

Background: Fucosidase releases terminal fucose from functionally diverse glycans.
Results: The first eukaryotic fucosidase crystal structures reveal two active-site conformations and a novel βγ-crystallin domain.
Conclusion: Catalytically essential glutamate in glycoside hydrolase family 29 (GH29) fucosidases is structurally conserved despite locally poor sequence conservation.
Significance: Conformational flexibility involving the general acid/base Glu exists in GH29 fucosidases across different life domains.
Keywords: Crystal Structure, Crystallin, Glycoprotein, Molecular Dynamics, Substrate Specificity, Fucosidase
Abstract
The secreted glycoside hydrolase family 29 (GH29) α-l-fucosidase from plant pathogenic fungus Fusarium graminearum (FgFCO1) actively releases fucose from the xyloglucan fragment. We solved crystal structures of two active-site conformations, i.e. open and closed, of apoFgFCO1 and an open complex with product fucose at atomic resolution. The closed conformation supports catalysis by orienting the conserved general acid/base Glu-288 nearest the predicted glycosidic position, whereas the open conformation possibly represents an unreactive state with Glu-288 positioned away from the catalytic center. A flexible loop near the substrate binding site containing a non-conserved GGSFT sequence is ordered in the closed but not the open form. We also identified a novel C-terminal βγ-crystallin domain in FgFCO1 devoid of calcium binding motif whose homologous sequences are present in various glycoside hydrolase families. N-Glycosylated FgFCO1 adopts a monomeric state as verified by solution small angle x-ray scattering in contrast to reported multimeric fucosidases. Steady-state kinetics shows that FgFCO1 prefers α1,2 over α1,3/4 linkages and displays minimal activity with p-nitrophenyl fucoside with an acidic pH optimum of 4.6. Despite a retaining GH29 family fold, the overall specificity of FgFCO1 most closely resembles inverting GH95 α-fucosidase, which displays the highest specificity with two natural substrates harboring the Fucα1-2Gal glycosidic linkage, a xyloglucan-derived nonasaccharide, and 2′-fucosyllactose. Furthermore, FgFCO1 hydrolyzes H-disaccharide (lacking a +2 subsite sugar) at a rate 103-fold slower than 2′-fucosyllactose. We demonstrated the structurally dynamic active site of FgFCO1 with flexible general acid/base Glu, a common feature shared by several bacterial GH29 fucosidases to various extents.
Introduction
Fucose (Fuc),2 although relatively low in abundance in the biosphere, has many important functions. In mammals l-Fuc-containing conjugates in the form of glycans, glycoproteins, or glycolipids in particular are involved in forming structural determinants of ABO blood group antigens, mediating host-microbe interactions, leukocyte-endothelial adhesion, fertilization and embryonic development, and signal transduction via O-fucosylation directly on Ser and Thr of specific types of protein modules as well as implicated in various human disease states including cancer, inflammation, and fucosidosis (1–4). In plants fucosylation appears to have two distinct functions. The first function is analogous to the function of fucosylation in mammal, serving a regulatory function. Fucosylation is believed to be involved in cell to cell communication in plants (5) and in reproductive development during flowering (6). Fucose appears to also have a second, structural function in plants in the form of terminal side chain modifications of cell wall polysaccharides including hemicellulose xyloglucan and pectin. Two allelic l-fucose-deficient Arabidopsis thaliana mutants (mur1-1 and 1-2) are dwarfed, and their rosette leaves do not grow normally due to reduced borate cross-linking of cell wall pectin rhamnogalacturonan II (7). Fucose has also been identified in the xyloglucan of Arabidopsis (8), apple fruit, potato (9), and many other plants (4). Xyloglucan is postulated to bind to the surface of cellulose fibrils and assist in their organization in the cell wall (10, 11). Computer simulations predict that fucosylation of xyloglucan prevents self-aggregation of the xyloglucan chains (12). The requirement for fucosylation on xyloglucan in growth and development does not seem to be absolute, as Arabidopsis mur2 mutants lacking fucosyltransferase appear to grow normally despite having no fucosylation in its xyloglucan (13). Besides the aforementioned biological distribution and functions of fucose, l-Fuc is also present in brown algal polysaccharide fucoidan as the main-chain and side-chain residues with substantial amounts of sulfation and acetylation modifications (14), bacterial and fungal exopolysaccharides (3, 4), bacterial lipopolysaccharides (3, 4), and non-mammalian animals (3, 4).
α-l-Fucosidases are exoglycosidases that catalyze the hydrolysis of α-linked l-fucose (Fuc) from the non-reducing end of glycans (15, 16). Depending on the specificity of α-l-fucosidases, they actively hydrolyze a variety of terminal fucosyl linkages typically in the form of α1,2 linkages to d-galactose (Gal), α1,3/4/6 linkages to N-acetylglucosamine (GlcNAc), α1,3 linkages to d-glucose (Glc) that are present in different natural substrates like milk oligosaccharides, plant cell wall polysaccharides, various glycoproteins, or glycolipids harboring various A, B, H, and Lewis blood group antigens or their analogs (15–24). In addition, α-l-fucosidase isolated from the marine mollusk Pecten maximus has been reported to efficiently release fucose without extensive depolymerization of fucoidan from Ascophyllum nodosum (24), which contains alternating Fucα1-4Fuc and Fucα1-3Fuc backbone units (14). On the other hand, an exofucosidase from abalone (Haliotis gigantea) liver has been demonstrated to catalyze complete hydrolysis of fucoidan from brown alga Ecklonia cava (25).
α-l-Fucosidases occur mainly in two glycosyl hydrolase (GH) families 29 and 95 (see the CAZy website) (26) (Table 1). GH29 enzymes retain glycosidases, whereas GH95 enzymes are inverting glycosidases (27–32). Enzymes in GH29 exist in all domains of life, whereas GH95 members have not been found in animals (15, 26). Crystal structural studies have shown that GH29 enzymes have a conserved catalytic active site formed at one end of a (β/α)8 triosephosphate isomerase (TIM) barrel domain, whereas GH95 enzymes have their active site formed by an (α/α)6 helical barrel domain (22, 23, 27–29). Besides GH29 and GH95 enzymes, A. thaliana also expresses a novel lipase-like fucosidase named AtFXG1, which has not been classified in CAZy (33).
TABLE 1.
Comparison of GH29 and GH95 families of α-fucosidases
| GH29 | GH95 | |
|---|---|---|
| Active-site folda | (β/α)8 | (α/α)6 |
| Hydrolysis mechanismb | Retaining | Inverting |
| Preferred fucosyl linkagesc | α1,2/3/4/6 | α1,2 |
| pNP-Fuc activityc,d | High/low | None |
| Taxonomic distributione | All kingdoms | Absent in animals |
d Different GH29 fucosidases show specificities toward various natural linkages. pNP-Fuc activity is reported to be high for most GH29 fucosidases except plant and bacterial GH29 α1,3/4-fucosidases or FgFCO1.
GH29 family fucosidases have diverse substrate specificities. Human GH29 fucosidases, FUCA1 and FUCA2, are both active on α1,2/3/4/6-linked fucosyl substrates as well as pNP-Fuc (20, 21, 31, 34). FUCA1 is a lysosomal enzyme whose deficiency causes fucosidosis, a disease characterized by progressive mental and motor deterioration (2). FUCA2 is a secreted enzyme and is essential for Helicobacter pylori adhesion during infection of gastric cancer cells (34). In contrast, GH29 enzyme Bt2970 from the enteric bacterium Bacteroides thetaiotaomicron is much more active on pNP-Fuc than on natural α-fucosyl linkages (22). On the other hand, several GH29 fucosidases from plant and bacteria prefer α1,3/4-linked fucosyl substrates to pNP-Fuc (19, 22, 35).
Paper et al. (17) characterized two α-fucosidases from the plant pathogenic fungi Fusarium graminearum (FgFCO1) and Fusarium oxysporum (FoFCO1). The former enzyme was active on xyloglucan-derived fucosylated oligosaccharide but not on pNP-Fuc, and the latter enzyme had the opposite specificity. Based on overall amino acid sequence similarity, GH29 α-fucosidases from fungi could be grouped into two subfamilies with FgFCO1 and FoFCO1 belonging to different subfamilies (17). Furthermore, Sakurama et al. (22, 23) classified plant, bacterial, and animal GH29 enzymes into two subfamilies, A and B, with a conserved Trp in the latter forming a +1 sugar binding subsite specific for α1,3/4-linked fucosyl substrates. However, without more characterized structures of both eukaryotic and prokaryotic GH29 enzymes, a general structure-based classification of GH29 subfamilies and understanding of their diverse specificities is not yet possible.
Crystal structural (22, 23, 27, 28) and site-directed mutagenesis (30–32, 36) studies of GH29 enzymes from various organisms have led to the assignment of the key catalytic residues, including a highly conserved nucleophile Asp residue and a structurally conserved general acid/base Glu residue. However, the poor sequence conservation near the real catalytic Glu residues across different species has made it impractical to assign the key general acid/base Glu solely based on sequence alignment (15, 23, 27, 31, 32, 36). Studies with the mechanism-based inhibitor 2-fluoro-fucosyl and analysis of site-directed mutants have confirmed the retaining catalytic mechanism (Fig. 1) (27, 28, 30–32). Specific conformational changes of the fucopyranose ring during catalysis via a 1C4 ↔ 3H4 ↔ 3S1 pathway with a possible 3H4 oxocarbonium-like transition state have been proposed based on crystal structures of the Michaelis complex, covalent intermediate, and computational modeling of free l-Fuc (Fig. 1) (27, 28, 37).
FIGURE 1.
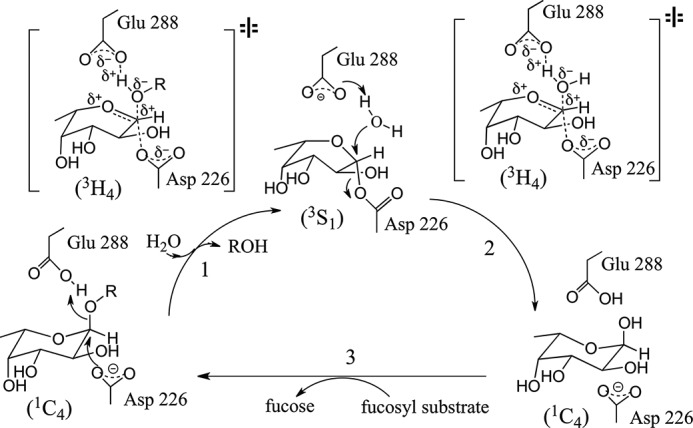
Catalytic mechanism proposed for FgFCO1. The catalytic cycle starts with formation of a fucosyl-enzyme intermediate by nucleophilic attack from Asp-226 on C1 of fucose with concomitant leaving of the free aglycon (ROH) stabilized by a proton donated from the general acid Glu-288. The fucosyl-enzyme intermediate is then hydrolyzed to yield an enzyme-product complex with the aided deprotonation of water by general base Glu-288. The replacement of the product Fuc by fucosylated substrate completes the catalytic cycle. The conformation change of the Fuc moiety during catalysis follows a 1C4-3S1-1C4 pathway with a possible 3H4 oxocarbonium-like transition state (intermediates and proposed transition states are based on crystal structures PDB 1HL9 and 2WVS) (26, 27, 37). The proposed transition state has an sp2-hybridized carbon center with oxygen atoms of nucleophilic and leaving group on each side of the plane. The other active-site residues like His-126, His-127, His-171, and Lys-272 of FgFCO1 may also be involved in stabilization of the transition state via hydrogen bonding interactions.
To date, structures of fucosidases from retaining GH29 and inverting GH95 families have been determined for multiple bacterial species but not yet for eukaryotic or archael members according to CAZy and Protein Data Bank (PDB) databases. In the current work we report the x-ray crystal structures of two active-site conformations of a GH29 α-l-fucosidase (FgFCO1) from the plant-pathogenic fungus F. graminearum (synonym Gibberella zeae). Furthermore, we identify 2′-fucosyllactose (2′-FL, Fig. 2) as an effective substrate for FgFCO1.
FIGURE 2.
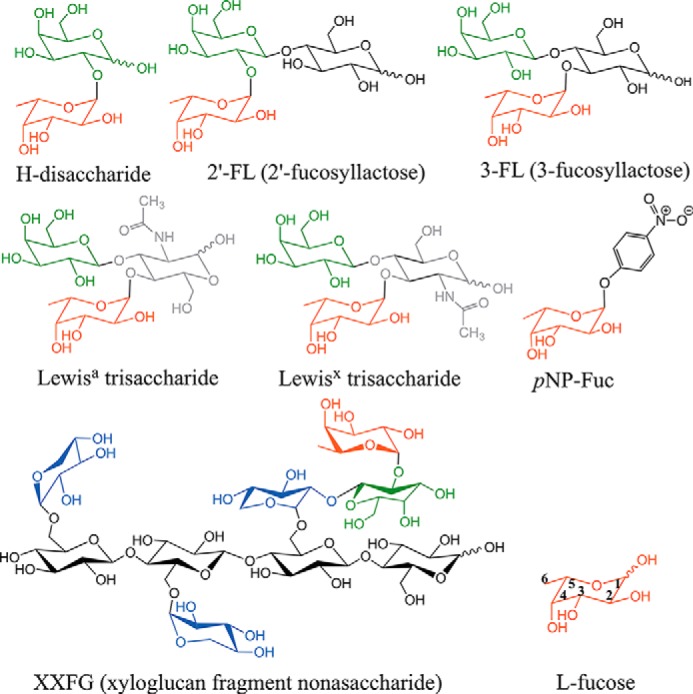
Structures of fucosylated substrates and products relevant to this study. l-Fucose (Fuc) is shown in red, d-galactose (Gal) is in green, d-xylose (Xyl) is in blue, d-glucose (Glc) is in black, and N-acetyl-d-glucosamine NAG (GlcNAc) is in gray. The XXFG nonasaccharide structure is drawn in stereo in analogy to the structures of the xyloglucan fragment XLLG in complex with GH16-family endo-xyloglucanase (PDB codes 2VH9 and 1UMZ) (77, 78). The shorthand nomenclature for xyloglucan oligosaccharides XXFG and XLLG (the latter not shown) is based on McDougall and Fry (76). The backbone of xyloglucan is β1,4-linked d-glucose. G stands for Glc, X is for Xylα1-6Glc, L is for Galβ11-22Xylα1-6Glc, and F is for Fucα1-2Galβ1-2Xylα1-6Glc. pNP-Fuc stands for p-nitrophenyl-α-l-fucoside; 2′-fucosyllactose, Fucα1-2Galβ1-4Glc; 3-fucosyllactose (3-FL), Galβ1-4(Fucα1-3)Glc. Lewisx trisaccharide is Galβ1-4(Fucα1-3)GlcNAc, and Lewisa trisaccharide is Galβ1-3(Fucα1-4)GlcNAc.
EXPERIMENTAL PROCEDURES
pNP-α-l-Fuc, xylan, and 2-hydroxyethyl cellulose were purchased from Sigma. Fucosylated oligosaccharides were obtained from V-Labs (Covington, LA). Xyloglucan was purchased from Megazyme International (Dublin, Ireland). Polyethylene glycol monomethyl ether 2000 (PEG 2000 MME) as a 50% (w/v) solution and individual reagent #24 (0.1 m Tris, pH 8.0, 30% (w/v) PEG MME 2000) from PEGRx 1 screen were both obtained from Hampton Research (Aliso Viejo, CA). Free Fuc was assayed with the K-FUCOSE kit from Megazyme International (Dublin, Ireland). SYPRO orange dye (5000× concentrate) was from Invitrogen. All other chemicals and reagents were purchased from Sigma or Fisher and were used without further purification.
Cloning, Expression, and Purification of FgFCO1
The coding region of FgFCO1 (GenBankTM AFR68935) including its native signal peptide was cloned into pPICZAα (Invitrogen) without the C-terminal Myc/His6 tags and expressed in Pichia pastoris (17). The enzyme was purified using gel filtration and ion exchange chromatography. The purified protein was buffer-exchanged into 25 mm Tris, pH 7.5, and stored at a concentration of 17–19 mg/ml at 4 °C.
Cloning, Expression, and Purification of the Crystallin Domain
Crystallin domain of FgFCO1 was graciously provided by Dr. Philip Gao from the University of Kansas, Protein Production Facility (Lawrence, KS). The DNA including the coding region of the crystallin domain of FgFCO1 was ordered from IDT and cloned into pTBSG vectors to introduce either a His6 or His6-MBP (maltose-binding protein)-cleavable tag at the N terminus. Fusion proteins were expressed in Escherichia coli with isopropyl 1-thio-β-d-galactopyranoside induction and purified by nickel-nitrilotriacetic acid affinity chromatography. Final products crystallin (Cryst)-His and Cryst-MBP both contained residues 501–585 of FgFCO1 with three extra residues (SNA) at the N terminus after cleavage of the corresponding fusion tags by TEV protease.
Deglycosylation of FgFCO1
FgFCO1 was predicted to have three N-glycosylation sites on Asn-187, Asn-280, and Asn-401 (here residues numbered for the protein without signal peptide). Because proteins expressed in P. pastoris can be abnormally N-glycosylated, we deglycosylated FgFCO1 by treatment with Endo H or peptide N-glycosidase F (both from New England Biolabs, Ipswich, MA). FgFCO1 (17–19 mg/ml in 25 mm Tris, pH 7.5), Endo H (500,000 units/ml in 20 mm Tris HCl, 50 mm NaCl, 5 mm Na2EDTA, pH 7.5), and 500 mm sodium citrate, pH 5.5, were mixed in a 10:1:1 volume ratio. FgFCO1 in 25 mm Tris, pH 7.5, was directly mixed with peptide N-glycosidase F at a volume ratio of 10:1. The reactions were incubated at room temperature (22 ± 2 °C) for 24–48 h. Deglycosylated fucosidase was either directly used for crystallization without further purification or stored at 4 °C. Aliquots of samples for mass spectral analysis were frozen at −20 °C and thawed immediately before analysis.
Mass and Sequence Analyses of FgFCO1
MALDI-TOF mass spectrometry was performed with an Applied Biosystem 4700 instrument in linear mode at the Mass Spectrometry/Proteomics Facility of the Biotechnology Center at the University of Wisconsin-Madison. The N terminus was sequenced by Edman degradation using an Applied Biosystems 494 Procise Protein Sequencer/140C Analyzer at the Iowa State University Protein Facility.
Crystallization, Diffraction Data Acquisition, and Structure Determination
Initial crystallization trials in the PEGRx HT screen via the sitting-drop vapor diffusion method at 20 °C yielded crystals under several conditions with Endo H-treated FgFCO1. FgFCO1 as expressed in P. pastoris or after treatment with peptide N-glycosidase F failed to grow crystals with the same sets of screens, and an overall trend of higher solubility was observed for these highly glycosylated forms of the enzyme compared with Endo H-deglycosylated fucosidase. Endo H-treated FgFCO1 was crystallized by the batch method by mixing 1 μl of 14–16 mg/ml Endo H-treated fucosidase solution in 25 mm Tris buffer, pH 7.5, with 1 μl of precipitant solution containing 0.1 m Tris, pH 8.0, and either 30% or 40% (w/v) PEG MME 2000. The crystallization drops were manually set up in batch mode using MRC 2 Well crystallization plates (Hampton Research) without reservoir solution, sealed with Crystal Clear sealing film (Hampton Research), and incubated at 20 °C.
Rectangular plate or block-shaped crystals grew after 2–3 days with the longest dimension reaching 100–500 μm. Ligand soaking was tried with Fuc and various fucosylated substrates at a final concentration of 1–50 mm including pNP-Fuc, 2′-FL, H-disaccharide (Fucα1-2Gal), 3-fucosyllactose, Lewisa trisaccharide (Galβ1-3(Fucα1-4)GlcNAc), and Lewisx trisaccharide (Galβ1-4(Fucα1-3)GlcNAc) with crystals grown under either 30% or 40% PEG MME 2000 conditions. Crystals were cryoprotected by transferring into MiTeGen's LV CryoOil (MiTeGen, Ithaca, NY) and flash-frozen in liquid nitrogen.
Diffraction data were collected at Argonne National Laboratory on the LS-CAT 21-ID-G beamline for the open form (30% PEG MME 2000 condition) using a wavelength of 0.97856 Å and a MAR 300 CCD detector and the 21-ID-F beamline for the closed form (40% PEG MME 2000 condition) using a wavelength of 0.97872 Å and a MAR 225 detector. Data sets were collected to a resolution of 1.56 Å (apo) or 1.38 Å (Fuc-bound) for the open form and a resolution of 1.4 Å for the apo closed form. The data were indexed, integrated, and scaled using HKL-2000 (38). The apo open form of structure of FgFCO1 was determined by molecular replacement using AutoMR (Phaser) (39) and Autobuild programs (40) of the PHENIX suite (41) with monomer polypeptide coordinates without ligands or water using Thermotoga maritima GH29 fucosidase as the search model (PDB code 1HL8) (27). The apo closed form was determined by molecular replacement with the apo open form as the search model. The first two-to-four N-terminal residues were disordered in all three FgFCO1 structures. The remaining residues including the C-terminal Ile-585 were successfully built into the structures based on clear electron density except for the GGSFT loop region (residues 391–395) in the open forms where little electron density was observed for Gly-392 and Ser-393. For this poor electron density region in the open form structures, the loop was built based on weak backbone electron density and by relying on favorable side-chain rotamers and favorable geometry statistics after real-space refinement in COOT (42). This loop region was clearly ordered in the closed form, which gives confidence in the connectivity of the polypeptide chain and that the poor density for the corresponding residues of the loop in the open form was not due to proteolysis.
Two copies of the protein were found in the asymmetric unit displaying an overall crystallographic pseudo-C2 symmetry. Ligand soaking was successful only when we used l-Fuc at 50 mm and crystals grown in 30% PEG MME 2000. On the other hand, crystals grown in 40% PEG MME 2000 failed to take up Fuc or other ligands in the trials but instead always showed strong density for Tris and glycerol occupying the Fuc binding site. In the Fuc-bound open form, one Fuc ligand was added to each active site of the two protein molecules with COOT based on the clear electron density in the Fo − Fc omit map. In the apo closed form a Tris and a glycerol were built into each active site following the same method, with one of the active sites showing density for an additional glycerol that was also built in. N-Glycosylation GlcNAc and mannose residues were also built into all three structures based on clear electron density in the Fo − Fc omit map. Alternative conformations of ligands or polypeptide were also built in and adjusted based on electron density, and the occupancies of different conformers were refined in PHENIX. The structures were completed with alternating rounds of manual model building with COOT and refinement with PHENIX. Water was added and updated during refinement.
The final structures were refined to the same resolution limit as in data collection with favorable Rcryst and Rfree values (Table 2). Model quality was assessed using MolProbity (43). All the information on data collection and refinement statistics is summarized in Table 2. All structures in the figures were rendered using PyMOL (44). Protein interfaces in the structures were analyzed by PDBePISA (45).
TABLE 2.
Statistics for x-ray data collection and structural refinement
Values in parentheses are for the highest resolution shell.
| Statistic | Open form | Open form with Fuc | Closed form |
|---|---|---|---|
| Protein Data Bank ID code | 4PSP | 4PSR | 4NI3 |
| Spacegroup | P1 | P1 | P1 |
| Cell dimensions | |||
| a, b, c (Å) | 53.9, 75.9, 80.8 | 54.2, 76.0, 81.0 | 53.5, 75.5, 80.4 |
| α, β, γ (°) | 105.9, 107.2, 106.8 | 105.7, 107.3, 106.9 | 105.6, 107.2, 106.7 |
| Wavelength (Å) | 0.9786 | 0.9786 | 0.9787 |
| Resolution of data collection (Å) | 50.0-1.59 (1.59-1.56) | 50.0-1.38 (1.40-1.38) | 50.0-1.40 (1.42-1.40) |
| No. of reflections (measured/unique) | 575,637/148,010 | 833,789/214,983 | 736,291/198,786 |
| Completeness % (Å) | 96.9 (94.4) | 96.1 (93.2) | 95.4 (92.0) |
| Redundancy | 3.9 (3.1) | 3.9 (3.0) | 3.7 (3.5) |
| Rsyma | 0.062 (0.45) | 0.062 (0.62) | 0.051 (0.56) |
| I/σb | 21.5 (2.6) | 16 (1.8) | 18 (2.3) |
| Resolution range in refinement (Å) | 41.24-1.56 | 43.28-1.38 | 37.93-1.40 |
| No. of unique reflections (work/test) | 148,007/7,414 | 214,972/10,714 | 198,748/1,905 |
| Rcrystc | 13.5 (20.2) | 13.8 (23.6) | 14.5 (22.0) |
| Rfreed | 16.9 (24.8) | 16.6 (27.2) | 16.7 (29.3) |
| Mean coordinate errore (Å) | 0.14 | 0.14 | 0.13 |
| Rmsd bond length (Å) | 0.009 | 0.011 | 0.01 |
| Rmsd bond angles (°) | 1.2 | 1.4 | 1.2 |
| Average B value (Å2) (overall/protein/waters/ligand) | 19.7/17.4/31.3/27.2 | 19.3/17.0/31.6/25.3 | 21.7/19.9/33.3/27 |
| No. of non-hydrogen atoms | 11,810 | 11,877 | 11,227 |
| No. of protein atoms | 9,752 | 9,807 | 9,548 |
| No. of waters | 1,786 | 1,765 | 1,402 |
| No. of ligands and sugars | 6 Tris, 4 glycerol, 7 GlcNAc, 9 mannose 3 Na+ | 2Fuc, 6 Tris, 4 glycerol, 7 GlcNAc, 9 mannose, 3 Na+ | 7 Tris, 4 glycerol, 7 GlcNAc, 8 mannose 3 Na+ |
| Ramachandran Statisticsf (%) | 97.55, 2.37, 0.08 | 97.80, 2.20, 0 | 97.66, 2.34, 0 |
a Rsym = Σhkl Σi |Ii(hkl) − 〈I(hkl)〉|/Σhkl ΣiIi(hkl), where Ii(hkl) is the intensity of an individual measurement of the symmetry related reflection, and 〈I(hkl)〉 is the mean intensity of the symmetry related reflections.
b I/σ is defined as the ratio of averaged value of the intensity to its standard deviation.
c Rcryst = Σhkl‖Fobs| − |Fcalc‖/Σhkl|Fobs|, where Fobs and Fcalc are the observed and calculated structure-factor amplitudes.
d Rfree was calculated as Rcryst using randomly selected small fractions of the unique reflections (5%, open form; 1%, closed form) that were omitted from the structure refinement.
e Mean coordinate error was calculated based on maximum likelihood.
f Ramachandran statistics indicate the percentage of residues in the most favored, additionally allowed and outlier regions of the Ramachandran diagram as defined by MOLPROBITY.
Small Angle X-ray Scattering (SAXS) Data Acquisition and Analysis
SAXS data were collected on a Bruker-Axs Nanostar small angle scattering spectrometer equipped with a Bruker Turbo rotating copper anode x-ray source generator and a Vantec-2000 detector at the National Magnetic Resonance Facility of University of Wisconsin, Madison. Measurements were carried out at room temperature for protein samples in 50 mm sodium acetate, pH 5.0, and loaded into a 1-mm capillary at a wavelength of 1.54 Å and exposure time 6–9 h. The scattering intensity was obtained by subtracting the background buffer scattering from the sample scattering. Little concentration dependence of scattering intensity was observed for different protein concentrations (1.25, 2.5, 5 mg/ml). Further SAXS data analysis was performed for protein samples at 2.5 mg/ml using ATSAS 2.5.2 package (46). SAXS data were processed with GNOM 4.5a program (47) to calculate P(r) plot. A Guinier plot was generated using AutoRg program (46). The GNOM output was then used with DAMMIF 1.1.2 (48) to calculate 15 independent ab initio dummy atom models. The mean value of normalized spatial discrepancy of the models was 0.692 with a S.D. of 0.034. The models were averaged using DAMAVER 5.0 (49), filtered with DAMFILT 5.0, and superimposed onto the crystal structure of FgFCO1 with SUPCOMB 2.3 (50). Theoretical scattering curves based on crystal structures were fit to experimental SAXS data using CRYSOL 2.8.2 (51). The momentum transfer, i.e. the modulus of the scattering vector, is denoted as s = 4πsin(θ)/λ, where 2θ is the scattering angle, and λ is the x-ray wavelength. Non-covalently bound ligands and water molecules were removed from the crystal structures during calculation and structure comparison.
Steady-state Kinetic Assays and pH Dependence
FgFCO1 at a concentration of 0.1–10 μm was mixed with various substrates (100 μm–5 mm) in 500-μl reactions. The substrate solutions were preincubated at 37 °C for 5–10 min, and the reactions were sampled every 10–20 min over 4 h. The buffer was 50 mm sodium acetate, pH 5, for all fucosylated oligosaccharides, and different buffers were used for pNP-Fuc. For reactions with pNP-Fuc, aliquots of the reaction were mixed with 200 mm borate, pH 9.8, and the absorbance was measured at 400 nm. Calculation of p-nitrophenol concentration used an extinction coefficient of 18 mm−1 cm−1 (30). For reactions with fucosylated oligosaccharides, formation of free Fuc was measured using the Megazyme K-FUCOSE kit.
We observed a linear dependence of initial reaction rate versus substrate concentration and nonsaturating behavior with all the active substrates tested up to 1 mm, including 2′-FL, pNP-Fuc, and H-disaccharide. Each reaction was repeated three times, and the average used to calculate kcat/Km S.D. were <20%.
Thermofluor Assay and Tm Determination
Thermal denaturation reactions of FgFCO1 were performed using an iCycler equipped with the MyiQ real-time PCR detection system (Bio-Rad) (52). The system was precalibrated before the run. Fucosidase at 1–2 μm was mixed with SYPRO orange (diluted to a final concentration of 15× from a 5000× stock solution), and 25 μl of each sample was transferred to the bottom of the well of an iQ96-well PCR plate and sealing with optical clear Microseal B Adhesive Seal (Bio-Rad) that was compatible with the fluorescent optical system. The plate was preincubated in the iCycler at 20 °C before raising the temperature from 20 °C to 95 °C. Fluorescence was scanned at 1 °C increments and dwell times >10 s. The excitation and emission wavelengths were 485 and 575 nm with 30- and 20-nm bandwidths, respectively. The negative first derivative of relative fluorescence, −d(RFU)/dt, was plotted against T with Excel software to determine the melting temperature (Endo H). Only one negative peak in the plot was observed for each sample of FgFCO1, indicating a concerted melting of the enzyme following a two-state model. The pH dependence of Endo H was performed for FgFCO1 isolated from P. pastoris in the indicated buffers and indicated pH values. Each condition was run in triplicate.
Affinity Gel Electrophoresis of Crystallin Domain of FgFCO1
Native polyacrylamide gels (6%) were polymerized in the absence or presence of 0.1% of different soluble polysaccharides including xyloglucan, xylan, and 2-hydroxyethyl cellulose. Bovine serum albumin (BSA), Sigma) was included in the gels as an internal standard. FgFCO1 crystallin or BSA (each 5 ug) was loaded on the gels. Electrophoresis was performed for 1.5 h at 4 °C in a Mini-PROTEAN system (Bio-Rad) at a constant voltage of 150 V. The gels were stained with Coomassie Blue. Binding of crystallins to soluble polysaccharides was tested following published procedures (53). The migration distances of the crystallin and BSA reference were defined as the distance between the major band of the protein and the bottom of the corresponding loading well. Relative mobilities for the crystallins were calculated as the migration distance of the crystallin major band divided by the migration distance of the reference major band. Measurement of the migration distance was performed directly on the gels and had an accuracy limit of 0.1 cm.
Bioinformatics
Protein molecular weight and extinction coefficients at 280 nm were calculated with ProtParam (54). N-Glycosylation sites were predicted with NetNGlyc 1.0. The signal peptide was predicted with SignalP version 4.1 (55). DALI was used to find structural homologs in the PDB (56). BLASTP was used to search for homologous sequences in the PDB and NCBI non-redundant sequence databases (57). Pairwise structural alignment was performed by PDBeFold (58). Sequence alignments were performed with ClustalW2 (59).
RESULTS AND DISCUSSION
Overall Crystal and Solution Structures of FgFCO1
The crystal structures of FgFCO1 were determined and refined to resolutions of 1.56 Å (apo, open form), 1.38 Å (Fuc-bound, open form), and 1.40 Å (apo, closed form). All of these structures belong to the P1 space group and have nearly the same unit cell dimensions (Table 2). The overall structure of FgFCO1 consists of three domains including an N-terminal conserved (β/α)8 TIM barrel domain forming the active site followed by a conserved β-sandwich domain, typical of GH29 fucosidases, and a non-conserved C-terminal βγ-crystallin domain (Figs. 3a and 4, a and b). The enzyme shows N-glycosylation on three Asn residues as predicted. Both open and closed forms display the same overall folds (Fig. 3b) with difference limited to active-site conformations (Fig. 3c and 3d).
FIGURE 3.

Overall crystal structures and two active-site conformations of FgFCO1. a, the overall structure of FgFCO1 (the open form) consisting of an N-terminal (β/α)8 TIM barrel domain forming the active site (white) and two β-sandwich domains (orange and magenta). The two linker loops between domains were colored yellow and purple, respectively. The secondary structures are represented as schematics. Fuc (spheres) and post-translational N-glycosylation (sticks) are color-coded with carbon (green) oxygen (red), and nitrogen (blue). Three glycosylated Asn residues are labeled. b, pseudo-C2 symmetry of two FgFCO1 fucosidase molecules in the asymmetric unit in both the closed form (pink) and the open form (white). c, the alignment of the active sites of two conformations shows that the general acid/base Glu-288 adopts two different orientations with concomitant conformational change of Arg-289 and the adjacent loop containing Phe-394. Nucleophile Asp-226 and most other active-site residues within 5 Å of the bound fucose show minimal movement between the open and closed forms. d, electron density maps are shown for the amino acids near the active site of closed (top) and open (bottom) forms of the enzyme. Glycerol (GOL) and Tris (TRS) are identified in the closed form, and l-fucose (Fuc) was observed in the open form. The 2Fo − Fc map (blue) is contoured at 2σ around the protein residues, and the Fo − Fc omit map (red), calculated with the ligands omitted, is contoured at 3σ around the ligands.
FIGURE 4.

Structure alignments of GH29 α-l-fucosidases and homologs of crystallin domains. a, FgFCO1 (white, open; pink, closed), T. maritima (green) (27), and B. thetaiotaomicron (cyan) (28) fucosidases show conserved N-terminal (β/α)8 TIM barrel and β-sandwich domains and a non-conserved crystallin domain in FgFCO1. Fucose bound in FgFCO1 is shown as spheres. b, crystallin domains of FgFCO1 (pink) and human eye γS-crystallin (blue) (70) differ from M. xanthus Protein S (yellow) (68), and an F. johnsoniae GH64 enzyme (gold) (69). The calcium binding motif (N/D)(N/D)XX(T/S) is conserved in the crystallin domains of M. xanthus Protein S and an F. johnsoniae GH64 enzyme but not FgFCO1 or human eye γS-crystallin. Calcium ions are shown as spheres. c, stereo view of the active sites of fucosidases. Amino acid residues near active sites are labeled with the one-letter code and the residue number for FgFCO1 (after which are non-conserved residues of T. maritima and B. thetaiotaomicron enzymes). d, stereo view of backbones (ribbon) and conserved general acid/base Glu of fucosidases. Structures are aligned based on conserved nucleophile Asp and hydrophobic residue Trp/Phe (sticks), the latter interacting with C4, C5, and C6 of fucose (spheres). Non-conserved long GGSFT-loops of FgFCO1 (open and closed) are highlighted as a schematic. Two conformations of α1,3/4-l-fucosidase BiAfcB involving general acid/base Glu from B. longum subsp. Infantis (blue, open; orange, closed) are also shown in d (19, 23). FUC, l-fucose; GOL, glycerol; TRS, Tris. Structures were aligned by PyMOL (a, c, and d) or PDBeFold (b).
The closest structural homologues to FgFCO1 in PDB are bacterial fucosidases from T. maritima and B. thetaiotaomicron with Cα r.m.s.d. values of 1.8 and 2.4 Å, 391 and 379 residues aligned, respectively (PDB codes 1HL8 and 2WVV, Fig. 4a) (27, 28). The corresponding sequence identities with FgFCO1 are 26% (PDB code 1HL8) and 20% (PDB code 2WVV). The next closest homologues are GH29 α1,3/4-fucosidases from Bacteroides longum subsp. infantis and B. thetaiotaomicron, with Cα r.m.s.d. values of 2.9, 2.9, and 3.5 Å of 299, 305, and 304 residues aligned, respectively (PDB codes 3MO4, 3EYP, and 3GZA) (19, 61).3 Both the overall folds of the N-terminal domains and active site residues are well conserved between FgFCO1 and bacterial homologues (Fig. 4, a and c). Active-site alternative conformations involving the structurally conserved general acid/base Glu residue are found in both FgFCO1 and B. longum subsp. infantis α1,3/4-l-fucosidase (PDB code 3MO4, open; PDB code 3UES, closed; Fig. 4d) (19, 23). The characteristic structural features of FgFCO1 different from the reported structures of prokaryotic fucosidases are the presence of the C-terminal βγ-crystallin domain, N-glycosylation, a flexible loop containing GGSFT (residues 391–395), and several active-site substitutions including His-171, which replaces a Tyr, and Lys-272, which replaces an Arg (Fig. 4).
The closest sequence homologues to FgFCO1 in the NCBI non-redundant database are mostly putative GH29 α-fucosidases from plant-pathogenic fungi such as Fusarium pseudograminearum, F. oxysporum, Colletotrichum graminicola, and Magnaporthe oryzae. Overall sequence identities range from 65 to 97%. Weaker homologues are present in various prokaryotes, especially species of Streptomyces in the Actinobacteria (overall identities 48–54%). The characteristic features near the active site of FgFCO1, including the flexible GGSFT-loop and His-171, are highly conserved in these close homologues. Low homology orthologues (sequence identities 23–28%) from various animals including human FUCA1 and FUCA2 lack these features.
Two copies of the enzyme were found per asymmetric unit showing an overall pseudo-C2 symmetry (Fig. 3b). PDBePISA analysis of the protein interfaces in the structures did not reveal any specific interactions that could result in the formation of stable quaternary structures. We further confirmed the monomeric state of FgFCO1 in solution by comparing the experimental SAXS data and the theoretical scattering curves of the crystal structures of FgFCO1 (Fig. 5a). The ab initio SAXS envelope also fits the monomeric crystal structures of FgFCO1 well (Fig. 5b). Despite different oligomeric states being reported for many prokaryotic and eukaryotic GH29 enzymes (16, 20, 23, 24, 27, 36), we did not find any evidence for oligomerization.
FIGURE 5.

Solution small angle x-ray scattering confirms monomeric structure of FgFCO1. a, p(r) plot. b, Guinier plot. c, fit of experimental SAXS data with theoretical scattering curves for monomer and crystallographic pseudo-dimer calculated with CRYSOL 2.8.2. s = 4πsin(θ)/λ. Both open and closed dimer structures fit poorly (the latter not shown). d, alignment of solution SAXS envelope (cyan dummy atom model) generated by DAMFILT with open (white) and closed (pink) crystal structures of FgFCO1 (secondary structures are shown as schematics and N-glycosylation sugars as sticks). Non-covalently bound ligands and water molecules were removed from the crystal structures during calculation and structure comparison. SAXS data of Endo H-treated FgFCO1 are shown. SAXS data of as-isolated FgFCO1 (not shown) also evidently fit better to monomer structure over dimer structure, showing more heterogeneity than the Endo H-treated sample possibly due to higher and varied degree of glycosylation (Rg values of 27.29 ± 4.87 Å from the Guinier plot and 27.66 Å from the P(r) plot, and Dmax of 95.53 Å; CRYSOL experimental and theoretical Rg values of 27.16 Å and 27.18 Å, and χ of 1.770 for fitting of open monomer structure).
At current resolution of the SAXS experiment (i.e. ∼17 Å in real space), we found the spatial organization of the adjacent domains of FgFCO1 to be very similar at solution equilibrium to that in crystals, although the remaining differences between SAXS envelope and the crystal structures might indicate possible restricted movement in solution (Fig. 5b). Interestingly, the theoretical scattering curve of the open form of FgFCO1 fits the experimental SAXS data somewhat better than that of the closed conformation (Fig. 5a). Accordingly, the calculated SAXS envelope aligns slightly better to the open form than the closed form, with normalized spatial discrepancy values of 0.889 and 0.895, respectively (Fig. 5b). Counterintuitively, the flexible GGSFT loop of the open crystallographic form lies further outside the aligned SAXS envelope than the closed form despite an overall statistically better fit of the former in both real space and reciprocal space. This may indeed suggest the disorder of this loop in solution under the experimental conditions near catalytic pH optimum and in the absence of ligands.
Active-site Conformational Flexibility of FgFCO1
We solved two conformational forms of the enzyme, “open” and “closed,” by using different crystal growth conditions. The structures differ mainly in the active-site loop conformation and orientation of the conserved general acid/base Glu-288 (Fig. 3, c and d). In the open Fuc-bound form, Glu-288 has its side chain disordered and positioned more than 7 Å away from the O-1 hydroxyl group of bound Fuc corresponding to the glycosidic oxygen (the apo open form shows the same conformation as the Fuc-bound). In contrast, the closed form has clear electron density for the Glu-288 side-chain carboxylic group, which is oriented in proximity to where substrate can be bound at the active site. Thus, the closed form is catalytically more competent than the open form, with the latter likely representing an unreactive state.
A close examination of the loop region where Glu-288 resides shows a significant change in the loop conformation involving WERG (residues 287–290) from the open form to the closed form. We also found that a loop containing GGSFT (residues 391–395) is disordered in the open form, i.e. it has high individual B factors and low electron density but is ordered in the closed form, where it forms a partial cap over the entrance to the substrate binding pocket. The concerted conformational change in the WERG and GGSFT loops suggest a possible interaction between these two loops. Indeed, we found that in the open form, the side chains of Arg-289 and Phe-394 on these two loops are stacked together with possible van der Waals and cation-π interactions (62). On the other hand, in the closed form, the conserved general acid/base Glu-288 interacts with Phe-394, and Trp-287 interacts with Gly-392 via possible van der Waals interactions, whereas Arg-289 no longer interacts with Phe-394 but interacts instead with conserved Trp-311. Subtle movement of Trp-311 between the open and closed conformations may potentially affect substrate affinity via stacking interactions (Fig. 3c). In addition, Phe-394 and Phe 227 in the closed conformation may form an additional subsite near the fucose binding site and consequently enhance catalytic specificity.
Although it is unclear why we were able to observe crystal structures of two conformational states of FgFCO1 under similar crystal growth conditions, we speculate this is due to molecular crowding effects at different concentrations of PEG MME 2000. Another possible mechanism of observing two conformational forms of FgFCO1 is ligand-induced conformational change due to binding of Tris at the +1 subsite of FgFCO1 in the closed form but not in the open form (Fig. 3, c and d). Glycerol and Tris are substrate mimics of carbohydrate-active enzymes. It is not uncommon to find Tris or glycerol binding at active sites of glycosidases as reviewed by Roberts and Davies (63). Other examples in the GH29 family include B. thetaiotaomicron enzyme Bt2970 complexed with Tris (28) as well as B. thetaiotaomicron α1,3/4-fucosidase Bt2192 complexed with two glycerol molecules at the −1 and +2 subsites (36). However, we cannot exclude the possibility that the closed conformation under molecular crowding enhances affinity of Tris to +1 subsite as a concerted event. Molecular crowding at the higher concentration of PEG MME 2000 might stabilize the GGSFT loop structure, whose ordering is concomitant with the conformational change of the active-site WERG loop.
The structural flexibility of active-site loops, including the loop containing the conserved catalytic acid/base Glu, is not uncommon. α1,3/4-l-Fucosidase BiAfcB from B. longum subsp. infantis shows a conformational change of its active-site loop and reorientation of the conserved catalytic acid/base Glu-217 upon substrate or inhibitor binding (Fig. 4d) (19, 23). Furthermore, the crystal structure of T. maritima fucosidase has a disordered loop region (residues 269–274) downstream of the conserved catalytic acid/base Glu-266 (equivalent to Glu-288 of FgFCO1), suggesting structural flexibility (27). In addition, B. thetaiotaomicron fucosidase (Bt2970) reorients general acid/base Glu-288 in complex with the preferred substrate, pNP-Fuc (28). Thus, active-site structural flexibility and conformational change involving the general acid/base residue appears to be a common property of GH29 fucosidases with functional relevance. However, a general comparison cannot be made in the absence of available structures for animal and plant fucosidases.
Post-translational Modifications of FgFCO1
Different from prokaryotic fucosidases, eukaryotic fucosidases are subject to post-translational modifications like glycosylation and signal peptide removal. Cleavage of signal peptide of P. pastoris-expressed FgFCO1 was confirmed by N-sequencing as AKADGPYEATWESTD, which represented a final product of 585 residues. Three N-glycosylation sites of FgFCO1, including Asn residues 187, 280, and 401, were predicted. Crystal structures of the Endo H-treated FgFCO1 showed N-glycans on all three predicted sites (Fig. 3a). The N-glycans on Asn-280 of the TIM barrel domain retained a chitobiose core and high mannose type substitutions, GlcNAcβ1-4GlcNAcβ1-4(Manα1-6)Manα1-3Manα1-2Manα1-2Man, possibly protected from Endo H by the βγ-crystallin domain. The other two N-glycosylation sites Asn-187 and Asn-401 were each attached to one GlcNAc residue, indicative of Endo H cleavage.
Mass spectrometry confirmed that FgFCO1 expressed in P. pastoris was heterogeneous due to glycosylation. The molecular weight of FgFCO1 without the N-terminal signal peptide was calculated to be 65,635 Da for 585 residues. Endo H treatment shifted the single major mass peak from 69,828 to 67,758 Da with a minor shoulder at 66,269 Da (Fig. 6). Peptide N-glycosidase-F treatment failed to deglycosylate FgFCO1 as evidenced by the persistence of the major peak at 69,842 Da, but Endo H caused a reduction in mass by 2,070 Da. This difference corresponded to ∼13 anhydrohexose units, taking 162.1 Da as the molecular weight of repeating 1-anhydromannose unit for high mannose type N-glycans typical for P. pastoris (64). This indicates that Endo H can partially deglycosylate FgFCO1 to give the 67,758-Da major form and fully deglycosylate FgFCO1 to leave only monomers of N-linked GlcNAc on the protein, corresponding to the 66,269-Da minor form.
FIGURE 6.

Mass spectra of FgFCO1. Normalized relative intensities were plotted for each sample. PNGase F, peptide N-glycosidase F.
Based on the conservation of N-glycosylation pathways between filamentous fungi and yeast (64, 65), endogenous FgFCO1 secreted by F. graminearum may possibly also be glycosylated. We found the total carbohydrate content of FgFCO1 expressed from P. pastoris to be ∼6% based on the major peak identified by mass spectrometry. Human fucosidases FUCA1 and FUCA2 are also known to be N-glycosylated (66, 67). The liver fucosidase was reported to contain Man, GlcNAc, sialic acid, and Glc contents with a total of ∼7% carbohydrates by weight (67). Similarly, The N-glycan contents of FgFCO1 are expected to be mainly Man and GlcNAc due to a lack of the processing machineries for the attachment of sialic acid in yeast (64). Like human fucosidases, we did not find evidence for O-glycosylation on FgFCO1. Human fucosidases FUCA1 (P04066.4) and FUCA2 (Q9BTY2.2) were both predicted to contain three potential N-glycosylation sites. In particular, one of the experimentally verified N-glycosylation sites of FUCA1 (Asn-382; Ref. 66) was conserved in FgFCO1 (Asn-401) (Fig. 3a).
We did not observe an effect of Endo H deglycosylation on the catalytic efficiency with small fucosylated substrates (2′-fucosyllactose and pNP-fucoside) or on thermal stability (data not shown). On the other hand, crystallization trials showed considerably higher solubility of the glycosylated than the deglycosylated FgFCO1.
C-terminal βγ-Crystallin Domain
BLASTP of the non-conserved C-terminal β-sandwich domain sequence of FgFCO1 (residues 501–585) against the Protein Data Bank gave as the top hit the N-terminal domain of Protein S from Myxococcus xanthus (PDB code 1NPS) domain sequence identity 31%), a protein found on the surface of spores (68). Structure-based search of the FgFCO1 β-sandwich domain by PDBeFold and DALI showed high structural homology to several proteins annotated as βγ-crystallins among which were the M. xanthus Protein S (C-α r.m.s.d. 1.21 Å of 82 residues by PDBeFold), a βγ-crystallin domain of a GH64 family enzyme from Flavobacterium johnsoniae (PDB code 3HZB, C-α r.m.s.d. 0.95 Å of 81 residues, sequence identity 24%) (69), and also the C-terminal domain of human eye γS-crystallin (PDB code 1HA4, C-α r.m.s.d. 1.88 Å of 77 residues, sequence identity 25%) (70) (Fig. 6b). However, the calcium binding motif (N/D)(N/D)XX(S/T)S characteristic of Protein S and F. johnsoniae βγ-crystallin was absent in the crystallin domain of FgFCO1 (68, 69). Consistent with this, calcium ions were not found in the crystal structures of FgFCO1 nor did adding calcium shift the Tm of the enzyme ([Ca2+] between 5 μm and 1 mm; data not shown). The absence of a calcium binding motif was also reported in the human eye γS-crystallin and many other βγ-crystallins (69, 70).
A broad distribution of homologues of the βγ-crystallin domain of FgFCO1 was found in many families of glycoside hydrolases. These include fungal and bacterial α-fucosidases (domain identities: 25–92%, with the highest identity for a putative F. pseudograminearum protein, GenBankTM accession number EKJ77323.1) and various other bacterial glycoside hydrolases acting on a variety of substrates (domain identities: 25–41%). The latter include glycoside hydrolase family 5, 16, 19, 28, 54, 64, 76, and 81 (classified in CAZy database). Most of these glycoside hydrolases contain the (N/D)(N/D)XX(S/T)S motif of the crystallin domain except the GH29 fucosidases and a putative GH54 α-l-arabinofuranosidase B family protein (GenBankTM ADO68190.1), both of which remove terminal sugars from branched glycans. Thus we propose a broad distribution of βγ-crystallin domains in various families of glycoside hydrolases. Further bioinformatic and biochemical studies are necessary to reveal the functional relevance of these two subfamilies of βγ-crystallin either with or without calcium binding motifs, and their effects on specific enzymatic activities. The exact biological function of FgFCO1 βγ-crystallin domain is not yet known. Because the FgFCO1 crystallin domain is structurally too distant from the active site to affect its activity, its potential function may be to regulate the expression, folding, or trafficking of the protein itself or to interact with physiological substrates (e.g. fucosylated glycans or proteins) or to bind to endogenous or foreign partners for attachment or assembly functions. We did not find evidence of binding of synthesized crystallin domain of FgFCO1 to xyloglucan, xylan, or 2-hydroxyethyl cellulose by affinity gel electrophoresis (Fig. 7, see Table 3).
FIGURE 7.

Affinity gel electrophoresis of crystallin domain of FgFCO1.
TABLE 3.
Gel mobility of crystallin domain of FgFCO1 in the presence of soluble polysaccharides
HEC, 2-hydroxyethyl cellulose.
| Polysaccharide | Migration distance |
Relative mobility |
|||
|---|---|---|---|---|---|
| BSA | Cryst-His | Cryst-MBP | Cryst-His | Cryst-MBP | |
| cm | cm | ||||
| Control | 4.9 | 4.5 | 4.5 | 0.918a | 0.92a |
| Xyloglucan | 4.7 | 4.3 | 4.3 | 0.914 | 0.914 |
| HEC | 4.8 | 4.4 | 4.4 | 0.916 | 0.916 |
| Xylan | 4.8 | 4.3 | 4.3 | 0.896a | 0.896a |
a The small difference (2.4%) in the relative mobility of crystallins between xylan and control gels was within the accuracy limit of measurement and did not indicate binding of crystallins to polysaccharides.
Substrate Specificity and Function of FgFCO1
FgFCO1 prefers 1,2-linked over 1,3/4-linked fucosyl substrates and exhibits little or no tendency to saturation with various substrates including pNP-Fuc up to 1 mm. This is consistent with the reported Km (9.8 mm) of pNP-Fuc with FgFCO1 (17). Due to the nonsaturating behavior of FgFCO1 with various substrates tested, only the second order reaction coefficients (kcat/Km) were calculated (Table 4). The chemical structures of substrates under study are shown in Fig. 2. Although we were not able to purify sufficient amount of XXFG to perform steady-state kinetic studies, we nonetheless found that FgFCO1 showed the highest activity with its structural analog 2′-FL within all the substrates tested. FgFCO1 showed minimal catalytic activity with pNP-Fuc (kcat/Km 12 m−1s−1). This is significantly slower than most reported GH29 enzymes with α1,2-linkage activity that have kcat/Km values with pNP-Fuc on the order of 103–107 m−1s−1 (17, 20, 30–32, 34, 36) but similar to GH29 family α1,3/4-fucosidases from plants or bacteria that were reported to be inactive with pNP-Fuc (19, 22, 35). However, in contrast to α1,3/4-fucosidases, we found that FgFCO1 was completely inactive against 3-fucosyllactose and Lewisa trisaccharide even with overnight incubation. Lewisx trisaccharide was also a poor substrate with minimal detectable activity, a significant portion of which could be attributed to substrate instability.
TABLE 4.
Kinetic parameters of FgFCO1 for substrate hydrolysis
H-D, H-disaccharide; 3-FL, 3-fucosyllactose (see Fig. 2).
| Substrate | pNP-Fuc | 2′-FL | H-D | 3-FL | Lewisa trisaccharide | Lewisx trisaccharide |
|---|---|---|---|---|---|---|
| kcat/Km (m−1s−1)a | 12 | 625 | 0.55 | ND | NDb | –c |
a Steady-state kinetics were performed at 37 °C in 50 mm sodium acetate, at pH 5. The standard deviation of kcat/Km was less than 20%.
b ND, no detectable activity.
c Not stable in solution.
Diversity of substrate specificities among GH29 enzymes is not uncommon. Human α-fucosidases were reported to be active on both pNP-Fuc and various 1,2/3/4/6-linked fucosyl substrates (20, 21). The B. thetaiotaomicron enzyme Bt2970, on the other hand, showed minimal activity with various natural substrates but high activity with pNP-Fuc (22). The overall low activity of FgFCO1 with both pNP-Fuc and α1,3/4-linked fucosyl substrates, unlike most other reported GH29 fucosidases, demands a structural basis. However, its active site shows high structural homology to the fucosidases from T. maritima and B. thetaiotaomicron (27, 28). In addition to the active-site structural flexibility of FgFCO1 involving general acid/base Glu-288 and the non-conserved GGSFT loop, several non-conserved residues, His-171, Lys-272, Tyr-32, and Asp-303, at the active site may also affect the enzyme's catalytic efficiency.
Unexpectedly, the overall substrate specificity spectrum of FgFCO1 (a member of GH29 family retaining fucosidases) was found to be most similar to AtFuc95A (A. thaliana GH95 family inverting fucosidase), which was also reported to be active toward xyloglucan (kcat/Km ∼ 30 m−1s−1) and the xyloglucan oligosaccharide XXFG (18). AtFuc95A is also active with 2′-FL (kcat/Km ∼ 208 M−1s−1, at 37 °C, pH 5), which is comparable with FgFCO1 (625 m−1s−1) (18). In addition, A. thaliana GH95 fucosidase (AtFuc95A) is inactive with pNP-Fuc or 1,3/4/6-linked fucosyl substrates with overall substrate specificity and an optimum pH of 5, similar to FgFCO1. As discussed above, both GH29 and GH95 fucosidases catalyze the specific hydrolysis of α-fucosyl linkages despite different active-site folds (22, 23, 27–29) and opposite overall retention or inversion of the anomeric configuration during catalysis (27–32). F. graminearum also encodes two hypothetical GH95 family enzymes, FG04734.1 and FG11516.1, based on a BLASTP search using AtFuc95A sequence (NP_195152.2) (18) as the query. However, unlike FgFCO1, a GH95 fucosidase was not detected by comparative proteomics analysis in the secretome of F. graminearum under the same growth conditions by which FgFCO1 was induced (71).
FgFCO1 showed a 103-fold decrease in kcat/Km against H-disaccharide compared with 2′-FL (Fig. 2), suggesting the importance of a +2 site sugar unit for efficient catalysis. Interestingly, human serum fucosidase (FUCA2) also shows higher activity with 2′-FL (1.8 × 104 m−1s−1) than H-disaccharide (2.8 × 103 m−1s−1) with a somewhat narrower range of rate variation compared with FgFCO1 (<7-fold, consistent with the broader substrate specificity of human fucosidase) (20). The fact that FgFCO1 shows specificity against various substrates suggests that the glycosylation step (i.e. formation of the fucosyl-enzyme intermediate) is at least partially rate-limiting and responsible for low activity with pNP-Fuc.
Although the physiological function and substrate of FgFCO1 are not yet known, several observations point to its possible role in plant cell wall deconstruction or infection. First, FgFCO1 actively releases fucose from xyloglucan fragment XXFG (17). XXFG is not only a plant cell wall structural unit but also a plant signaling molecule whose fucose moiety is essential to its antiauxin activity (72, 73). Second, FgFCO1 is induced specifically by growth of F. graminearum on media containing various plant cell wall materials (71). Third, F. graminearum is one of the world's most serious pathogens of plants (74). Nonetheless, other fucosylated substrates like pectin, glycoproteins, or even fungal or bacterial exopolysaccharide are not to be excluded.
pH Dependence of Activity and Stability of FgFCO1
We performed steady-state kinetic assays with the model substrate pNP-Fuc to determine the pH dependence of catalytic efficiency of FgFCO1. Despite the overall low reactivity of the enzyme with pNP-Fuc, the reaction rate coefficient kcat/Km did show strong bell-shaped pH dependence with an optimum at 4.6. The acid dissociation constants were pKa1 = 3.47 ± 0.16 and pKa2 = 5.73 ± 0.13 (Fig. 8a). kcat/Km is influenced by all the reaction steps up to the first irreversible catalytic step and thus likely includes the formation of the fucosyl-enzyme intermediate, which involves nucleophilic attack by conserved Asp-226 and stabilization of the negative charge accumulated on the leaving group in the transition state by conserved general acid Glu-288 (Fig. 1). Thus, acid dissociation constants pKa1 (3.47) and pKa2 (5.78) are assigned to nucleophile Asp-226 and general acid Glu-288, respectively. The overall pH dependence of kcat/Km of FgFCO1 in reaction with pNP-Fuc is similar to the T. maritima enzyme (pHoptimum = 5.0, pKa1 = 3.8, and pKa2 = 6.1) with somewhat acidic shifts (30). Human fucosidases also have pH optima between 4.5 and 5.5 (20, 21). However, the B. thetaiotaomicron enzyme shows very different parameters, with a pHoptimum = 7.4, pKa1 = 6.7, and pKa2 = 8.1 (75). On the other hand, the fucosidase of Sulfolobus solfataricus shows a peculiar pH dependence with an optimum around pH 4–5 and an additional peak of activity at pH 8.6 (32). This indicates that GH29 family fucosidases differ not only in substrate specificities but also ionization properties of catalytically or structurally important residues.
FIGURE 8.
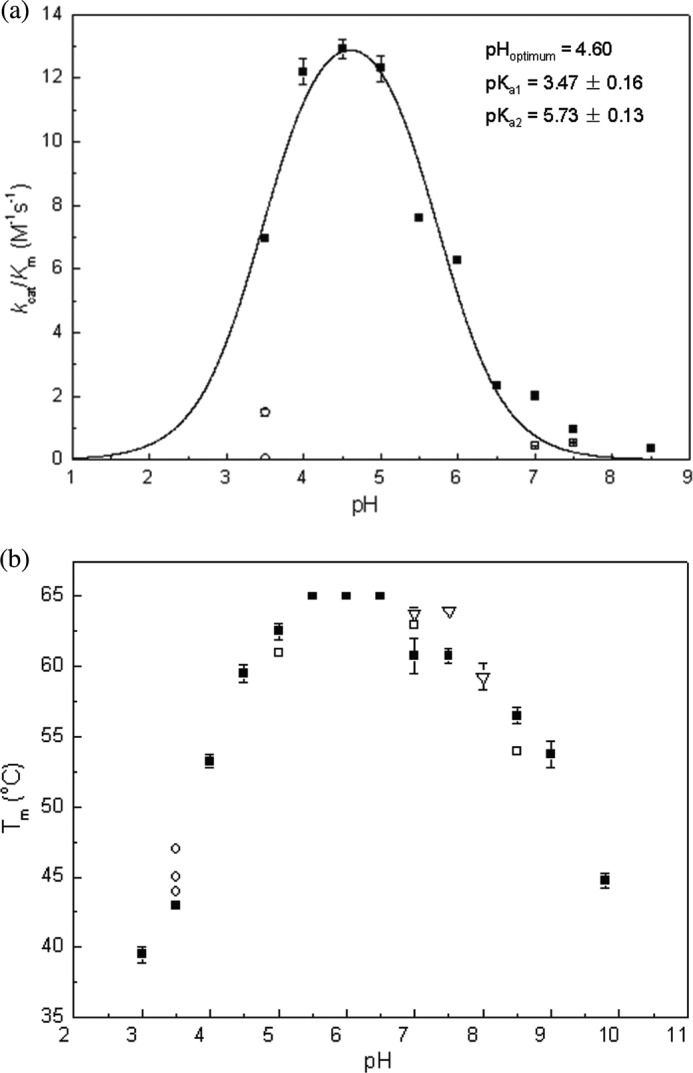
pH dependence of kcat/Km (a) and melting temperature Endo H (b) of FgFCO1. All steady-state reactions were performed with pNP-Fuc at 37 °C. The kinetic data (filled squares) were fit to kcat/Km = (kcat/Km)max/(1 + 10pKa1 − pH + 10pH − pKa2). Error bars indicate S.D. a, ■, 50 mm buffers of sodium citrate, pH 3, sodium acetate (pH 3.5–5.5), MES (pH 6–6.5), HEPES (pH 7–7.5), and Bicine (pH 8.5); □, 50 mm Tris at pH 7 and pH 7.5, respectively; ○, 10 mm citrate and 40 mm sodium acetate (upper circle), 50 mm citrate (lower circle) at pH 3.5. b, ■, the same as in a; □, the same buffer system as ■ plus 100 mm NaCl; ○, 1 mm citrate and 50 mm sodium acetate (lower), 10 mm citrate and 40 mm sodium acetate (middle), 50 mm citrate (upper) at pH 3.5; ▿, 50 mm Tris (pH 7–8).
Buffers had a dramatic effect on the activity of FgFCO1. Citrate and Tris at 50 mm were strongly inhibitory (Fig. 8a). Citrate showed concentration-dependent inhibition in the range 10–50 mm (Fig. 8a), indicating an inhibition constant Ki in the mm range. This is likely due to competitive binding at the active site between the inhibitor and the poor substrate pNP-Fuc (reported Km = 9.8 mm) (17). In particular, inhibition by Tris is consistent with the crystallographic structure that showed Tris occupying the putative +1 sugar binding subsite (Fig. 3, c and d). Citrate and Tris with their multivalent carboxymethyl or hydroxymethyl groups are structural analogues of sugars like Fuc. However, although many studies on fucosidase activity from various other organisms have employed citrate as the buffer in the acidic pH range, no inhibitory effect of citrate at 50–100 mm has previously been reported. Nevertheless, our results suggest that Tris and citrate should be used with caution as buffers in the study of activities of glycoside hydrolases or other carbohydrate active enzymes. Roberts and Davies (63) have reviewed the evidence that Tris is a common glycosidase inhibitor and proposed that the positively charged amino center partially mimics the oxocarbenium ion-like transition state, and the flexible alkyl hydroxyl moieties mimic the oxygen atoms of sugars.
The pH dependence of the melting temperature (Endo H) of FgFCO1 was measured (Fig. 8b). The enzyme was relatively thermostable over a wide range of pH 4–9 (Endo H > 52 °C) with the highest Endo H (65 °C) at pH 5.5–6.5. At pH values below 3.5, the enzyme was not stable and also lost activity rapidly. Interestingly, we found that citrate increased the Endo H of FgFCO1 in a concentration-dependent manner by up to 3 °C (at 50 mm citrate). Similarly, we also found that 50 mm Tris increased the Endo H by 2 °C at pH 7 and 7.5 (in comparison to 50 mm HEPES). In addition, salt (100 mm NaCl) also affected the thermal stability of the enzyme, albeit in a pH-dependent manner, increasing the Endo H by 2 °C at pH 7 (in 50 mm HEPES) while decreasing the Endo H by 1.5 °C at pH 5 (in 50 mm sodium acetate) and by 3 °C at pH 8.5 (in 50 mm Bicine).
Conclusions
We solved the crystal structures of F. graminearum GH29 α-fucosidase in two active-site conformations involving different orientations of the structurally conserved general acid/base Glu. We identified characteristic features near the active site including the GGSFT flexible loop and His-171 and a novel βγ-crystallin domain. SAXS scattering revealed the monomeric state of the enzyme at solution equilibrium with overall shape very similar to the crystal structure. Despite the generally conserved GH29 family fold of FgFCO1, its overall catalytic specificity most closely resembles that of GH95 fucosidases. Analogous to the active site dynamics involving conserved general acid/base glutamate observed in fungal GH29 fucosidase FgFCO1, the GH3 family retaining β-N-acetylglucosaminidases NagZ from Salmonella typhimurium and Bacillus subtilis, which exo-act on the terminal GlcNAc sugar from internalized bacterial cell wall peptidoglycan fragments, was also reported to show substantial active site structural plasticity (60). In the latter case a highly mobile loop that contains a proposed general acid/base His drives substrate distortion to facilitate catalysis. Considering the family diversity and broad distribution of glycoside hydrolases, the prevalence of structural dynamics and its functional consequences await elucidation by structural biology and complementary methods. Crystal structures of human fucosidases are desirable to elucidate the structural basis of substrate specificity, assign general acid/base Glu, and provide structural insights into molecular pathology of fucosidosis.
Acknowledgments
We thank the staff at the LS-CAT beamline for help and advice on data collection. Use of the Advanced Photon Source was supported by the United States Department of Energy, Office of Science, Office of Basic Energy Sciences under Contract DE-AC02-+06CH11357. Use of the LS-CAT Sector 21 was supported by the Michigan Economic Development Corp. and Michigan Technology Tri-Corridor under Grant 085P1000817. We also thank Dr. Sam Butcher and Dr. Jordan Burke of the Biochemistry Department, University of Wisconsin-Madison, for generously collecting SAXS data and advising on data analysis. This study made use of the National Magnetic Resonance Facility at Madison, which is supported by National Institutes of Health Grants P41RR02301 (Biomedical Research Training Program/Center for Research Resources) and P41GM66326 (NIGMS). Additional equipment was purchased with funds from the University of Wisconsin, the National Institutes of Health Grants Grant RR02781 and RR08438, National Science Foundation Grants DMB-8415048, OIA-9977486, and BIR-9214394, and the United States Department of Agriculture. We thank Dr. Johnnie Walker and Dr. Brian Fox from the University of Wisconsin-Madison for advice on affinity gel electrophoresis assays. We thank Dr. Philip Gao from University of Kansas for graciously synthesizing crystallin domain of FgFCO1. Dr. Cao is especially thankful to Dr. Craig Bingman, Kate Helmich, and Bob Smith from the University of Wisconsin-Madison for valuable advice on crystallography and thermofluor assays and Dr. Ragothaman Yennamalli and Fengbin Wang from Rice University for helpful comments. We thank the Mass Spectrometry/Proteomics Facility of the Biotechnology Center at UW-Madison for the MALDI TOF analysis and the Iowa State University Protein Facility for the N-terminal sequencing. We acknowledge use of facilities acquired in part by the National Institutes of Health Protein Structure Initiative Grant U54 GM079401.
This work was supported by the United States Department of Energy Great Lakes Bioenergy Research Center, Office of Science, Biological, and Environmental Research Grant DE-FC02-07ER64494, the National Institutes of Health Protein Structure Initiative Grant U01GM098248, and by Rice University.
The atomic coordinates and structure factors (codes 4PSP, 4PSR, and 4NI3) have been deposited in the Protein Data Bank (http://wwpdb.org/).
J. B. Bonanno, J. Freeman, K. T. Bain, S. Hu, R., Romero, S., Wasserman, J. M., Sauder, S. K. Burley, S. C. Almo, and New York SGX Research Center for Structural Genomics, PDB ID: 3EYP.
- Fuc
- l-fucose
- FgFCO1
- F. graminearum fucosidase
- GH
- glycoside hydrolase
- Gal
- d-galactose
- pNP-Fuc
- p-nitrophenyl-α-l-fucoside
- 2′-FL
- 2′-fucosyllactose
- PDB
- Protein Data Bank
- PEG 2000 MME
- PEG monomethyl ether 2000
- Endo H
- endo-β-N-acetylglucosaminidase H
- SAXS
- small angle x-ray scattering
- Bicine
- N,N-bis(2-hydroxyethyl)glycine
- H-disaccharide
- Fucα1-2Gal
- Endo H
- endoglycosidase H
- r.m.s.d.
- root mean square deviation
- TIM
- triose phosphate isomerase.
REFERENCES
- 1. Becker D. J., Lowe J. B. (2003) Fucose: biosynthesis and biological function in mammals. Glycobiology 13, 41R–53R [DOI] [PubMed] [Google Scholar]
- 2. Willems P. J., Seo H. C., Coucke P., Tonlorenzi R., O'Brien J. S. (1999) Spectrum of mutations in fucosidosis. Eur. J. Hum. Genet. 7, 60–67 [DOI] [PubMed] [Google Scholar]
- 3. Vanhooren P. T., Vandamme E. J. (1999) l-Fucose, occurrence, physiological role, chemical, enzymatic, and microbial synthesis. J. Chem. Technol. Biotechnol. 74, 479–497 [Google Scholar]
- 4. Ma B., Simala-Grant J. L., Taylor D. E. (2006) Fucosylation in prokaryotes and eukaryotes. Glycobiology 16, 158R–184R [DOI] [PubMed] [Google Scholar]
- 5. Fitchette-Lainé A. C., Gomord V., Cabanes M., Michalski J. C., Saint Macary M., Foucher B., Cavelier B., Hawes C., Lerouge P., Faye L. (1997) N-Glycans harboring the Lewis a epitope are expressed at the surface of plant cells. Plant J. 12, 1411–1417 [DOI] [PubMed] [Google Scholar]
- 6. Joly C., Léonard R., Maftah A., Riou-Khamlichi C. (2002) α4-Fucosyltransferase is regulated during flower development: increases in activity are targeted to pollen maturation and pollen tube elongation. J. Exp. Bot. 53, 1429–1436 [DOI] [PubMed] [Google Scholar]
- 7. O'Neill M. A., Eberhard S., Albersheim P., Darvill A. G. (2001) Requirement of borate cross-linking of cell wall rhamnogalacturonan II for Arabidopsis growth. Science 294, 846–849 [DOI] [PubMed] [Google Scholar]
- 8. Zablackis E., Huang J., Müller B., Darvill A. G., Albersheim P. (1995) Characterization of the cell-wall polysaccharides of Arabidopsis thaliana leaves. Plant Physiol. 107, 1129–1138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Vincken J. P., Wijsman A. J., Beldman G., Niessen W. M., Voragen A. G. (1996) Potato xyloglucan is built from XXGG-type subunits. Carbohydr. Res. 288, 219–232 [DOI] [PubMed] [Google Scholar]
- 10. Nishitani K. (1997) The role of endoxyloglucan transferase in the organization of plant cell walls. Int. Rev. Cytol. 173, 157–206 [DOI] [PubMed] [Google Scholar]
- 11. Hayashi T., Kaida R. (2011) Functions of xyloglucan in plant cells. Mol. Plant 4, 17–24 [DOI] [PubMed] [Google Scholar]
- 12. Levy S., York W. S., Stuike-Prill R., Meyer B., Staehelin L. A. (1991) Simulations of the static and dynamic molecular conformations of xyloglucan. The role of the fucosylated sidechain in surface-specific side-chain folding. Plant J. 1, 195–215 [PubMed] [Google Scholar]
- 13. Vanzin G. F., Madson M., Carpita N. C., Raikhel N. V., Keegstra K., Reiter W. D. (2002) The mur2 mutant of Arabidopsis thaliana lacks fucosylated xyloglucan because of a lesion in fucosyltransferase AtFUT1. Proc. Natl. Acad. Sci. U.S.A. 99, 3340–3345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kusaykin M., Bakunina I., Sova V., Ermakova S., Kuznetsova T., Besednova N., Zaporozhets T., Zvyagintseva T. (2008) Structure, biological activity, and enzymatic transformation of fucoidans from the brown seaweeds. Biotechnol. J. 3, 904–915 [DOI] [PubMed] [Google Scholar]
- 15. Intra J., Perotti M. E., Pavesi G., Horner D. (2007) Comparative and phylogenetic analysis of α-l-fucosidase genes. Gene 392, 34–46 [DOI] [PubMed] [Google Scholar]
- 16. Johnson S. W., Alhadeff J. A. (1991) Mammalian α-l-fucosidases. Comp. Biochem. Physiol. B 99, 479–488 [DOI] [PubMed] [Google Scholar]
- 17. Paper J. M., Scott-Craig J. S., Cavalier D., Faik A., Wiemels R. E., Borrusch M. S., Bongers M., Walton J. D. (2013) α-Fucosidases with different substrate specificities from two species of Fusarium. Appl. Microbiol. Biotechnol. 97, 5371–5380 [DOI] [PubMed] [Google Scholar]
- 18. Léonard R., Pabst M., Bondili J. S., Chambat G., Veit C., Strasser R., Altmann F. (2008) Identification of an Arabidopsis gene encoding a GH95 α1,2-fucosidase active on xyloglucan oligo- and polysaccharides. Phytochemistry 69, 1983–1988 [DOI] [PubMed] [Google Scholar]
- 19. Sela D. A., Garrido D., Lerno L., Wu S., Tan K., Eom H. J., Joachimiak A., Lebrilla C. B., Mills D. A. (2012) Bifidobacterium longum subsp. infantis ATCC 15697 α-fucosidases are active on fucosylated human milk oligosaccharides. Appl. Environ. Microbiol. 78, 795–803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. DiCioccio R. A., Barlow J. J., Matta K. L. (1982) Substrate specificity and other properties of α-l-fucosidase from human serum. J. Biol. Chem. 257, 714–718 [PubMed] [Google Scholar]
- 21. Dawson G., Tsay G. (1977) Substrate specificity of human α-l-fucosidase. Arch. Biochem. Biophys. 184, 12–23 [DOI] [PubMed] [Google Scholar]
- 22. Sakurama H., Tsutsumi E., Ashida H., Katayama T., Yamamoto K., Kumagai H. (2012) Differences in the substrate specificities and active-site structures of two α-l-fucosidases (glycoside hydrolase family 29) from Bacteroides thetaiotaomicron. Biosci. Biotechnol. Biochem. 76, 1022–1024 [DOI] [PubMed] [Google Scholar]
- 23. Sakurama H., Fushinobu S., Hidaka M., Yoshida E., Honda Y., Ashida H., Kitaoka M., Kumagai H., Yamamoto K., Katayama T. (2012) 1,3–1,4-α-l-fucosynthase that specifically introduces Lewis a/x antigens into type-1/2 chains. J. Biol. Chem. 287, 16709–16719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Berteau O., McCort I., Goasdoué N., Tissot B., Daniel R. (2002) Characterization of a new α-l-fucosidase isolated from the marine mollusk Pecten maximus that catalyzes the hydrolysis of α-l-fucose from algal fucoidan (Ascophyllum nodosum). Glycobiology 12, 273–282 [DOI] [PubMed] [Google Scholar]
- 25. Tanaka K., Sorai S. (1970) Hydrolysis of fucoidan by abalone liver α-l-fucosidase. FEBS Lett. 9, 45–48 [DOI] [PubMed] [Google Scholar]
- 26. Cantarel B. L., Coutinho P. M., Rancurel C., Bernard T., Lombard V., Henrissat B. (2009) The carbohydrate-active EnZymes database (CAZy): an expert resource for glycogenomics. Nucleic Acids Res. 37, D233–D238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sulzenbacher G., Bignon C., Nishimura T., Tarling C. A., Withers S. G., Henrissat B., Bourne Y. (2004) Crystal structure of Thermotoga maritima α-l-fucosidase. Insights into the catalytic mechanism and the molecular basis for fucosidosis. J. Biol. Chem. 279, 13119–13128 [DOI] [PubMed] [Google Scholar]
- 28. Lammerts van Bueren A., Ardèvol A., Fayers-Kerr J., Luo B., Zhang Y., Sollogoub M., Blériot Y., Rovira C., Davies G. J. (2010) Analysis of the reaction coordinate of α-l-fucosidases: a combined structural and quantum mechanical approach. J. Am. Chem. Soc. 132, 1804–1806 [DOI] [PubMed] [Google Scholar]
- 29. Nagae M., Tsuchiya A., Katayama T., Yamamoto K., Wakatsuki S., Kato R. (2007) Structural basis of the catalytic reaction mechanism of novel 1,2-α-l-fucosidase from Bifidobacterium bifidum. J. Biol. Chem. 282, 18497–18509 [DOI] [PubMed] [Google Scholar]
- 30. Tarling C. A., He S., Sulzenbacher G., Bignon C., Bourne Y., Henrissat B., Withers S. G. (2003) Identification of the catalytic nucleophile of the family 29 α-l-fucosidase from Thermotoga maritima through trapping of a covalent glycosyl-enzyme intermediate and mutagenesis. J. Biol. Chem. 278, 47394–47399 [DOI] [PubMed] [Google Scholar]
- 31. Liu S. W., Chen C. S., Chang S. S., Mong K. K., Lin C. H., Chang C. W., Tang C. Y., Li Y. K. (2009) Identification of essential residues of human α-l-fucosidase and tests of its mechanism. Biochemistry 48, 110–120 [DOI] [PubMed] [Google Scholar]
- 32. Cobucci-Ponzano B., Mazzone M., Rossi M., Moracci M. (2005) Probing the catalytically essential residues of the α-l-fucosidase from the hyperthermophilic archaeon Sulfolobus solfataricus. Biochemistry 44, 6331–6342 [DOI] [PubMed] [Google Scholar]
- 33. de La Torre F., Sampedro J., Zarra I., Revilla G. (2002) AtFXG1, an Arabidopsis gene encoding α-l-fucosidase active against fucosylated xyloglucan oligosaccharides. Plant Physiol. 128, 247–255 [PMC free article] [PubMed] [Google Scholar]
- 34. Liu T. W., Ho C. W., Huang H. H., Chang S. M., Popat S. D., Wang Y. T., Wu M. S., Chen Y. J., Lin C. H. (2009) Role for α-l-fucosidase in the control of Helicobacter pylori-infected gastric cancer cells. Proc. Natl. Acad. Sci. U.S.A. 106, 14581–14586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zeleny R., Leonard R., Dorfner G., Dalik T., Kolarich D., Altmann F. (2006) Molecular cloning and characterization of a plant α1,3/4-fucosidase based on sequence tags from almond fucosidase I. Phytochemistry 67, 641–648 [DOI] [PubMed] [Google Scholar]
- 36. Shaikh F. A., Lammerts van Bueren A., Davies G. J., Withers S. G. (2013) Identifying the catalytic acid/base in GH29 α-l-fucosidase subfamilies. Biochemistry 52, 5857–5864 [DOI] [PubMed] [Google Scholar]
- 37. Sinnott M. L. (1990) Catalytic mechanisms of enzymic glycosyl transfer. Chem. Rev. 90, 1171–1202 [Google Scholar]
- 38. Otwinowski Z., Minor W. (1997) Processing of x-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326 [DOI] [PubMed] [Google Scholar]
- 39. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Terwilliger T. C., Grosse-Kunstleve R. W., Afonine P. V., Moriarty N. W., Zwart P. H., Hung L.-W., Read R. J., Adams P. D. (2008) Iterative model building, structure refinement, and density modification with the PHENIX AutoBuild wizard. Acta Crystallogr. D Biol. Crystallogr. 64, 61–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L.-W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., Zwart P. H. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Emsley P., Lohkamp B., Scott W. G., Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Chen V. B., Arendall W. B., 3rd, Headd J. J., Keedy D. A., Immormino R. M., Kapral G. J., Murray L. W., Richardson J. S., Richardson D. C. (2010) MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 66, 12–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. DeLano W. L. (2014) The PyMOL Molecular Graphics System, Version 1.7, Schrodinger, LLC, New York [Google Scholar]
- 45. Krissinel E., Henrick K. (2007) Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 372, 774–797 [DOI] [PubMed] [Google Scholar]
- 46. Petoukhov M. V., Franke D., Shkumatov A. V., Tria G., Kikhney A. G., Gajda M., Gorba C., Mertens H. D. T., Konarev P. V., Svergun D. I. (2012) New developments in the ATSAS program package for small-angle scattering data analysis. J. Appl. Crystallogr. 45, 342–350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Svergun D. I. (1992) Determination of the regularization parameter in indirect-transform methods using perceptual criteria. J. Appl. Crystallogr. 25, 495–503 [Google Scholar]
- 48. Franke D., Svergun D. I. (2009) DAMMIF, a program for rapid ab initio shape determination in small-angle scattering. J. Appl. Crystallogr. 42, 342–346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Volkov V. V., Svergun D. I. (2003) Uniqueness of ab-initio shape determination in small-angle scattering. J. Appl. Crystallogr. 36, 860–864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Kozin M. B., Svergun D. I. (2001) Automated matching of high and low resolution structural models. J. Appl. Crystallogr. 34, 33–41 [Google Scholar]
- 51. Svergun D. I., Barberato C., Koch M. H. J. (1995) CRYSOL: a program to evaluate x-ray solution scattering of biological macromolecules from atomic coordinates. J. Appl. Crystallogr. 28, 768–773 [Google Scholar]
- 52. Pantoliano M. W., Petrella E. C., Kwasnoski J. D., Lobanov V. S., Myslik J., Graf E., Carver T., Asel E., Springer B. A., Lane P., Salemme F. R. (2001) High density miniaturized thermal shift assays as a general strategy for drug discovery. J. Biomol. Screen. 6, 429–440 [DOI] [PubMed] [Google Scholar]
- 53. Tomme P., Boraston A., Kormos J. M., Warren R. A., Kilburn D. G. (2000) Affinity electrophoresis for the identification and characterization of soluble sugar binding by carbohydrate binding modules. Enzyme Microb. Technol. 27, 453–458 [DOI] [PubMed] [Google Scholar]
- 54. Gasteiger E., Hoogland C., Gattiker A., Duvaud S., Wilkins M. R., Appel R. D., Bairoch A. (2005) Protein identification and analysis tools on the ExPASy Server in The Proteomics Protocols Handbook (Walker J. M., ed.) pp. 571–607, Humana Press Inc., Totowa, NJ [Google Scholar]
- 55. Petersen T. N., Brunak S., von Heijne G., Nielsen H. (2011) SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat. Methods 8, 785–786 [DOI] [PubMed] [Google Scholar]
- 56. Holm L., Rosenström P. (2010) Dali server: conservation mapping in 3D. Nucleic Acids Res. 38, W545–W549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Altschul S. F., Madden T. L., Schäffer A. A., Zhang J., Zhang Z., Miller W., Lipman D. J. (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Krissinel E., Henrick K. (2004) Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Crystallogr. D Biol. Crystallogr. 60, 2256–2268 [DOI] [PubMed] [Google Scholar]
- 59. Larkin M. A., Blackshields G., Brown N. P., Chenna R., McGettigan P. A., McWilliam H., Valentin F., Wallace I. M., Wilm A., Lopez R., Thompson J. D., Gibson T. J., Higgins D. G. (2007) ClustalW and ClustalX Version 2.0. Bioinformatics 23, 2947–2948 [DOI] [PubMed] [Google Scholar]
- 60. Bacik J. P., Whitworth G. E., Stubbs K. A., Vocadlo D. J., Mark B. L. (2012) Active site plasticity within the glycoside hydrolase NagZ underlies a dynamic mechanism of substrate distortion. Chem. Biol. 19, 1471–1482 [DOI] [PubMed] [Google Scholar]
- 61. Guillotin L., Lafite P., Daniellou R. (2014) Unraveling the substrate recognition mechanism and specificity of the unusual glycosyl hydrolase family 29 BT2192 from Bacteroides thetaiotaomicron. Biochemistry 53, 1447–1455 [DOI] [PubMed] [Google Scholar]
- 62. Ma J. C., Dougherty D. A. (1997) The cation-π interaction. Chem. Rev. 97, 1303–1324 [DOI] [PubMed] [Google Scholar]
- 63. Roberts S. M., Davies G. J. (2012) The crystallization and structural analysis of cellulases (and other glycoside hydrolases): strategies and tactics. Methods Enzymol. 510, 141–168 [DOI] [PubMed] [Google Scholar]
- 64. Wildt S., Gerngross T. U. (2005) The humanization of N-glycosylation pathways in yeast. Nat. Rev. Microbiol. 3, 119–128 [DOI] [PubMed] [Google Scholar]
- 65. Deshpande N., Wilkins M. R., Packer N., Nevalainen H. (2008) Protein glycosylation pathways in filamentous fungi. Glycobiology 18, 626–637 [DOI] [PubMed] [Google Scholar]
- 66. Chen R., Jiang X., Sun D., Han G., Wang F., Ye M., Wang L., Zou H. (2009) Glycoproteomics analysis of human liver tissue by combination of multiple enzyme digestion and hydrazide chemistry. J. Proteome Res. 8, 651–661 [DOI] [PubMed] [Google Scholar]
- 67. Alhadeff J. A., Freeze H. (1977) Carbohydrate composition of purified human liver α-l-fucosidase. Mol. Cell. Biochem. 18, 33–37 [DOI] [PubMed] [Google Scholar]
- 68. Wenk M., Baumgartner R., Holak T. A., Huber R., Jaenicke R., Mayr E. M. (1999) The domains of protein S from Myxococcus xanthus: structure, stability, and interactions. J. Mol. Biol. 286, 1533–1545 [DOI] [PubMed] [Google Scholar]
- 69. Aravind P., Mishra A., Suman S. K., Jobby M. K., Sankaranarayanan R., Sharma Y. (2009) The βγ-crystallin superfamily contains a universal motif for binding calcium. Biochemistry 48, 12180–12190 [DOI] [PubMed] [Google Scholar]
- 70. Purkiss A. G., Bateman O. A., Goodfellow J. M., Lubsen N. H., Slingsby C. (2002) The x-ray crystal structure of human γS-crystallin C-terminal domain. J. Biol. Chem. 277, 4199–4205 [DOI] [PubMed] [Google Scholar]
- 71. Paper J. M., Scott-Craig J. S., Adhikari N. D., Cuomo C. A., Walton J. D. (2007) Comparative proteomics of extracellular proteins in vitro and in planta from the pathogenic fungus Fusarium graminearum. Proteomics 7, 3171–3183 [DOI] [PubMed] [Google Scholar]
- 72. York W. S., Darvill A. G., Albersheim P. (1984) Inhibition of 2,4-dichlorophenoxyacetic acid-stimulated elongation of pea stem segments by a xyloglucan oligosaccharide. Plant Physiol. 75, 295–297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. McDougall G. J., Fry S. C. (1989) Structure-activity relationships for xyloglucan oligosaccharides with antiauxin activity. Plant Physiol. 89, 883–887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Dean R., Van Kan J. A., Pretorius Z. A., Hammond-Kosack K. E., Di Pietro A., Spanu P. D., Rudd J. J., Dickman M., Kahmann R., Ellis J., Foster G. D. (2012) The Top 10 fungal pathogens in molecular plant pathology. Mol. Plant. Pathol. 13, 414–430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Lammerts van Bueren A., Popat S. D., Lin C. H., Davies G. J. (2010) Structural and thermodynamic analyses of α-l-fucosidase inhibitors. Chembiochem. 11, 1971–1974 [DOI] [PubMed] [Google Scholar]
- 76. Fry S. C., York W. S., Albersheim P., Darvill A., Hayashi T., Joseleau J. P., Kato Y., Lorences E. P., Maclachlan G. A., McNeil M., Mort A. J., Reid J. S. G., Seitz H. U., Selvendran R. R., Voragen A. G. J., White A. R. (1993) An unambiguous nomenclature for xyloglucan-derived oligosaccharides. Physiol. Plant. 89, 1–3 [Google Scholar]
- 77. Mark P., Baumann M. J., Eklöf J. M., Gullfot F., Michel G., Kallas A. M., Teeri T. T., Brumer H., Czjzek M. (2009) Analysis of nasturtium Endo HNXG1 complexes by crystallography and molecular dynamics provides detailed insight into substrate recognition by family GH16 xyloglucan endo-transglycosylases and endo-hydrolases. Proteins 75, 820–836 [DOI] [PubMed] [Google Scholar]
- 78. Johansson P., Brumer H., 3rd, Baumann M. J., Kallas A. M., Henriksson H., Denman S. E., Teeri T. T., Jones T. A. (2004) Crystal structures of a poplar xyloglucan endotransglycosylase reveal details of transglycosylation acceptor binding. Plant Cell 16, 874–886 [DOI] [PMC free article] [PubMed] [Google Scholar]