

Key Points
Prothrombinase bound in the vicinity of vascular damage is distributed away from platelets and is largely found on activated endothelium.
The activated endothelium plays an unexpectedly important role in supporting prothrombinase assembly and function at the site of damage.
Abstract
The membrane-dependent interaction of factor Xa (FXa) with factor Va (FVa) forms prothrombinase and drives thrombin formation essential for hemostasis. Activated platelets are considered to provide the primary biological surface to support prothrombinase function. However, the question of how other cell types may cooperate within the biological milieu to affect hemostatic plug formation remains unaddressed. We used confocal fluorescence microscopy to image the distribution of site-specific fluorescent derivatives of FVa and FXa after laser injury in the mouse cremaster arteriole. These proteins bound to the injury site extend beyond the platelet mass to the surrounding endothelium. Although bound FVa and FXa may have been present on the platelet core at the nidus of the injury, bound proteins were not evident on platelets adherent even a small distance from the injury site. Manipulations to drastically reduce adherent platelets yielded a surprisingly modest decrease in bound FXa and FVa with little impact on fibrin formation. Thus, platelets adherent to the site of vascular injury do not play the presumed preeminent role in supporting prothrombinase assembly and thrombin formation. Rather, the damaged/activated endothelium and possibly other blood cells play an unexpectedly important role in providing a procoagulant membrane surface in vivo.
Introduction
Vascular damage and inflammatory processes allow for platelet adhesion and the exposure of tissue factor to blood triggering the blood coagulation response. The resulting series of proteolytic activation steps on the surface of activated cells culminates in thrombin formation and the conversion of soluble fibrinogen to insoluble fibrin.1 Binding of clotting factors to membranes facilitates assembly of the intrinsic Xase and prothrombinase enzyme complexes and profoundly increases the rate of cleavage of their cognate biological substrates, thereby localizing enhanced product formation to the site of vascular damage.2 To mitigate unwanted clot formation, assembly of the intrinsic Xase and prothrombinase complexes requires cell activation and exposure of phosphatidylserine on the outer leaflet of the cell membrane.3-5 Although activated platelets, microparticles, leukocytes, endothelial, and even red cells support their assembly and function to varying degrees, it is the activated platelet surface that is, by far, considered the preeminent site for the function of these enzyme complexes.4-10 This paradigm is largely based on extrapolations from in vitro studies with isolated platelets in the absence of key participants that could further contribute to clot formation within the vasculature. A weakness in this extrapolation lies in predicting the contribution of different cells to function on the basis of measurements performed using “optimal” agonist concentrations to stimulate various cell types with little knowledge of the agonist concentrations encountered in flowing blood after vascular damage. Consequently, it remains uncertain how other activated cells, in addition to platelets, contribute to thrombin generation in vivo after different types of vascular injury.
Mouse models and advances in intravital video microscopy to visualize the dynamics of thrombus formation in vivo provide new opportunities to test hypotheses within the context of the authentic injured vasculature.11-13 Work using laser-induced injury to the mouse cremasteric arteriole has revealed surprising findings that challenge otherwise accepted ideas about thrombus formation. The findings point to a significant role for other activated cells, in addition to platelets, in supporting fibrin formation. This is revealed by significant amounts of fibrin deposition after laser injury, despite large reductions in adherent platelets using a pharmacologic approach or mice that are deficient in protease-activated receptor (PAR)4.14-16 Although this could be related to the type of injury or other factors, these findings warrant a more detailed evaluation of the cellular site(s) of coagulation enzyme complex formation in vivo.
Here, we generated site-specific labeled fluorescent species of coagulation factor Va (FVa) and factor Xa (FXa) with well-established properties to advance our understanding of coagulation enzyme function and the relative contribution of blood cells, and the vasculature to the regulation of clot formation in vivo. Surprisingly, the data show that the distribution of FVa and FXa at the site of vascular injury, to a large extent, is on the damaged endothelium with minor contributions from platelets. Furthermore, a significant reduction of adherent platelets at the core injury site had little impact on the total amount of prothrombinase or fibrin detected. These results challenge long-standing models of blood coagulation in which activated platelets are the primary site for prothrombinase assembly. Thus, although activated platelets can support robust thrombin generation in vitro, this role in vivo may be much more limited than previously appreciated.
Methods
Reagents
Benzamidine, 4-amidinophenylmethanesulfonyl fluoride hydrochloride, and bovine serum albumin were purchased from Sigma. N-ethylmaleimide, maleimide derivatives of Alexa and N-hydroxysuccinimidyl derivatives of Alexa probes were purchased from Life Technologies. All tissue culture reagents were purchased from Life Technologies, except insulin-transferrin-sodium selenite (Roche Applied Science). Integrilin (eptifibatide) was obtained from Merck.
Proteins
Recombinant FX (rFX), activated rFX (rFXa), and a partial B-domain-less form of human FV (FV-810) were prepared, purified, and characterized as previously described.17,18 Molecular weights and extinction coefficients (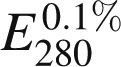 ) of the various proteins have been previously reported.19 A FV-810 variant with Cys585, Cys1960, and Cys2113 replaced by Ser, Tyr, and Ala, respectively (FV-810SYA) was generated with the QuikChange site-directed mutagenesis kit (Agilent) using appropriate complementary oligonucleotides containing the desired mutations. FV-810SYA reacted with Alexa488-malemide for 1 hour at pH 7.0 to yield Alexa488- FV-810SYA. Free dye was removed by gel filtration as previously described.20 FXa was inactivated with acetothioacetyl-EGR-CH2-Cl and reacted with an Alexa-maleimide probe (Alexa-488 or Alexa-647) after thioester hydrolysis and purified to yield Alexa-inactivated FXa (FXai) as previously described.20 Rat anti-mouse CD41 antibody (clone MWReg 30) prepared as a F(ab)2 fragment was obtained from BD Bioscience (San Jose, CA). Mouse anti-human fibrin monoclonal antibody (clone 59D8), which cross reacts with mouse fibrin has been previously described.21,22 These antibodies were conjugated with Alexa555 or Alexa488 using the Alexa Fluor Protein Labeling Kit, according to the manufacturer’s instructions (Invitrogen, Carlsbad, CA). Annexin V was labeled with amine reactive Alexa488 and repurified as described.23
) of the various proteins have been previously reported.19 A FV-810 variant with Cys585, Cys1960, and Cys2113 replaced by Ser, Tyr, and Ala, respectively (FV-810SYA) was generated with the QuikChange site-directed mutagenesis kit (Agilent) using appropriate complementary oligonucleotides containing the desired mutations. FV-810SYA reacted with Alexa488-malemide for 1 hour at pH 7.0 to yield Alexa488- FV-810SYA. Free dye was removed by gel filtration as previously described.20 FXa was inactivated with acetothioacetyl-EGR-CH2-Cl and reacted with an Alexa-maleimide probe (Alexa-488 or Alexa-647) after thioester hydrolysis and purified to yield Alexa-inactivated FXa (FXai) as previously described.20 Rat anti-mouse CD41 antibody (clone MWReg 30) prepared as a F(ab)2 fragment was obtained from BD Bioscience (San Jose, CA). Mouse anti-human fibrin monoclonal antibody (clone 59D8), which cross reacts with mouse fibrin has been previously described.21,22 These antibodies were conjugated with Alexa555 or Alexa488 using the Alexa Fluor Protein Labeling Kit, according to the manufacturer’s instructions (Invitrogen, Carlsbad, CA). Annexin V was labeled with amine reactive Alexa488 and repurified as described.23
Animals
Hemophilia B (HB) mice on the Balb/c strain have been previously described and backcrossed for more than 5 generations.24,25 Wild-type (wt)–Balb/c mice were purchased from Jackson Laboratory (Bar Harbor, ME). The endothelial-specific receptor tyrosine kinase (Tie2)-green fluorescent protein (GFP) mice on the FVB/N background were obtained from Jackson Laboratory (Bar Harbor, ME). These mice express GFP under the control of the Tie2 promoter.26 To obtain HB-Tie2-GFP mice, Tie2-GFP mice were crossed with HB mice (Balb/c) and bred for more than 5 generations. For all experiments, mice were between 8 to12 weeks of age and weighed 25 to 30 g. Experimental approval was obtained from the Children’s Hospital of Philadelphia Institutional Animal Care and Use Committee.
Real-time in vivo imaging of thrombus formation
Evaluation of hemostasis after laser injury to the cremaster muscle has been previously described and our specific experimental system has been detailed.12,22,27 Different combinations of Alexa488-FV-810SYA (600 µg/kg), FXai (80 µg/kg) tethered to either Alexa488 or Alexa647, and Alexa555-labeled rat anti-CD41 F(ab)2 or Alexa647-labeled anti-fibrin antibody21 were administered via a jugular vein cannula. Based on empirical studies, antibodies were infused at 0.2 mg/kg for the anti-CD41 Fab fragment and 0.1 mg/kg for the anti-fibrin antibody. All proteins were infused in 20 mM N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid, 0.15 M NaCl, 2 mM CaCl2, 0.1% polyethylene glycol 8000, and pH 7.4 (assay buffer). Due to space limitations, we restricted the presentation of findings to a limited combination of probes. However, to rule out potential overlap between emission spectra of different fluorescent probes, fluorescent derivatives of FVa, FXai, anti-CD41, and anti-fibrin were used singly, as well as in multiple combinations to ensure the final results were equivalent. We also used combinations of proteins individually labeled with different Alexa probes to ensure that the conclusions were independent of the combination of probes used to image the different components present with different abundance.
Cremasteric arterioles of 30 to 50 µm were selected for study and visualized using an Olympus BX61WI upright fluorescence microscope with ×60 water immersion lenses, equipped with Yokogawa CSU-X1 spinning disk confocal scanner. Vascular injury was induced using a pulse-nitrogen dye laser applied through the microscope objective. Brightfield and fluorescence images were collected over the course of 3 to 4 minutes at 4 Hz, and the data were analyzed using Slidebook version 5 software (Intelligent Imaging Innovations, Denver, CO). Three-dimensional (3D) optical images were captured with a confocal microscope equipped with a CoolSnap HQ CCD digital camera (Photometrics, Tucson, AZ) and 3D reconstruction was performed using Volocity Software (PerkinElmer, Waltham, MA).
Statistics
Data were expressed as median ± standard error of the mean and were compared by nonparametric, two-tailed P value Wilcoxon matched-pairs test.
Results
Site-specific labeling of coagulation factors
Human FVa has 4 free Cys residues, 2 in the heavy chain (539 and 585) and 2 in the light chain (1960 and 2113)28 with Cys539 most amenable to modification.29-31 We prepared a B-domainless FVa variant (FV-810SYA) that is constitutively active and has a single, free cysteine at 539 (supplemental Figure 1A, available on the Blood Web site). Mutagenesis of the other Cys residues to Ser, Tyr, Ala was based on sequence alignment analysis, which revealed that these amino acids are found at positions 585, 1960, and 2113 in other species. As with FV-810, the variant was processed by thrombin to FVa (supplemental Figure 1B). As expected, FV-810SYA was labeled with Alexa488-maleimide within the heavy chain (supplemental Figure 1B), whereas FV-810 was labeled on both chains (data not shown). Pre-treatment of FV-810 (data not shown) or FV-810SYA with N-ethylmaleimide eliminated further labeling consistent with Cys-specific modification at residue 539 (supplemental Figure 1A). Preparative labeling of FV-810SYA with Alexa488-maleimide, and repurification and analysis by absorbance measurements revealed a stoichiometry of approximately 1:1. Procoagulant specific activity of the labeled material was comparable to unlabeled FV-810SYA, FV-810, and rFVa (data not shown). Based on this functional equivalence, the unlabeled and labeled forms of FV-810SYA are interchangeably referred to as FVa in this manuscript. We also used rFXa bearing Alexa488 or Alexa647 tethered to the active site with a peptidyl chloromethyl ketone (EGR-cmk). The resulting Alexa488-FXai or Alexa647-FXai derivatives are inactive, but they retain the ability to assemble into prothrombinase with a similar affinity to active FXa.32
Hemostatic plug formation in wt and FV-810SYA-treated HB mice
We have previously reported clot formation after laser injury in the cremasteric arterioles of hemophilic mice that could be corrected by preinfusion of human FVa to levels comparable to those seen after reconstitution with either FVIII or FIX.27 Therefore, it follows that some or all of the infused FVa localized at the site of the growing hemostatic plug must be functional and complexed with endogenous FXa to generate thrombin. These observations form the basis for our experimental design to use HB mice as a platform to image the constituents of prothrombinase in the developing clot. This approach was chosen to minimize the confounding possibility that a significant fraction of FVa or FXa localized at the site of injury may be nonproductively or adventitiously bound in a way that is irrelevant to thrombin formation.
To examine whether this model resembles hemostatic plug formation in wt animals, we compared the kinetics of platelet and fibrin accumulation after laser injury in wt mice with HB mice preinfused with unlabeled FV-810SYA. Platelet and fibrin accumulation after laser injury were comparable in both sets of animals. Platelet accumulation was initially more robust in HB mice preinfused with FV-810SYA, but differences were not apparent after approximately 1.5 minutes (Figure 1A). The kinetics of fibrin deposition was equivalent in the 2 models (Figure 1B). No obvious differences were evident in platelet or fibrin accumulation between the 2 groups of animals when evaluated at 30 second intervals (P > .1). Consistent with prior findings,12,33 both models showed fibrin deposition adjacent to and even extending into the vessel wall (supplemental Video 1A-B). Spatially, fibrin colocalized with only a portion of the deposited platelets within a core region at the site of microscopic laser injury. The remaining platelets were more extensively distributed around this core with no accumulating fibrin. Comparable kinetics of clot formation and its spatial organization in wt animals and in HB mice infused with unlabeled FV-810SYA further validates the use of the latter approach to assess the spatiotemporal distribution of prothrombinase constituents.
Figure 1.

Hemostatic plug formation in wt and HB mice with FV-810SYA. Quantitative analysis of platelet (A) and fibrin accumulation (B) over time after laser injury to cremasteric arterioles in wt mice (n = 5; 23 thrombi), and HB mice treated with FV-810SYA (n = 5; 22 thrombi; 600 µg/kg). The kinetics of platelet and fibrin accumulation were examined by monitoring the fluorescent intensity at the site of the injury using an Alexa555-labeled rat anti- CD41 F(ab)2 and Alexa488-labeled anti-fibrin antibodies. Median fluorescence intensity (MFI) for platelets (A) and fibrin (B) fluorescence are plotted vs time. For both platelets and fibrin accumulation, there is no statistically significant difference (P > .1) in MFI (evaluated at 30-second intervals) between wt mice and HB + FV-810SYA by the Wilcoxon rank-sum test.
FVa and FXa imaged individually after laser-induced injury
The distribution of bound FVa relative to platelets after laser injury in HB mice was visualized after infusion of Alexa488-FV-810SYA and Alexa555-labeled rat anti-CD41 F(ab)2 directed against platelets. A rapid and robust fluorescence signal was detected denoting accumulation of Alexa488-FV-810SYA (green) and platelets (red) at the site of vascular damage (see Figure 2 for graphical image). Representative digital composite fluorescence and brightfield images at various time points after injury illustrate that bound Alexa488-FV-810SYA at the core injury site colocalized with adherent platelets, as well as being seemingly bound to the vessel wall both upstream and downstream from the injury site where the platelets were not observed (Figure 2A and supplemental Video 2A). The distribution of bound FXa was assessed in separate experiments after coinfusion of unlabeled FV-810SYA, Alexa647-FXai, and the Alexa555-labeled platelet antibody. Alexa647-FXai also produced a rapid and robust fluorescence signal revealing its extended distribution beyond the core injury site, which was seemingly bound to the vessel wall at regions where the platelets were not detected (Figure 3A and supplemental Video 3A). The kinetics of Alexa488-FV-810SYA (Figure 2B) and Alexa647-FXai (Figure 3B) accumulation at the site of injury were similar and in-line with the expectation that these proteins are complexed with each other. The kinetics of platelet accumulation in both experiments was similar indicating that the procoagulant stimulus and overall injuries were consistent (Figures 2C and 3C).
Figure 2.

Direct imaging of FVa and platelets. (A) Digital composite fluorescence and brightfield images of representative thrombi in HB mice infused with Alexa488-FV-810SYA (600 µg/kg) at 10, 40, 70, 100, 130, and 190 seconds after injury; Alexa488-FV-810SYA (green), platelets (red), and overlap between FV-810SYA and platelets (yellow). Bar represents 10 µm. (B-C) Kinetics of Alexa488-FV-810SYA (n = 4 mice; 20 thrombi) and platelets (n = 4 mice; 20 thrombi) fluorescence accumulation over time. (D) Confocal imaging of Alexa488-FV-810SYA and platelets 4 minutes after laser injury, merged or presented individually; step size = 1 µm in z-plane; total section 30 µm. Images depict different orientations of the same thrombus. (D) Upper panel: horizontal orientation; lower panel: 45 degrees rotated. Bar represents 30 µm. Platelet accumulation was monitored using Alexa555-labeled rat anti- CD41 F(ab)2 antibody.
Figure 3.

Direct imaging of FXa and platelets. (A) Digital composite fluorescence and brightfield images of representative thrombi in HB mice infused with unlabeled FV-810SYA (600 µg/kg) and labeled Alexa647-FXai (80 µg/kg) at 10, 40, 70, 100, 130, and 190 seconds after injury; FXai (cyan), platelets (red), and overlap between FXai and platelets (white). (B-C) Kinetics of Alexa647-FXai (n = 5 mice; 22 thrombi) and platelets (n = 5 mice; 22 thrombi) fluorescence accumulation over time. (D) Confocal imaging of Alexa647-FXai and platelets 4 minutes after laser injury, merged or presented individually; step size = 1 µm in z-plane; total section 30 µm. Images depict different orientations of the same thrombus. (D) Upper panel: horizontal orientation; lower panel: 90 degrees to the right. Bar represents 30 µm. Platelet accumulation was monitored using Alexa555-labeled rat anti- CD41 F(ab)2 antibody.
The 2-dimensional (2D) images suggest only partial colocalization of platelets and FXa or FVa at the site of the growing hemostatic plug. To overcome the potentially misleading feature of 2D images, 3-dimensional confocal images were obtained approximately 4 minutes postinjury by the collection of 30 µm image sections acquired at 1 µm intervals across the vessel diameter. Representative 3D images of labeled platelets and Alexa488-FV-810SYA (Figure 2D), again, show that FVa is more broadly distributed than adherent platelets localized to the core injury site (supplemental Video 2B). Similar results with labeled Alexa647-FXai also confirm the interpretation of the 2D images indicating that a substantial fraction of the enzyme is found away from the core injury site on vascular surfaces (Figure 3D and supplemental Video 3B). It also appears that a fraction of platelets at the injury site do not have exogenously added FVa or FXa bound, suggesting these platelets are not activated sufficiently to support coagulation factor binding.
Thus, bound FVa and FXa can be individually imaged with high sensitivity after laser injury. The imaging studies yield the tentative conclusion that while prothrombinase may be bound to a subset of platelets at the core injury site, a significant fraction of bound FVa and FXa appear to be localized to the activated endothelium or perhaps other blood cells (eg, neutrophils) or microparticles adherent to the region surrounding the injury site. Because hemostatic plug formation in the hemophilic animals requires the infusion of FVa and the assembly of prothrombinase, at least part of the FVa visualized in the vicinity of the thrombus must be functional rather than adventitiously bound.
Simultaneous imaging of FVa and FXa at the site of injury
The unexpectedly broad distribution of FVa and FXa imaged individually was further pursued in studies designed to simultaneously detect bound FVa, FXai, and adherent platelets. This was accomplished by coinfusion of Alexa488-FV-810SYA, Alexa647-FXai, and Alexa555-labeled platelet antibody prior to laser-induced injury in HB animals. A single snapshot image approximately 3 minutes postinjury from 1 of these experiments shows the broad distribution of Alexa488-FV-810SYA and Alexa647-FXai beyond the platelet core at the injury site. Although the proteins were concentrated at the platelet core, they were also found both upstream and downstream from the site of microscopic injury (Figure 4A-B and supplemental Video 4). Quantitative analysis of fluorescence intensity using a constant grid size (red box) at the core injury site revealed that the kinetics of Alexa488-FV-810SYA and Alexa647-FXai accumulation were equivalent and mirrored that of platelets for the first minute postinjury. Subsequently, platelet accumulation decreased while Alexa488-FV-810SYA and Alexa647-FXai continued to accumulate. In contrast, although minimal fluorescence associated with adherent platelets was detected upstream from the core injury site, substantial amounts of Alexa488-FV-810SYA and Alexa647-FXai were present. These data provide the quantitative basis for the suggestion that a major fraction of FV-810SYA and FXai bound in the vicinity of the injury site is found on cellular surfaces other than those provided by adherent/activated platelets. The fact that this is most prominent at a distance from the site of microscopic injury suggests that laser injury or components released from cells in the local microenvironment contribute to cellular activation, perhaps facilitating the exposure of phosphatidylserine required for clotting factor binding.
Figure 4.

Imaging of prothrombinase constituents. Digital composite fluorescence and brightfield images of a representative thrombus acquired 3 minutes postinjury in HB mice infused with labeled Alexa488-FV-810SYA (green; 600 µg/kg), platelet antibody (red), and Alexa647-FXai (cyan; 80 µg/kg); yellow (FV-810SYA and platelets) or white (FV-810SYA, FXai, and platelets) represent colocalization. (A) Red box covers core injury site; (B) red box covers upstream of injury. (A-B) Bar represents 10 µm. Time course fluorescence intensity for Alexa488-FVa-810SYA (green), Alexa647-FXai (cyan), and platelets (red) either at the injury site (C) or upstream of the injury (D). Platelet accumulation was monitored using Alexa555-labeled rat anti- CD41 F(ab)2 antibody.
This suggestion was further pursued using labeled annexin V to reveal phosphatidylserine exposure after laser injury.23,34 Coinfusion of Alexa488-annexin V, Alexa647-FXai, and Alexa555-labeled platelet antibody prior to laser-induced injury and imaging yielded excellent agreement between the distribution of annexin V and FXai illustrated by 3D confocal images acquired approximately 4 minutes postinjury (supplemental Figure 2 and supplemental Video 8). The remarkable agreement in the distribution of bound annexin V and FXai, particularly in areas distant from the platelet core, supports the conclusion that phosphatidylserine exposure resulting from cell activation in areas surrounding the platelet core contributes to the binding of significant amounts of FXa and FVa.
Abrogation of platelet adhesion and activation
The wide distribution of bound FVa and FXai in the area of vascular damage extending beyond the platelet core raises the possibility that the activated endothelial surface contributes in a substantive manner to thrombin formation beyond the contribution of the adherent and activated platelets. We tested this possibility using pharmacologic and genetic approaches to impair platelet adhesion at the injury site. In the first approach, HB mice were treated with integrilin to block the binding functions of αIIbβ3, reversibly inhibit platelet aggregation, and reduce their adhesion to the injury site.35 Coinfusion of Alexa488-FV-810SYA, Alexa647-FXai, and Alexa555-labeled platelet antibody prior to challenge with laser injury in the animals not pretreated with integrilin reproduced earlier findings (Figure 5A-B and supplemental Video 5A). Prior treatment with integrilin greatly reduced platelets at the core injury site, but without altering the kinetics and distribution of Alexa488-FV-810SYA and Alexa647-FXai while the total amount of these proteins was reduced approximately 50% (Figure 5A-B and supplemental Video 5B). These conclusions were further supported by 3D reconstructions from confocal images obtained 4 minutes postinjury (Figure 5C and supplemental Video 5C-D). The broadly distributed profile of bound Alexa488-FV-810SYA and Alexa647-FXai was mostly unchanged after the large reduction in adherent platelets after integrilin treatment. These findings could be reproduced using a genetic approach with Par4−/− mice. Analysis of normalized fluorescence from Alexa488-FV-810, Alexa647-FXai,, and platelets from multiple thrombi at approximately 3 minutes show that platelets at the injury site were substantially reduced, although the constituents of prothrombinase were not (Figure 6A).
Figure 5.

Effect of integrilin on distribution of prothrombinase constituents. (A) Kinetics of Alexa488-FV-810SYA (600 µg/kg), Alexa647-FXai (80 µg/kg), and (B) platelet fluorescence accumulation over time in the presence (n = 5 mice; 23 thrombi) or absence (n = 5 mice; 22 thrombi) of integrilin. (C) Confocal imaging of labeled platelets, Alexa488-FV-810SYA, and Alexa647-FXai 240 seconds after laser injury, merged or presented individually; step size = 1 µm in z-plane; total section 30 µm. (C) Without integrilin (top); with integrilin (bottom). Platelet accumulation was monitored using Alexa555-labeled rat anti-CD41 F(ab)2 antibody.
Figure 6.

Effect of integrilin on distribution and accumulation of fibrin. (A) Normalized fluorescence intensity of, Alexa488-FV-810SYA (green), Alexa647-FXai (cyan), platelets (red) and fibrin (blue) over volume from multiple thrombi (n = 3-7 mice; 14-27 thrombi per group) generated at approximately 4 minutes after injury in HB or Par 4−/− mice infused with labeled-FV-810SYA FVa (600 µg/kg), Alexa647-FXai (80µg/kg), antibodies against platelets, or fibrin with or without integrilin. Normalized fluorescence intensity was not acquired for FV-810SYA and FXai for Par4−/− mice in the presence of integrilin (NA). NA, not acquired. (B) Kinetics of fibrin accumulation over time in Par 4−/− mice (n = 3 mice; 15 thrombi) or HB mice (n = 5 mice; 22 thrombi) infused with labeled Alexa488-FV-810SYA (600 µg/kg) in the presence or absence of integrilin (10 mg/kg). (C) Confocal imaging of labeled platelets, Alexa488-FV-810SYA, and labeled fibrin 240 seconds after laser injury, merged or presented individually; step size = 1 µm in z-plane; total section 30 µm. (C) Without integrilin (top); with integrilin (bottom). Platelet accumulation was monitored using Alexa555-labeled rat anti-CD41 F(ab)2 antibody and fibrin was detected with Alexa647-labeled anti-fibrin antibody. (C) Bar represents 30 μm.
The pharmacologic or genetic reduction of platelets also had essentially no impact on fibrin deposition (Figure 6). This was evident after analysis of total fibrin 3 minutes postinjury in both integrilin treated HB mice or in Par4−/− mice (± integrilin; Figure 6A). In addition to total fibrin, the kinetics of fibrin accumulation was also unchanged (Figure 6B). The 2D and 3D image analysis shows that the fibrin is only partly associated with platelets and FVa, and appears more distal to these components in either the absence or presence of integrilin (Figure 6C and supplemental Video 6).
Prothrombinase distribution on the endothelial surface
To strengthen the conclusion that activated endothelium contributes phosphatidylserine to support the binding of prothrombinase constituents, we further examined the distribution of adherent platelets and bound Alexa647-FXai after laser injury in HB mice, but without the infusion of FV-810SYA. Supplemental Video 7A-B shows identical movies, but only 1 detector channel is turned on to image platelets or Alexa647-FXai. In the absence of coinfused FV-810SYA, no platelet fluorescence at the injury site was seen in HB animals (supplemental Video 7A). In contrast, in a parallel movie (supplemental Video 7B), a strong signal was detected for Alexa647-FXai, indicating that a damaged vascular surface, likely representing the activated endothelium, prominently supports the binding of Alexa647-FXai, even when adherent platelets were undetectable.
The more striking visual evidence for this conclusion was sought from HB mice that selectively express GFP in endothelial cells under control of the Tie2 promoter to illuminate the endothelial layer (HB-Tie2-GFP; Figure 7 and Supplemental Video 7C). Confocal imaging and 3D reconstruction after laser injury, which was performed after infusion of Alexa647-FXai and the Alexa555-labeled platelet antibody, either with or without unlabeled FV-810SYA, was used to assess the relationship between bound FXai and the endothelium. In the absence of FV-810SYA, Alexa647-FXai binds to the damaged endothelium, even when platelets were effectively absent. Although adherent platelets were observed after FV-810SYA infusion, the relationship between bound Alexa647-FXai and the illuminated endothelial surface was not obviously different. These findings with HB/Tie2-GFP mice point to a prominent role played by the endothelium surrounding the injury site in supporting enzyme complex assembly and the eventual formation of fibrin.
Figure 7.

Spatial distribution of platelets and FXai in HB-Tie2-GFP mice. Confocal images of endothelium defined by GFP fluorescence (green) Alexa647-FXai (cyan) and Alexa555-anti-CD41 (red) at approximately 3 minutes without (-) FV-810SYA or after infusion of unlabeled FV-810SYA. Step size = 2 µm in z-plane, total sections = 20 µm. Bar represents 30 µm.
Discussion
Current ideas defining the cellular control of the hemostatic response to vascular damage center on the adherent and activated platelet as the primary biological surface for the assembly and function of the prothrombinase and intrinsic Xase complexes of coagulation.36 This view is rooted in the ability of isolated platelets to support the assembly and function of prothrombinase and intrinsic Xase after activation with physiologically relevant agonists, the intimate association of activated platelets with fibrin within the thrombus, and the established observation of bleeding in humans with thrombocytopenia or platelet disorders.37 However, given the key structural contributions of fibrin and aggregated/adherent platelets to thrombus stability, it does not necessarily or logically follow that bleeding seen when a reduction in platelet function/level reflects their primary contribution to prothrombinase assembly, thrombin formation, and resulting fibrin deposition. The conjecture that the activated platelet is the primary biological surface for thrombin formation needs further tempering by the unknown agonist concentration profiles during the response to vascular damage in flowing blood.33 Thus, although platelets may be readily activated by “optimal” concentrations of agonists to support coagulation enzyme complex function under contrived conditions in vitro, their extent of activation within and around the evolving thrombus may be insufficient to allow their implied primary and robust contribution to prothrombinase or intrinsic Xase function.
In this study, we circumvent these limitations by examining the distribution of prothrombinase in the area of vascular damage after laser injury to the cremaster arteriole in the living mouse. Although prothrombinase may be present on a limited subset of platelets present at the core of the thrombus, the striking observation is that both FXa and FVa are much more broadly distributed on the vessel wall in the area surrounding the injury site, but lacking adherent platelets. We surmise that this extended distribution reflects their binding to activated or damaged endothelial cells found in the vicinity of vascular damage. It is also possible that there are contributions from microparticles and neutrophils, as these membranous surfaces have been proposed to contribute to thrombus formation.9,38 Such activated surfaces, other than those provided by adherent platelets, must support thrombin formation in a robust way as genetic or pharmacologic manipulations to yield large reductions in platelets adherent to the injury site produced modest effects on the distribution of the bound constituents of prothrombinase and only minimal decreases in fibrin deposition. This synopsis outlines our observations that question the preeminent role ascribed to the surface of the activated platelet in supporting the assembly and function of prothrombinase in vivo.
At issue is whether surfaces that play an especially important role in supporting functional prothrombinase assembly and thrombin formation are somehow obscured for reasons of sensitivity. This would require that the specific activity of bound prothrombinase is highly dependent on cell type or just not governed by physical principles as established in purified systems with synthetic membranes.39,40 Although there is no sound evidence to support either possibility, we are unable to directly address this question. Instead, we have used a variety of alternative strategies to deplete the bulk of adherent platelets to partially address this concern. Nevertheless, it remains possible that a small subset of activated platelets present at the core of the thrombus support the binding of FXa and FVa in a way that is essential for the hemostatic response. It is for this reason that we phrase our conclusions in terms of the broad distribution of bound FXa and FVa, both at the core of the injury site where platelets are present, as well as more extended regions where adherent platelets are not detected. Thereby, suggesting a far more substantial role than previously anticipated for other activated cells highlighted by the activated endothelium rather than just the platelets in supporting the assembly and function of prothrombinase. The recent documentation that more extensively activated platelets are found in a limited core region of the thrombus adjacent to the vessel wall and the localization of annexin V binding to this limited region warrant this type of circumspect conclusion.33,41
Our findings question the functional demarcation of procoagulant function attributed to platelets (and possibly other blood cells such as neutrophils and microparticles) and the anticoagulant role played by the endothelium in selectively supporting the activation of protein C.42 Although venous endothelial cells in culture have long been known to support prothrombinase assembly and thrombin formation, the biological significance of these observations has been uncertain.43 Our findings in the cremaster arteriole now show that activated/damaged endothelial cells immediately surrounding the site of vascular injury likely play a very important role in facilitating prothrombinase assembly and the subsequent formation of thrombin essential for the hemostatic response. The key observation is that it is only those endothelial cells immediately surrounding the injury that exhibit this function with no bound FVa or FXa detectable on the vessel wall further upstream or downstream from the site of damage. We speculate that our findings arise from 2 principles: the high sensitivity of endothelial cells to activation in the physiological milieu and the likely limited sensitivity of platelets to activation under these conditions. The former is supported by the documented ability of laser light to increase intracellular Ca2+ in the target endothelial cell, as well as cells immediately surrounding it in culture.15 The latter principle is surprising given the accepted propensity of platelets to activation.44 It is nevertheless consistent with the finding that only a limited number of platelets at the core of the thrombus actually express P-selectin on their surface.33
The significance of laser-induced damage in the mouse cremaster arteriole to the different forms of vascular injury relevant to normal or pathological thrombus formation in humans is arguable. However, close parallels between the response to laser damage and a microscopic piercing injury to the blood vessel done with a micropipette, implicates the laser injury model as most relevant to hemostasis rather than pathological thrombus formation.33 The latter, by its nature, is more complicated and typically involves several abnormalities. The laser injury model and our findings may not accurately portray the relative contributions of the activated endothelium vs platelets to coagulation enzyme complex assembly under pathological conditions such as plaque rupture or disseminated intravascular coagulation. Our conclusions also need modulation in terms of the specific injury model that we have used with qualifiers that the outcome may be dependent on vessel size, in the venous vs arterial system and type of injury.45-47 Differences with the cremaster laser injury model in different injury scenarios could also yield alternative outcomes in terms of prothrombinase distribution and contribution of platelets to thrombin generation. Interestingly, however, Cooley et al48,49 have established parallels between some of our observations and the response to electrolytic injury in large veins. Pharmacologic inhibition of platelet adhesion had only a minor effect on fibrin levels at the injury site, again highlighting the importance of cells other than platelets contributing to thrombin generation. The finding that such effects were not as striking in electrolytic injury to large arteries further justifies carefully qualified interpretation of our findings. Relating the outcome in the different mouse models to human physiology and pathology remains a future challenge to the field.
In contrast to the widely accepted idea that the activated platelet is the primary biological surface for the assembly and function of the prothrombinase, we now show a prominent contribution for the activated endothelium adjacent to the site of damage in these processes. Although presently, we have not studied the distribution of the components of intrinsic Xase, it seems possible that similar conclusions also may apply to FIXa and FVIIIa. The contribution of different cell types to the function of clotting factor complexes likely adds an underappreciated dimension of control to thrombus formation or to limiting thrombus formation in vivo by partitioning reactants and products to different surfaces.
Acknowledgments
The authors thank Michael Boltz for assistance with molecular biology and Dr Matthew Bunce for preparation of some proteins.
This work was supported in part by grants from the National Institutes of Health, National Heart, Lung, and Blood Institute (P01 HL-74124, RC1 HL-099467, and NIH-National Center for Research Resources 1S10RR26716).
Footnotes
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
The online version of this article contains a data supplement.
There is an Inside Blood Commentary on this article in this issue.
Authorship
Contribution: L.I. conducted research; and L.I., S.K., and R.M.C. analyzed data and wrote the paper.
Conflict-of-interest disclosure: R.M.C. receives licensing fees and research funding from Pfizer. The remaining authors declare no competing financial interests.
Correspondence: Rodney M. Camire, The Children's Hospital of Philadelphia, Division of Hematology, 5018 Colket Translational Research Building, 3501 Civic Center Blvd, Philadelphia, PA 19104; e-mail: rcamire@mail.med.upenn.edu.
References
- 1.Furie B, Furie BC. Mechanisms of thrombus formation. N Engl J Med. 2008;359(9):938–949. doi: 10.1056/NEJMra0801082. [DOI] [PubMed] [Google Scholar]
- 2.Mann KG, Nesheim ME, Church WR, Haley P, Krishnaswamy S. Surface-dependent reactions of the vitamin K-dependent enzyme complexes. Blood. 1990;76(1):1–16. [PubMed] [Google Scholar]
- 3.Mann KG, Krishnaswamy S, Lawson JH. Surface-dependent hemostasis. Semin Hematol. 1992;29(3):213–226. [PubMed] [Google Scholar]
- 4.Monroe DM, Hoffman M, Roberts HR. Platelets and thrombin generation. Arterioscler Thromb Vasc Biol. 2002;22(9):1381–1389. doi: 10.1161/01.atv.0000031340.68494.34. [DOI] [PubMed] [Google Scholar]
- 5.Heemskerk JW, Mattheij NJ, Cosemans JM. Platelet-based coagulation: different populations, different functions. J Thromb Haemost. 2013;11(1):2–16. doi: 10.1111/jth.12045. [DOI] [PubMed] [Google Scholar]
- 6.Bouchard BA, Tracy PB. Platelets, leukocytes, and coagulation. Curr Opin Hematol. 2001;8(5):263–269. doi: 10.1097/00062752-200109000-00001. [DOI] [PubMed] [Google Scholar]
- 7.Bevers EM, Comfurius P, van Rijn JL, Hemker HC, Zwaal RF. Generation of prothrombin-converting activity and the exposure of phosphatidylserine at the outer surface of platelets. Eur J Biochem. 1982;122(2):429–436. doi: 10.1111/j.1432-1033.1982.tb05898.x. [DOI] [PubMed] [Google Scholar]
- 8.Stern D, Nawroth P, Handley D, Kisiel W. An endothelial cell-dependent pathway of coagulation. Proc Natl Acad Sci USA. 1985;82(8):2523–2527. doi: 10.1073/pnas.82.8.2523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bouchard BA, Tracy PB. The participation of leukocytes in coagulant reactions. J Thromb Haemost. 2003;1(3):464–469. doi: 10.1046/j.1538-7836.2003.00089.x. [DOI] [PubMed] [Google Scholar]
- 10.Whelihan MF, Mann KG. The role of the red cell membrane in thrombin generation. Thromb Res. 2013;131(5):377–382. doi: 10.1016/j.thromres.2013.01.023. [DOI] [PubMed] [Google Scholar]
- 11.Rosen ED, Raymond S, Zollman A, et al. Laser-induced noninvasive vascular injury models in mice generate platelet- and coagulation-dependent thrombi. Am J Pathol. 2001;158(5):1613–1622. doi: 10.1016/S0002-9440(10)64117-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Falati S, Gross P, Merrill-Skoloff G, Furie BC, Furie B. Real-time in vivo imaging of platelets, tissue factor and fibrin during arterial thrombus formation in the mouse. Nat Med. 2002;8(10):1175–1181. doi: 10.1038/nm782. [DOI] [PubMed] [Google Scholar]
- 13.Furie B, Furie BC. Thrombus formation in vivo. J Clin Invest. 2005;115(12):3355–3362. doi: 10.1172/JCI26987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vandendries ER, Hamilton JR, Coughlin SR, Furie B, Furie BC. Par4 is required for platelet thrombus propagation but not fibrin generation in a mouse model of thrombosis. Proc Natl Acad Sci USA. 2007;104(1):288–292. doi: 10.1073/pnas.0610188104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Atkinson BT, Jasuja R, Chen VM, Nandivada P, Furie B, Furie BC. Laser-induced endothelial cell activation supports fibrin formation. Blood. 2010;116(22):4675–4683. doi: 10.1182/blood-2010-05-283986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kroh HK, Panizzi P, Tchaikovski S, et al. Active site-labeled prothrombin inhibits prothrombinase in vitro and thrombosis in vivo. J Biol Chem. 2011;286(26):23345–23356. doi: 10.1074/jbc.M111.230292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Camire RM. Prothrombinase assembly and S1 site occupation restore the catalytic activity of FXa impaired by mutation at the sodium-binding site. J Biol Chem. 2002;277(40):37863–37870. doi: 10.1074/jbc.M203692200. [DOI] [PubMed] [Google Scholar]
- 18.Toso R, Camire RM. Removal of B-domain sequences from factor V rather than specific proteolysis underlies the mechanism by which cofactor function is realized. J Biol Chem. 2004;279(20):21643–21650. doi: 10.1074/jbc.M402107200. [DOI] [PubMed] [Google Scholar]
- 19.Zhu H, Toso R, Camire RM. Inhibitory sequences within the B-domain stabilize circulating factor V in an inactive state. J Biol Chem. 2007;282(20):15033–15039. doi: 10.1074/jbc.M701315200. [DOI] [PubMed] [Google Scholar]
- 20.Buddai SK, Toulokhonova L, Bergum PW, Vlasuk GP, Krishnaswamy S. Nematode anticoagulant protein c2 reveals a site on factor Xa that is important for macromolecular substrate binding to human prothrombinase. J Biol Chem. 2002;277(29):26689–26698. doi: 10.1074/jbc.M202507200. [DOI] [PubMed] [Google Scholar]
- 21.Weiler-Guettler H, Christie PD, Beeler DL, et al. A targeted point mutation in thrombomodulin generates viable mice with a prethrombotic state. J Clin Invest. 1998;101(9):1983–1991. doi: 10.1172/JCI2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Neyman M, Gewirtz J, Poncz M. Analysis of the spatial and temporal characteristics of platelet-delivered factor VIII-based clots. Blood. 2008;112(4):1101–1108. doi: 10.1182/blood-2008-04-152959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tait JF, Gibson D, Fujikawa K. Phospholipid binding properties of human placental anticoagulant protein-I, a member of the lipocortin family. J Biol Chem. 1989;264(14):7944–7949. [PubMed] [Google Scholar]
- 24.Lin HF, Maeda N, Smithies O, Straight DL, Stafford DW. A coagulation factor IX-deficient mouse model for human hemophilia B. Blood. 1997;90(10):3962–3966. [PubMed] [Google Scholar]
- 25.Kung SH, Hagstrom JN, Cass D, et al. Human factor IX corrects the bleeding diathesis of mice with hemophilia B. Blood. 1998;91(3):784–790. [PubMed] [Google Scholar]
- 26.Motoike T, Loughna S, Perens E, et al. Universal GFP reporter for the study of vascular development. Genesis. 2000;28(2):75–81. doi: 10.1002/1526-968x(200010)28:2<75::aid-gene50>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- 27.Schlachterman A, Schuettrumpf J, Liu JH, et al. Factor V Leiden improves in vivo hemostasis in murine hemophilia models. J Thromb Haemost. 2005;3(12):2730–2737. doi: 10.1111/j.1538-7836.2005.01639.x. [DOI] [PubMed] [Google Scholar]
- 28.Xue J, Kalafatis M, Silveira JR, Kung C, Mann KG. Determination of the disulfide bridges in factor Va heavy chain. Biochemistry. 1994;33(44):13109–13116. doi: 10.1021/bi00248a021. [DOI] [PubMed] [Google Scholar]
- 29.Krishnaswamy S, Mann KG. The binding of factor Va to phospholipid vesicles. J Biol Chem. 1988;263(12):5714–5723. [PubMed] [Google Scholar]
- 30.Krishnaswamy S, Russell GD, Mann KG. The reassociation of factor Va from its isolated subunits. J Biol Chem. 1989;264(6):3160–3168. [PubMed] [Google Scholar]
- 31.Yegneswaran S, Mesters RM, Fernández JA, Griffin JH. Prothrombin residues 473-487 contribute to factor Va binding in the prothrombinase complex. J Biol Chem. 2004;279(47):49019–49025. doi: 10.1074/jbc.M406645200. [DOI] [PubMed] [Google Scholar]
- 32.Betz A, Krishnaswamy S. Regions remote from the site of cleavage determine macromolecular substrate recognition by the prothrombinase complex. J Biol Chem. 1998;273(17):10709–10718. doi: 10.1074/jbc.273.17.10709. [DOI] [PubMed] [Google Scholar]
- 33.Stalker TJ, Traxler EA, Wu J, et al. Hierarchical organization in the hemostatic response and its relationship to the platelet-signaling network. Blood. 2013;121(10):1875–1885. doi: 10.1182/blood-2012-09-457739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tait JF, Smith C, Wood BL. Measurement of phosphatidylserine exposure in leukocytes and platelets by whole-blood flow cytometry with annexin V. Blood Cells Mol Dis. 1999;25(5-6):271–278. doi: 10.1006/bcmd.1999.0254. [DOI] [PubMed] [Google Scholar]
- 35.Jasuja R, Furie B, Furie BC. Endothelium-derived but not platelet-derived protein disulfide isomerase is required for thrombus formation in vivo. Blood. 2010;116(22):4665–4674. doi: 10.1182/blood-2010-04-278184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Haynes LM, Bouchard BA, Tracy PB, Mann KG. Prothrombin activation by platelet-associated prothrombinase proceeds through the prethrombin-2 pathway via a concerted mechanism. J Biol Chem. 2012;287(46):38647–38655. doi: 10.1074/jbc.M112.407791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bennett JS. Overview of aquired and inherited clinical platelet disorders. In: Marder VJ, Aird WC, Bennett JS, Schulman S, White GC, editors. Hemostasis and Thrombosis. Philadelphia: Lippincott Williams & Wilkins; 2012. pp. 751–756. [Google Scholar]
- 38.Zwicker JI, Trenor CC, III, Furie BC, Furie B. Tissue factor-bearing microparticles and thrombus formation. Arterioscler Thromb Vasc Biol. 2011;31(4):728–733. doi: 10.1161/ATVBAHA.109.200964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Krishnaswamy S. Coagulation Enzymology. In: Marder VJ, Aird WC, Bennett JS, Schulman S, White GC, editors. Hemostasis and Thrombosis. Philadelphia: Lippincott Williams & Wilkins; 2012. pp. 41–51. [Google Scholar]
- 40.Tracy PB. Regulation of thrombin generation at cell surfaces. Semin Thromb Hemost. 1988;14(3):227–233. doi: 10.1055/s-2007-1002782. [DOI] [PubMed] [Google Scholar]
- 41.Hayashi T, Mogami H, Murakami Y, et al. Real-time analysis of platelet aggregation and procoagulant activity during thrombus formation in vivo. Pflugers Arch. 2008;456(6):1239–1251. doi: 10.1007/s00424-008-0466-9. [DOI] [PubMed] [Google Scholar]
- 42.Esmon CT, Esmon NL. The link between vascular features and thrombosis. Annu Rev Physiol. 2011;73:503–514. doi: 10.1146/annurev-physiol-012110-142300. [DOI] [PubMed] [Google Scholar]
- 43.Hackeng TM, van ’t Veer C, Meijers JC, Bouma BN. Human protein S inhibits prothrombinase complex activity on endothelial cells and platelets via direct interactions with factors Va and Xa. J Biol Chem. 1994;269(33):21051–21058. [PubMed] [Google Scholar]
- 44.Bennett JS. Overview of megakaryocyte and platelet biology. In: Marder VJ, Aird WC, Bennett JS, Schulman S, White GC, editors. Hemostasis and Thrombosis. Philadelphia: Lippincott Williams & Wilkins; 2012. pp. 341–348. [Google Scholar]
- 45.Kretz CA, Vaezzadeh N, Gross PL. Tissue factor and thrombosis models. Arterioscler Thromb Vasc Biol. 2010;30(5):900–908. doi: 10.1161/ATVBAHA.108.177477. [DOI] [PubMed] [Google Scholar]
- 46.Denis CV, Dubois C, Brass LF, Heemskerk JW, Lenting PJ Biorheology Subcommittee of the SSC of the ISTH. Towards standardization of in vivo thrombosis studies in mice. J Thromb Haemost. 2011;9(8):1641–1644. doi: 10.1111/j.1538-7836.2011.04350.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Whinna HC. Overview of murine thrombosis models. Thromb Res. 2008;122(Suppl 1):S64–S69. doi: 10.1016/S0049-3848(08)70022-7. [DOI] [PubMed] [Google Scholar]
- 48.Cooley BC, Herrera AJ. Cross-modulatory effects of clopidogrel and heparin on platelet and fibrin incorporation in thrombosis. Blood Coagul Fibrinolysis. 2013;24(6):593–598. doi: 10.1097/MBC.0b013e3283602a03. [DOI] [PubMed] [Google Scholar]
- 49.Cooley BC. Platelet integrin β3 deletion or pharmacologic blockade augments fibrin in large-vein thrombosis. [abstract] J Thromb Haemost. 2011;9:618. Abstract P-WE-312. [Google Scholar]