

Abstract
Objective:
In India, various groups have studied different regions to find out deletion pattern of dystrophin gene. We have investigated its deletion pattern among Duchenne/Becker muscular dystrophy (D/BMD) patients across Gujarat. Moreover, in this study we also correlate the same with reading frame rule. However, we too consider various clinicopathological features to establish as adjunct indices when deletion detection fails.
Materials and Methods:
In this pilot study, a total of 88 D/BMD patients consulting at our centers in Gujarat, India were included. All patients were reviewed on basis of their clinical characteristics, tested by three primer sets of 10-plex, 9-plex, and 7-plex polymerase chain reaction (PCR) for genetic analysis; whereas, biochemical indices were measured using automated biochemical analyzers.
Results:
The diagnosis of D/BMD was confirmed by multiplex-PCR (M-PCR) in D/BMD patients. A number of 65 (73.86%) out of 88 patients showed deletion in dystrophin gene. The exon 50 (58.46%) was the most frequent deletion found in our study. The mean age of onset of DMD and BMD was 4.09 ± 0.15 and 7.14 ± 0.55 years, respectively. In patients, mean creatine phosphokinase (CPK), lactate dehydrogenase (LDH), and myoglobin levels were elevated significantly (P < 0.05) in comparison to controls. Addition to CPK, LDH and myoglobin are good adjunct when deletion detection failed. These data are further in accordance with world literature when correlated with frame rule.
Conclusion:
The analysis has been carried out for the first time for a total of 88 D/BMD patients particularly from Gujarat, India. More research is essential to elucidate specific mutation pattern in association with management and therapies of proband.
Keywords: Duchenne/Becker muscular dystrophy, dystrophin gene, exon deletion
Introduction
In this genomic epoch, populace has restored their health and avoided unnecessary therapeutics by facilitation of proper diagnosis. Muscular dystrophy is a group of genetically determined muscular disorders, which have been largely classified by the clinical features.[1] Majority of muscular dystrophies are caused by bouleversement of different components of the dystrophin glycoprotein complex (DGC) that is critical for the integrity of skeletal muscle fibers.[2] Dystrophin, 427 kDa protein product of the dystrophin gene, is one of the largest component of the DGC,[3] it is absent or severely truncated in a devastating Duchenne muscular dystrophy (DMD) (OMIM #310200), while decreased levels in milder allelic form of Becker muscular dystrophy (BMD) (OMIM #300376). DMD and the allelic BMD are the most common forms of muscular dystrophy in humans and together termed as dystrophinopathies.[4] DMD alone accounts for approximately 80% of all the myopathies, with an incidence of around 1 in 3,500 live born males.[5] X-linked recessive D/BMD caused by mutation/s of the dystrophin gene (OMIM #300377) is located at locus Xp21.2. The dystrophin gene remains the only known human metagene, spanning 2.4 Mb, transcript into a 14 kb mRNA, and contains 79 exons.[6] In DMD, affected boys are usually wheelchair-bound by the age of 13 years and die early in their 3rd decade of life. On the other hand, BMD is associated with a later age of onset and a slower clinical progression.[7] The incidence of BMD is around 1 in 18,500 live births of males.[8] Since last 2 decades, analyses of both the dystrophin gene and protein have improved the diagnosis of D/BMD.[9,10] Knowing the exact mutation in a proband, today one can determine the possibility of a life span up to their 4th decade.[7] Therefore, it is important to prognose and/or diagnose the disorder as earliest. Large reorganizations in the gene are found in about two-thirds of DMD patients. Deletion accounts for 65%, 5-8% by duplications, and the remaining caused by point mutations and small insertion.[11] Several methods are available for detection of deletion pattern. Here we have used the three multiplex polymerase chain reaction (M-PCR) sets developed by Chamberlain et al.,[12] Beggs et al.,[13,14] and Kunkel et al.,[15] which are the most commonly used for deletion detection. These methods can deal out 90-98% deletions detection in male patients.
Reading frame rule explains the phenotypic differences between DMD and BMD in 90% of cases.[16] In DMD, translation termination (out-frame) results in a loss or truncation of dystrophin, while BMD patients carry mainly frame maintaining (in-frame) mutation, which leads to partial function of protein and hence results in a milder phenotype.[17] The understanding of this mechanism has a potential role for development of specific management or treatment of proband.
Historically, determination of the activity of some muscle elements has been drawn to be potent evidence for neuromuscular diseases and in monitoring their progression.[18] However, literatures that describe and compare the muscle components in this disorder are few. In D/BMD, absence or truncation of dystrophin component leads to disruption of myocyte organization caused by genetic defects in the machinery of it, which in turn leads to progressive fiber damage and degeneration of muscle fibers;[19] it is thought to be a key molecular event in the pathology. Along with, myocyte activity would cause physical damage to the sarcolemma, which could result in abnormal egress of various muscle components as well.[20]
The aim of the study is to examine the burden of D/BMD in Gujarat, based on the application of M-PCR as a potential diagnostic tool. We have also attempted to investigate patients with help of various clinicopathological characteristics, including creatine phosphokinase (CPK), lactate dehydrogenase (LDH), myoglobin, Gowers’ maneuver, calf hypertrophy, scoliosis, and state of ambulation indices; first time in Gujarat population.
Materials and Methods
Sample collection
The study was approved by the institutional ethics board. All boys with D/BMD were reviewed who had consulted at our centers in Gujarat, India between the periods of 2011 to 2013 for their clinicopathological condition. BMD patients were classified on the basis of their muscle weakness and ability to walk after the age of 12 years. Patients found to have any abnormal condition like recent injury, infection, and trauma were not considered in this study. Information regarding use of any medication and therapies taken at the time of blood collection were also considered. Fifty age-matched controls were included. Informed consent was given in writing before sample collection by the subject or the parents of individuals <18 years.
Biochemical indices
Serum CPK, LDH, and myoglobin levels were measured by using the fully automated biochemical analyzers on Cobas Integra 400 (Roche) and Architect i2000 System (Abbott). The reference range of CPK, LDH, and myoglobin levels were settled at 38.00-174.00 U/l, 135.00-225.00 U/l, and 0.0-110.0 ng/ml, respectively.
Genetic analysis
Genomic DNA was extracted from peripheral blood lymphocytes by phenol-chloroform extraction method.[21] M-PCR for deletion detection were performed by three separate PCR reactions sets, the Chamberlain et al.,[12] modified by Beggs et al.,[13] (exon: 45, 48, 19, 17, 51, 8, 12, 44, 4, and 46) allowed amplification of exon 46 plus the original set; Beggs et al.,[14] (exon: Dp427m, 3, 43, 50, 13, 6, 47, 60, and 52) and Kunkel et al.,[15] (exon: 49, Dp427c, 16, 41, 32, 42, and 34) allowed screening for deletions of 26 exons, using the condition recommended at the Leiden muscular dystrophy pages. D/BMD exonic deletions were checked by reading-frame checker 1.9.[22]
Statistical methods
Data are presented as mean ± standard error (SE). Data were analyzed using GraphPad Prism 5.0 statistical software. Significance was assumed at a probability value of P < 0.05.
Results
Clinical finding
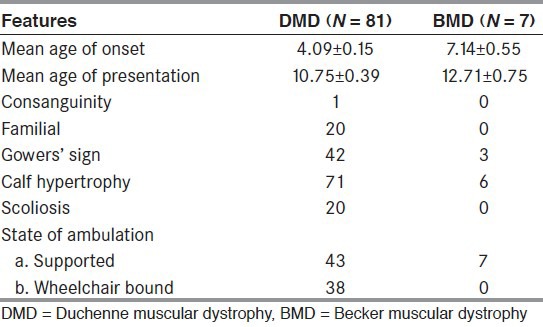
Of 88 patients (81 cases of DMD and seven with BMD phenotype), 16 cases had a positive family history with 20 (22.73%) of innate cases. The overviews of clinical symptoms are described in Table 1. All boys presented with complain of repeated fall and particularly with lower limb muscle weakness and calf hypertrophy. Majority patients had also confirmed positive Gower's sign.
Table 1.
Clinical indices of DMD and BMD patients
Investigations
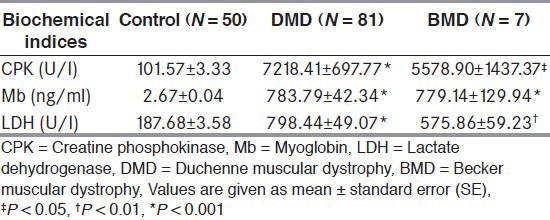
CPK, myoglobin, and LDH levels were significantly (P < 0.05) elevated in all patients compared to controls. The biochemical indices are portrayed in Table 2.
Table 2.
Biochemical indices
Genetic findings
Of 88 cases clinically suspected D/BMD boys, the diagnoses of D/BMD were confirmed by M-PCR in 65 (73.86%) patients. Out of 65 probands, 62 (95.38%) DMD and three (4.62%) BMD were identified. Majority of patients (58, 89.23%) showed deletions in the downstream region of the gene. The frequency of individual exon deletions is presented in [Figure 1]. Exon 50 (58.46%) was one of the most commonly observed deletion. Deletions started with exon 45 (24.62%), which was frequent as seen in our study. The highest deletion rate was the del 45-52 (10.77%) in seven DMD probands. The pattern of mutations found in the Gujarat population is illustrated in [Figure 2]. Every DMD patient showed out-frame deletion, while one patient with exceptional del 41-43 with out-frame deletion showed BMD phenotype and 3.08% (one DMD and one with BMD phenotype) could not be classified as in/out-frame [Figure 3]. One of the longest deletion include exon 3-44 was found in BMD proband. In our study cohort, out of 20 familial probands, two sibs and one proband of nonconsanguineous marriages were not confirmed any deletion pattern with M-PCR assay.
Figure 1.
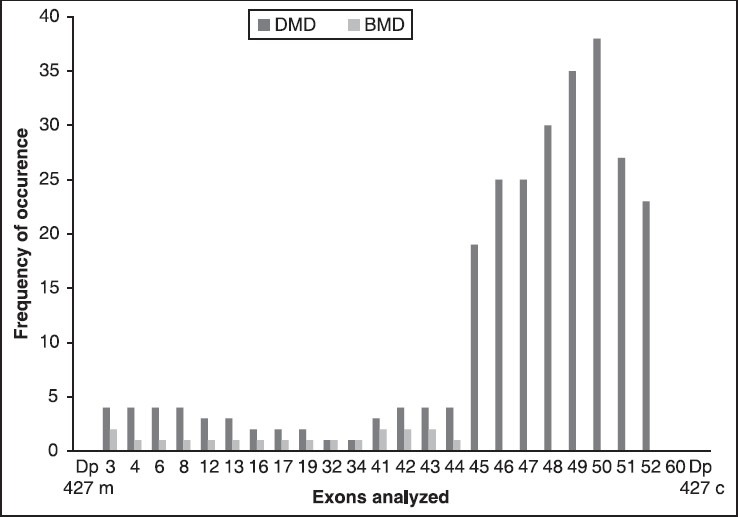
Graph representing frequency of individual exon deletions
Figure 2.
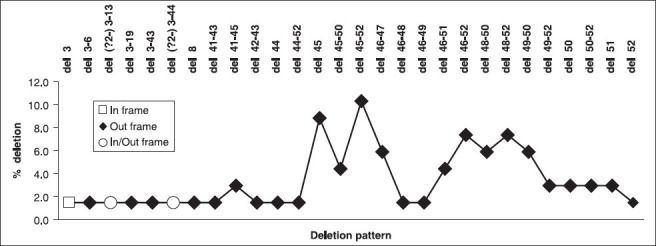
Deletion pattern in the dystrophin gene in Duchenne/Becker muscular dystrophy (D/BMD) probands
Figure 3.
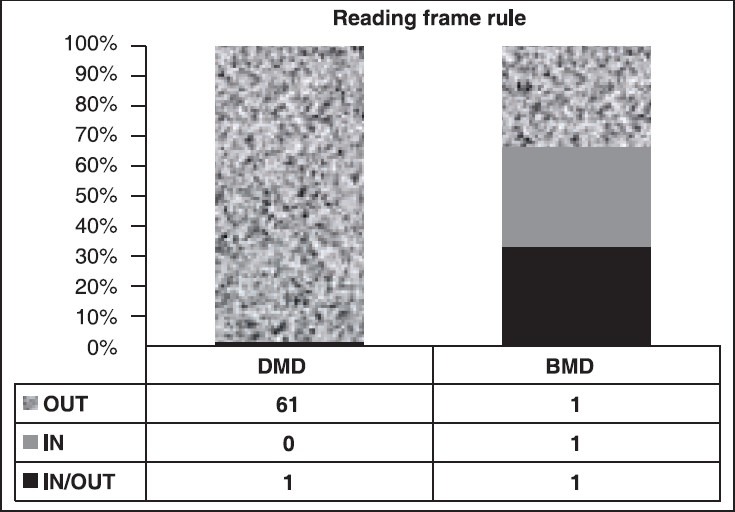
Graph analyzing the frame rule
Discussion
The importance of deletion mutations in the dystrophin gene causing D/BMD phenotype has been reported worldwide. This study specifically looks at the distribution spectrum of deletion in the proximal and distal ‘hot spot’ regions of the dystrophin gene in view of clinicopathological condition among Gujarati patients by using M-PCR in this study.
In our study, we have included 88 Gujarati patients from Gujarat, India [Figure 4]. The deletion rate was 73.86% (65/88), in agreement to the frequency reported earlier among western Indian studies.[23,24] The deletion rates in the distal and proximal hotspot regions were 89.23 and 10.77%, respectively. The majority of probands (80.00%, 52/65) had deletions in the central rod domain between exon 45-52 and out of these 24.62% (16/65) deletions started with exon 45. We also found to be the most frequent deletion of exon 50 (58.46%) at the central deletion hot spot, which also coincided with previous western Indian studies.[23] Our observation concluded that this part of the gene is more deletion prone in Gujarat population. However, M-PCR failed to diagnose duplication as well as deletion outside the hotspot region. This included 26.14% (23/88) patients who were clinically confirmed as D/BMD.
Figure 4.
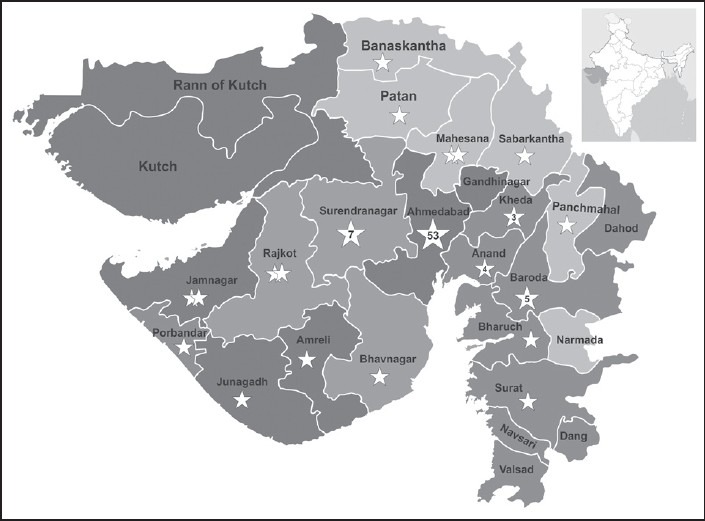
Demographic distribution of studied Duchenne/Becker muscular dystrophy (D/BMD) patients across Gujarat, India
The reading frame rule is initially postulated by Monaco et al., 1988.[16] Mutations that disturb the frame leads to DMD, while maintaining frame would lead to milder BMD phenotype.[25] In our study, applying the reading frame rule for prediction of deletion mutation characteristics, we found 1.54% (1/65) exceptions, as an out-frame deletion in one of the BMD patient that is del 41-43 Figure 3. One DMD and one with BMD phenotype showed exon deletions between 3-13 and 3-44, respectively could not be confirmed in-frame mutations due to exon 2 (not included in the study); which gives alternate out-frame deletion patterns. However, our results are in agreement with world literatures[25,26] except for a north Indian study.[27]
In BMD, various studies from India and abroad have documented deletions in the major “hot spot” region between exons 45-53.[14,17,25,26,27] However in our data, we found seven clinically suspected BMD patients, out of which 42.86% (3/7) were identified by M-PCR. Out of which 66.67% (2/3) deletions started from 5’ region of the gene. Moreover, a largest deletion (del 3-44) was found in one of the BMD proband. We have also noticed phenotypic variability in our patients who have shared even identical deletion pattern. Hence, there is no apparent correlation between the size and/or location of the deletion and the severity or succession of the condition.[26,27]
Further we have analyzed some muscle components profile in clinically suspected D/BMD patients. Various muscle components may have some implications[18,28] for making decision when the deletion is not found in patients. Results for various biochemical indices for D/BMD are presented in Table 2. On basis of these data, the following observation can be made:
In our study highest level of CPK was 34676.07 U/L (≈200X) in one of the DMD proband.
It was also suggested that along with CPK,[29] studied indices like myoglobin and LDH are an essential adjunct in the prognosis and management of D/BMD patients.
Comparing the age with these indices, we confirmed that there is a gradual decrease in these parameters with advancing age. It might be as a result of the progressive elimination of dystrophic muscle fibers.
We further observed that there was no significant correlation of these elevated indices with the gene deletion pattern [Figure 5], severity as well as disease progression.
Figure 5.
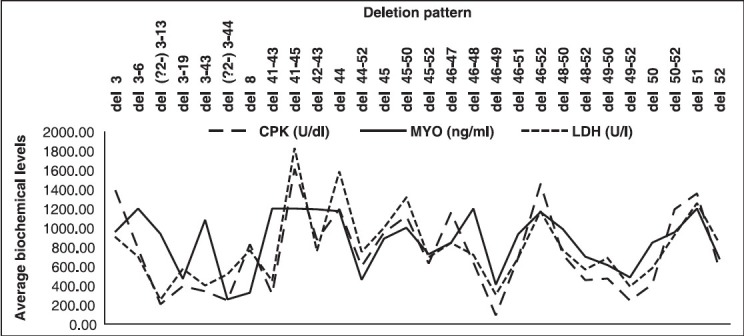
Graph representing deletion versus average biochemical levels in D/BMD probands
In sum, the study was carried out in a limited number of Gujarati D/BMD patients. The method of M-PCR is doubtless tool for diagnoses; even though not rule out every mutation in patients. Thus, we ought to use alternative method like multiplex ligation-dependent probe amplification (MLPA) or direct sequencing for diagnosis as well as for carrier recognition. Location and size of deletion mutations found in the dystrophin gene does not clearly indicate any correlation with severity of D/BMD. Biochemical indices probably do not have potential role in diagnosis even though it can be capable enough for screening and management of patients. This study strongly emphasizes a need for further investigation into the genotype/phenotype aspects of the Gujarati D/BMD population in order to provide better diagnostic, prognostic, prenatal services, and counseling to the suffering families.
Acknowledgment
Financial assistance from Gujarat State Biotechnology Mission (GSBTM), Gandhinagar is gratefully acknowledged. We would also like to thank Indian Muscular Dystrophy Society (IMDS), Ahmedabad and patients for their assistance and cooperation, respectively.
Footnotes
Source of Support: Financial assistance from Gujarat State Biotechnology Mission (GSBTM), Gandhinagar
Conflict of Interest: None declared
References
- 1.Mercuri E, Muntoni F. Muscular dystrophies. Lancet. 2013;381:845–60. doi: 10.1016/S0140-6736(12)61897-2. [DOI] [PubMed] [Google Scholar]
- 2.Cohn RD, Campbell KP. Molecular basis of muscular dystrophies. Muscle Nerve. 2000;23:1456–71. doi: 10.1002/1097-4598(200010)23:10<1456::aid-mus2>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- 3.Ervasti JM. Dystrophin, its interactions with other proteins, and implications for muscular dystrophy. Biochim Biophys Acta. 2007;1772:108–17. doi: 10.1016/j.bbadis.2006.05.010. [DOI] [PubMed] [Google Scholar]
- 4.Flanigan KM, Dunn DM, von Niederhausern A, Soltanzadeh P, Gappmaier E, Howard MT, et al. United Dystrophinopathy Project Consortium, Weiss RB. Mutational spectrum of DMD mutations in dystrophinopathy patients: Application of modern diagnostic techniques to a large cohort. Hum Mutat. 2009;30:1657–66. doi: 10.1002/humu.21114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Emery AE. Population frequencies of inherited neuromuscular diseases-a world survey. Neuromuscul Disord. 1991;1:19–29. doi: 10.1016/0960-8966(91)90039-u. [DOI] [PubMed] [Google Scholar]
- 6.Den Dunnen JT, Grootscholten PM, Dauwerse JG, Walker AP, Monaco AP, Butler R, et al. Reconstruction of the 2.4 Mb human DMD gene by homologous YAC recombination. Hum Mol Genet. 1992;1:19–28. doi: 10.1093/hmg/1.1.19. [DOI] [PubMed] [Google Scholar]
- 7.Bushby K, Finkel R, Birnkrant DJ, Case LE, Clemens PR, Cripe L, et al. DMD Care Considerations Working Group. Diagnosis and management of Duchenne muscular dystrophy, part 1: Diagnosis, and pharmacological and psychosocial management. Lancet Neurol. 2010;9:77–93. doi: 10.1016/S1474-4422(09)70271-6. [DOI] [PubMed] [Google Scholar]
- 8.Bushby KM, Thambyayah M, Gardner-Medwin D. Prevalence and incidence of Becker muscular dystrophy. Lancet. 1991;337:1022–4. doi: 10.1016/0140-6736(91)92671-n. [DOI] [PubMed] [Google Scholar]
- 9.Koenig M, Hoffman EP, Bertelson CJ, Monaco AP, Feener C, Kunkel LM. Complete cloning of the Duchenne muscular dystrophy (DMD) cDNA and preliminary genomic organization of the DMD gene in normal and affected individuals. Cell. 1987;50:509–17. doi: 10.1016/0092-8674(87)90504-6. [DOI] [PubMed] [Google Scholar]
- 10.Kunkel LM, Monaco AP, Hoffman E, Koenig M, Feener C, Bertelson C. Molecular studies of progressive muscular dystrophy (Duchenne) Enzyme. 1987;38:72–5. doi: 10.1159/000469192. [DOI] [PubMed] [Google Scholar]
- 11.Takeshima Y, Yagi M, Okizuka Y, Awano H, Zhang Z, Yamauchi Y, et al. Mutation spectrum of the dystrophin gene in 442 Duchenne/Becker muscular dystrophy cases from one Japanese referral center. J Hum Genet. 2010;55:379–88. doi: 10.1038/jhg.2010.49. [DOI] [PubMed] [Google Scholar]
- 12.Chamberlain JS, Gibbs RA, Ranier JE, Nguyen PN, Caskey CT. Deletion screening of the Duchenne muscular dystrophy locus via multiplex DNA amplification. Nucleic Acids Res. 1988;16:11141–56. doi: 10.1093/nar/16.23.11141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Beggs AH, Hoffman EP, Snyder JR, Arahata K, Specht L, Shapiro F, et al. Exploring the molecular basis for variability among patients with Becker muscular dystrophy: Dystrophin gene and protein studies. Am J Hum Genet. 1991;49:54–67. [PMC free article] [PubMed] [Google Scholar]
- 14.Beggs AH, Koenig M, Boyce FM, Kunkel LM. Detection of 98% of DMD/BMD gene deletions by polymerase chain reaction. Hum Genet. 1990;86:45–8. doi: 10.1007/BF00205170. [DOI] [PubMed] [Google Scholar]
- 15.Kunkel LM, Snyder JR, Beggs AH, Boyce FM, Feener CA. Searching for dystrophin gene deletions in patients with atypical presentations. In: Lindsten J, Petterson U, editors. Etiology of Human Diseases at the DNA Level. New York: Raven Press; 1991. pp. 51–60. [Google Scholar]
- 16.Monaco AP, Bertelson CJ, Liechti-Gallati S, Moser H, Kunkel LM. An explanation for the phenotypic differences between patients bearing partial deletions of the DMD locus. Genomics. 1988;2:90–5. doi: 10.1016/0888-7543(88)90113-9. [DOI] [PubMed] [Google Scholar]
- 17.Dastur RS, Gaitonde PS, Khadilkar SV, Nadkarni JJ. Becker muscular dystrophy in Indian patients: Analysis of dystrophin gene deletion patterns. Neurol India. 2008;56:374–8. doi: 10.4103/0028-3886.40961. [DOI] [PubMed] [Google Scholar]
- 18.Munsat TL, Baloh R, Pearson CM, Fowler W., Jr Serum enzyme alterations in neuromuscular disorders. JAMA. 1973;226:1536–43. [PubMed] [Google Scholar]
- 19.Blake DJ, Weir A, Newey SE, Davies KE. Function and genetics of dystrophin and dystrophin-related proteins in muscle. Physiol Rev. 2002;82:291–329. doi: 10.1152/physrev.00028.2001. [DOI] [PubMed] [Google Scholar]
- 20.Rowland LP. Biochemistry of muscle membranes in Duchenne muscular dystrophy. Muscle Nerve. 1980;3:3–20. doi: 10.1002/mus.880030103. [DOI] [PubMed] [Google Scholar]
- 21.Sambrook J, Russel DW. 3rd ed. New York: Cold Spring Harbor Laboratory Press; 2001. Molecular cloning: A laboratory manual. [Google Scholar]
- 22.Den Dunnen JT. Leiden muscular dystrophy pages. Center for human and clinical genetics, Leiden university medical center DMD exonic deletions/duplications reading frame checker 1.9. [Last updated on 2009 Mar 03]. Available from: http://www.dmd.nl/
- 23.Nadkarni JJ, Dastur RS, Viswanathan V, Gaitonde PS, Khadilkar SV. Duchenne and Becker muscular dystrophies: An Indian update on genetics and rehabilitation. Neurol India. 2008;56:248–53. doi: 10.4103/0028-3886.43442. [DOI] [PubMed] [Google Scholar]
- 24.Khalap NV, Joshi VP, Ladiwalla U, Khadilkar SV, Mahajan SK. A report on higher frequency of DMD gene deletion in the Indian subcontinent. Indian J Hum Genet. 1997;3:117–20. [Google Scholar]
- 25.Murugan S, Chandramohan A, Lakshmi BR. Use of multiplex ligation-dependent probe amplification (MLPA) for Duchenne muscular dystrophy (DMD) gene mutation analysis. Indian J Med Res. 2010;132:303–11. [PubMed] [Google Scholar]
- 26.Aartsma-Rus A, Van Deutekom JC, Fokkema IF, Van Ommen GJ, Den Dunnen JT. Entries in the Leiden Duchenne muscular dystrophy mutation database: An overview of mutation types and paradoxical cases that confirm the reading-frame rule. Muscle Nerve. 2006;34:135–44. doi: 10.1002/mus.20586. [DOI] [PubMed] [Google Scholar]
- 27.Pandey GS, Kesari A, Mukherjee M, Mittal RD, Mittal B. Re-evaluation of reading frame-shift hypothesis in Duchenne and Becker muscular dystrophy. Neurol India. 2003;51:367–9. [PubMed] [Google Scholar]
- 28.Rosalki SB. Serum enzymes in disease of skeletal muscle. Clin Lab Med. 1989;9:767–81. [PubMed] [Google Scholar]
- 29.Swaminathan B, Shubha GN, Shubha D, Murthy AR, Kiran Kumar HB, Shylashree S, et al. Duchenne muscular dystrophy: A clinical, histopathological and genetic study at a neurology tertiary care center in Southern India. Neurol India. 2009;57:734–8. doi: 10.4103/0028-3886.59468. [DOI] [PubMed] [Google Scholar]