

Abstract
The α-helix is a ubiquitous secondary structural element that is almost exclusively observed in proteins when stabilized by tertiary or quaternary interactions. However, beginning with the unexpected observations of α-helix formation in the isolated C-peptide in ribonuclease A, there is growing evidence that a significant percentage (0.2%) of all proteins contain isolated stable single α-helical domains (SAH). These SAH domains provide unique structural features essential for normal protein function. A subset of SAH domains contain a characteristic ER/K motif, composed of a repeating sequence of ∼4 consecutive glutamic acids followed by ∼4 consecutive basic arginine or lysine (R/K) residues. The ER/K α-helix, also termed the ER/K linker, has been extensively characterized in the context of the myosin family of molecular motors and is emerging as a versatile structural element for protein and cellular engineering applications. Here, we review the structure and function of SAH domains, as well as the tools to identify them in natural proteins. We conclude with a discussion of recent studies that have successfully used the modular ER/K linker for engineering chimeric myosin proteins with altered mechanical properties, as well as synthetic polypeptides that can be used to monitor and systematically modulate protein interactions within cells.
Keywords: Biosensor, Fluorescence Resonance Energy Transfer (FRET), Myosin, Protein Structure, Protein-Protein Interaction, ER/K Alpha-Helix, ER/K Linker, Single Alpha-helical Domain (SAH), Coiled-coil
Coiled-coil or Single α-Helical (SAH) Domain?
Until recently, SAH2 domains in natural proteins were predicted by secondary structure prediction algorithms to form a coiled-coil, in part due to the high concentration of charged and polar residues that are also the hallmark of the coiled-coil motif (1). Among the multiple folds in globular proteins that stabilize α-helices, the coiled-coil motif has been extensively characterized and in general is the most predictable form of tertiary protein structure (2). In the coiled-coil motif, two or more α-helices are individually stabilized by sequence-specific packing at consensus hydrophobic patches. Extensive studies have elicited general sequence and structure rules that govern coiled-coil interactions. Briefly, the amino acid sequence of each α-helix in a coiled-coil is divided into heptads (7 residues) that form nearly two complete α-helical turns and span 1.05 nm along the helical axis. Each amino acid in the heptad is described by its relative position, moving from the N to C terminus, using the nomenclature abcdefg. In canonical dimeric coiled-coils, the a and d positions radiate away from the core of the α-helix, 60° apart and offset by 0.45 nm along the helix length, and are typically occupied by aliphatic hydrophobic residues, whereas polar residues comprise the rest of the positions. As the heptad is repeated, it forms a continuous hydrophobic patch located along a single face of the α-helix compatible with a tight intermolecular interaction between polypeptides with the same or similar heptad pattern (2). However, often polar or charged residues occupy the a and d positions, leading to local destabilization of the coiled-coil motif (2–4). By extension, when most or all of the a and d sites in consecutive heptads are occupied by polar residues, individual α-helices cannot be mutually stabilized through hydrophobic packing. In this event, the polypeptide either will exist as a monomeric random coil or in some cases will form an SAH domain. The SAH domain is a stable, monomeric, extended α-helix that is encoded by its primary amino acid sequence and exists in polar solvent independent of tertiary interactions with other protein motifs.
Stable Synthetic Alanine-based α-Helices
The rules governing the helicity of isolated peptides have been extensively studied. We highlight select studies of particular interest, and refer to Ref. 5 for a more comprehensive review. Prior to work on synthetic peptides from the Baldwin laboratory, including one derived from ribonuclease A (6–8), peptides shorter than 20 amino acids were not expected to show measurable helix formation in aqueous solution based on the statistical Zimm-Bragg model, which focuses on the propagation of spontaneous helicity and does not account for long distance electrostatic interactions between side chains (9). Marqusee et al. (6) found that a synthetic peptide, 16 residues long, containing primarily alanine residues sparsely interspersed with a single Glu or Lys for solubilization, had high helical content (up to 80%) in aqueous solution as measured by CD. This study highlighted the inherent helix-forming potential of alanine in the absence of electrostatic interactions between side chains. In a separate study, Marqusee and Baldwin (7) synthesized peptides containing 16 residues with three pairs of a Glu and a Lys separated by either 3 (i, i + 3) or 4 (i, i + 4) alanines. These peptides were synthesized, in part, to characterize the influence of electrostatic interactions between Glu and Lys on α-helix formation. Both peptides were soluble and monomeric in aqueous solution and had detectable helical content as determined by CD. However, the (i, i + 4) (i.e. (EAAAK)3) spacing yielded significantly higher helicity when compared with (i, i + 3), presumably due to the preferred rotamer configurations of the Glu and Lys side chains. Interestingly, the measured helicity was preserved at extremes of pH (from 2 to 12) and at high concentrations of NaCl, suggesting that the electrostatic interactions were primarily derived from salt bridges (H-bonded ionic interactions) rather than direct ionic interactions. The helix-stabilizing effects of E-K interactions on an alanine backbone were subsequently extended to D-K, E-R, and D-R pairs (10). A subsequent study with alanine-based peptides also defined the role of capping residues at the N and C termini in stabilizing isolated α-helices. In general, α-helix formation is aided by negatively charged residues at the N terminus and positively charged residues at the C terminus (11). This observation is consistent with an interaction of appropriately charged capping residues with the dipole moment of the protein α-helix, which is directed from its C to N terminus. Further, in the context of peptides, the charged residues at the ends can also undergo stabilizing electrostatic interactions with the free NH2 and COOH groups (11). These studies of synthetic alanine-based peptides provided precedence and laid the groundwork for identifying and predicting the stability of isolated α-helices.
The (EAAAK)n motif was subsequently incorporated into various synthetically engineered polypeptides. Arai et al. (12) evaluated the utility of (EAAAK)n (n = 2–5) motifs as rigid spacing linkers between a pair of green fluorescent protein variants (EBFP and EGFP). The linkers did not interfere with EBFP and EGFP folding as evaluated from their fluorescence spectra. FRET between EBFP and EGFP decreased, whereas helicity increased with linker length, suggesting that stabilization of the α-helix increased the spacing between the fluorescent proteins fused to the ends of the linker. In a subsequent study, the conformations of the (EAAAK)n linkers were evaluated by small angle x-ray scattering (SAXS). Short linkers (n ≤ 3) showed multimerization, whereas longer linkers (n ≥ 4) remain monomeric even at high concentrations (>25 μm). The radius of gyration increased with linker length and is higher than flexible unstructured linkers (GGGGS)n of the same length (13). Together, these studies support the use of the EAAAK linkers as extended spacers between polypeptides. Utilizing this structural property, the EAAAK linker has been employed to increase expression (14) and bioactivity (15) of fusion proteins. However, of concern for the use of tethering generic peptides with this particular linker, the EAAAK motif has been reported to have autocleavage properties at pH 6–7 (16). Regardless, this SAH domain both demonstrates the feasibility and the highlights potential utility of a modular genetic element to control intramolecular spacing between protein domains.
Stability of ER/K α-Helices
In a parallel vein, helix formation through E-K interactions, independent of alanine, was examined by Lyu et al. (17) with 18-amino acid peptides with either (E2K2)4 or (E4K4)4 repeats. Although the composition was exactly the same for both peptides, CD and 1H NMR data showed that the E4K4 peptide has 65% helical content, whereas the E2K2 peptide is essentially a random coil. This is consistent with studies in alanine peptides discussed earlier, wherein (i, i + 4) interactions, as in E4K4, stabilize the helical conformation, whereas the (i, i + 2) spacing in E2K2 cannot facilitate these ionic interactions (Fig. 1, b and c). These results were independent of peptide concentration in a range of 20–250 μm, indicating that oligomerization was not a factor in augmenting helicity. Additionally, pH and salt titrations showed that both direct ionic (E-K) and salt bridge interactions (H-bonded) likely contribute to the stability of E4K4 helices. This is in contrast to (EAAAK)n, which appears to be primarily stabilized by salt bridge interactions (7). Free energy calculations yielded 0.5 kcal/mol stabilization for each (i, i + 4) interaction in E4K4, and the contribution from each interaction is proposed to stabilize the isolated α-helical conformation.
FIGURE 1.

Glu and Arg/Lys side chain interactions stabilize a monomeric α-helix in solution. a, the primary amino acid sequence of the ER/K motif in Kelch motif family protein from Trichomonas vaginalis. Positively charged residues (Arg and Lys) are shown in blue, negatively charged Glu is depicted in red, polar residues are in black, and hydrophobic residues are in green. Note the recurrence of Glu and R/K spaced at (i, i + 4) intervals. b, a pinwheel diagram representing the spacing of residues along an α-helix. The (i, i + 4) spacing in the ER/K motif positions amino acids 40° apart, with a distance of 0.6 nm along the helical backbone. (Pinwheel adapted from Ref. 22.) c, ionic interactions and H-bonded salt bridges occur between Glu and R/K side chains with (i, i + 4) spacing. Top panel, top view down the backbone from the N terminus of one heptad of (E4K4)2 peptide from an MD simulation (18). Bottom panel, a 90° rotation visualizing the entire 16-amino acid peptide with a E-K interaction highlighted. d, a representative snap shot from a Monte Carlo simulation of the Kelch motif family protein ER/K α-helix (as in a (29)) highlighting the extended α-helical conformation in a large polypeptide (∼30 kDa).
The mechanisms that underlie the stability of E4K4 helices are also evident in molecular dynamics (MD) simulations. Sivaramakrishnan et al. (18) performed a replica exchange MD simulation on a (E4K4)2 peptide (Fig. 1c). Starting from either a random coil or a fully formed α-helix, both simulations converged on an α-helix. Thermal melting curves derived from the MD simulations matched previous experimentally measured helicities of this peptide (17). The simulations revealed dynamic and continuous interactions between side chains, with preferential i − 4 and i + 3 interactions centered on E(i) residues and i − 3 and i + 4 interactions centered on K(i) residues (Fig. 1c). These computations parallel experimental measurements by Olson et al. (19), who measured the influence of E-R side chain spacing on helicity. Monitoring the distances between the side chains in MD simulations, it was estimated that 45% of the time, direct ionic interactions occur between Glu and Lys, whereas solvent-separated salt bridges occur 37% of the time. This observation is consistent with the presence of both ionic and salt bridge interactions in E4K4 inferred from the pH dependence of helicity (17). Additionally, it was observed that the bulky side chains were able to partially shield backbone hydrogen bonds from the polar solvent. Thus, the stability of the α-helical core appears to arise from both shielded backbone hydrogen bonds and ER/K side chain interactions. In this regard, the ER/K motif has been likened to a tensegrity structure that is stabilized by the juxtaposition of “contractile” (backbone hydrogen bonds) and “tensile” (side chain) interactions (20). Overall, the observations in synthetic ER/K peptides have important structural implications in natural proteins as discussed in the next section.
Identification and Characterization of SAH Domains in Natural Proteins
Although the (EAAAK)n motif was the first identified to form SAH domains, SAH domains identified in natural proteins to date more closely resemble the ER/K motif (i.e. (E4(R/K)4)n). Smooth muscle caldesmon contains an ∼150-residue stretch in its central region that is essentially repeating segments of KAEEEKKAAEEK (21). Wang et al. (22) extensively characterized a 285-residue fragment of caldesmon that encompasses this ER/K stretch. CD revealed ∼55% helicity, which is consistent with a near continuous 150-amino acid α-helical region. The sedimentation profile of this polypeptide in ultracentrifugation experiments suggests a monomeric species over a wide range of protein concentrations (0.1–3.5 mg/ml). Rotary shadowed EMs revealed rods with an average length of 35 nm that is near the predicted length of an extended 150-residue α-helix. The rod thickness in EMs was significantly less than coiled-coils from tropomyosin or the myosin rod segment, again suggesting a single extended α-helix. In addition to stabilizing the α-helix, Wang et al. (22) proposed that the regularly spaced salt bridges may protect this α-helix from proteolytic cleavage. Caldesmon interacts with both actin and myosin through distinct domains located at the ends of its SAH domain. Although muscle caldesmon serves to inhibit the actin-activated ATPase activity of myosins, it does not inhibit the actin-myosin interaction (23). Therefore, the extended single α-helix in caldesmon likely serves as a spacer to allosterically modify this interaction. However, not all SAH domains identified are expected to function solely as spacers, for example the human programmed cell death 5 (PDCD5) protein.
The N-terminal 26-amino acid fragment (GSADEELEALRRQRLAELQAKHGDPG) of PDCD5 was demonstrated by Liu et al. (24) to form a single α-helix by both CD and NMR spectroscopic measurements (Fig. 2c). Deletion of the N-terminal α-helix of PDCD5 significantly attenuated the apoptosis-promoting effects triggered by serum withdrawal. Based on the differential nuclear translocation of full-length and truncated PDCD5, Liu et al. (24) propose a role for this SAH domain in the nuclear targeting of PDCD5.
FIGURE 2.
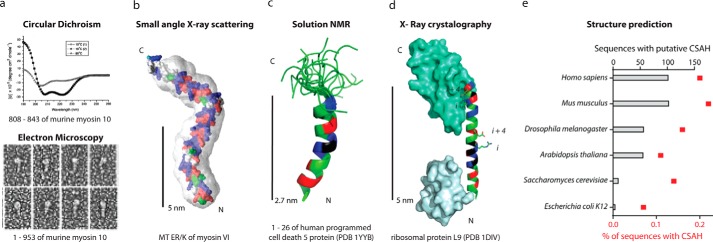
SAH domains have been observed in natural peptides or full-length proteins with multiple structural techniques. a, from Ref. 1, the putative coiled-coil region of myosin 10 forms an SAH domain. Top, CD of a peptide from murine myosin 10 has a canonical α-helical spectrum at 10 °C with a negative ellipticity at 222 nm (before and after heating to 80 °C) and a random coil spectra (with loss of ellipticity at 222 nm) at 80 °C. Bottom, rotary shadowed transmission electron microscopy of recombinant murine myosin 10 fragments. b, from Ref. 18, the predicted structure of the medial tail (MT) ER/K region (residues 916–981) of myosin VI docked into its SAXS envelope. c, from Ref. 24, an alignment of 20 solution states, determined by NMR, for the programed cell death 5 protein residues 1–26 following 1H-15N HSQC. PDB, Protein Data Bank. d, from Ref. 25, the x-ray crystal structure of ribosomal protein L9. e, from Ref. 32, the number and percentage of total sequences of putative CSAH domains identified from the primary amino acid sequences of the indicated organisms listed in Swiss-Prot and TrEMBL databanks utilizing the CSAH server. b–d, residues in SAH domains are colored as in Fig. 1.
The first high resolution structure of an SAH domain was revealed in a crystal structure of the Bacillus stearothermophilus ribosomal protein L9 (25). This protein contained a rigid and fully extended 34-amino acid linker region between two compact globular domains (Fig. 2d). A subsequent study synthesized the corresponding peptide and found that the peptide was primarily monomeric at concentrations up to 1 mm in analytical ultracentrifugation studies and primarily (∼70%) α-helical as determined by CD spectroscopy, a much higher degree of helicity than was expected based on its primary sequence (PANLKALEAQKQKEQRQAAEELANAKKLKEQLEK) (26). Further, they found that the helicity of the peptide could be disrupted by moderate salt concentrations (<500 mm NaCl), indicative of ionic side chain interactions contributing to the net stability of the α-helix. Although this peptide is far from the ideal E4K4 motif, it does have several predicted (i, i + 4) electrostatic interactions. The single α-helix was postulated to act as a rigid spacer between two globular domains of the L9 ribosomal protein such that they are properly positioned to bind RNA. In addition to its structural role in the intact proteins, the authors also suggest that the folding of the α-helix may initiate and stabilize the folding of the two domains at its ends. Specifically, if the α-helix does initiate the folding process, then the folding of the two domains at the ends will be interdependent.
The most extensively characterized SAH domains to date are the ER/K α-helices found in myosin X and VI. A study by Knight et al. (1) investigated a putative coiled-coil motif in murine myosin X that was highly enriched in charged residues in the a and d positions of the heptad repeats. The authors identified this as a peptide that did not conform with typical coiled-coil motifs and coined the term SAH to describe an isolated stable single α-helix (Fig. 2a). The 36-residue peptide studied (RQLLAEKRELEEKKRREEEKKREEEERERERAQR) resembles an ideal ER/K motif. Using 1H NMR, they found that the N-terminal 6 residues form a random coil, whereas all other residues in the peptide are α-helical. Using analytical ultracentrifugation, they determined the peptide to be monomeric at concentrations up to 700 μm. Surprisingly, they found that the α-helical content, as determined by CD, is less sensitive to higher salt concentrations than a synthetic 19-amino acid E4K4 peptide, presumably due to more stable electrostatic interactions. The study also investigated the SAH domain in the context of a nearly intact myosin X, which included a directly N-terminal putative coiled-coil region (120 amino acids total), by rotary shadowing and negative stain EM (Fig. 2a). They found that 90% of the peptide was monomeric, whereas 10% appeared dimerized. Further, in the dimeric population, only a small portion of the α-helical region appeared to be interacting. The “head” region of the entire myosin was 15 nm longer than expected (34.7 nm versus an expected 18.4 nm), corresponding to the expected length of 18 nm if the entire 120-amino acid region were in an extended α-helix. Finally, this study made predictions for two additional SAH domains in regions putatively described as coiled-coil regions in myosin VI and MyoM (Dictyostelium myosin) based on the ER/K motif in their primary amino acid sequence, both of which were subsequently confirmed.
Spink et al. (27) demonstrated that an ER/K motif was necessary for the large (36-nm) step size of dimeric myosin VI. The myosin VI medial tail was previously proposed to form a coiled-coil (28) based on a prediction of the PAIRCOILS algorithm. Spink et al. (27) showed that a polypeptide derived from the medial tail failed to demonstrate a cooperative melting profile characteristic of coiled-coil domains. Using a combination of spectroscopic approaches, the medial tail was found to be monomeric even at high concentrations (>200 μm). SAXS reconstructions revealed an extended conformation (∼10 nm) consistent with the medial tail as an ER/K motif (Fig. 2b). Forced dimerization of the medial tail, by insertion of a canonical coiled-coil at its N terminus, substantially diminished the step size and processivity of myosin VI. The rigidity of this ER/K α-helix is evident in its ability to extend the mechanical stroke of myosin VI as it resists an external force applied by optical tweezers (29). By pairing SAXS and optical trapping measurements of this and another naturally occurring ER/K motif (10 and 30 nm), the persistence length of the ER/K motif based on an ideal worm-like chain was estimated to be 15 nm (29) (Fig. 1d).
Predicting SAH Domains from Primary Sequence
Until recently, the identification of the SAH domain was limited to the handful of natural proteins where it had been biochemically characterized. Searching specifically for the minimal ER/K motif (E4(R/K)4)2, Sivaramakrishnan et al. (18) identified 123 distinct proteins, which had an average of 80% conformity to this motif, in 137 organisms ranging from archaea to humans. Peckham et al. (30) sought to identify SAH domains by examining sequences predicted to be coiled-coils based on their high charge density, but that failed to have hydrophobic residues in the a and d sites. After a manual screen of putative coiled-coil domains that were homologous to the bona fide SAH domain in murine myosin X, they found that up to 4% of proteins classified as coiled-coils might indeed be SAH domains instead. This study provides an upper bound of 0.5% of all proteins in the human database that contain an SAH domain (30).
Suveges et al. (31) took a more systematic and analytical approach to identify charged SAH domains. They generated two conceptually different computational models, one a scoring function that identifies characteristic (i, i + 4) or (i, i + 3) salt bridges (SCAN4CSAH), and the second one a fast Fourier transform approach that finds like-charged residues ∼1 heptad apart (FT_CHARGE). These methods detected several putative SAH domains by cross-examining the Swiss-Prot database for regions larger than 40 amino acids (31). Both search databases are available at the charged SAH (CSAH) server. Applied to the UniProt database, they provide a conservative estimate of 0.2% of all proteins in an organism as containing CSAH structural motifs (Fig. 2e). Interestingly, Homo sapiens were identified to have the highest number of CSAH-containing sequences in their genome. Of almost 300,000 proteins identified in this search, only one had a published high resolution structure, suggestive of the difficulty of obtaining x-ray crystal structures of CSAH domains. The identified sequences had high overlap with servers, identifying unstructured regions, and coiled-coil motifs. The authors postulate that this motif may be rapidly evolving and that single charge mutations may lead to fine tuning of sequences between SAH, coiled-coil, and disordered segments (32). As an alternative to differentiating between SAH and coiled-coil domains, Sunitha et al. (3) have developed a new computational tool termed COILCHECK+. This web interface informs the user of the strength of a potential coiled-coil interaction at the interface region based on the relative density of both the charged residues and the characteristic repeat pattern of hydrophobic residues. Although these bioinformatics screens require additional experimental verification, it appears that SAH domains are ubiquitous.
Applications for Modular ER/K Motifs in Protein and Cellular Engineering
The ER/K motif has readily found applications in protein engineering. Three separate studies have used chimeric approaches to investigate the interplay of ER/K α-helix mechanical properties on myosin function (Fig. 3). Baboolal et al. (33) demonstrated that an ER/K α-helix can function as a lever arm in myosin V by extending a single myosin stroke nearly as efficiently as its native rigid calmodulin-stabilized lever arm (Fig. 3a). However, in contrast to the native lever, the ER/K α-helix was not able to coordinate the chemomechanical cycles of the two myosin heads within a single dimer, possibly due to its lower bending rigidity. Nagy and Rock (34) demonstrated that the ER/K α-helix from myosin X was necessary and sufficient to engineer preferential processive movement of both myosin X and myosin V on fascin-linked actin bundles. Engineering additional flexibility into the native myosin X abolishes selectivity for actin bundles, suggesting that the ER/K α-helix selectively biases the orientation of the myosin heads within a dimer (Fig. 3b). Hariadi et al. (35) found that ensembles of myosin VI but not myosin V undergo linear directed movement on a dense cellular actin meshwork. Stochastic simulations revealed that myosin lever arm rigidity alone was sufficient to dictate the skewness of movement patterns on cellular actin networks. Swapping a portion of the myosin V lever with the ER/K α-helix from myosin VI was sufficient to linearize myosin V trajectories and vice versa (Fig. 3c). Together, these studies bridge the mechanical properties of ER/K α-helices with specific functions in myosin.
FIGURE 3.

The ER/K α-helix is a modular genetic motif that can be used to create myosin chimeras with altered mechanical properties. a, Baboolal et al. (33) created a myosin V (MyoV) chimera containing the putative ER/K α-helix from Dictyostelium myosin M. Left, the power stroke distances of WT myosin V, myosin V with truncation of two or four calmodulin stabilized IQ domains, and a chimera of myosin V with two native IQ domains and a 16.8-nm ER/K α-helix. Right, an extended and rigid ER/K α-helix can propagate force generated in the myosin catalytic domain to facilitate long processive steps on actin filaments. b, Nagy and Rock (34) generated multiple chimeras between myosin V and myosin X (MyoX) to assess structural elements that allow myosin X to preferably move on fascin-actin bundles. Left, representative chimeras that were used to identify that in myosin X, the ER/K α-helix and not the motor domain or step size dictates processive movement on fascin-actin bundles. Right, insertion of unstructured Gly-Ser-Gly residues between the SAH and IQ domains of myosin X disrupts preferential processivity on fascin-actin bundles. The ER/K α-helix alters the orientation of the motor domain, allowing it to favorably bind actin sites uniquely presented in fascin-actin bundles. c, Hariadi et al. (35) generated chimeras swapping the ER/K α-helix from myosin VI (MyoVI) with the IQ domains from myosin V while investigating the collective movement of multiple myosins tethered together. Left, multiple myosin V proteins, but not myosin VI, display meandering trajectories while traversing actin meshworks. Swapping regions of the lever arm containing the ER/K α-helix can reverse this phenomenon. Right, the IQ domains are likely more rigid than the ER/K α-helix, such that the inter-myosin force can selectively alter the accessibility of actin binding sites for the less rigid myosin VI.
In a radically different approach to protein engineering, the ER/K linker has also been used to tether and dictate the effective concentration of intramolecular protein-protein interactions (Fig. 4a). Sivaramakrishnan and Spudich (36) engineered a single polypeptide sensor containing an ER/K linker with an N-terminal calmodulin (CaM) and CFP variant attached by a flexible (GSG)2 linker and a C-terminal YFP variant attached with a (GSG)2 linker to a peptide known to dimerize with the Ca2+ bound CaM. The calcium-induced intramolecular interaction between CaM and its binding partner was detected by changes in FRET between CFP and YFP variants. In the absence of calcium, no significant FRET was detected with ER/K linkers of 73 amino acids or more (corresponding to >10 nm in length along the α-helical backbone). A dramatic increase in FRET was observed upon the addition of calcium. Unexpectedly, calcium-stimulated FRET was independent of the concentration of sensor at levels below the bimolecular dissociation constant, indicating that an intramolecular interaction was bringing the FRET pair on either end of the ER/K linker into close proximity. Competitive inhibition of FRET with increasing concentrations of unlabeled CaM was used to quantify the effective concentration of the intramolecular interaction. Regardless of the bimolecular dissociation constant, the effective concentration decreased by about an order of magnitude for each additional 10 nm of ER/K linker. Essentially, changing ER/K linker length from 10 to 30 nm altered the effective concentration of the intramolecular interaction from 10 μm to 100 nm (Fig. 4c) (for reference, the effective concentration of a 60-residue unstructured linker is ∼100 μm (37)). Further, similar results are observed when CaM and the CaM binding peptide are replaced by different interacting peptides (Fig. 4b). This trend is in contrast to the expected behavior of an ideal worm-like chain. One possible explanation is that a sensor in the closed state may introduce unfavorable conformations of the ER/K linker, which could increase the off-rate of the CaM-peptide interaction. However, the measured off-rate was found to be independent of ER/K linker length. The authors proposed a structural interpretation of these observations by suggesting that the ER/K linker undergoes rare stochastic breaks in helicity that create pivot points to facilitate interactions between the ends. The frequency of stochastic breaks scales linearly with α-helix length (α L). However, the breaks are unlikely to be spatially coordinated, resulting in far fewer conformations (α 1/L2) that bring the ends in close enough proximity to precipitate CaM-peptide interactions. Although this interpretation needs further testing, it is consistent with a previous SAXS study suggesting that certain α-helical peptides exhibit breaks along their length (38). The use of the ER/K linker to modulate protein-protein interactions was termed Systematic Protein Affinity Strength Modulation (SPASM). A key feature of the SPASM approach is its modularity, and the individual protein/peptide/fluorophore domains can be easily exchanged with basic molecular biology tools in most research laboratories.
FIGURE 4.

The ER/K α-helix dictates the effective concentration of peptides attached to its distal ends and can be used for protein/cellular engineering applications. a, top, The ER/K α-helix adopts an extended conformation in the absence of an interaction between polypeptides fused to its ends. Bottom, interaction between peptides stabilizes the closed conformation of the ER/K α-helix, which is detected by a reporter system (e.g. FRET between CFP and YFP). b, from Ref. 40, an example of the schematic depicted in a, in which the interacting FAK, FERM, and kinase domains, as well as the fluorescent protein FRET pair CFP and YFP, are separated by a disordered GSG linker or by ER/K linkers with extended lengths of 10–20 and 30 nm. Fluorescence emission of these polypeptides was monitored at concentrations significantly lower than the bimolecular dissociation constant for the kinase-FERM domain interaction; FRET is assessed by the characteristic increase in fluorescence at 525 nm. c, Sivaramakrishnan and Spudich (36) found that the effective concentration of the intramolecular interaction was dependent on the ER/K α-helix length. Longer ER/K α-helix length leads to a lower effective concentration. d–f, sample applications, some experimentally demonstrated (*) and others conceptual, of a modular ER/K linker in protein engineering. d, left, reporting protein-protein interactions using fluorescent protein FRET reporters between conditionally interacting protein/peptide pairs, as demonstrated by Malik et al. (39) investigating G protein-coupled receptor-G protein interactions. Top right, ER/K linker with flanking FRET reporters inserted between domains of a multidomain protein as reported by Swanson et al. (41) investigating protein kinase C. Middle right, tethering a bimolecular fluorescence complementation (BiFC) (42) pair to fluorescent protein with an ER/K α-helix, places the fluorescent protein beyond FRET distance to allow for quantification of expression levels of the sensor. Bottom right, a non-fluorescent readout of a conditional protein-protein interaction, for example, enzymatic activity of a split tobacco etch virus (TEV) protease, or deriving antibiotic resistance from a split β-lactamase (43). These approaches allow for increased stringency of detection by increasing the ER/K α-helix length, while controlling for stoichiometry of interacting proteins. e, left, ER/K α-helix length modulates protein-protein interactions to control the activity resulting from the interaction. For instance, an activity that is dependent on two proteins interacting can occur more or less frequently depending on the length of the ER/K α-helix as demonstrated by Ritt et al. (40) investigating the intramolecular interaction between FERM and kinase domains of FAK. Top right, the ER/K α-helix can be used to generate single polypeptide actuators, using inducible protein interaction pairs. For instance, the optogenetically controlled dimeric dronpa fluorescent proteins (44) can be used to modulate autoinhibition of a catalytic domain. Bottom right, the ER/K α-helix can be used to control co-recruitment of peptides to an intermolecular complex. Although the initial interaction will be dependent on polypeptide concentration, recruitment of the second peptide tethered by the ER/K linker will be dependent on the linker length. f, ER/K α-helices can be used to engineer structural scaffolds from polypeptides. A schematic of one such design is depicted in which the size of the structure can be adjusted by the length of the ER/K α-helix.
The SPASM approach has since been used to engineer protein-protein interactions in live cells. Single polypeptide FRET sensors based on SPASM have been used to detect G protein-selective conformations of G protein-coupled receptors (39) (Fig. 4d). Modulating effective concentration with varying ER/K linker length has been used to modify the enzymatic activity of focal adhesion kinase (FAK) and dissect the differential effects of kinase activity and domain-domain interactions in controlling cellular migration (40) (Fig. 4e). The ER/K linker provides control over the stoichiometry of expression, with minimal FRET in the absence of an intramolecular interaction. This feature has been used to understand the pH dependence of FAK regulation, while correcting for the effects of pH on the fluorescence levels of GFP variants. Additionally, the ER/K linker has found utility in modulating domain-domain interactions in multidomain proteins, which has yielded a coarse configuration of both intramolecular and intermolecular interactions in protein kinase C (41) (Fig. 4d). Beyond these studies, we propose that the ER/K linker may be readily used in conjunction with other synthetic protein tools, including split proteins and inducible protein interactions, as a means to study individual interactions or to engineer cellular systems (Fig. 4, d–f).
Conclusions
The SAH domain is a structural element found in numerous proteins, where it appears to operate as a semi-rigid structural element that tethers globular domains. Although it has only been extensively characterized in a few natural proteins, its sequence specifications and structural properties allow for its identification distinct from coiled-coils. The ER/K motif is a subset of SAH domains that have been extensively characterized through studies of the myosin family of molecular motors. ER/K α-helices, encoded by this motif, have already found applications for monitoring or systematically modulating protein interactions. The modularity of the ER/K motif makes it a versatile protein structural element in an expanding toolbox of technologies that are being deployed to dissect an integrated cellular signaling network. In addition to informing cellular function, sensors developed on an ER/K platform have direct applications for identifying new small molecule therapeutics.
Acknowledgment
We thank Mike Ritt for manuscript review.
This work was supported, in whole or in part, by National Institutes of Health Grants 1DP2 CA186752-01 and 1-R01-GM-105646-01-A1 and the American Heart Association (AHA) Scientist Development Grant Award (13SDG14270009) (to S. S.).
- SAH
- single α-helical domain
- CSAH
- charged SAH
- EBFP
- enhanced blue fluorescent protein
- EGFP
- enhanced green fluorescent protein
- SAXS
- small angle x-ray scattering
- MD
- molecular dynamics
- PDCD5
- programmed cell death 5
- CaM
- calmodulin
- SPASM
- Systematic Protein Affinity Strength Modulation
- FAK
- focal adhesion kinase
- Myo
- myosin.
REFERENCES
- 1. Knight P. J., Thirumurugan K., Xu Y., Wang F., Kalverda A. P., Stafford W. F., 3rd, Sellers J. R., Peckham M. (2005) The predicted coiled-coil domain of myosin 10 forms a novel elongated domain that lengthens the head. J. Biol. Chem. 280, 34702–34708 [DOI] [PubMed] [Google Scholar]
- 2. Woolfson D. N. (2005) The design of coiled-coil structures and assemblies. Adv. Protein Chem. 70, 79–112 [DOI] [PubMed] [Google Scholar]
- 3. Sunitha M. S., Nair A. G., Charya A., Jadhav K., Mukhopadhyay S., Sowdhamini R. (2012) Structural attributes for the recognition of weak and anomalous regions in coiled-coils of myosins and other motor proteins. BMC Res. Notes 5, 530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kreuzer S. M., Elber R. (2013) Coiled-coil response to mechanical force: global stability and local cracking. Biophys. J. 105, 951–961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Scholtz J. M., Baldwin R. L. (1992) The mechanism of α-helix formation by peptides. Annu. Rev. Biophys. Biomol. Struct. 21, 95–118 [DOI] [PubMed] [Google Scholar]
- 6. Marqusee S., Robbins V. H., Baldwin R. L. (1989) Unusually stable helix formation in short alanine-based peptides. Proc. Natl. Acad. Sci. U.S.A. 86, 5286–5290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Marqusee S., Baldwin R. L. (1987) Helix stabilization by Glu−…Lys+ salt bridges in short peptides of de novo design. Proc. Natl. Acad. Sci. U.S.A. 84, 8898–8902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bierzynski A., Kim P. S., Baldwin R. L. (1982) A salt bridge stabilizes the helix formed by isolated C-peptide of RNase A. Proc. Natl. Acad. Sci. U.S.A. 79, 2470–2474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zimm B. H., Bragg J. K. (1959) Theory of the phase transition between helix and random coil in polypeptide chains. J. Chem. Phys. 31, 526–535 [Google Scholar]
- 10. Huyghues-Despointes B. M., Scholtz J. M., Baldwin R. L. (1993) Helical peptides with three pairs of Asp-Arg and Glu-Arg residues in different orientations and spacings. Protein Sci. 2, 80–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Doig A. J., Baldwin R. L. (1995) N- and C-capping preferences for all 20 amino acids in α-helical peptides. Protein Sci. 4, 1325–1336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Arai R., Ueda H., Kitayama A., Kamiya N., Nagamune T. (2001) Design of the linkers which effectively separate domains of a bifunctional fusion protein. Protein Eng. 14, 529–532 [DOI] [PubMed] [Google Scholar]
- 13. Arai R., Wriggers W., Nishikawa Y., Nagamune T., Fujisawa T. (2004) Conformations of variably linked chimeric proteins evaluated by synchrotron X-ray small-angle scattering. Proteins 57, 829–838 [DOI] [PubMed] [Google Scholar]
- 14. Amet N., Lee H. F., Shen W. C. (2009) Insertion of the designed helical linker led to increased expression of Tf-based fusion proteins. Pharm. Res. 26, 523–528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bai Y., Shen W. C. (2006) Improving the oral efficacy of recombinant granulocyte colony-stimulating factor and transferrin fusion protein by spacer optimization. Pharm. Res. 23, 2116–2121 [DOI] [PubMed] [Google Scholar]
- 16. Wu Y. J., Fan C. Y., Li Y. K. (2009) Protein purification involving a unique auto-cleavage feature of a repeated EAAAK peptide. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 877, 4015–4021 [DOI] [PubMed] [Google Scholar]
- 17. Lyu P. C., Gans P. J., Kallenbach N. R. (1992) Energetic contribution of solvent-exposed ion pairs to α-helix structure. J. Mol. Biol. 223, 343–350 [DOI] [PubMed] [Google Scholar]
- 18. Sivaramakrishnan S., Spink B. J., Sim A. Y., Doniach S., Spudich J. A. (2008) Dynamic charge interactions create surprising rigidity in the ER/K α-helical protein motif. Proc. Natl. Acad. Sci. U.S.A. 105, 13356–13361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Olson C. A., Spek E. J., Shi Z., Vologodskii A., Kallenbach N. R. (2001) Cooperative helix stabilization by complex Arg-Glu salt bridges. Proteins 44, 123–132 [DOI] [PubMed] [Google Scholar]
- 20. Spudich J. A., Sivaramakrishnan S. (2010) Myosin VI: an innovative motor that challenged the swinging lever arm hypothesis. Nat. Rev. Mol. Cell Biol. 11, 128–137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bryan J., Imai M., Lee R., Moore P., Cook R. G., Lin W. G. (1989) Cloning and expression of a smooth muscle caldesmon. J. Biol. Chem. 264, 13873–13879 [PubMed] [Google Scholar]
- 22. Wang C. L., Chalovich J. M., Graceffa P., Lu R. C., Mabuchi K., Stafford W. F. (1991) A long helix from the central region of smooth muscle caldesmon. J. Biol. Chem. 266, 13958–13963 [PMC free article] [PubMed] [Google Scholar]
- 23. Lash J. A., Sellers J. R., Hathaway D. R. (1986) The effects of caldesmon on smooth muscle heavy actomeromyosin ATPase activity and binding of heavy meromyosin to actin. J. Biol. Chem. 261, 16155–16160 [PubMed] [Google Scholar]
- 24. Liu D., Yao H., Chen Y., Feng Y., Chen Y., Wang J. (2005) The N-terminal 26-residue fragment of human programmed cell death 5 protein can form a stable α-helix having unique electrostatic potential character. Biochem. J. 392, 47–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hoffman D. W., Davies C., Gerchman S. E., Kycia J. H., Porter S. J., White S. W., Ramakrishnan V. (1994) Crystal structure of prokaryotic ribosomal protein L9: a bi-lobed RNA-binding protein. EMBO J. 13, 205–212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kuhlman B., Yang H. Y., Boice J. A., Fairman R., Raleigh D. P. (1997) An exceptionally stable helix from the ribosomal protein L9: implications for protein folding and stability. J. Mol. Biol. 270, 640–647 [DOI] [PubMed] [Google Scholar]
- 27. Spink B. J., Sivaramakrishnan S., Lipfert J., Doniach S., Spudich J. A. (2008) Long single α-helical tail domains bridge the gap between structure and function of myosin VI. Nat. Struct. Mol. Biol. 15, 591–597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Rock R. S., Ramamurthy B., Dunn A. R., Beccafico S., Rami B. R., Morris C., Spink B. J., Franzini-Armstrong C., Spudich J. A., Sweeney H. L. (2005) A flexible domain is essential for the large step size and processivity of myosin VI. Mol. Cell 17, 603–609 [DOI] [PubMed] [Google Scholar]
- 29. Sivaramakrishnan S., Sung J., Ali M., Doniach S., Flyvbjerg H., Spudich J. A. (2009) Combining single-molecule optical trapping and small-angle x-ray scattering measurements to compute the persistence length of a protein ER/K α-helix. Biophys. J. 97, 2993–2999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Peckham M., Knight P. J. (2009) When a predicted coiled coil is really a single [small α]-helix, in myosins and other proteins. Soft Matter. 5, 2493–2503 [Google Scholar]
- 31. Süveges D., Gáspári Z., Tóth G., Nyitray L. (2009) Charged single α-helix: a versatile protein structural motif. Proteins 74, 905–916 [DOI] [PubMed] [Google Scholar]
- 32. Gáspári Z., Süveges D., Perczel A., Nyitray L., Tóth G. (2012) Charged single α-helices in proteomes revealed by a consensus prediction approach. Biochim. Biophys. Acta 1824, 637–646 [DOI] [PubMed] [Google Scholar]
- 33. Baboolal T. G., Sakamoto T., Forgacs E., White H. D., Jackson S. M., Takagi Y., Farrow R. E., Molloy J. E., Knight P. J., Sellers J. R., Peckham M. (2009) The SAH domain extends the functional length of the myosin lever. Proc. Natl. Acad. Sci. U.S.A. 106, 22193–22198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Nagy S., Rock R. S. (2010) Structured post-IQ domain governs selectivity of myosin X for fascin-actin bundles. J. Biol. Chem. 285, 26608–26617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hariadi R. F., Cale M., Sivaramakrishnan S. (2014) Myosin lever arm directs collective motion on cellular actin network. Proc. Natl. Acad. Sci. U.S.A. 111, 4091–4096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sivaramakrishnan S., Spudich J. A. (2011) Systematic control of protein interaction using a modular ER/K α-helix linker. Proc. Natl. Acad. Sci. U.S.A. 108, 20467–20472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Robinson C. R., Sauer R. T. (1998) Optimizing the stability of single-chain proteins by linker length and composition mutagenesis. Proc. Natl. Acad. Sci. U.S.A. 95, 5929–5934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zagrovic B., Jayachandran G., Millett I. S., Doniach S., Pande V. S. (2005) How large is an α-helix? Studies of the radii of gyration of helical peptides by small-angle X-ray scattering and molecular dynamics. J. Mol. Biol. 353, 232–241 [DOI] [PubMed] [Google Scholar]
- 39. Malik R. U., Ritt M., DeVree B. T., Neubig R. R., Sunahara R. K., Sivaramakrishnan S. (2013) Detection of G protein-selective G protein-coupled receptor (GPCR) conformations in live cells. J. Biol. Chem. 288, 17167–17178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ritt M., Guan J. L., Sivaramakrishnan S. (2013) Visualizing and manipulating focal adhesion kinase regulation in live cells. J. Biol. Chem. 288, 8875–8886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Swanson C. J., Ritt M., Wang W., Lang M. J., Narayan A., Tesmer J. J., Westfall M., Sivaramakrishnan S. (2014) Conserved modular domains team up to latch-open active PKCα. J. Biol. Chem. 289, 17812–17829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kerppola T. K. (2008) Bimolecular fluorescence complementation (BiFC) analysis as a probe of protein interactions in living cells. Annu. Rev. Biophys. 37, 465–487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Shekhawat S. S., Ghosh I. (2011) Split-protein systems: beyond binary protein-protein interactions. Curr. Opin. Chem. Biol. 15, 789–797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Zhou X. X., Chung H. K., Lam A. J., Lin M. Z. (2012) Optical control of protein activity by fluorescent protein domains. Science 338, 810–814 [DOI] [PMC free article] [PubMed] [Google Scholar]