, Khan S, Safe S. Activation of nuclear TR3 (NR4A1) by a diindolylmethane analog induces apoptosis and proapoptotic genes in pancreatic cancer cells and tumors. Carcinogenesis. 2011;32:836–842. doi: 10.1093/carcin/bgr040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Lee SO, Abdelrahim M, Yoon K, Chintharlapalli S, Papineni S, Kim K, Wang H, Safe S. Inactivation of the orphan nuclear receptor TR3/Nur77 inhibits pancreatic cancer cell and tumor growth. Cancer Res. 2010;70:6824–6836. doi: 10.1158/0008-5472.CAN-10-1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Lee MA, Park GS, Lee HJ, Jung JH, Kang JH, Hong YS, Lee KS, Kim DG, Kim SN. Survivin expression and its clinical significance in pancreatic cancer. BMC Cancer. 2005;5:127. doi: 10.1186/1471-2407-5-127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Bhanot U, Heydrich R, Möller P, Hasel C. Survivin expression in pancreatic intraepithelial neoplasia (PanIN): steady increase along the developmental stages of pancreatic ductal adenocarcinoma. Am J Surg Pathol. 2006;30:754–759. doi: 10.1097/00000478-200606000-00013. [DOI] [PubMed] [Google Scholar]
- 145.Liang B, Song X, Liu G, Li R, Xie J, Xiao L, Du M, Zhang Q, Xu X, Gan X, et al. Involvement of TR3/Nur77 translocation to the endoplasmic reticulum in ER stress-induced apoptosis. Exp Cell Res. 2007;313:2833–2844. doi: 10.1016/j.yexcr.2007.04.032. [DOI] [PubMed] [Google Scholar]
- 146.Chen HZ, Wen Q, Wang WJ, He JP, Wu Q. The orphan nuclear receptor TR3/Nur77 regulates ER stress and induces apoptosis via interaction with TRAPγ. Int J Biochem Cell Biol. 2013;45:1600–1609. doi: 10.1016/j.biocel.2013.04.026. [DOI] [PubMed] [Google Scholar]
- 147.Lin B, Kolluri SK, Lin F, Liu W, Han YH, Cao X, Dawson MI, Reed JC, Zhang XK. Conversion of Bcl-2 from protector to killer by interaction with nuclear orphan receptor Nur77/TR3. Cell. 2004;116:527–540. doi: 10.1016/s0092-8674(04)00162-x. [DOI] [PubMed] [Google Scholar]
- 148.Green DR, Reed JC. Mitochondria and apoptosis. Science. 1998;281:1309–1312. doi: 10.1126/science.281.5381.1309. [DOI] [PubMed] [Google Scholar]
- 149.Fayard E, Auwerx J, Schoonjans K. LRH-1: an orphan nuclear receptor involved in development, metabolism and steroidogenesis. Trends Cell Biol. 2004;14:250–260. doi: 10.1016/j.tcb.2004.03.008. [DOI] [PubMed] [Google Scholar]
- 150.Lazarus KA, Wijayakumara D, Chand AL, Simpson ER, Clyne CD. Therapeutic potential of Liver Receptor Homolog-1 modulators. J Steroid Biochem Mol Biol. 2012;130:138–146. doi: 10.1016/j.jsbmb.2011.12.017. [DOI] [PubMed] [Google Scholar]
- 151.Krylova IN, Sablin EP, Moore J, Xu RX, Waitt GM, MacKay JA, Juzumiene D, Bynum JM, Madauss K, Montana V, et al. Structural analyses reveal phosphatidyl inositols as ligands for the NR5 orphan receptors SF-1 and LRH-1. Cell. 2005;120:343–355. doi: 10.1016/j.cell.2005.01.024. [DOI] [PubMed] [Google Scholar]
- 152.Benod C, Carlsson J, Uthayaruban R, Hwang P, Irwin JJ, Doak AK, Shoichet BK, Sablin EP, Fletterick RJ. Structure-based discovery of antagonists of nuclear receptor LRH-1. J Biol Chem. 2013;288:19830–19844. doi: 10.1074/jbc.M112.411686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Holmstrom SR, Deering T, Swift GH, Poelwijk FJ, Mangelsdorf DJ, Kliewer SA, MacDonald RJ. LRH-1 and PTF1-L coregulate an exocrine pancreas-specific transcriptional network for digestive function. Genes Dev. 2011;25:1674–1679. doi: 10.1101/gad.16860911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.von Figura G, Morris JP, Wright CV, Hebrok M. Nr5a2 maintains acinar cell differentiation and constrains oncogenic Kras-mediated pancreatic neoplastic initiation. Gut. 2014;63:656–664. doi: 10.1136/gutjnl-2012-304287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Flandez M, Cendrowski J, Cañamero M, Salas A, del Pozo N, Schoonjans K, Real FX. Nr5a2 heterozygosity sensitises to, and cooperates with, inflammation in KRas(G12V)-driven pancreatic tumourigenesis. Gut. 2014;63:647–655. doi: 10.1136/gutjnl-2012-304381. [DOI] [PubMed] [Google Scholar]
- 156.Petersen GM, Amundadottir L, Fuchs CS, Kraft P, Stolzenberg-Solomon RZ, Jacobs KB, Arslan AA, Bueno-de-Mesquita HB, Gallinger S, Gross M, et al. A genome-wide association study identifies pancreatic cancer susceptibility loci on chromosomes 13q22.1, 1q32.1 and 5p15.33. Nat Genet. 2010;42:224–228. doi: 10.1038/ng.522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Rizzato C, Campa D, Giese N, Werner J, Rachakonda PS, Kumar R, Schanné M, Greenhalf W, Costello E, Khaw KT, et al. Pancreatic cancer susceptibility loci and their role in survival. PLoS One. 2011;6:e27921. doi: 10.1371/journal.pone.0027921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Li D, Duell EJ, Yu K, Risch HA, Olson SH, Kooperberg C, Wolpin BM, Jiao L, Dong X, Wheeler B, et al. Pathway analysis of genome-wide association study data highlights pancreatic development genes as susceptibility factors for pancreatic cancer. Carcinogenesis. 2012;33:1384–1390. doi: 10.1093/carcin/bgs151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Benod C, Vinogradova MV, Jouravel N, Kim GE, Fletterick RJ, Sablin EP. Nuclear receptor liver receptor homologue 1 (LRH-1) regulates pancreatic cancer cell growth and proliferation. Proc Natl Acad Sci USA. 2011;108:16927–16931. doi: 10.1073/pnas.1112047108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Murtaugh LC. Putting GWAS to the functional test: NR5A2 and pancreatic cancer risk. Gut. 2014;63:535–536. doi: 10.1136/gutjnl-2013-305030. [DOI] [PubMed] [Google Scholar]
- 161.Krishnan V, Elberg G, Tsai MJ, Tsai SY. Identification of a novel sonic hedgehog response element in the chicken ovalbumin upstream promoter-transcription factor II promoter. Mol Endocrinol. 1997;11:1458–1466. doi: 10.1210/mend.11.10.9992. [DOI] [PubMed] [Google Scholar]
- 162.Okamura M, Kudo H, Wakabayashi K, Tanaka T, Nonaka A, Uchida A, Tsutsumi S, Sakakibara I, Naito M, Osborne TF, et al. COUP-TFII acts downstream of Wnt/beta-catenin signal to silence PPARgamma gene expression and repress adipogenesis. Proc Natl Acad Sci USA. 2009;106:5819–5824. doi: 10.1073/pnas.0901676106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.You LR, Lin FJ, Lee CT, DeMayo FJ, Tsai MJ, Tsai SY. Suppression of Notch signalling by the COUP-TFII transcription factor regulates vein identity. Nature. 2005;435:98–104. doi: 10.1038/nature03511. [DOI] [PubMed] [Google Scholar]
- 164.Rosa A, Brivanlou AH. A regulatory circuitry comprised of miR-302 and the transcription factors OCT4 and NR2F2 regulates human embryonic stem cell differentiation. EMBO J. 2011;30:237–248. doi: 10.1038/emboj.2010.319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Yamazaki T, Suehiro J, Miyazaki H, Minami T, Kodama T, Miyazono K, Watabe T. The COUP-TFII variant lacking a DNA-binding domain inhibits the activation of the Cyp7a1 promoter through physical interaction with COUP-TFII. Biochem J. 2013;452:345–357. doi: 10.1042/BJ20121200. [DOI] [PubMed] [Google Scholar]
- 166.Kruse SW, Suino-Powell K, Zhou XE, Kretschman JE, Reynolds R, Vonrhein C, Xu Y, Wang L, Tsai SY, Tsai MJ, et al. Identification of COUP-TFII orphan nuclear receptor as a retinoic acid-activated receptor. PLoS Biol. 2008;6:e227. doi: 10.1371/journal.pbio.0060227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Lin FJ, Qin J, Tang K, Tsai SY, Tsai MJ. Coup d’Etat: an orphan takes control. Endocr Rev. 2011;32:404–421. doi: 10.1210/er.2010-0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Polvani S, Tarocchi M, Tempesti S, Mello T, Ceni E, Buccoliero F, D’Amico M, Boddi V, Farsi M, Nesi S, et al. COUP-TFII in pancreatic adenocarcinoma: clinical implication for patient survival and tumor progression. Int J Cancer. 2014;134:1648–1658. doi: 10.1002/ijc.28502. [DOI] [PubMed] [Google Scholar]
- 169.Suzuki T, Moriya T, Darnel AD, Takeyama J, Sasano H. Immunohistochemical distribution of chicken ovalbumin upstream promoter transcription factor II in human tissues. Mol Cell Endocrinol. 2000;164:69–75. doi: 10.1016/s0303-7207(00)00242-2. [DOI] [PubMed] [Google Scholar]