

Summary
Dolichol is an obligate carrier of glycans for N-linked protein glycosylation, O-mannosylation, and GPI anchor biosynthesis. Cis-prenyltransferase (cis-PTase) is the first enzyme committed to the synthesis of dolichol. However, the proteins responsible for mammalian cis-PTase activity have not been delineated. Here we show that Nogo-B receptor (NgBR) is a subunit required for dolichol synthesis in yeast, mice and man. Moreover, we describe a family with a congenital disorder of glycosylation caused by a loss of function mutation in the conserved C terminus of NgBR-R290H and show that fibroblasts isolated from patients exhibit reduced dolichol profiles and enhanced accumulation of free cholesterol identically to fibroblasts from mice lacking NgBR. Mutation of NgBR-R290H in man and orthologs in yeast proves the importance of this evolutionarily conserved residue for mammalian cis-PTase activity and function. Thus, these data provides a genetic basis for the essential role of NgBR in dolichol synthesis and protein glycosylation.
Introduction
Nogo B receptor (NgBR) was identified via expression cloning as a protein that interacts with the N terminus of Nogo-B also called reticulon-4b (Miao et al., 2006). NgBR is a polytypic membrane protein, and its C terminal domain shares significant homology with two gene products; NUS1, a gene in yeast required for survival and N-glycosylation (Harrison et al., 2011; Yu et al., 2006) and cis-prenyltransferases (cis-PTase) including genes in yeast, RER22 and SRT1, a human ortholog, hCIT (also called dehydrodolichol diphosphate synthase, DHDDS) and bacterial undecaprenyl pyrophosphate synthase (uppS) (Sato et al., 1999; Schenk et al., 2001; Surmacz and Swiezewska, 2011). In lower organisms, single subunit cis-PTases such as UPPS catalyze the condensation reactions of isopentenyl pyrophosphate (IPP) with farnesyl pyrophosphate (FPP) to synthesize linear polyprenyl pyrophosphate with specific chain lengths. Polyprenyl pyrophosphate is dephosphorylated into polyprenol and then reduced by a polyprenol reductase to produce dolichol (Cantagrel et al., 2010). In mammals, the relative contribution of Nus1/NgBR versus Rer2/Srt1/hCIT to cis-PTase activity and dolichol synthesis are unknown since loss of function of each grouping of genes results in reduced glycosylation.
Congenital disorders of glycosylation (CDG) are genetic diseases that represent an extremely broad spectrum of clinical presentations due to defects in several steps of protein glycosylation. Recently, there have been several reports of genetic defects in the dolichol biosynthetic pathway such as mutations in DHDDS/hCIT and SRD5A3 (Cantagrel et al., 2010; Kasapkara et al., 2012; Zelinger et al., 2011; Zuchner et al., 2011). DHDDS-CDG is associated with inherited retinitis pigmentosa, a disorder causing retinal degeneration and DHDDS-CDG patients did not show the other typical CDG symptoms. SRD5A3-CDG affects the final step in dolichol synthesis. Its clinical features are typical for CDG type 1 glycosylation disorders including psychomotor retardation, ocular malformations, cerebellar hypoplasia, skin lesions, and facial dysmorphism.
Here, we characterize the dolichol biosynthesis pathway in mice and yeast and demonstrate the necessity of both hCIT and NgBR for dolichol biosynthesis. In addition, we describe a unique congenital disorder of glycosylation caused by a mutation in NgBR, a conserved subunit of cis-PTase. Patients harboring a R290H mutation of NgBR have congenital scoliosis, profound psychomotor retardation, refractory epilepsy and macular lesions showing retinitis pigmentosa. Thus, hCIT/NgBR heteromers are essential, conserved components of the machinery necessary for glycosylation reactions in mammals.
Results and Discussion
Targeted disruption of NgBR causes early embryonic lethality in vivo and defective cis-PTase activity and cholesterol levels in isolated fibroblasts
To examine the physiological significance of NgBR, we generated a conditional knockout mouse (Figures. S1A-C). The NgBR knockout allele (NgBRΔ) was generated by crossing NgBR conditional allele (NgBRf) with a protamine Cre driver expressed in the male germ line (O'Gorman et al., 1997). Heterozygous NgBR mice (NgBRΔ/+) appeared normal and intercrosses between NgBRΔ/+ showed no viable homozygous mice (NgBRΔ/Δ) (Figure 1A). To determine when lethality occurred, timed pregnancies of NgBRΔ/+ breeding were examined at embryonic day (E) 6.5 and E7.5. No NgBRΔ/Δ embryos were identified at these time points indicating post-implantation embryonic lethality before E6.5 (Figures 1B, C). Next, we established mouse embryonic fibroblasts (MEF) cultured from NgBRf/f mice using an inducible Cre-loxP system. Reduced expression of NgBR in the tamoxifen inducible NgBR knockout (NgBR iKO) MEF cells was confirmed by PCR and Western blotting for mRNA and protein levels, respectively (Figure S1D). NgBR iKO MEFs showed accumulation of free cholesterol as determined by filipin staining (Figure 1D), decreased cis-PTase activity in isolated membranes (Figure 1E), and mannose incorporation into protein (Figure 1F). Transduction of cells with lentiviral human NgBR rescues the increase in free cholesterol (Harrison et al., 2009) and the decrease in mannose incorporation (Figures 1E and F). Furthermore, we exposed cells to lovastatin, an inhibitor of 3-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) reductase, the rate-limiting enzyme in the synthesis of isoprenoids and measured cell viability using an MTT assay. NgBR iKO MEF cells were significantly more sensitive to lovastatin than control MEF cells (Figure1G). Since defects in protein glycosylation can induce the unfolded protein response (UPR), activation of the UPR pathway in WT and NgBR iKO MEF cells was examined by real-time RT-PCR for marker genes of the pathway including, Bip, Chop and Chac (Figure1H). All three genes were markedly increased in NgBR iKO MEFs implying that defects in dolichol synthesis and protein glycosylation were activating the UPR pathway of ER stress. Thus NgBR is essential for early development and cis-PTase activity in vivo.
Figure 1. Characterization of NgBR knockout mouse embryos and fibroblasts.

(A) Genotype obtained from the progeny of heterozygous mating. No NgBRΔ/Δ embryo was detected. (B) Embryo resorption frequencies during post-implantation development. Resorption sites were apparent at E7.5 among ∼25% decidua. (C) Representative decidua of E 7.5 embryo resorption sites analyzed. Decidua were obtained from NgBRΔ/+ x NgBRΔ/+ breeding. Decidua with embryo contained normally developed E7.5 embryo (insert). Presumptive NgBRΔ/Δ decidua exhibit implanted site for embryo without evident embryonic material (arrowhead). (D) Filipin staining and quantitative representation for mouse embryonic fibroblast. Filipin staining was performed 48hrs after Lenti-NgBR transduction into NgBR iKO MEF cells. U18666A was used as a positive control for inhibition of cholesterol trafficking. (E) Microsomal cis-PTase activity assay for NgBR iKO MEF. 83% reduced enzyme activity was detected in NgBR iKO MEF compare to control. (F) [2-3H]-mannose-labeling of proteins in mouse embryonic fibroblasts. Tunicamycin (Tm) treatment was used as a control. (G) Statin sensitivity measured by MTT assay. Cell viability was determined by MTT assay after 16 h exposure with various concentration of lovastatin (1 to 80 μM). Cell viability was calculated by following equation: MTT OD value of treated sample/MTT OD value of non-treated sample. (H) Real-time RT-PCR for UPR pathway genes. Relative mRNA expression to control show increased expression. Data are representative of at least 3 experiments. *p < 0.05. Data are mean +/− standard error. Also see Figure S1.
Heteromeric organization of cis-prenyltransfease is conserved between fungi and mammals
The eukaryotic cis-PTase was initially presumed to be homodimer based on studies of UPPS of E. coli and M. luteus (Fujihashi et al., 2001; Guo et al., 2005). Our previous work demonstrated that hCIT or NgBR were necessary for cis-PTase activity (Harrison et al., 2011). However, we did not provide unequivocal evidence that NgBR is indispensable for enzymatic complex formation and activity. To definitively dissect the roles of NgBR and hCIT as components of cis-PTase activity, we characterized the yeast orthologs (in S.cerevisiae and S.pombe) of NgBR and hCIT using genetic and biochemical approaches. We hypothesized that baker's yeast may have two heteromeric cis-PTase complexes: Nus1-Rer2 and Nus1-Srt1. To test this, we generated a triple deletion strain, nus1Δ, rer2Δ, srt1Δ, expressing the homomeric cis-PTase from Giardia lamblia (GlcisPT) to support growth (Grabinska et al., 2010). Indeed, we were able to isolate yeast cells lacking chromosomal copies of NUS1, RER2, SRT1 genes but bearing instead GlcisPT on a plasmid with a URA3 marker (Figure 2A). To examine conservation of heteromeric structure of cis-PTases, the triple deletion strain was transformed with MET15 and LEU2 plasmids bearing the cDNAs indicated in Figure 2A. As expected, viable strains were obtained after expressing NUS1/RER2 or NUS1/SRT1 as positive controls (Figure 2A). In addition, NUS1 is also compatible with Sprer2 or hCIT, Spnus1 with Sprer2, Spnus1with hCIT and NgBR with hCIT only. Reverse-phase TLC of polyprenols generated from membranes isolated from wild type BY4742 or transformed mutant cells revealed that human and S. pombe enzymes synthesize polyprenols similar to that in their parental organisms (Figure 2B). However, the size of the dominant polyprenol pyrophosphate synthesized by the hybrid enzymes varied implying that the different gene products (NgBR/Nus1 and hCIT/Rer2/Srt1) determined polyprenol chain length.
Figure 2. Mammalian and Fungi cis-PTase is heteromer consisting of NgBR/Nus1 and hCIT/Rer2/Srt1 orthologs.

(A) The rer2Δ, srt1Δ, nus1Δ triple deletion strain expressing G. lamblia cis-PTase from URA3 plasmid was transformed with the respective plasmids as indicated. The cells were streaked onto complete plates (YPD) or synthetic complete medium containing 1% 5-fluoroorotic acid (FOA). The Ura3 protein, which is expressed from the URA3 marker present in the plasmids, converts FOA to toxic 5-fluorouracil. The viable combination of genes was marked with asteriks. (B) In vitro cisPTase assay. Reverse-phase TLC of polyprenols from membranes prepared from wild type BY4742 (WT) or triple mutant expressing the respective plasmids for hybrid cis-PTases supporting growth of the triple knockout strain. (c-f) Reverse-phase TLC of polyprenols from cis-PTase activity assay by Nus1/Rer2 (C), Nus1/Srt1 (D), SpNus1/SpRer2 (E) or NgBR/hCIT (F) expressed in IVT. Assays were done according to standard conditions using 20 ul of IVT for cis-PTase activity. Reaction products were extracted according and developed on HPTLC RF18 plate. Expression of the HA or myc-tagged proteins was verified by Western blotting of the reaction mix. coT, co-translation. Data are representative of at least 3 experiments.
To examine the enzymatic activities of the gene products, we used in vitro translation (IVT) followed by cis-PTase assays on the above combinations of the cis-PTase components. Enzymes present in the IVT mixture were able to incorporate 14C-IPP into short prenols up to 6 units (Figure 2C, first column) but were unable to synthesize longer chain polyprenyl pyrophosphates. Expression of Nus1, Rer2 or mixtures of IVT Nus1 with Rer2 products in equal amounts (as shown by Western blotting in the bottom panel) did not catalyze formation of polyprenols. Interestingly, only co-translation of Nus1 with Rer2 and its orthologs in S.pombe or humans, formed an active cis-PTase complex producing prenols of expected lengths (Figures 2C-F, lane 5) indicating that both proteins are required for a functional enzyme (Figure 2D-F). Collectively, the data support the heteromeric structure of mammalian and yeast cis-PTase and suggests that eukaryotic cis-PTase is assembled during translation since only co-translation, but not mixing of the proteins, yields active enzyme. Taken together, these data provide a clear rationale for the role of RER2/NUS1 and related genes in dolichol biosynthesis and advance our understanding of this important pathway.
A mutation on NgBR causes congenital scoliosis, severe neurological impairment, refractory epilepsy, hearing deficit and visual impairment
Recently, exome sequencing of individual families with symptoms of a congenital disorder of glycosylation (CDG) has led to the discovery of mutations in DHDDS (hCIT) and SRD5A3, genes involved in the early steps of polyprenol synthesis (Cantagrel et al., 2010; Kasapkara et al., 2012; Zelinger et al., 2011; Zuchner et al., 2011). DHDDS-CDG is associated with inherited retinitis pigmentosa, a disorder causing retinal degeneration and SRD5A3-CDG patients exhibit psychomotor retardation, ocular malformations, cerebellar hypoplasia, skin lesions, and facial dysmorphism. In our clinic, a family of Roma origin (Figure 3A) composed of healthy unrelated parents and four siblings were examined and two siblings presented with congenital scoliosis, severe neurological impairment, refractory epilepsy, hearing deficit and visual impairment with discrete bilateral macular lesions.
Figure 3. Identification of a conserved mutation in the NgBR Gene in patients with a constellation of symptoms consistent with a glycosylation disorder.

(A) Pedigree of the Czech family. Black symbols denote affected individuals, open symbols denote unaffected parents and siblings. M/+ denotes presence (M) or absence (+) of the mutation as defined by Sanger sequencing. (B) Dilated fundus photograph in proband II.4. reveals a granular yellow-white lesion in the fovea, pale optic nerve and retinal vessels with signs of attenuation. (C) Chromatograms of NgBR genomic DNA sequences showing identified mutations in the family. (left panel) Sequence of the unaffected individual, (middle panel) sequence showing heterozygous mutation c.G869>A in the heterozygous carrier, and (right panel) sequence showing homozygous mutation c.G869>A in one of the probands. (D) Schematic representation of NgBR showing the protein primary structure, location of the p.R290H mutation and amino acid sequence alignment of the C-terminal part of the protein. SA, putative signal anchor; TM, putative transmembrane domain. The amino acid residues are color-coded, small amino acids are red, acidic in blue, basic in magenta and hydroxyl with amine in green. Also see Table S1
Proband II.3 was born at term with intrauterine growth retardation. Muscle hypotonia was present since birth and congenital scoliosis and developmental delay were observed since early infancy. Tonic-clonic seizures, refractory epilepsy and recurrent attacks of “status epilepticus” developed from age 11 months. Microcephaly (3rd centile), failure to thrive (< 3rd centile), regression of psychomotor development, severe axial hypotonia and acral spasticity develop after discharge. Routine laboratory tests were unremarkable and cholesterol level was within reference range. The boy died at age of 29 months. Histopathological findings in autopsy tissue revealed non-specific neuronal loss in brain cortex and cerebellum. Similarly to his brother proband II.4 had generalized hypotonia, congenital scoliosis and significant delay in motor milestones. Refractory epilepsy started at age of 7 months and he has been hospitalized several times with severe seizures. He lost any social interaction and he displays no spontaneous movements. At the age of 4 years, he has microcephaly (0.6th centile), failure to thrive (<5th centile) and marked hypertrichosis. He has severe axial hypotonia, acral spasticity with preserved deep tendon reflexes, pseudobulbar palsy, and central visual and hearing impairment. MRI of the brain revealed severe cortical atrophy. A complete dilated fundus examination including color fundus photography was performed under general anesthesia. At the age of 31 months, except for an opacity located in the inferior half of the right cornea, there were bilaterally no other anterior segment abnormalities and the vitreous was optically clear. There were no bone spicule pigmentations but diffuse retinal pigment epithelium mottling could be observed bilaterally. Optic nerves appeared paler and retinal vessels narrower. Repeated examination at the age of 4 years documented a development of bilateral macular lesion showing foveal hyperautoflorescence (Figure 3B).
The exomes of parents and both affected probands were sequenced and searched for genetic variants in the internal exome database, the Exome Variant Server and 1000 genomes databases and only four such variants were discovered; three are located in the autozygous region identified on chromosome 6 and one on chromosome 21 (Table S1). Corresponding genes were evaluated based on their potential contribution to the clinical phenotypes and a homozygous missense mutation c.869G>A in the NUS1 (NM_138459 or NgBR was found. The c.869G>A mutation was confirmed by Sanger sequencing and the affected probands are homozygous for this mutation, whereas their parents and healthy siblings are heterozygous (Figure 3C). The mutation encodes for amino acid exchange, p.Arg290His (R290H), which is located in the evolutionarily conserved C-terminal domain of NgBR (Figure 3D) and is predicted to affect protein function with a score of 0.00 according to SIFT and to be damaging using Polyphen. This mutation was not reported in dbSNP, 1000 Genomes and the Exome variant server, and was not listed in our internal exome database (> 250 exomes). Targeted genotyping of genomic DNA from 255 individuals of Roma origin identified two additional heterozygous carriers of the c.869G>A mutation. Even though the identity and relation status of these two carriers is unknown, this finding suggests, that the congenital disorder of glycosylation caused by a loss of function mutation of NgBR may be relatively frequent among the European Roma population.
NgBR R290H mutation triggers defects in cellular cholesterol trafficking and dolichol biosynthesis
To characterize the NgBR R290H mutation, fibroblasts were isolated from control and NgBR R290H patients and examined the levels of NgBR mRNA, protein, and interaction of NgBR with hCIT (Figure S3A-C). We did not observe any significant differences in the migration of NgBR on SDS-PAGE or the levels of NgBR protein compared to WT (Figure S1B). This suggests that the translation and the subsequent processing of mutant NgBR protein were not altered by the presence of the mutation. NgBR was isolated as a protein that interacted with reticulon 4B, also called Nogo-B (Miao et al., 2006). Therefore, we examined whether Nogo-B levels and its interaction with NgBR were altered in carriers of the NgBR mutation. The levels of Nogo-B, its interaction with NgBR and the localization of Nogo-B were not different (Figure S3D-F). Next, we assessed three aspects of NgBR function, free cholesterol levels, cis-PTase activity and glycosylation. WT cells had little filipin positive free cholesterol whereas treatment with U18666A to induce an NPC phenotype (Cenedella, 2009) increased free cholesterol (Figure 4A and quantified in the right panel). In contrast NgBR R290H mutant cells exhibited increased accumulation of free cholesterol similar to cells where NgBR was silenced (Harrison et al., 2009). Additionally, cis-PTase activity (Figure 4B) and mannose incorporation into proteins (Figure 4C) was markedly lower in NgBR R290H fibroblasts compared to control. We also examined defective glycosylation of proteins in patient fibroblasts by Western blotting for two known glycoproteins, LAMP-1 and ICAM-1 (He et al., 2012; Xiang et al., 2013). Both LAMP-1 and ICAM-1 were hypo-glycosylated in the patient fibroblasts (Figure 4D).Thus, the NgBR R290H mutant is a loss of function mutation that affects cis-PTase function of NgBR without disrupting complex formation with hCIT or Nogo-B. The reduced cis-PTase activity in fibroblasts was manifested as altered dolichol profiles in the urine or serum as assessed by mass spectrometry of all carriers of the R290H mutation (Figure S4), as recently described for patients harboring loss of function mutations in DHDDS(Wen et al., 2013).
Figure 4. NgBR R290H mutation causes defects in cellular cholesterol trafficking and the dolichol biosynthesis pathway.

(A) Filipin staining and quantitative representation for human dermal fibroblast cells from patients (II.3 and II.4). U18666A was used as a positive control for inhibition of cholesterol trafficking. (B) Microsomal cis-PTase activity using isolated membrane from fibroblasts. Compare to wild type, less than 20% of activity was detected in the patient cells. (C) [2-3H]-mannose-labeling of proteins. Cells were incubated with [2-3H]-mannose for 4hrs and TCA precipitated proteins were counted by scintillation. Tunicamycin (Tm) treatment was used as a control for loss of [2-3H]-mannose incorporation into newly synthesized proteins. *p < 0.05. Data are mean +/− standard error, with n=4 from 3 independent experiments. (D) Western blot analysis of LAMP-1 and ICAM-1 levels in patient fibroblasts. Total lysates were analyzed, and the loading controls Nogo-B and Hsp90 shown. Also see Figure S2
Amino acid at 4th position from the C-terminus of NgBR is a functionally and evolutionary conserved residue
Alignment of NgBR orthologs from distantly related eukaryotic organisms reveals a high degree of conservation at the C-terminus with arginine or asparagine present at the fourth position from the C-terminus (Figure 5A). To test the evolutionary conservation of this position, hCIT was expressed with NgBR or NgBR R290H in the nus1Δ, rer2Δ, srt1Δ. triple knockout strain. Cells expressing the NgBR R290H allele have lower cis-PTase activity (Figure 5B), overall polyprenols (Figure 5C) and dolichol levels measured by MS (Figure 5D). In addition, we analyzed S. cerevisiae and S. pombe Nus1 mutants to determine the importance of the amino acid conservation at the 4th position from the C-terminus in NgBR orthologs. S. cerevisiae Nus1 belongs to group of fungi and plants NgBR orthologs bearing asparagine instead of arginine while S. pombe Nus1 encodes arginine at position 255 corresponding to the R290 in human NgBR. Therefore, we compared cis-PTase activity of S. cerevisiae nus1Δ strain expressing wild type Nus1, Nus1-N372H (mimicking NgBR R290H mutation) as well as Nus1-N372R. Also, we expressed wild type SpNus1 or SpNus1-R255H in nus1Δ fission yeast strain. Mutation of the same position in Nus1 in S. cerevisiae (Figures 5E and F) and S. pombe (Figures 5G and H) resulted in a similar loss of function. Interestingly, the N372R allele of Nus1 from S. cerevisiae affects only the chain length of the product (Figures 5F) but not the rate of incorporation of IPP (Figures 5E).
Figure 5. Characterization of NgBR/NUS1 mutation in S. cerevisiea and S. pombe.

(A) Amino acid alignment of C-terminus of NgBR orthologs. Arginine or asparagine is present at the fourth position from the C-terminus. (B) cis-PTase activity measurements and (D) total dolichol level measured from rer2Δ, srt1Δ, nus1Δ S.cerevisiea strain expressing hCIT and NgBR or NgBR-R290H by mass spectrometry (C) reverse phase TLC separation of cis-PTase products from rer2Δ, srt1Δ, nus1Δ triple deletion S.cerevisiea strain expressing the indicated constructs. About 15 % of dolichol was detected in the NgBR mutant expressing cells compare to wild type NgBR. cis-PTase activity (E) and reverse phase TLC separation (F) in S.cerevisiea nus1Δ strain expressing the indicated constructs. cis-PTase activity (G) and TLC separation of the products (H) in S. pombe nus1Δ strain expressing the indicated constructs. Mutated form of NgBR or Nus1 expressing cells show reduced cis-PTase activity. Not only reduced cis-PTase product but also shortened chain length was detected in the mutated form of protein expressed cells. (I) cis-PTase activity in rer2Δ, srt1Δ, nus1Δ S.cerevisiea strain expressing NgBR and hCIT indicated constructs. Samples were not dephosphorylated prior TLC analysis. Data are +/− standard error. *p < 0.05. Also see Figure S3
Recently, altered ratios of plasma and urinary dolichols was observed in retinitis pigmentosa patients carrying the K42E mutation in DHDDS/hCIT (Wen et al., 2013). To compare the influence of NgBR R290H and hCIT K42E mutations on cis-PTase activity, we expressed hCIT or hCIT K42E with NgBR or NgBR R290H in the S. cerevisiae triple knockout strain and measured enzyme activity. Introduction of this mutant into triple knockout strain expressing WT NgBR reduced steady state cis-PTase activity to a similar extent as NgBR R290H expressed with WT hCIT (Figure 5I) and combining the mutations reduced activity further demonstrating epistasis of the gene products.
NgBR and its orthologs are essential genes and NgBR/hCIT heteromers are responsible for dolichol synthesis in mammalian cells (Figure 6A and B). Based on previous work, NgBR can interact with hCIT, NPC2 and Nogo-B (Figure 6B; numbered 1-3). The interaction with NPC2 was identified by an independent broad based screening strategy (Harrison et al., 2009). Genetic evidence for the importance of this interaction stems from data showing that NgBR knockout MEF and patient fibroblasts harboring the R290H mutation exhibit increased free cholesterol levels. Since NPC2 is a soluble glycoprotein (Naureckiene et al., 2000) and glycosylation of NPC2 is important for its function (Chikh et al., 2004), it is feasible that in addition to a direct stabilizing effect of NgBR on NPC2, mutant NgBR can influence NPC2 glycosylation due to reduced cis-PTase activity contributing to this phenotype (Figure 6B). The interaction of NgBR with Nogo-B does not impact cis-PTase activity or cellular cholesterol content and may influence intracellular signaling pathways.
Figure 6. Functions of the NgBR/hCIT complex in cellular metabolism.
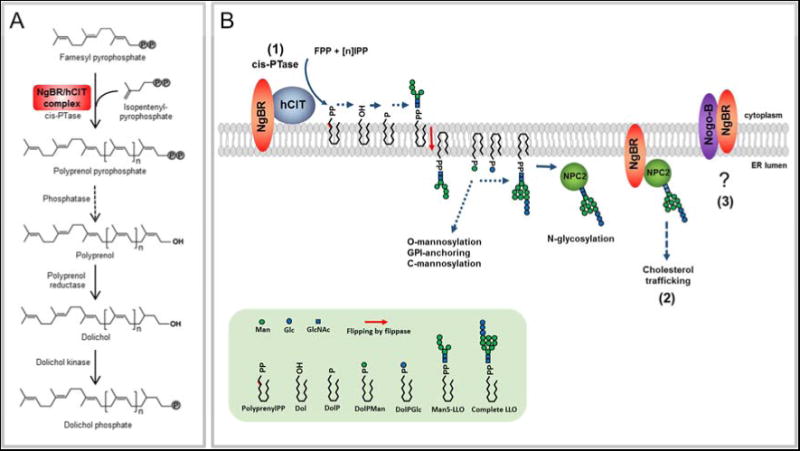
(A) The NgBR and hCIT complex promotes cis-PTase activity. NgBR/hCIT catalyzes the condensation of isopentenyl pyrophosphate with farnesyl pyrophosphate to generate a polyprenol pyrophosphate. Polyprenol diphosphate is dephosphorylated by unidentified phosphatase and reduced by polyprenol reductase (SRD5A3) to form dolichol. Finally, dolichol is phosphorylated by dolichol kinase prior to the synthesis of dolichol linked sugars required for glycosylation pathways. (B) (1) NgBR and hCIT assembly is essential for cis-PTase activity generating polyprenol pyrophosphate on the cytoplasmic leaflet of the ER membrane. Polyprenol pyrophosphate serves as an intermediate in synthesis of dolichol linked saccharides. Dolichol-pyrophosphate tetradecasaccharide (LLO) is indispensable for protein N-glycosylation reactions. Dolichol-phosphate mannose (DolPMan) is also involved in O-mannosylation, GPI-anchor synthesis and C-mannosylation. (2) NgBR influences cholesterol trafficking by directly interacting with NPC2 and indirectly via modifying NPC2 N-glycosylation. (3) The interaction between Nogo-B and NgBR does not influence glycosylation or cholesterol trafficking and the function of this interaction remains to be clarified.
Little is known about the function of dolichol species in vivo besides its role as a glycan carrier, although in vitro evidence suggests that dolichol can modulate biophysical properties of membranes and serve as a cellular antioxidant (Surmacz and Swiezewska, 2011). Patients carrying a mutation in NgBR demonstrated altered ratios of dolichol in urine and in blood. Although altered dolichol chain length ratios are important biomarkers in patients with mutations in hCIT/DHDDS and NgBR, alterations in dolichol chain length are unlikely to exert a dominant effect since lipid linked oligosaccharides built on as short as 11 dolichol units seem to be efficient substrates in N-glycosylation reactions (Grabinska 2010, Rush 2010). However, the overall lower dolichol content of cell membranes not only directly affects glycosylation but can impair membrane structure and in turn, affect multiple cellular processes including sterol biosynthesis. In summary, the development of a knockout strain of mice, the establishment of a NgBR/hCIT reconstitution system in yeast and the discovery of a highly conserved mutation in NgBR mutation in humans will assist in the further characterization of the cellular functions of this essential polyisoprene lipid.
Methods
Generation of NgBR mouse embryonic fibroblasts
NgBRf/f was crossed with NgBR+/Δ; R26CreER (Badea et al., 2003), and primary MEFs were prepared from E 13.5 embryos. Each MEF line was derived from an individual embryo. Isolated MEF were immortalized using an SV40-large T expressing retrovirus obtained from Genecoepiea (LP-SV40T-LV105-0205) according to the manufacturer's protocol. Immortalized cells were maintained in DMEM with 1% penicillin/streptomycin containing 10% FBS. Genotype of control MEF and NgBR iKO MEF used in this study is NgBRf/+ and NgBR+/Δ; R26CreER. Both cell lines were treated with 1 μM 4-hydroxytamoxifen (Sigma) for more than 5 days to induce Cre recombination. mRNA or protein expression level was confirmed for each experiment.
Filipin staining
Filipin staining was performed as previously described (Harrison et al., 2009). In brief, cells were fixed in 4% PFA for 10 min and permeablized in 0.1% TritonX-100 for 5 min. Cells were then incubated with 50μg/ml concentration of filipin (Sigma, F4767) for 1 hr. As a positive control for induction of cholesterol accumulation, cells were treated for 8 hr with 1 μM U18666A (EMD Biosciences). Relative intensity of filipin staining was quantified by calculating average pixel intensity using Adobe Photoshop according to the equation: average filipin intensity = total intensity above low threshold/number of pixels above low threshold (Pipalia et al., 2006)
Microsomal cis-PTase activity assay
For mammalian cells, cude microsomes were prepared as described before (Rush et al., 2010) with minor modification. cis-PTase activity in mammalian cells was assayed as described before (Harrison et al., 2011) with minor modification. For S. cerevisiae and S. pombe, membrane fraction was prepared as described before (Szkopinska et al., 1997). cis-PTase assay using yeast membranes was performed as described (Szkopinska et al., 1997) with minor modifications. For a detailed description, please see the Extended Experimental Procedures.
[2-3H]-mannose incorporation
D-[2-3H]-mannose (15–30 Ci/mmol) was purchased from Perkin-Elmer. Cells were grown in 6 well dishes until 80–90% confluent. Growth medium was replaced and incubated for 1hr with glucose-free DMEM supplemented with 0.1 mg/ml glucose and 5% dialyzed FCS. 5 μg/ml of tunicamycin was added to media a control for inhibition of N-glycosylation. After 1 h, 20 uCi/ml [2-3H]-mannose was added and incubated for 4hr at 37°C. Then cells were washed with PBS, lysed in RIPA buffer. Cell lysates were then subjected to precipitation of proteins with 10%TCA for 1 h on ice. Precipitates resuspended in 6M Urea/SDS buffer and were counted by scintillation.
Co-immunoprecipitation and western blot analysis
Cos7 cell was transfected using lipofectamine 2000 (Invitrogen) according to manufacturer's protocol and harvested 48 hr after transfection. Cells were collected and lysed in IP buffer (IP buffer: 50mM HEPES, 150mM NaCl, 1mM EDTA, 1% Triton X-100, 1.5 mg/ml protease inhibitor cocktail). Lysates were cleared at 12000 rpm and 20μl of anti-HA agarose (Pierce) was used to pulldown the HA-tagged protein from 1mg of cell lysate. After incubation 2 hour at 4°C, agarose were washed with IP buffer, resuspended in SDS loading buffer and boiled for 5mini before western bloting.
Quantitative RT-PCR
Cells were collected in RLT buffer (Qiagen). Total RNA wea extracted using the RNeasy mini kit (Qiagen) and 500 ng of total RNA was transcribed using superscript First-Strand synthesis sytem with oligo dT primers (Invitrogen). Quantitative RT-PCR was performed using iQ SYBR Green Supermix (Bio-Rad) for the detection of fluorescence during amplification. Gapdh was used as an internal control. Expression of target genes was normalized to that of Gapdh using the comparative deltaCT-method. Data are presented in relative expression to control ± SEM.
Human subjects and DNA analysis
The family of Roma origin was ascertained at the Department of Pediatrics of the First Faculty of Medicine, Charles University in Prague. Investigations were approved by Institutional Review Board, and were conducted according to the Declaration of Helsinki principles. Written, informed consent was obtained from all subjects. Participants provided urine and venous blood. Skin biopsy was obtained from both affected individuals and skin fibroblasts were cultured according to standard protocols. Autopsy material has been collected from deceased proband II.3. Genomic DNA was isolated from blood using standard technology and analyzed as described in Extended Experimental Procedures.
Yeast strains, plasmids and culture methods
S. cerevisiae strains used in these studies include: KG404-16B (nus1Δ/pNEV-GlcisPT), KG405 (rer2Δ, srt1Δ, nus1Δ,/pNEV-GlcisPT) and BY4742 and their derivatives. S. pombe strain used was KGSP16 (Spnus1Δ REP42GW-GlcisPT) For Yeast culture methods and detailed information about the strains, please see the Extended Experimental Procedures.
Analysis of dolichols by LC-MS
Serum and urine from family members and healthy unrelated controls were collected according standard protocols. The samples were frozen at −80°C until the lipid extraction and the analysis. The lipid fraction was isolated from membranes isolated of S. cervisiae (2 mg of proteins) or fibroblast (0.5 mg of proteins) as described before (Grabinska et al., 2005) and dolichol content was analyze by liquid chromatography and mass spectrometry (LC-MS)(Guan and Eichler, 2011; Wen et al., 2013). For a detailed description, please see the Extended Experimental Procedures.
Supplementary Material
Highlights.
Loss of NgBR in mice causes early embryonic lethality and defects in glycosylation.
Eukaryotic cis-PTase is heteromer composed of NgBR and hCIT.
Mutations of NgBR cause defects in cholesterol trafficking and dolichol biosynthesis.
NgBR mutation in humans causes a new congenital disorder of glycosylation.
Acknowledgments
We would like to thank Dr. Patrick Lusk (Department of Cell Biology, Yale School of Medicine) for giving us access to dissection microscope for yeast tetrad separation and Marcela Michaličková (General Faculty Hospital Prague) for ophthalmologic investigations. This work was supported by Grants R01 HL64793, R01 HL61371, R01 HL081190, RO1 HL096670, and P01 HL70295 from the National Institutes of Health to WCS. Z.G. and the mass spectrometry facility in the Department of Biochemistry, Duke University Medical Center were supported by a LIPID MAPS glue grant (GM-069338) from the National Institutes of Health. S.K.et al. were supported by the by Charles University institutional programs PRVOUK-P24/LF1/3, UNCE 204011 and SVV2014/260 022; and by BIOCEV – Biotechnology and Biomedicine Centre of the Academy of Sciences and Charles University (CZ.1.05/1.1.00/02.0109), from the European Regional Development Fund. Specific support was provided by grants NT13116-4/2012 and NT12166-5/2011 from the Ministry of Health of the Czech Republic
Footnotes
Author Contributions: EJP and KAG equally contributed to all aspects of this paper; EJP characterized NgBR deficient mice and performed all mammalian cell based studies, cloning/plasmid construction, and KAG developed yeast strains and cisPTase characterization in vivo and in vitro. Both authors contributed to writing and editing of the manuscript. ZG and RW contributed to MS analysis of urinary and serum dolichol levels and writing of the manuscript. VS, HH, KH, VB, JS, NO, HH, TH, JZ, and HH contributed to all aspects of patient sample acquisition, isolation of patient genotypes, exome sequencing and cell isolation. LJ performed high resolution imaging of Nogo-B in patient cells. WCS, EJP, KAG and SK were responsible concept development and preparation of manuscript. SK was responsible for clinical and diagnostic assessment of affected patients and over-seeing the genetic aspects of the study, and WCS was responsible for overall integration and execution of the scientific approaches.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Badea TC, Wang Y, Nathans J. A noninvasive genetic/pharmacologic strategy for visualizing cell morphology and clonal relationships in the mouse. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2003;23:2314–2322. doi: 10.1523/JNEUROSCI.23-06-02314.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantagrel V, Lefeber DJ, Ng BG, Guan Z, Silhavy JL, Bielas SL, Lehle L, Hombauer H, Adamowicz M, Swiezewska E, et al. SRD5A3 is required for converting polyprenol to dolichol and is mutated in a congenital glycosylation disorder. Cell. 2010;142:203–217. doi: 10.1016/j.cell.2010.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cenedella RJ. Cholesterol synthesis inhibitor U18666A and the role of sterol metabolism and trafficking in numerous pathophysiological processes. Lipids. 2009;44:477–487. doi: 10.1007/s11745-009-3305-7. [DOI] [PubMed] [Google Scholar]
- Chikh K, Vey S, Simonot C, Vanier MT, Millat G. Niemann-Pick type C disease: importance of N-glycosylation sites for function and cellular location of the NPC2 protein. Molecular genetics and metabolism. 2004;83:220–230. doi: 10.1016/j.ymgme.2004.06.013. [DOI] [PubMed] [Google Scholar]
- Fujihashi M, Zhang YW, Higuchi Y, Li XY, Koyama T, Miki K. Crystal structure of cis-prenyl chain elongating enzyme, undecaprenyl diphosphate synthase. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:4337–4342. doi: 10.1073/pnas.071514398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grabinska K, Sosinska G, Orlowski J, Swiezewska E, Berges T, Karst F, Palamarczyk G. Functional relationships between the Saccharomyces cerevisiae cis-prenyltransferases required for dolichol biosynthesis. Acta biochimica Polonica. 2005;52:221–232. [PubMed] [Google Scholar]
- Grabinska KA, Cui J, Chatterjee A, Guan Z, Raetz CR, Robbins PW, Samuelson J. Molecular characterization of the cis-prenyltransferase of Giardia lamblia. Glycobiology. 2010;20:824–832. doi: 10.1093/glycob/cwq036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan Z, Eichler J. Liquid chromatography/tandem mass spectrometry of dolichols and polyprenols, lipid sugar carriers across evolution. Biochimica et biophysica acta. 2011;1811:800–806. doi: 10.1016/j.bbalip.2011.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo RT, Ko TP, Chen AP, Kuo CJ, Wang AH, Liang PH. Crystal structures of undecaprenyl pyrophosphate synthase in complex with magnesium, isopentenyl pyrophosphate, and farnesyl thiopyrophosphate: roles of the metal ion and conserved residues in catalysis. The Journal of biological chemistry. 2005;280:20762–20774. doi: 10.1074/jbc.M502121200. [DOI] [PubMed] [Google Scholar]
- Harrison KD, Miao RQ, Fernandez-Hernando C, Suarez Y, Davalos A, Sessa WC. Nogo-B receptor stabilizes Niemann-Pick type C2 protein and regulates intracellular cholesterol trafficking. Cell metabolism. 2009;10:208–218. doi: 10.1016/j.cmet.2009.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison KD, Park EJ, Gao N, Kuo A, Rush JS, Waechter CJ, Lehrman MA, Sessa WC. Nogo-B receptor is necessary for cellular dolichol biosynthesis and protein N-glycosylation. The EMBO journal. 2011;30:2490–2500. doi: 10.1038/emboj.2011.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He P, Ng BG, Losfeld ME, Zhu W, Freeze HH. Identification of intercellular cell adhesion molecule 1 (ICAM-1) as a hypoglycosylation marker in congenital disorders of glycosylation cells. The Journal of biological chemistry. 2012;287:18210–18217. doi: 10.1074/jbc.M112.355677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasapkara CS, Tumer L, Ezgu FS, Hasanoglu A, Race V, Matthijs G, Jaeken J. SRD5A3-CDG: a patient with a novel mutation. European journal of paediatric neurology : EJPN : official journal of the European Paediatric Neurology Society. 2012;16:554–556. doi: 10.1016/j.ejpn.2011.12.011. [DOI] [PubMed] [Google Scholar]
- Miao RQ, Gao Y, Harrison KD, Prendergast J, Acevedo LM, Yu J, Hu F, Strittmatter SM, Sessa WC. Identification of a receptor necessary for Nogo-B stimulated chemotaxis and morphogenesis of endothelial cells. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:10997–11002. doi: 10.1073/pnas.0602427103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naureckiene S, Sleat DE, Lackland H, Fensom A, Vanier MT, Wattiaux R, Jadot M, Lobel P. Identification of HE1 as the second gene of Niemann-Pick C disease. Science. 2000;290:2298–2301. doi: 10.1126/science.290.5500.2298. [DOI] [PubMed] [Google Scholar]
- O'Gorman S, Dagenais NA, Qian M, Marchuk Y. Protamine-Cre recombinase transgenes efficiently recombine target sequences in the male germ line of mice, but not in embryonic stem cells. Proceedings of the National Academy of Sciences of the United States of America. 1997;94:14602–14607. doi: 10.1073/pnas.94.26.14602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pipalia NH, Huang A, Ralph H, Rujoi M, Maxfield FR. Automated microscopy screening for compounds that partially revert cholesterol accumulation in Niemann-Pick C cells. Journal of lipid research. 2006;47:284–301. doi: 10.1194/jlr.M500388-JLR200. [DOI] [PubMed] [Google Scholar]
- Rush JS, Matveev S, Guan Z, Raetz CR, Waechter CJ. Expression of functional bacterial undecaprenyl pyrophosphate synthase in the yeast rer2{Delta} mutant and CHO cells. Glycobiology. 2010;20:1585–1593. doi: 10.1093/glycob/cwq107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato M, Sato K, Nishikawa S, Hirata A, Kato J, Nakano A. The yeast RER2 gene, identified by endoplasmic reticulum protein localization mutations, encodes cis-prenyltransferase, a key enzyme in dolichol synthesis. Molecular and cellular biology. 1999;19:471–483. doi: 10.1128/mcb.19.1.471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schenk B, Rush JS, Waechter CJ, Aebi M. An alternative cis-isoprenyltransferase activity in yeast that produces polyisoprenols with chain lengths similar to mammalian dolichols. Glycobiology. 2001;11:89–98. doi: 10.1093/glycob/11.1.89. [DOI] [PubMed] [Google Scholar]
- Surmacz L, Swiezewska E. Polyisoprenoids - Secondary metabolites or physiologically important superlipids? Biochemical and biophysical research communications. 2011;407:627–632. doi: 10.1016/j.bbrc.2011.03.059. [DOI] [PubMed] [Google Scholar]
- Szkopinska A, Grabinska K, Delourme D, Karst F, Rytka J, Palamarczyk G. Polyprenol formation in the yeast Saccharomyces cerevisiae: effect of farnesyl diphosphate synthase overexpression. Journal of lipid research. 1997;38:962–968. [PubMed] [Google Scholar]
- Wen R, Lam BL, Guan Z. Aberrant dolichol chain lengths as biomarkers for retinitis pigmentosa caused by impaired dolichol biosynthesis. Journal of lipid research. 2013;54:3516–3522. doi: 10.1194/jlr.M043232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang Y, Zhang X, Nix DB, Katoh T, Aoki K, Tiemeyer M, Wang Y. Regulation of protein glycosylation and sorting by the Golgi matrix proteins GRASP55/65. Nature communications. 2013;4:1659. doi: 10.1038/ncomms2669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu L, Pena Castillo L, Mnaimneh S, Hughes TR, Brown GW. A survey of essential gene function in the yeast cell division cycle. Molecular biology of the cell. 2006;17:4736–4747. doi: 10.1091/mbc.E06-04-0368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zelinger L, Banin E, Obolensky A, Mizrahi-Meissonnier L, Beryozkin A, Bandah-Rozenfeld D, Frenkel S, Ben-Yosef T, Merin S, Schwartz SB, et al. A missense mutation in DHDDS, encoding dehydrodolichyl diphosphate synthase, is associated with autosomal-recessive retinitis pigmentosa in Ashkenazi Jews. American journal of human genetics. 2011;88:207–215. doi: 10.1016/j.ajhg.2011.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuchner S, Dallman J, Wen R, Beecham G, Naj A, Farooq A, Kohli MA, Whitehead PL, Hulme W, Konidari I, et al. Whole-exome sequencing links a variant in DHDDS to retinitis pigmentosa. American journal of human genetics. 2011;88:201–206. doi: 10.1016/j.ajhg.2011.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.