

Abstract
The fusion between ERG coding sequences and the TMPRSS2 promoter is the most prevalent in prostate cancer (CaP). The presence of two main types of TMPRSS2-ERG fusion transcripts in CaP specimens, Type I and Type II, prompted us to hypothesize that the cumulative actions of different ERG variants may impact CaP development/progression. Using TMPRSS2-ERG3 (Type I) and TMPRSS2-ERG8 (Type II) expression vectors, we determined that the TMPRSS2- ERG8 encoded protein is deficient in transcriptional regulation compared to TMPRSS2-ERG3. Co-transfection of vectors resulted in decreased transcriptional regulation compared to TMPRSS2-ERG3 alone, suggesting transdominance of ERG8. Expression of exogenous ERG8 protein resulted in a decrease in endogenous ERG3 protein levels in TMPRSS2-ERG positive VCaP cells, with a concomitant decrease in C-MYC. Further, we showed a physical association between ERG3 and ERG8 in live cells by the bimolecular fluorescence complementation assay, providing a basis for the observed effects. Inhibitory effects of TMPRSS2-ERG8 on TMPRSS2- ERG3 were also corroborated by gene expression data from human prostate cancers, which showed a positive correlation between C-MYC expression and TMPRSS2-ERG3/TMPRSS2- ERG8 ratio. We propose that an elevated TMPRSS2-ERG3/TMPRSS2-ERG8 ratio results in elevated C-MYC in CaP, providing a strong rationale for the biomarker and therapeutic utility of ERG splice variants, along with C-MYC.
Keywords: ERG, Splice variants, Prostate cancer, Dominant negative, C-MYC
INTRODUCTION
Genetic rearrangements involving DNA sequences from different chromosomes or intrachromosomal regions have been extensively documented in various cancers, including prostate cancer (CaP) [1]. It has been reported that male hormone dependent ETS-related transcription factors play causal roles in CaP, as a result of rearrangements. Hence, gene fusions involving the ETS family have potential value in cancer diagnosis, prognosis, and therapy [2-5]. Of these, overexpression of the ETS-related gene (ERG) [6, 7], resulting from the fusion of ERG coding sequences to the androgen-responsive TMPRSS2 gene [8], represents the most common subtype among ETS fusions, with a prevalence of approximately 50% in clinically localized prostate cancers [1-4, 9-12]. In addition, studies evaluating the expression of ERG in epithelial cells of matched benign and malignant prostate cells from a large patient cohort indicate that CaP cells harboring TMPRSS2-ERG fusion show overexpression of ERG in 60-70% of patients [10]. This genomic rearrangement is now established as one of the most common mechanisms of oncogenic activation in CaP [3-5]. ERG has also been originally implicated in Ewing's sarcoma and acute myeloid leukemia [13-15].
The multi-exon structure of ERG is transcribed to nine different splice variants by a combination of alternative transcription initiation, mRNA splicing, and transcription termination [6, 7, 16]. These variants can be divided into near full length (lacking 32 N-terminal amino acids) Type I ERG, containing the DNA-binding domain (DBD), with the embedded nuclear localization signal (NLS) and Type II ERG, a truncated form lacking DBD/NLS coding sequences [17]. The analysis of CaP associated fusion transcripts has revealed the presence of multiple splice variants, potentially exhibiting different biological activities and correlating with different tumor phenotypes [12, 13, 17-22]. Along these lines, our laboratory cloned and sequenced the relatively abundant full length TMPRSS2-ERG cDNAs from a pool of mRNAs from six TMPRSS2-ERG positive prostate tumors. In addition to the expected near full length fusion transcripts, we also identified mRNA splice variants of fusion transcripts lacking the C- terminus of ERG beyond exon 12 (Owczarek nomenclature), which was replaced by sequences resulting in the addition of 4 and 70 unique amino acids in TEPC1 and ERG8, respectively [16, 23]. The translated products of most of the various ERG transcripts have been shown to function as oncoproteins retaining the ETS domain with transforming activity [6, 14, 24, 25]. In addition, ERG, similar to other members of the ETS family, has been described as a mediator of mitogenic signals, such as mitogen activator protein kinases [26].
The relationship between the ERG transcripts and prognosis of CaP, however, is not clear. Analyses have shown that a specific variant is associated with an aggressive form of disease [27], while Hermans et al. [9] showed a favorable prognosis of CaP with another variant. This scenario highlights the need to evaluate the specific function of the different ERG-encoded proteins in the context of CaP. This knowledge will further our understanding of ERG towards its clinical utility, including patient stratification, treatment monitoring, and therapeutic targeting of CaP. In this regard, we hypothesized that the variants may function either additively, synergistically, or in a dominant negative fashion due to potential interactions, or competition, between the different splice forms. Such an interaction may modify the physiological responses of ERG protein, which include transcriptional activation, cell growth, differentiation, and apoptosis. To address this, we utilized the two most abundant prototypic variants described in CaP tumors, designated TMPRSS2-ERG3 (Type I) and TMPRSS2-ERG8 (Type II), and assessed their effects alone and in combination, using in vitro cell culture models. Our data show that TMPRSS2-ERG8 exhibited a dominant negative effect over TMPRSS-ERG3 mediated transcriptional regulation. Furthermore, the demonstration of a physical interaction between the variants may provide the basis for the functional antagonism. In addition, analysis of the ratio of Type I/Type II ERG in relation with the prevalent oncogene C-MYC may be of value as a biomarker in prostate cancer diagnosis and prognosis.
RESULTS
ERG encoded proteins localize to distinct regions within cells
For functional studies, TMPRSS2-ERG3 and TMPRSS2-ERG8 coding sequences were cloned into a pIRES-EGFP eukaryotic expression vector. A schematic representation of both constructs, in comparison to wild type ERG3 (wt-ERG3), is presented in Figure 1a. In order to examine the subcellular localization of ERG proteins, TMPRSS2-ERG3 and TMPRSS2-ERG8 coding sequences were fused to GFP and RFP tags at the C-termini, respectively. The expression of tagged proteins was verified in HEK293 cells by immunoblot analysis, using the CPDR ERG mouse monoclonal antibody, 9FY [5, 28]. Since 9FY recognizes an epitope present at the N-terminus, both TMPRSS2-ERG3 and TMPRSS2-ERG8 can be detected in the molecular weight range of 53 kDa and 38 kDa, respectively. Alternatively, TMPRSS2-ERG3-GFP and TMPRSS2-ERG8-RFP revealed chimeric proteins in the range of 79 and 64 kDa, respectively (Figure 1b). The subcellular localization of proteins was examined by microscopy, upon transfection of TMPRSS2-ERG3-GFP and TMPRSS2-ERG8-RFP vectors in HEK293 cells. The backbone plasmid vector was transfected into cells as a negative control. As expected, ERG3, which contains the DBD and NLS, was localized to the nuclear compartment of cells. In contrast, the NLS-lacking ERG8 protein was detected in the cytoplasm (Figure 1c). The results demonstrate that the majority of ERG3 and ERG8 proteins exhibit distinct subcellular localizations in cells.
Figure 1. TMPRSS2-ERG splice variants and subcellular localization of the respective proteins.

(A) Schematic representation of TM-ERG3, TM-ERG8 and wild type ERG3 proteins. NTD, N- terminal domain; PNT, Pointed domain; CAE, Central alternative exons; CD, Central domain; ETS, DNA binding domain; CTD, C-terminal domain; U, Unique 70 amino acids (B) Expression of TM-ERG3 and TM-ERG8 proteins in HEK293 cells. Cell lysate, separated on a SDS-PAGE gel and transferred to PVDF membrane, was probed with ERG specific 9FY monoclonal antibody. TM-ERG3 and TM-ERG3-GFP correspond to 53 and 79 kDa proteins, respectively. TM-ERG8 and TM-ERG8-RFP correspond to 38 and 64 kDa proteins, respectively. Glyceraldehyde 3- phosphate dehydrogenase, GAPDH, was used as an internal control. (C) Subcellular localization of TM-ERG3-GFP and TM-ERG-RFP proteins in HEK293 cells. ERG3 was localized to the nucleus while ERG8 was found predominantly in the cytoplasm. DAPI was used for staining the nuclei.
ERG8 interferes with the transcriptional regulatory function of ERG3
The effect of ERG on downstream regulatory processes was examined using a reporter construct in which the mouse myocyte enhancer factor 2 (mef2c) sequence was cloned upstream of the luciferase gene. Cells were transfected with the luciferase construct alone and also in combination with wt-ERG3, TMPRSS2-ERG3, or TMPRSS2-ERG8. A mutant TMPRSS2-ERG3 construct, containing an inactivating N-terminal +1 frame shift mutation, served as a negative control. While cells transfected with reporter construct by itself showed baseline luciferase activity, co-transfection with either the wtERG3 or TMPRSS2-ERG3 activated the transcription mediated by the mef2c reporter. Transfection with either TMPRSS2-ERG8 or mutTMPRSS2-ERG3, on the other hand, did not lead to an increase of luciferase activity (Figure 2a). Considering the presence of multiple TMPRSS2-ERG transcriptional variants in human prostate tumors, we also evaluated the effect of TMPRSS2-ERG8 on TMPRSS2-ERG3 function. For this purpose, the expression constructs were co-transfected along with the luciferase reporter construct such that TMPRSS2- ERG3 was kept constant, while TMPRSS2-ERG8 was transfected in increasing concentrations, in HEK293 cells. The results showed a loss of the transcriptional activator function of TMPRSS2- ERG3 with the addition of TMPRSS2-ERG8. The addition of either 20 ng or 40 ng of the TMPRSS2-ERG8 expression plasmid in the co-transfection assay exhibited around 50% reduction in luciferase activity in comparison to TM-ERG3 alone (Figure 2b). This suggests that there is an interference of TMPRSS2-ERG3 mediated activation by TMPRSS2-ERG8.
Figure 2. Transcriptional activation of downstream targets by TMPRSS2-ERG splice variants.
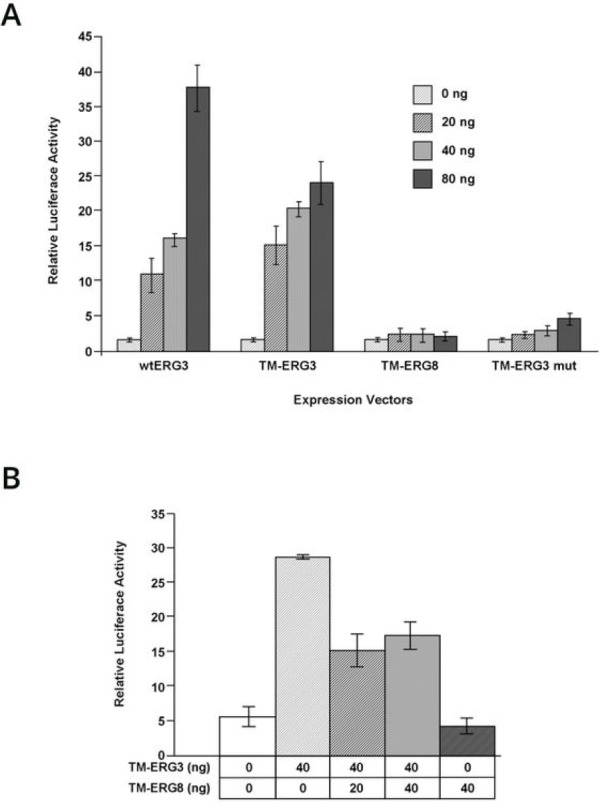
(A) Modulation of expression of a luciferase reporter construct containing mef2c enhancer. The reporter construct was co-transfected with 0, 20, 40 and 80 ng of vectors expressing wtERG3, TM-ERG3, TM-ERG8 or TM-ERG3 mutant. Each assay was repeated a minimum of 3 times. The data from a representative experiment is presented here. The extent of activation is presented in the form of relative luciferase activity, which increased with increasing concentrations of vector for both wtERG3 and TM-ERG3. TM-ERG8 and TM-ERG3mut resulted in baseline levels of activity. (B) Transdominant effect of TM-ERG8 on transcriptional activation mediated by TM- ERG3. A fixed amount of luciferase reporter (40 ng) and TM-ERG3 (40 ng) were co-transfected along with an increasing amount of TM-ERG8 (0-40 ng) into HEK293 cells. A decrease in relative luciferase activity was observed with increased TM-ERG8.
Ectopic expression of ERG8 in VCaP cells influences the endogenous level of ERG3
Next, we analyzed the effect of TMPRSS2-ERG8 expression in VCaP cells, which are known to harbor fusions involving ERG and TMPRSS2 genes. VCaP cells were transfected with the TMPRSS2- ERG8 vector in increasing concentrations (0, 2, 4, 6 μg), along with an empty vector in order to maintain the quantity of transfected plasmid DNA constant in each sample. Results revealed that as ectopically expressed ERG8 protein levels were increased, protein level of endogenous ERG3 was decreased, as shown in immunoblot assays (Figure 3a). Earlier studies from our laboratory showed that ERG3 binds to the C-MYC P2 promoter downstream elements and activates the expression of C-MYC [29]. Based on this, we evaluated the effect of ERG8 on C-MYC expression in VCaP cells. The results showed that C-MYC protein level also exhibited a decreasing trend similar to ERG3 protein level. The quantitation of signals in the immunoblot by Odyssey software, revealed the following: i) There was an increase in TMPRSS2-ERG8 expression corresponding to the amount of DNA transfected; ii) The endogenous TMPRSS2- ERG3 registered a 32% decrease in protein level in cells transfected with 6 μg of TMPRSS2- ERG8 DNA.; iii) The endogenous C-MYC showed a trend similar (38% decrease) to that of endogenous TMPRSS2-ERG3. Furthermore, the analysis at the RNA level by qPCR showed that both TMPRSS2-ERG3 and C-MYC remained unchanged, suggesting the modulation by TMPRSS2-ERG8 is at the protein level (Figure 3b).
Figure 3. Effect of ectopic expression of TMPRSS2-ERG8 in prostate cancer derived VCaP cells.
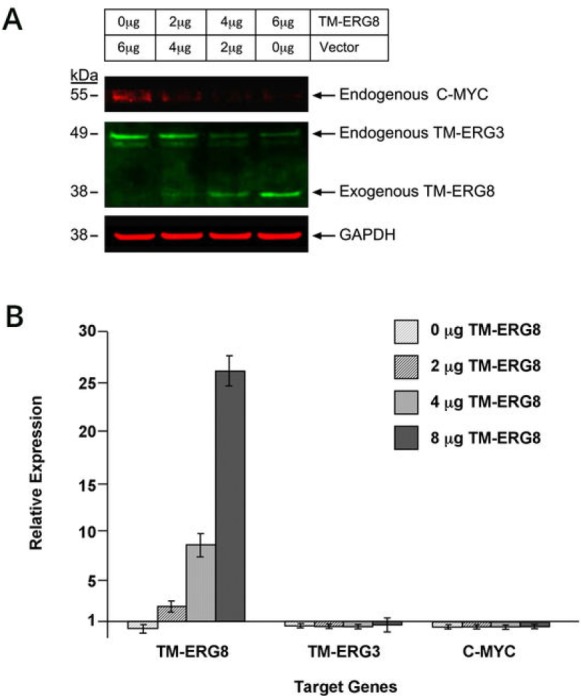
(A) Ectopic expression of TM-ERG8 affects the level of endogenous TM-ERG3 and C- MYC proteins in VCaP cells. Cells were transfected with different amounts of TM-ERG8 as indicated, lysed, and analyzed for protein expression of TM-ERG3 and C-MYC. Signals detected were quantitated using Odyssey software (data not shown). A decrease in endogenous TM- ERG3 protein and C-MYC protein was evident with the expression of TM-ERG8. The experiment was repeated three times. (B) Analysis of the effect of TM-ERG8 on endogenous TM-ERG3 and C-MYC at the RNA level. Quantitative RT-PCR was performed using RNA from VCaP cells transfected with TM-ERG8. Both TM-ERG3 and C-MYC remained at baseline levels of expression.
Bimolecular fluorescence complementation assay shows interaction between splice variants
To address the underlying basis for the interference of TMPRSS2-ERG3 mediated functions by TMPRSS2-ERG8, we assessed the potential interaction between ERG3 and ERG8 proteins through fluorescence microscopy and flow cytometry using the bimolecular fluorescence complementation (BiFC) assay in live cells. This assay involves a chimeric Venus protein which emits fluorescence upon dimerization in live cells (Figure 4a). For this purpose, we generated constructs containing TMPRSS2-ERG3 and TMPRSS2-ERG8 coding sequences fused to either N- or C-terminal coding sequences of the Venus reporter (Figure 4b). HIV-1 Vpr constructs were used as positive controls, as Vpr has been well characterized and shown to oligomerize [30]. As expected, we observed that VN-Vpr and VC-Vpr constructs co-transfected in HEK293 cells produced fluorescent signal, which was found to be localized in the nuclear region. In addition, co-transfection of VN-ERG3 and VC-ERG3 resulted in fluorescence, also observed in the nucleus. Similar results were seen for VN-ERG8 and VC-ERG8 co-transfection in which fluorescence was observed in the cytoplasm. Furthermore, co-transfection of VN-ERG3 and VC-ERG8, as well as VC-ERG3 and VN-ERG8, resulted in fluorescent protein in the nucleus, indicating a direct interaction between protein products of the two splice variants (Figure 4c). The transfection of two VN- constructs or the two VC- constructs together, as negative controls, showed no fluorescence in cells. In addition, a VN- or VC- construct transfected by itself also resulted in no fluorescence. The results from transfected cells independently analyzed by flow cytometry were consistent with the results obtained by microscopy (Figure 4d). The Vpr positive control cell population contained 10.9% cells positive for fluorescence (Figure 4d-iii). The positive control for ERG3 resulted in 17.9% positive cells (Figure 4d-iv), and for ERG8 resulted in 5.4% positive cells (Figure 4d-v). As predicted, co-transfection of VN-ERG3 and VC-ERG8, as well as VC-ERG3 and VN-ERG8, showed 10.1% and 12.7% cells positive for fluorescence, respectively (Figure 4d-vi and vii). Negative controls displayed ≤ 0.3% base line positivity (Figure 4d-i and ii). Thus, BiFC assay demonstrated interaction between ERG3 and ERG8 variants in live cells.
Figure 4. Analysis of the interaction between TMPRSS2-ERG3 and TMPRSS2-ERG8 by BiFC assay.

(A) Schematic representation of the bimolecular fluorescence complementation assay. (B) Schematic representation of the chimeric proteins containing N- or C-terminal regions of the Venus gene and coding sequences of TM-ERG splice variants. (C) Demonstration of the interaction between TM-ERG3 and TM-ERG8 proteins in live HEK293 cells. Yellow fluorescence is visible upon reconstitution of the Venus protein, resulting from the interaction of splice variants. Cells were visualized by microscopy using Leica, DMIRE2. (D) Quantitation of the fluorescence was performed by flow cytometry: i, Venus N-terminal constructs transfected together (negative control); ii, Venus C-terminal constructs transfected together (negative control); iii, VN- and VC- constructs for Vpr transfected together (positive control); iv, VN- and VC- constructs for TM-ERG3 transfected together; v. VN- and VC- constructs for TM- ERG8 transfected together; vi, VN:TM-ERG3 and VC:TM-ERG8 constructs transfected together; vii, VN:TM-ERG8 and VC:TM-ERG3 constructs transfected together; viii, Graphical representation of flow cytometry results.
Clinicopathological analysis of patient data reveals a correlation between C-MYC and ERG3/ERG8 ratios
The intriguing observation that altered ERG expression can significantly affect C-MYC levels in the VCaP cells prompted us to re-examine Type I/Type II ratios and C- MYC expression from our previously published qRT-PCR data from laser capture micro- dissected (LCM) prostate cancer cells [10, 17]. Our previous report showed that increased Type I (ERG3)/Type II (ERG8) ratio is associated with a higher Gleason sum and poorly differentiated phenotype. In contrast, a decrease in Type I/Type II ERG ratio was associated with favorable clinical-pathologic data [17]. Consistent with these observations, we also noted a significant correlation between Type I/Type II ERG ratio and C-MYC mRNA levels (rho = 0.37, P = 0.013) within the same specimens (Table 1). In comparison, we also analyzed the relationship between PCA3 expression levels (a gene up-regulated in CaP patients, though not an ERG transcriptional target) and Type I/Type II ERG ratio, which did not show any correlation (rho = -0.06, P = 0.682; Table 1). We then compared the differences in ratios of Type I/Type II ERG across quartile groups of C-MYC gene expression, and found a trend in which groups with higher levels of C- MYC expression had higher Type I/Type II ERG ratios, (P = 0.0894). PCA3 analysis across quartile groups again revealed that higher gene expression levels did not correlate to higher Type I/Type II ERG ratio (P = 0.842). Further, from a biological standpoint, we compared the Type I/Type II ERG ratio between down-regulated C-MYC (<0.5 fold) vs. up-regulated C-MYC (>2 fold). This analysis showed that up-regulated C-MYC had higher Type I/Type II ERG ratio compared to down-regulated C-MYC which shifted even closer towards significance (P = 0.056; Table 2).
Table 1. Spearman's correlation analysis of CaP oncogenes with Type I/Type II ERG ratio.
| Gene Expression | Type I/Type II ratio | |||
|---|---|---|---|---|
| N | rho | P | ||
| C-MYC fold difference | 45 | 0.37 | *0.013 | |
| PCA3 fold difference | 51 | -0.06 | 0.682 | |
P < 0.05 was considered statistically significant.
Spearman's correlation analysis reveals that increased C-MYC expression correlates with higher Type I/II ERG ratios. This correlation is not noted for analysis with PCA3
Table 2. Correlation of C-MYC gene expression with Type I/Type II ERG ratio.
| Biological Breakdown | Type I/Type II ratio | ||
|---|---|---|---|
| N | Median | P | |
| C-MYC fold difference: | (45) | 0.37 | 0.063 |
| **0.056 | |||
| < 0.5 fold (down-regulation) | 8 | 0.28 | ** |
| 0.5-2 fold (relatively unchanged) | 23 | 0.46 | |
| > 2.0 fold (up-regulation) | 14 | 0.72 | |
Trending towards significance. P < 0.05 was considered significant.
As C-MYC gene expression is found to be down-regulated in CaP patients, Type I/II ERG ratio is low. When C-MYC gene expression is up-regulated in CaP patients, Type I/II ERG ratio is found to be high.
DISCUSSION
Genomic rearrangements of oncogenes are established features in several human cancers and have been recognized for decades [1, 3, 5]. The involvement of fusion genes in prostate cancer has been defined by Tomlins et al. [4] and consistently reported by others [3, 5, 17-19, 23]. The fusion between androgen regulated TMPRSS2 and ERG genes is present in ~50% of prostate cancer patients in Western countries [3, 4, 17, 29, 31]. Analysis of fusion gene transcripts from prostate cancer patients by several investigators has shown the presence of multiple forms containing varying lengths of TMPRSS2 (promoter and exon 1 and 2) and ERG. The predominant form noted involves the fusion of TMPRSS2 exon 1 and ERG exon 4 (Tomlins nomenclature)/exon 8 (Owczarek nomenclature) [4, 16, 23]. Recent studies have shown that TMPRSS2-ERG expression is associated with an aggressive CaP phenotype [18, 32] while others noted that splice variants exhibit varied level of transforming activity [12]. Interestingly, the splice variants have been suggested to play both a positive and negative role in the prognosis of CaP [10, 33]. In addition, Wang et al. showed that the presence of the various isoforms led to increased proliferation/invasion, while knockdown led to decreased ectopic tumor size [19]. However, the functional significance of the individual transcripts, as well as their effect on each other, was not evaluated. Here, we have characterized the most prevalent prototypic fusion transcripts (TMPRSS2-ERG3 and TMPRSS2-ERG8) that were originally identified in our previous report [17].
As a prelude to functional evaluation, we initially characterized the synthesis and subcellular localization of the proteins encoded by TMPRSS2-ERG3 and TMPRSS2-ERG8. The proteins exhibited distinct subcellular localization; ERG3 localized predominantly in the nucleus and ERG8 exhibited cytoplasmic distribution. These distinct biological properties substantiated that TMPRSS2-ERG3 and TMPRSS2-ERG8 were appropriate fusion variants for investigating the functional significance of splice variants, as well as examining their influence on one another.
The transcriptional regulatory function of proteins encoded by these splice variants was explored through use of the enhancer from the mouse mef2c gene which has been shown to be active in the vascular endothelium during embryogenesis [34] and in adulthood where endogenous ERG protein is highly expressed [21, 35, 36]. We utilized the mef2c vascular endothelial enhancer sequence, containing a cluster of four conserved binding sites for ETS factors, linked to the luciferase reporter, to assess the transcriptional regulatory function of ERG splice variants. Both full length (wtERG3) and the N-terminus truncated Type I ERG protein (TMPRSS2-ERG3) activated the transcription from the mef2c driven luciferase reporter construct in HEK293 cells. Consistent with the absence of the NLS in the Type II ERG product, the ERG8 protein did not alter the basal promoter activity of the reporter. However, co-expression of TMPRSS2-ERG3 and increasing concentrations of TMPRSS2-ERG8 expression vectors showed an inhibition of TMPRSS2-ERG3-mediated activation of the mef2c reporter. The results suggested that TMPRSS2-ERG exhibited a dominant negative effect on the function mediated by TMPRSS2-ERG.
Based on these findings, we then used VCaP cells which express both TMPRSS2-ERG variants, allowing us to carry out studies mimicking the in vivo context of prostate tumors. Specifically, these cells express high levels of TMPRSS2-ERG8 (copy number) in comparison to TMPRSS2-ERG3 and also respond to androgens. Hence, introduction of exogenous TMPRSS2- ERG8 into VCaP cells enabled us to assess its effect on the endogenous ERG3 level. An increase in expression of ERG8 resulted in a decrease in endogenous ERG3 protein in VCaP cells. Remarkably, C-MYC protein levels mirrored the decrease in ERG3 protein levels. Based on this, we suggest that ERG3, a transcriptional activator of C-MYC, is concomitantly regulated by ERG8. Interestingly, while an alteration at the protein level was observed with the addition of TMPRSS2-ERG8, such an effect was not evident at the RNA level. It should be noted that ERG8 lacks the DBD and embedded NLS, but retains the SAM-pointed and protein-protein interaction domain. Therefore, it is suggested that elevated levels of ERG8 likely result in a dominant negative effect due to dimerization-mediated quenching of functional ERG3. This was alluded to in the analysis performed by Zammarchi et al. [12], though not explored further as to biological function.
In order to further investigate the mechanistic basis of the transdominant effect between splice variants, it was important to elucidate whether there is a direct interaction between the two isoforms. The BiFC assay, utilizing a chimeric Venus protein, allowed us to examine this directly in live cells through fluorescence microscopy. Venus constructs that contained either the N-terminus or C-terminus coding sequences of the Venus protein, linked to either TMPRSS2- ERG3 or TMPRSS2-ERG8 coding sequences were first evaluated, as they are known to homodimerize [33, 37]. As expected, cells were found to fluoresce, either in the nuclear region, as in the case with TMPRSS2-ERG3 or in the cytoplasmic region, as in the case with TMPRSS2- ERG8 alone. The extent of fluorescence positive cells upon expression of chimeric TMPRSS2- ERG3 and TMPRSS2-ERG8 supports the presence of interaction domains at the N- and C- termini of ERG3 protein. When cells were co-transfected with both splice variants together, we again observed fluorescence in cells, indicating heterodimerization between the two splice variants in live cells. It is interesting to note, that while the subcellular localization of Type I and Type II ERG was found to differ, heterodimers containing ERG3 and ERG8 were observed within the nuclear compartment of cells. This suggests that protein-protein interactions are likely to take place upon synthesis in the cytoplasm, after which they are transported to the nucleus. As ERG8 does not contain the NLS, it is unlikely to be transported to the nucleus on its own.
Based on this observation, we envision a scenario in which both homodimers and heterodimers may exist. Thus, it is reasonable to expect that in the context of an altered ratio containing high TMPRSS2-ERG8 over TMPRSS2-ERG3, there is a likelihood that heterodimers may predominate over the homodimers of ERG3. This possibility will lead to a diminished function of the TMPRSS2-ERG3 variant. Thus, our experiments involving co-transfection of plasmids expressing TMPRSS2-ERG3 and TMPRSS2-ERG8 variants along with mef2c driven luciferase reporter construct showed a diminished activation, in comparison to cells transfected with TMPRSS2-ERG3 alone. There is a possibility that heterodimers containing TMPRSS2- ERG3 and TMPRSS2-ERG8 may preferentially be subjected to degradation resulting in a decreased level of TMPRSS2-ERG3 in VCaP cells. However, further investigation of this is necessary.
It has been shown that C-MYC amplification increases as carcinogenesis progresses and it is strongly associated with higher histopathological grades and Gleason's scores as well as with earlier disease progression and earlier cancer death [27, 38]. C-MYC is also a known downstream target of TMPRSS2-ERG3 [23, 26, 29, 39, 40], however its relationship to Type I/II ERG ratio has not yet been examined. We therefore explored clinical patient data specifically for the direct relation of Type I/II ERG ratio and C-MYC gene expression levels. Indeed, it was observed that when oncogenic C-MYC was found to be up-regulated, the Type I/II ERG ratio was also high, corroborating with earlier data in which a high Gleason sum and poor overall patient prognosis were associated with an increased Type I/II ERG ratio. C-MYC levels would also be increased in this scenario, which could be involved in progression of the disease state. The results of this analysis represent a case for the use of the ratio of Type I/Type II ERG, in conjunction with C-MYC gene expression levels, to serve as potential prognostic markers for treatment of prostate cancer patients.
Based on these findings we propose a mechanistic model in which Type II ERG (TMPRESS2-ERG8) regulates the activity of Type I ERG (TMPRSS2-ERG3), in a dominant negative manner (Figure 5). As TMPRSS2-ERG8 is increased, TMPRSS2-ERG3 is decreased and the downstream events regulated by TMPRSS2-ERG3 are also likely inhibited. This scenario is also supported by the data on C-MYC, which has been shown as a regulatory target of TMPRSS2- ERG3. In accordance with this, an increased TMPRSS2-ERG8 correlates with a lower level of both TMPRSS2-ERG3 and C-MYC.
Figure 5. Mechanistic model of TM-ERG splice variants.
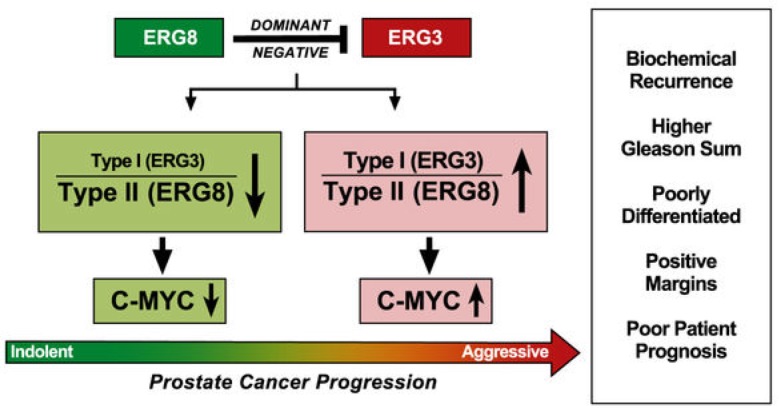
As levels of TM-ERG8 are increased, the overall ratio of Type I/Type II ERG is effectively decreased, which is associated with lower levels of C-MYC and a more favorable patient prognosis. When levels of TM-ERG8 are lower, the Type I/II ratio is effectively higher, which is found to be associated with higher levels of C- MYC and a more aggressive form of disease in patients.
Overall, studies presented here show that the interaction between Type I and Type II splice variants of TMPRSS2-ERG present in prostate tumor tissues may lead to functional antagonism of selected variants. We hypothesize that an altered ratio of high TMPRSS2-ERG8 in comparison to TMPRSS2-ERG3 may be beneficial to prostate cancer patients. The results further highlight the potential for the ratio of ERG splice variants in combination with C-MYC expression to be exploited for diagnostic and therapeutic applications against prostate cancer.
METHODS
Cell culture
The Vertebral-Cancer of the Prostate (VCaP) cells and human embryonic kidney (HEK) 293 cells were obtained from American Type Culture Collection (ATCC; Manasses, VA, USA). VCaP and HEK293 cells were cultured in Dulbecco's Modified Eagle Medium (DMEM; ATCC) supplemented with 10% fetal bovine serum (FBS; ATCC). All cells were cultured in humidified conditions, at 37°C with 5% CO2. Media changes occurred every other day, and cells were passaged as needed, when confluent.
Generation of expression vectors
TMPRSS2-ERG3, TMPRSS2-ERG8, and wt-ERG3 (Flag- tagged) constructs were cloned into pIRES-EGFP plasmid vector, as previously described [17]. In addition, we generated TMPRSS2-ERG3-GFP and TMPRSS2-ERG8-RFP constructs by cloning TMPRSS2-ERG3 and TMPRSS2-ERG8 sequences into pAcGFP1-N1 and pDsRed- Monomer-N1 plasmid vectors, respectively. The expression of proteins was verified in HEK293 cells. Cell lysates derived from transfected cells were detected by western blot using 9FY ERG MAb. For the BiFC assay, sequences encoding the N-terminus (residues 1 to 173, VN) or C- terminus (residues 155 to 238, VC) fragments of the Venus fluorescence protein were fused to TMPRSS2-ERG3 or TMPRSS2-ERG8 coding sequences via a 10 amino acid linker. Plasmids were generated in pcDNA3 through a commercial vendor (GenScript; Piscataway, NJ, USA).
Western blot
Cell pellets from transfection procedures were lysed in Mammalian Protein Extract Reagent (M-PER; Pierce/Thermo Scientific, Rockford, IL, USA). Following pre-cleaning by centrifugation, protein concentrations of cell lysates were determined by using Protein Assay Reagent (Bio-Rad, Hercules, CA, USA). Lysates equivalent to 25 μg of protein were separated on NuPAGE Bis-Tris (4-12%) gels (Invitrogen, Carlsbad, CA, USA) and transferred onto PVDF membranes. Membranes were blocked in Blocking Buffer (LI-COR, Lincoln, NE, USA) and incubated with specific antibodies against ERG splice variants (ERG MAb 9FY; Biocare Medical Inc., Concord, CA, USA), C-MYC (Epitomics, Burlingame, CA, USA) and GAPDH (Santa Cruz biotechnology, Santa Cruz, CA, USA). Membranes were washed in Tris-Buffered Saline + Tween 20 (TBST) before incubation with appropriate secondary antibodies (goat anti- Mouse IRDye 8000CW or goat anti-Rabbit IRDye 680CW, LI-COR). Signals of proteins detected were visualized and quantitatively measured using the Odyssey infra-red imaging scanner and software (LI-COR).
Quantitative RT-PCR
RNA was isolated from cell pellets from transfection procedures using RNeasy RNA Isolation Kit (Qiagen; Germantown, MD, USA). cDNA was reverse transcribed from RNA, and quantitative RT-PCR was performed using 500 pg of cDNA. The appropriate primers for ERG3, ERG8, and C-MYC (Table 3) were used with gene-specific TaqMan probes for quantitative evaluation of ERG3 (5'- FAM-ACTAGGCCAGATTTACCA - 3') and C-MYC (5'- FAM-ACCTTTTGCCAGGAGCCTGCCTCT - 3') and SYBR Green for quantitative evaluation of ERG8, in 96-well plates. Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was used as an internal control gene. Results were calculated as a function of 2(−ΔΔCt).
Table 3. Primers used for analysis of RNA.
| Gene | Primer Sequences | Product | RefSeq | |
|---|---|---|---|---|
| ERG3 | Forward Reverse |
5'-CAGTATATCCTGAAGCTACGCAAAGA-3' 5'-GGTCCAGGCTGATCTCCT-3' |
80 | NM182918.3 |
| ERG8 | Forward Reverse |
5'-GGTACGAAAACACCCCTGTG-3' 5'-CCAAATCAACAGAGGCAGAA-3' |
150 | AY204742.1 |
| C-MYC | Forward Reverse |
5'-ACCACCAGCAGCGACTCTGA-3' 5'-TCCAGCAGAAGGTGATCCAGACT-3' |
117 | NM002467.4 |
| GAPDH | Forward Reverse |
5'-GAGCCACATCGCTCAGACACC-3' 5'-GTAGTTGAGGTCAATGAAGGGGTC-3' |
147 | NM001289745.1 |
Forward and reverse primer sequences for TM- ERG3, TM-ERG8, C-MYC and GAPDH used in quantitative RT-PCR.
Subcellular localization of ERG splice variants
HEK293 cells were seeded onto poly-lysine coated 12 mm round cover glass slides at 1×105 cells per well in 12-well plates. Cells were co- transfected with 1 μg each TMPRSS2-ERG3-GFP and TMPRSS2-ERG8-RFP vectors using Lipofectamine 2000. After 72 hours, cells were washed with PBS, mounted in ProLongFade Gold (Invitrogen) anti-fade mounting reagent containing 4'6-diamidino-2-phenylindole (DAPI) for DNA staining. Cellular localization of ERG3-GFP and ERG8-RFP proteins were visualized by using an inverted Leica DMIRE2 microscope equipped with a QImaging Retiga-EX CCD camera (Surrey, BC, Canada), operated by OpenLab Software.
Dual Luciferase Assays
HEK293 cells were transfected to examine the regulatory efficiency of both TMPRSS2-ERG3 and TMPRSS2-ERG8 on the ETS-regulated promoter of the myocyte- specific enhancer factor 2, mef2c. The endothelial cell enhancer derived mef2c was excised from pmef2c-F7-3-lacZ (a kind gift from Dr. Brian L. Black, UCSF) [34] and cloned upstream of a luciferase reporter in pGL4.24[luc2P/minP] in the mef2c construct. Cells seeded at 2.5×104 cells per well in 48-well plates were co-transfected with the luciferase construct and either wt-ERG3, TMPRSS2-ERG3, TMPRSS2-ERG8, or a mutated TMPRSS2-ERG3 construct, along with a Renilla construct at various doses, using Lipofectamine 2000 (Invitrogen). Cells were lysed in situ 24 hours post-transfection, rocked for 15 minutes at room temperature, and centrifuged at 15,000 × g for 15 minutes to pellet the cell debris. Cell supernatants were loaded onto Blackwell 96-well plates and evaluated for luciferase activity using the Dual Luciferase Reporter Assay System (Promega, Madison, WI, USA).
Overexpression of TMPRSS2-ERG8 in VCaP cells
VCaP cells were transfected with TMPRSS2- ERG8 (pIRES-TMPRSS2-ERG8-EGFP) vector in increasing concentrations using Lipofectamine 2000. Briefly, VCaP cells were seeded at 2×106 cells in 100 mm plates. Cells were incubated overnight at 37°C to reach 50% confluency at the time of transfection. The next day, cells were transfected with TMPRSS2-ERG8 (0, 2, 4, or 6 μg) using Lipofectamine 2000. An empty vector (pIRES-EGFP) was co-transfected into the cells, in order to maintain the amount of transfected DNA constant in each condition (6μg), where necessary. Cells were harvested 72 hours post-transfection using cell scrapers, centrifuged at 5,000 × g to pellet the cells, washed twice with PBS, and lysed for further analysis.
Bimolecular Fluorescence Complementation Assay
HEK293 cells were seeded at 5×104 cells in 35mm glass bottom dishes coated with poly-D-lysine. Cells were co-transfected with 1 μg each of VN-ERG3 and VC-ERG8 or VC-ERG3 and VN-ERG8 vectors using Lipofectamine 2000. VN- and VC- plasmids of the same splice variant transfected together were used as a positive control and both VN- or VC- plasmids transfected together were used as a negative control. After 24 hours, cells were washed once with PBS and stained with Hoechst 33342 nuclear stain (Invitrogen). Cells were again washed and visualized for presence of yellow fluorescent protein, indicating interactions between the splice variants, using microscopy (Leica, DMIRE2). For quantification of fluorescent cells using flow cytometry, HEK293 cells were seeded at 1×106 cells in 100mm plates. Transfections were carried out as above. After 24 hours, cells were washed and stained with Hoechst 33342 nuclear stain. Cells were trypsinized, pelleted, washed, and resuspended in PBS + 2% FBS. The cell suspension was filtered into flow cytometry tubes. An unstained control and a Hoechst-only stained control were used to set up gates, and quantification was performed for each set of transfections.
Clinicopathological analysis of patient data
The prostate tissue specimens used in this study were obtained from radical prostatectomy procedures under an Institutional Review Board– approved protocol at Walter Reed Army Medical Center. Detailed methods of laser capture microdissection (LCM) of tumor and benign epithelial cells, as well as quantitative gene expression are described in an earlier report from our laboratory [17]. Quantitative RT-PCR was carried out to analyze the expression of Type I and Type II ERG splice variants and C-MYC in LCM selected prostate cancer cells. We then examined the Type I/Type II ERG ratio in relation to C-MYC and PCA3 gene expression levels through Spearman's correlation analysis. The differences of Type I/Type II ERG ratios across C-MYC and PCA3 quartile groups, as well as down-regulated C-MYC vs. up-regulated C-MYC, were compared by using a Kruskal-Wallis test. A P-value of <0.05 was considered statistically significant.
Acknowledgments
The authors would like to express sincere thanks to Dr. Brian L. Black at the University of California, San Francisco for generously providing us with the mef2c plasmid. We would also like to thank Dr. Velpandi Ayyavoo, at the University of Pittsburgh for kindly providing us with the Vpr Venus constructs. In addition, we would like to thank Ms. Natallia Mikhalkevich for her superb technical support in this study and Mr. Stephen Doyle for the artwork. The prostate tissue specimens used in this study were obtained from radical prostatectomy procedures under an IRB- approved protocol at Walter Reed Army Medical Center. This study was supported by the DoD/CDMRP/PCRP grant PC073614 and by the EDRN/NCI ACN12011-001-0. The authors declare no conflict of interest.
DoD Disclaimer
The opinions and assertions contained herein represent the personal views of the authors and are not to be construed as official or as representing the views of the Department of the Army, the Department of Defense, or the United States Government.
REFERENCES
- 1.Albertson DG, Collins C, McCormick F, Gray JW. Chromosome aberrations in solid tumors. Nature genetics. 2003;34(4):369–376. doi: 10.1038/ng1215. [DOI] [PubMed] [Google Scholar]
- 2.Mani RS, Iyer MK, Cao Q, Brenner JC, Wang L, Ghosh A, Cao X, Lonigro RJ, Tomlins SA, Varambally S, Chinnaiyan AM. TMPRSS2-ERG-mediated feed-forward regulation of wild-type ERG in human prostate cancers. Cancer research. 2011;71(16):5387–5392. doi: 10.1158/0008-5472.CAN-11-0876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kumar-Sinha C, Tomlins SA, Chinnaiyan AM. Recurrent gene fusions in prostate cancer. Nature reviews Cancer. 2008;8(7):497–511. doi: 10.1038/nrc2402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tomlins SA, Rhodes DR, Perner S, Dhanasekaran SM, Mehra R, Sun XW, Varambally S, Cao X, Tchinda J, Kuefer R, Lee C, Montie JE, Shah RB, Pienta KJ, Rubin MA, Chinnaiyan AM. Recurrent fusion of TMPRSS2 and ETS transcription factor genes in prostate cancer. Science. 2005;310(5748):644–648. doi: 10.1126/science.1117679. [DOI] [PubMed] [Google Scholar]
- 5.Rubin MA, Maher CA, Chinnaiyan AM. Common gene rearrangements in prostate cancer. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2011;29(27):3659–3668. doi: 10.1200/JCO.2011.35.1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rao VN, Papas TS, Reddy ES. erg, a human ets-related gene on chromosome 21:alternative splicing, polyadenylation, and translation. Science. 1987;237(4815):635–639. doi: 10.1126/science.3299708. [DOI] [PubMed] [Google Scholar]
- 7.Reddy ES, Rao VN, Papas TS. The erg gene: a human gene related to the ets oncogene. Proceedings of the National Academy of Sciences of the United States of America. 1987;84(17):6131–6135. doi: 10.1073/pnas.84.17.6131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lin B, Ferguson C, White JT, Wang S, Vessella R, True LD, Hood L, Nelson PS. Prostate-localized and androgen-regulated expression of the membrane-bound serine protease TMPRSS2. Cancer research. 1999;59(17):4180–4184. [PubMed] [Google Scholar]
- 9.Ernst T, Hergenhahn M, Kenzelmann M, Cohen CD, Bonrouhi M, Weninger A, Klaren R, Grone EF, Wiesel M, Gudemann C, Kuster J, Schott W, Staehler G, Kretzler M, Hollstein M, Grone HJ. Decrease and gain of gene expression are equally discriminatory markers for prostate carcinoma: a gene expression analysis on total and microdissected prostate tissue. The American journal of pathology. 2002;160(6):2169–2180. doi: 10.1016/S0002-9440(10)61165-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Petrovics G, Liu A, Shaheduzzaman S, Furusato B, Sun C, Chen Y, Nau M, Ravindranath L, Dobi A, Srikantan V, Sesterhenn IA, McLeod DG, Vahey M, Moul JW, Srivastava S. Frequent overexpression of ETS-related gene-1 (ERG1) in prostate cancer transcriptome. Oncogene. 2005;24(23):3847–3852. doi: 10.1038/sj.onc.1208518. [DOI] [PubMed] [Google Scholar]
- 11.Vanaja DK, Cheville JC, Iturria SJ, Young CY. Transcriptional silencing of zinc finger protein 185 identified by expression profiling is associated with prostate cancer progression. Cancer research. 2003;63(14):3877–3882. [PubMed] [Google Scholar]
- 12.Zammarchi F, Boutsalis G, Cartegni L. 5' UTR control of native ERG and of Tmprss2:ERG variants activity in prostate cancer. PloS one. 2013;8(3):e49721. doi: 10.1371/journal.pone.0049721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Clark J, Merson S, Jhavar S, Flohr P, Edwards S, Foster CS, Eeles R, Martin FL, Phillips DH, Crundwell M, Christmas T, Thompson A, Fisher C, Kovacs G, Cooper CS. Diversity of TMPRSS2-ERG fusion transcripts in the human prostate. Oncogene. 2007;26(18):2667–2673. doi: 10.1038/sj.onc.1210070. [DOI] [PubMed] [Google Scholar]
- 14.Sementchenko VI, Schweinfest CW, Papas TS, Watson DK. ETS2 function is required to maintain the transformed state of human prostate cancer cells. Oncogene. 1998;17(22):2883–2888. doi: 10.1038/sj.onc.1202220. [DOI] [PubMed] [Google Scholar]
- 15.Rahim S, Uren A. Emergence of ETS transcription factors as diagnostic tools and therapeutic targets in prostate cancer. American journal of translational research. 2013;5(3):254–268. [PMC free article] [PubMed] [Google Scholar]
- 16.Owczarek CM, Portbury KJ, Hardy MP, O'Leary DA, Kudoh J, Shibuya K, Shimizu N, Kola I, Hertzog PJ. Detailed mapping of the ERG-ETS2 interval of human chromosome 21 and comparison with the region of conserved synteny on mouse chromosome 16. Gene. 2004;324:65–77. doi: 10.1016/j.gene.2003.09.047. [DOI] [PubMed] [Google Scholar]
- 17.Hu Y, Dobi A, Sreenath T, Cook C, Tadase AY, Ravindranath L, Cullen J, Furusato B, Chen Y, Thangapazham RL, Mohamed A, Sun C, Sesterhenn IA, McLeod DG, Petrovics G, Srivastava S. Delineation of TMPRSS2-ERG splice variants in prostate cancer. Clinical cancer research : an official journal of the American Association for Cancer Research. 2008;14(15):4719–4725. doi: 10.1158/1078-0432.CCR-08-0531. [DOI] [PubMed] [Google Scholar]
- 18.Wang J, Cai Y, Ren C, Ittmann M. Expression of variant TMPRSS2/ERG fusion messenger RNAs is associated with aggressive prostate cancer. Cancer research. 2006;66(17):8347–8351. doi: 10.1158/0008-5472.CAN-06-1966. [DOI] [PubMed] [Google Scholar]
- 19.Wang J, Cai Y, Yu W, Ren C, Spencer DM, Ittmann M. Pleiotropic biological activities of alternatively spliced TMPRSS2/ERG fusion gene transcripts. Cancer research. 2008;68(20):8516–8524. doi: 10.1158/0008-5472.CAN-08-1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hermans KG, Boormans JL, Gasi D, van Leenders GJ, Jenster G, Verhagen PC, Trapman J. Overexpression of prostate-specific TMPRSS2(exon 0)-ERG fusion transcripts corresponds with favorable prognosis of prostate cancer. Clinical cancer research : an official journal of the American Association for Cancer Research. 2009;15(20):6398–6403. doi: 10.1158/1078-0432.CCR-09-1176. [DOI] [PubMed] [Google Scholar]
- 21.Mohamed AA, Tan SH, Mikhalkevich N, Ponniah S, Vasioukhin V, Bieberich CJ, Sesterhenn IA, Dobi A, Srivastava S, Sreenath TL. Ets family protein, erg expression in developing and adult mouse tissues by a highly specific monoclonal antibody. Journal of Cancer. 2010;1:197–208. doi: 10.7150/jca.1.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.FitzGerald LM, Agalliu I, Johnson K, Miller MA, Kwon EM, Hurtado-Coll A, Fazli L, Rajput AB, Gleave ME, Cox ME, Ostrander EA, Stanford JL, Huntsman DG. Association of TMPRSS2-ERG gene fusion with clinical characteristics and outcomes: results from a population-based study of prostate cancer. BMC cancer. 2008;8:230. doi: 10.1186/1471-2407-8-230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sreenath TL, Dobi A, Petrovics G, Srivastava S. Oncogenic activation of ERG: A predominant mechanism in prostate cancer. Journal of carcinogenesis. 2011;10:37. doi: 10.4103/1477-3163.91122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Oikawa T, Yamada T. Molecular biology of the Ets family of transcription factors. Gene. 2003;303:11–34. doi: 10.1016/s0378-1119(02)01156-3. [DOI] [PubMed] [Google Scholar]
- 25.Rainis L, Toki T, Pimanda JE, Rosenthal E, Machol K, Strehl S, Gottgens B, Ito E, Izraeli S. The proto-oncogene ERG in megakaryoblastic leukemias. Cancer research. 2005;65(17):7596–7602. doi: 10.1158/0008-5472.CAN-05-0147. [DOI] [PubMed] [Google Scholar]
- 26.Hart AH, Corrick CM, Tymms MJ, Hertzog PJ, Kola I. Human ERG is a proto- oncogene with mitogenic and transforming activity. Oncogene. 1995;10(7):1423–1430. [PubMed] [Google Scholar]
- 27.Sato H, Minei S, Hachiya T, Yoshida T, Takimoto Y. Fluorescence in situ hybridization analysis of c-myc amplification in stage TNM prostate cancer in Japanese patients. International journal of urology : official journal of the Japanese Urological Association. 2006;13(6):761–766. doi: 10.1111/j.1442-2042.2006.01399.x. [DOI] [PubMed] [Google Scholar]
- 28.Rastogi A, Tan SH, Banerjee S, Sharad S, Kagen J, Srivastava S, McLeod DG, Srivastava S, A S. ERG monoclonal antibody (ERG MAb, 9FY) in the diagnosis and biological stratification of prostate cancer: Delineation of minimal epitope, critical residues for binding and molecular basis of specificity. Monoclon Antib Immunodiagn Immunother. 2014. In press. [DOI] [PubMed]
- 29.Sun C, Dobi A, Mohamed A, Li H, Thangapazham RL, Furusato B, Shaheduzzaman S, Tan SH, Vaidyanathan G, Whitman E, Hawksworth DJ, Chen Y, Nau M, Patel V, Vahey M, Gutkind JS, et al. TMPRSS2-ERG fusion, a common genomic alteration in prostate cancer activates C-MYC and abrogates prostate epithelial differentiation. Oncogene. 2008;27(40):5348–5353. doi: 10.1038/onc.2008.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Venkatachari NJ, Walker LA, Tastan O, Le T, Dempsey TM, Li Y, Yanamala N, Srinivasan A, Klein-Seetharaman J, Montelaro RC, Ayyavoo V. Human immunodeficiency virus type 1 Vpr: oligomerization is an essential feature for its incorporation into virus particles. Virology journal. 2010;7:119. doi: 10.1186/1743-422X-7-119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Magi-Galluzzi C, Tsusuki T, Elson P, Simmerman K, LaFargue C, Esgueva R, Klein E, Rubin MA, Zhou M. TMPRSS2-ERG gene fusion prevalence and class are significantly different in prostate cancer of Caucasian, African-American and Japanese patients. The Prostate. 2011;71(5):489–497. doi: 10.1002/pros.21265. [DOI] [PubMed] [Google Scholar]
- 32.Hagglof C, Hammarsten P, Stromvall K, Egevad L, Josefsson A, Stattin P, Granfors T, Bergh A. TMPRSS2-ERG expression predicts prostate cancer survival and associates with stromal biomarkers. PloS one. 2014;9(2):e86824. doi: 10.1371/journal.pone.0086824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Carrere S, Verger A, Flourens A, Stehelin D, Duterque-Coquillaud M. Erg proteins, transcription factors of the Ets family, form homo, heterodimers and ternary complexes via two distinct domains. Oncogene. 1998;16(25):3261–3268. doi: 10.1038/sj.onc.1201868. [DOI] [PubMed] [Google Scholar]
- 34.De Val S, Anderson JP, Heidt AB, Khiem D, Xu SM, Black BL. Mef2c is activated directly by Ets transcription factors through an evolutionarily conserved endothelial cell-specific enhancer. Developmental biology. 2004;275(2):424–434. doi: 10.1016/j.ydbio.2004.08.016. [DOI] [PubMed] [Google Scholar]
- 35.Furusato B, Tan SH, Young D, Dobi A, Sun C, Mohamed AA, Thangapazham R, Chen Y, McMaster G, Sreenath T, Petrovics G, McLeod DG, Srivastava S, Sesterhenn IA. ERG oncoprotein expression in prostate cancer: clonal progression of ERG-positive tumor cells and potential for ERG-based stratification. Prostate cancer and prostatic diseases. 2010;13(3):228–237. doi: 10.1038/pcan.2010.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Miettinen M, Wang ZF, Paetau A, Tan SH, Dobi A, Srivastava S, Sesterhenn I. ERG transcription factor as an immunohistochemical marker for vascular endothelial tumors and prostatic carcinoma. The American journal of surgical pathology. 2011;35(3):432–441. doi: 10.1097/PAS.0b013e318206b67b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li R, Pei H, Watson DK. Regulation of Ets function by protein - protein interactions. Oncogene. 2000;19(55):6514–6523. doi: 10.1038/sj.onc.1204035. [DOI] [PubMed] [Google Scholar]
- 38.Hawksworth D, Ravindranath L, Chen Y, Furusato B, Sesterhenn IA, McLeod DG, Srivastava S, Petrovics G. Overexpression of C-MYC oncogene in prostate cancer predicts biochemical recurrence. Prostate cancer and prostatic diseases. 2010;13(4):311–315. doi: 10.1038/pcan.2010.31. [DOI] [PubMed] [Google Scholar]
- 39.King JC, Xu J, Wongvipat J, Hieronymus H, Carver BS, Leung DH, Taylor BS, Sander C, Cardiff RD, Couto SS, Gerald WL, Sawyers CL. Cooperativity of TMPRSS2-ERG with PI3-kinase pathway activation in prostate oncogenesis. Nature genetics. 2009;41(5):524–526. doi: 10.1038/ng.371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zong Y, Xin L, Goldstein AS, Lawson DA, Teitell MA, Witte ON. ETS family transcription factors collaborate with alternative signaling pathways to induce carcinoma from adult murine prostate cells. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(30):12465–12470. doi: 10.1073/pnas.0905931106. [DOI] [PMC free article] [PubMed] [Google Scholar]