

Background: FK506-binding protein 38 (FKBP38) inhibits the Sonic hedgehog (Shh) signaling pathway in mouse embryos, but the underlying molecular mechanism has remained unclear.
Results: ANKMY2 interacts with FKBP38 and acts downstream of FKBP38 to activate Shh signaling.
Conclusion: The FKBP38-ANKMY2 axis plays a key role in regulation of Shh signaling in vivo.
Significance: Our findings reveal a new mode of regulation of the Shh pathway.
Keywords: Cell Signaling, Hedgehog Signaling Pathway, Mouse, Prolyl Isomerase, Proteomics, Zebrafish
Abstract
Sonic hedgehog (Shh) is a secreted morphogen that controls the patterning and growth of various tissues in the developing vertebrate embryo, including the central nervous system. Ablation of the FK506-binding protein 38 (FKBP38) gene results in activation of the Shh signaling pathway in mouse embryos, but the molecular mechanism by which FKBP38 suppresses Shh signaling has remained unclear. With the use of a proteomics approach, we have now identified ANKMY2, a protein with three ankyrin repeats and a MYND (myeloid, Nervy, and DEAF-1)-type Zn2+ finger domain, as a molecule that interacts with FKBP38. Co-immunoprecipitation analysis confirmed that endogenous FKBP38 and ANKMY2 interact in the mouse brain. Depletion or overexpression of ANKMY2 resulted in down- and up-regulation of Shh signaling, respectively, in mouse embryonic fibroblasts. Furthermore, combined depletion of both FKBP38 and ANKMY2 attenuated Shh signaling in these cells, suggesting that ANKMY2 acts downstream of FKBP38 to activate the Shh signaling pathway. Targeting of the zebrafish ortholog of mouse Ankmy2 (ankmy2a) in fish embryos with an antisense morpholino oligonucleotide conferred a phenotype reflecting loss of function of the Shh pathway, suggesting that the regulation of Shh signaling by ANKMY2 is conserved between mammals and fish. Our findings thus indicate that the FKBP38-ANKMY2 axis plays a key role in regulation of Shh signaling in vivo.
Introduction
The Sonic hedgehog (Shh) signaling pathway is conserved among animals and plays key roles in embryonic development, in the maintenance of adult stem cells, and in cancer (1–3). In the absence of Shh, the signaling pathway is inactivated as a result of inhibition of the seven-transmembrane domain protein Smoothened (Smo) by the membrane protein Patched (Ptch) (4). The pathway is activated by the binding of Shh to and the consequent inactivation of Ptch (5, 6), which relieves the inhibition of Smo. Active Smo then signals to the cytoplasm, resulting in activation of the zinc finger transcription factors that control the output of the Shh pathway, which in vertebrates are the Gli proteins (Gli1, -2, and -3) (7). A unique feature of the vertebrate Shh pathway is that the primary cilium plays a central role in signal transduction (8, 9), with the initial signaling events at the membrane taking place in this organelle. Ptch is located at the base of the primary cilium (10), and the binding of Shh to Ptch results in the activation of Smo and its recruitment to the cilium (11). By an as yet unknown mechanism, active Smo in the cilium relays the Shh signal to the cytoplasm, resulting in the activation of Gli2 and Gli3, which control transcription of Shh target genes, such as those for Gli1 and Ptch1 (7).
FKBP38 is a member of the FK506-binding protein (FKBP)2 family (12, 13). It contains a transmembrane domain and a short juxtamembrane sequence at its COOH terminus, and it localizes predominantly to the mitochondrial outer membrane (14). We have previously shown that FKBP38 contributes to the localization of the antiapoptotic proteins Bcl-2 and Bcl-xL to mitochondria and thereby inhibits apoptosis (15). FKBP38 translocates from mitochondria to the endoplasmic reticulum during mitophagy, with this translocation being essential for the suppression of apoptosis (16). In addition, FKBP38 appears to regulate protein degradation by anchoring the 26 S proteasome to organellar membranes (17). It thereby affects the stability of both Bcl-2 and the prolyl 4-hydroxylase domain-containing enzyme PHD2, the latter of which regulates the hypoxia-inducible transcription factor HIF1α (18). Although FKBP38 has been found to be expressed in all mammalian tissues examined, it is especially abundant in the central nervous system, both in neurons and in glial cells (19). To investigate the physiological roles of FKBP38, we generated mice nullizygous for the FKBP38 gene. We found that the Fkbp38−/− mice died shortly after birth, manifesting a defect in neural tube closure (spina bifida) (20), whereas others showed that FKBP38 controls neural tube patterning primarily by acting in a cell-autonomous manner to prevent inappropriate activation of the Shh pathway by Gli2 (21). This role of FKBP38 is independent of the upstream pathway activator Smo but is dependent on the kinesin-2 motor subunit Kif3a, which participates in intraflagellar transport and cilium assembly.
We have now further investigated the physiological roles of FKBP38 by proteomic screening and thereby identified ANKMY2 (also known as ZMYND20) as a protein that interacts with FKBP38. Depletion or overexpression of ANKMY2 in mouse embryonic fibroblasts (MEFs) resulted in down- and up-regulation of Shh signaling, respectively. Our observation that depletion of both FKBP38 and ANKMY2 attenuated Shh signaling in these cells suggests that FKBP38 inhibits such signaling via suppression of the stimulatory effect of ANKMY2. Furthermore, the regulation of Shh signaling by ANKMY2 is conserved between mammals and fish, given that a morpholino oligonucleotide (MO) that targets the zebrafish ortholog of mouse Ankmy2 (ankmy2a) conferred a specific phenotype reflecting loss of function of the Shh pathway in zebrafish embryos. We thus propose that the FKBP38-ANKMY2 axis plays a key role in regulation of the Shh signaling pathway in vivo.
EXPERIMENTAL PROCEDURES
Construction of Plasmids
Construction of vectors encoding human or mouse FKBP38 or human FKBP52 was described previously (15), and cDNAs encoding FKBP38 mutants were generated by PCR. Complementary DNAs encoding mouse Shh (full-length or amino acids 1–197) were generated by PCR with Prime Star polymerase (Takara, Ohtsu, Japan) from a brain cDNA library; those encoding human or mouse ANKMY2 (ZMYND20) were generated from human kidney and mouse brain cDNA libraries; those encoding mouse ZMYND1, -2, -3, -4, -7, -8, -12, -14, -15, -18, -19, -21, or -23 were generated from a mouse brain cDNA library; and those encoding human ZMYND5, -10, -11, or -17 were generated with the use of cDNA preparations from HeLa, SH-SY5Y, or HEK293T cells. Human ZMYND6 (PHD2) and mouse ZMYND9 (USP19) cDNAs were kindly provided by Y. Minamishima (Keio University, Japan) and S. S. Wing (McGill University, Montreal, Canada), respectively. Complementary DNAs encoding zebrafish ANKMY2a and FKBP38 were generated by PCR from a zebrafish cDNA library. For the production of recombinant proteins, the cDNA for mouse ANKMY2 was subcloned into pGEX-6P (GE Healthcare) and pET-28 (Novagen, Madison, WI), and that for mouse FKBP38 was subcloned into pET-28. Otherwise, cDNAs were subcloned into the p3×FLAG-CMV-7.1 (Sigma), pEF-BOS-2×HA (kindly provided by S. Nagata, Kyoto University), pcDNA3 (Invitrogen), pEGFP (Clontech), pCS2P+ (Addgene, Cambridge, MA), or pMX-puro (kindly provided by T. Kitamura, Tokyo University) vectors.
Cell Culture, Transfection, and Assay of Shh Signaling
Plat-E, HeLa, and HEK293T cells were maintained in DMEM supplemented with 10% FBS (Invitrogen) and, in the case of Plat-E cells, with blasticidin (10 μg/ml). Primary MEFs were isolated as described previously (22, 23) and were immortalized by transfection with a plasmid containing SV40 genomic DNA (kindly provided by T. Akagi, KAN Research Institute, Kobe, Japan). Retroviral infection was performed as described previously (24), and cells were transfected with expression vectors with the use of the FuGENE HD reagent (Invitrogen). Shh was produced in HEK293T cells by transient transfection of an expression plasmid encoding mouse Shh (amino acids 1–197) for 24 h, after which the medium was replaced with Opti-MEM reduced serum medium (Invitrogen), and the cells were incubated for an additional 24 h for collection of Shh-containing culture supernatants. For assay of Shh signaling, confluent cell cultures were deprived of serum for 24 h by incubation in Opti-MEM reduced serum medium, exposed to mouse Shh for the indicated times, and then harvested for RT and real-time PCR as well as immunoblot analyses.
RT and Real-time PCR Analysis of Shh Pathway Activity
Total RNA isolated from MEFs with the use of Isogen (Nippon Gene, Tokyo, Japan) was subjected to RT with the use of a QuantiTect reverse transcription kit (Qiagen, Hilden, Germany), and the resulting cDNA was subjected to PCR with Power SYBR Green PCR Master Mix in an ABI-Prism 7000 sequence detection system (Applied Biosystems, Foster City, CA). The amounts of each target mRNA were calculated and normalized by that of Hprt mRNA as described previously (25). The PCR primer sequences (sense and antisense, respectively) were 5′-ATCACCTGTTGGGGATGCTGGAT-3′ and 5′-GGCGTGAATAGGACTTCCGACAG-3′ for mouse Gli1, 5′-ACTGTCCAGCTACCCCAATG-3′ and 5′-CATCATGCCAAAGAGCTCAA-3′ for mouse Ptch1, and 5′-GCCTAAGATGAGCGCAAGTTG-3′ and 5′-TACATGGCAGATGGCCACAG-3′ for mouse Hprt.
RNAi
Stealth siRNAs targeted to mouse Ankmy2 (Ankmy2-1, 5′-CCCAGGGACUGGAUAAAGAGCCAAA-3′; Ankmy2-2, 5′-CCACCCUGUCAAGAUCGUGAUGCUU-3′) or negative control duplexes (Invitrogen) were introduced into MEFs by reverse transfection with the use of Lipofectamine RNAiMax (Invitrogen).
Antibodies
Rabbit polyclonal antibodies to (anti-) FKBP38 (FK38N1) were generated in response to a hexahistidine (His6)-tagged recombinant protein comprising the NH2-terminal half of FKBP38 that was expressed in and purified from Escherichia coli (15). Rabbit polyclonal anti-ANKMY2 was generated in response to His6-tagged recombinant full-length mouse ANKMY2 that was expressed in and purified from E. coli. Mouse monoclonal anti-HSP90 was obtained from BD Biosciences; rabbit polyclonal anti-calnexin was from Stressgen (Victoria, Canada); rabbit monoclonal anti-Gli1 (V812) was from Cell Signaling (Beverly, MA); anti-FLAG epitope (mouse monoclonal M2 and rabbit polyclonal), mouse monoclonal anti-acetylated α-tubulin, and anti-γ-tubulin were from Sigma; mouse monoclonal anti-HA epitope was from Research Diagnostics (Flanders, NJ); mouse monoclonal anti-GST (M071-3) was from MBL (Nagoya, Japan); and mouse monoclonal anti-T7 tag was from Novagen.
Generation of Transgenic Mice
Animal experiments were performed in accordance with the guidelines of Kyushu University. For generation of transgenic mouse lines, a cDNA for mouse FKBP38 tagged at its NH2 terminus with His6-FLAG was subcloned into the XhoI site of the pPrPpE1/E2, E3sal plasmid (26, 27). The resulting vector was digested with NotI for linearization and removal of the backbone vector. The prepared DNA construct was then injected into fertilized eggs of the B6D2F1/Crlj (BDF1) background. Primary genotyping was performed by PCR and Southern blot analysis and was followed by immunoblot analysis with anti-FLAG. Examination of phenotypes was performed with mice of the C57BL/6J background. Wild-type (WT) littermates were studied as control mice.
Preparation of Protein Complexes by Dual Affinity Purification
Brains of mice stably expressing His6-FLAG-tagged mouse FKBP38 were disrupted with a Potter homogenizer in a solution containing 10 mm Tris-HCl (pH 8.0), 0.32 m sucrose, 5 mm EDTA, 1 mm Na3VO4, 25 mm NaF, aprotinin (10 μg/ml), leupeptin (10 μg/ml), and 1 mm PMSF. The homogenate was centrifuged at 1000 × g for 5 min at 4 °C to remove nuclei and nondisrupted cells, and the resulting supernatant was centrifuged at 20,000 × g for 1 h at 4 °C to isolate a membrane fraction (pellet). This pellet was solubilized with a solution containing 20 mm Tris-HCl (pH 8.0), 150 mm NaCl, 1.0% Triton X-100, 1 mm Na3VO4, 25 mm NaF, aprotinin (10 μg/ml), leupeptin (10 μg/ml), and 0.5 mm PMSF, and the insoluble material was removed by centrifugation at 37,000 × g for 1 h at 4 °C. The protein concentration of the resulting supernatant was determined with the Bradford assay (Bio-Rad), and this soluble membrane fraction was then incubated with rotation for 1 h at 4 °C with 40 μl of anti-FLAG (M2)-agarose affinity gel (Sigma)/mg of protein. The beads were washed three times with solubilization buffer, after which protein complexes were eluted from the beads by incubation for several min at 4 °C with 800 μl of the same buffer containing the FLAG peptide (Sigma) at 0.25 mg/ml. For the second affinity purification step, nickel-nitrilotriacetic acid (Ni-NTA)-agarose (ProBond resin, Invitrogen) was added to the eluate at one-half the volume of anti-FLAG (M2)-agarose used in the first step, and the mixture was incubated with rotation for 1 h at 4 °C. The beads were washed three times with solubilization buffer, after which protein complexes were eluted by incubation for several min at 4 °C with the same solution containing 300 mm imidazole. Protein identification by LC-MS/MS analysis was performed as described previously (25).
Immunoprecipitation and Immunoblot Analysis
Whole mouse brain was homogenized by the application of 10 strokes (900 rpm) with a Potter homogenizer in a solution containing 20 mm HEPES-NaOH (pH 7.6), 0.32 m sucrose, 1 mm EDTA, 1 mm Na3VO4, 25 mm NaF, aprotinin (10 μg/ml), leupeptin (10 μg/ml), and 1 mm PMSF. The homogenate was centrifuged twice at 1000 × g for 5 min at 4 °C, and the second supernatant was centrifuged at 100,000 × g for 1 h at 4 °C. The crude microsomal pellet was resuspended in a lysis buffer (40 mm HEPES-NaOH (pH 7.5), 150 mm NaCl, 10% glycerol, 0.5% Triton X-100, 1 mm Na3VO4, 25 mm NaF, aprotinin (10 μg/ml), leupeptin (10 μg/ml), 1 mm PMSF), incubated for 1 h at 4 °C, and then centrifuged again at 100,000 × g for 1 h at 4 °C to remove insoluble material. The resulting supernatant was subjected to immunoprecipitation for 1 h at 4 °C with protein G-Sepharose 4 Fast Flow (Amersham Biosciences) and either anti-FKBP38, anti-ANKMY2, rabbit anti-FLAG, or normal rabbit serum. The immunoprecipitates were washed three times with lysis buffer and then subjected to immunoblot analysis, as described previously (28). The images were scanned with a LAS-4000 (GE Healthcare) instrument. For analysis of transfected HEK293T cells, the cells were cultured for 1 day after transfection and then lysed by incubation for 10 min at 4 °C with lysis buffer. The lysates were centrifuged at 20,400 × g for 10 min at 4 °C, after which equal amounts of protein from the resulting supernatants were subjected either to immunoblot analysis directly or to immunoprecipitation for 1 h at 4 °C with rabbit anti-FLAG and protein G-Sepharose 4 Fast Flow followed by immunoblot analysis.
In Vitro Binding Assay
Recombinant GST-ANKMY2 and His6-T7-FKBP38 expressed in and purified from E. coli were mixed and then incubated at 4 °C for 1 h with rotation. After the addition of glutathione-Sepharose 4B beads (Amersham Biosciences), the mixture was incubated for an additional 1 h at 4 °C with rotation, the beads were washed twice, and the bound proteins were subjected to immunoblot analysis.
Immunofluorescence Analysis of Mouse Embryos
Mouse embryos at embryonic day 11.5 (E11.5) were fixed with 4% paraformaldehyde in PBS, exposed to 30% (w/v) sucrose in PBS, embedded in Tissue-Tek O.C.T. compound (Sakura, Tokyo, Japan), and cryosectioned at a thickness of 8 μm. Sections were rehydrated in PBS, permeabilized with 0.01% Triton X-100 in PBS containing 0.1% BSA, and incubated for 24 h at 4 °C with primary antibodies to ANKMY2. Immune complexes were detected with Alexa Fluor 488-conjugated goat secondary antibodies to rabbit IgG (Invitrogen) at a dilution of 1:500. Nuclei were stained with Hoechst 33258 (Wako, Osaka, Japan). Specimens were observed with a confocal microscope (LSM510 META, Carl Zeiss).
Immunofluorescence Analysis of Cells
HeLa cells grown on glass coverslips were transfected with the use of the FuGENE HD reagent and subsequently prepared for immunostaining. Confluent MEF cultures on glass coverslips were deprived of serum for 24 h by incubation in Opti-MEM reduced serum medium and subsequently prepared for immunostaining. In brief, cells were fixed for 10 min at room temperature with 4% formaldehyde in PBS and were then incubated for 2 h at room temperature first with primary antibodies in PBS containing 0.1% BSA and 0.1% saponin and then with Alexa Fluor 488- or Alexa Fluor 546-labeled goat secondary antibodies at a dilution of 1:2000. The cells were finally stained with Hoechst 33258, covered with a drop of GEL/MOUNT (Biomeda, Foster City, CA), and examined with a confocal microscope (LSM510 META) equipped with a ×63/1.4 oil immersion objective.
Injection of MOs
Antisense MOs (Gene Tools, Philomath, OR) were injected into zebrafish embryos at the one-cell stage according to standard protocols (29). Given that a high dose of MOs induces activation of the p53 pathway, we injected 2–5 ng of ankmy2a MO and 5 ng of p53 MO concomitantly. The MO sequences were 5′-ACATTTCATTTCATTACCTCATCCA-3′ for ankmy2a spl MO, 5′-CTCCTTTCTTTGGTGCAGACATTAT-3′ for ankmy2a MO, and 5′-GCGCCATTGCTTTGCAAGAATTG-3′ for p53 MO.
Cyclopamine Treatment
Cyclopamine (Sigma) was dissolved in DMSO. Early stage zebrafish embryos were manually dechorionated and treated with 10 nm cyclopamine (or 1% DMSO as a control) for 16 h in the dark at 28.5 °C.
In Situ Hybridization
Whole-mount zebrafish embryos were subjected to in situ hybridization under standard conditions with digoxigenin-labeled antisense RNA probes prepared from the zebrafish ankmy2a (FP102311, NM_199894), gli1 (CU855923, NM_178296), myoD (BX004812, NM_131262), nkx2.2 (BX927409, NM_131422), or fox2a (CU856140, NM_001083815) genes.
Immunohistochemical Staining of Zebrafish Embryos
Immunohistochemical staining of zebrafish embryos was performed as described previously (30). Primary antibodies included F59, which preferentially recognizes slow muscle fibers (reacting only weakly with fast muscle fibers) (31), and 4D9, which recognizes zebrafish Engrailed (32), both of which were obtained from the Developmental Studies Hybridoma Bank (University of Iowa, Iowa City, IA). Immune complexes were detected with the use of HISTOFINE (Nichirei, Tokyo, Japan).
Statistical Analysis
Quantitative data are presented as means ± S.D. and were analyzed with Student's t test. A p value of <0.05 was considered statistically significant.
RESULTS
Identification of FKBP38-associated Proteins by a Proteomics Approach
To investigate the physiological roles of FKBP38, we adopted a proteomics approach to identify proteins with which FKBP38 is physically associated in neural cells of the brain. We thus generated mice (Fkbp38TG mice) that express a transgene for FKBP38 tagged with His6 and FLAG epitopes at its NH2 terminus (His6-FLAG-FKBP38) under the control of the neuron-specific prion gene promoter (26, 27). Expression of the transgene in the brain of six independent Fkbp38TG lines was examined by immunoblot analysis (Fig. 1A). Animals of line 5 were chosen for most subsequent examinations because of their relatively high level of transgene expression. Spina bifida, the most prominent characteristic of newborn Fkbp38−/− mice, with a penetrance of 100% (20), was not observed in newborn Fkbp38−/− mice expressing the transgene (Fig. 1B), suggesting that His6-FLAG-FKBP38 effectively substituted for endogenous FKBP38 in the brain. The Fkbp38−/−;TG mice died immediately after birth, however, indicating that neither the lack of FKBP38 in the brain nor the presence of spina bifida is primarily responsible for the neonatal death of Fkbp38−/− mice. Proteins associated with His6-FLAG-FKBP38 were purified from lysates of the brain of Fkbp38TG mice by dual affinity chromatography with anti-FLAG and Ni-NTA resin followed by SDS-PAGE and silver staining (Fig. 1C). The gel was then sliced, the gel fragments were exposed to trypsin, and the generated peptides were subjected to LC-MS/MS analysis. ANKMY2, a protein with three ankyrin repeats and a MYND (myeloid, Nervy, and DEAF-1)-type Zn2+ finger domain, showed the highest level of sequence coverage among the FKBP38-associated proteins identified by this approach (Table 1). We therefore compared the expression of ANKMY2 and FKBP38 in various mouse tissues. Immunoblot analysis revealed that ANKMY2 is highly expressed in the central nervous system, whereas FKBP38 is relatively widely distributed (Fig. 1D). Immunoblot analysis with anti-ANKMY2 also revealed that ANKMY2 was present in similar amounts regardless of FKBP38 abundance among Fkbp38+/+, Fkbp38+/−, and Fkbp38−/− mouse embryos at E11.5 (Fig. 1E). We also confirmed that ANKMY2 is expressed in the neural tube of mouse embryos at E11.5, being especially abundant in the ventral region near the floor plate that is under the influence of Shh signaling (Fig. 1F).
FIGURE 1.

Identification of FKBP38-associated proteins by proteomics analysis of transgenic mice. A, whole brain lysates prepared from WT or six different lines of Fkbp38TG (harboring one allele of a His6-FLAG-tagged FKBP38 transgene) mice were subjected to immunoblot (IB) analysis with anti-FLAG, anti-FKBP38, and anti-calnexin (loading control). B, gross appearance of newborns of Fkbp38−/− (left) and Fkbp38−/−;TG (right) mice. Arrows, neural tube closure defects; arrowheads, normal neural tube closure. Scale bar, 5 mm. C, extracts of WT or Fkbp38TG mouse brain were subjected to dual affinity chromatography with anti-FLAG and Ni-NTA resin. The purified proteins were fractionated by SDS-PAGE and stained with silver. The arrowhead indicates the band corresponding to His6-FLAG-FKBP38. D, immunoblot analysis of various mouse tissues with anti-ANKMY2, anti-FKBP38, and anti-HSP90 (loading control). Asterisks, nonspecific bands; arrowheads, corresponding specific bands. E, lysates prepared from Fkbp38−/−, Fkbp38+/+, and Fkbp38+/− mouse embryos (E11.5) were subjected to immunoblot analysis with anti-ANKMY2, anti-FKBP38, and anti-HSP90. F, immunohistofluorescence analysis of a neural tube section from a WT mouse embryo (E11.5) with anti-ANKMY2. Nuclei were stained with Hoechst 33258. Dashed line, neural tube. FP, floor plate. Scale bar, 100 μm. G, extracts of WT or Fkbp38TG mouse brain were subjected to immunoprecipitation (IP) with anti-FLAG. The resulting precipitates, as well as a portion (5% of the input for immunoprecipitation) of the extracts, were subjected to immunoblot analysis with anti-ANKMY2, anti-FLAG, and anti-HSP90. H, WT mouse brain extract was subjected to immunoprecipitation with anti-ANKMY2, anti-FKBP38, or control rabbit IgG, after which the resulting precipitates, as well as a portion (3% of the input for immunoprecipitation) of the tissue extract, were subjected to immunoblot analysis with anti-FKBP38 and anti-ANKMY2. Asterisk and arrowhead, nonspecific and specific bands, respectively.
TABLE 1.
Identification of FKBP38-associated proteins by proteomics analysis
| Protein | Sequence coverage |
|---|---|
| % | |
| Ankyrin repeat- and MYND domain-containing protein 2 (ANKMY2) | 21.1 |
| T-complex protein 1, α subunit (TCPA) | 11.3 |
| T-complex protein 1, η subunit (TCPH) | 2.9 |
| T-complex protein 1, δ subunit (TCPD) | 2.4 |
FKBP38 Specifically Interacts with ANKMY2
To confirm the interaction between FKBP38 and ANKMY2, we performed a co-immunoprecipitation assay with lysates prepared from the brains of WT or Fkbp38TG mice. Immunoprecipitates prepared with anti-FLAG were subjected to immunoblot analysis with anti-FLAG and anti-ANKMY2 (Fig. 1G). ANKMY2 was detected in the immunoprecipitates prepared from Fkbp38TG mouse brain lysates. Similar analysis was performed to detect the potential interaction between the two endogenous proteins. Immunoprecipitates prepared from WT mouse brain extract with anti-ANKMY2 were subjected to immunoblot analysis with anti-FKBP38. Endogenous FKBP38 was co-immunoprecipitated with endogenous ANKMY2 (Fig. 1H). A reciprocal experiment in which immunoprecipitates prepared with anti-FKBP38 were subjected to immunoblot analysis with anti-ANKMY2 also revealed that endogenous ANKMY2 interacted with endogenous FKBP38 (Fig. 1H). Collectively, these results suggested that FKBP38 interacts with ANKMY2 in the brain under physiological conditions.
We next examined the specificity of the interaction between FKBP38 and ANKMY2. We thus first determined whether ANKMY2 interacts with FKBP52, the FKBP family protein most closely related to FKBP38 (15, 33). A co-immunoprecipitation assay with transfected HEK293T cells revealed that, whereas FLAG-FKBP38 interacted with HA-ANKMY2, FLAG-FKBP52 did not (Fig. 2A), suggesting that the interaction between FKBP38 and ANKMY2 is specific. Furthermore, we investigated whether FKBP38 interacts with other members of the MYND-type Zn2+ finger protein family. Immunoblot analysis of immunoprecipitates prepared with anti-FLAG from lysates of HEK293T cells expressing 20 different FLAG-tagged MYND-type Zn2+ finger proteins revealed that ANKMY2 (ZMYND20) associated with endogenous FKBP38 to the greatest extent, whereas ZMYND7, ZMYND10, and ZMYND14 interacted with FKBP38 to a moderate extent (Fig. 2B). Although PHD2 (ZMYND6) was previously shown to interact with FKBP38 via its MYND domain (18), such interaction occurred only at a low level, if at all, under our assay conditions. FLAG-tagged ZMYND11, -15, and -21 were expressed at only low levels in the transfected cells, possibly as a result of conformational constraints imposed by the FLAG tag or of instability of these proteins in HEK293T cells. Together, these results thus suggested that FKBP38 and ANKMY2 specifically associate with each other.
FIGURE 2.

Specificity of the interaction between FKBP38 and ANKMY2. A, extracts of HEK293T cells transiently transfected with expression vectors for 3×FLAG-tagged human FKBP38 or human FKBP52 (negative control) and for 2×HA-tagged human ANKMY2 were subjected to immunoprecipitation with anti-FLAG. The resulting precipitates, as well as a portion (2% of the input for immunoprecipitation (IP)) of the cell extracts, were subjected to immunoblot analysis (IB) with anti-HA, anti-FLAG, and anti-calnexin. B, extracts of HEK293T cells transiently transfected with expression vectors for 3×FLAG-tagged ZMYND proteins were subjected to immunoprecipitation with anti-FLAG. The resulting precipitates, as well as a portion (3% of the input for immunoprecipitation) of the cell extracts, were subjected to immunoblot analysis with anti-FKBP38, anti-FLAG, and anti-calnexin. C, His6-T7-tagged mouse FKBP38 was incubated with GST-tagged mouse ANKMY2 or GST (negative control), and the binding mixtures were then subjected to precipitation with glutathione-conjugated beads. The bead-bound proteins (pull-down), as well as a portion (10% of the input for precipitation) of the binding mixtures, were subjected to immunoblot analysis with anti-T7 and anti-GST.
To examine whether FKBP38 and ANKMY2 directly bind to each other, we performed an in vitro pull-down assay with recombinant GST-tagged ANKMY2 and His6-T7-tagged FKBP38 (Fig. 2C). GST-ANKMY2 bound to His6-T7-FKBP38, whereas GST alone did not, indicating that FKBP38 and ANKMY2 indeed associate directly with each other.
Molecular Dissection of the FKBP38-ANKMY2 Interaction
Given that the NH2 terminus of FKBP38 was shown to be necessary for interaction with PHD2 (ZMYND6) (18), we examined whether the binding of ANKMY2 to FKBP38 might also be dependent on this region of FKBP38. We constructed a mutant of human FKBP38 lacking 57 amino acids at the NH2 terminus (Fig. 3A), and we found that the amount of endogenous ANKMY2 associated with this FKBP38(ΔN57) mutant in HEK293T cells was greatly reduced compared with that associated with the WT protein (Fig. 3B). This observation thus suggested that the interaction of FKBP38 with ANKMY2 is dependent on its NH2-terminal domain.
FIGURE 3.
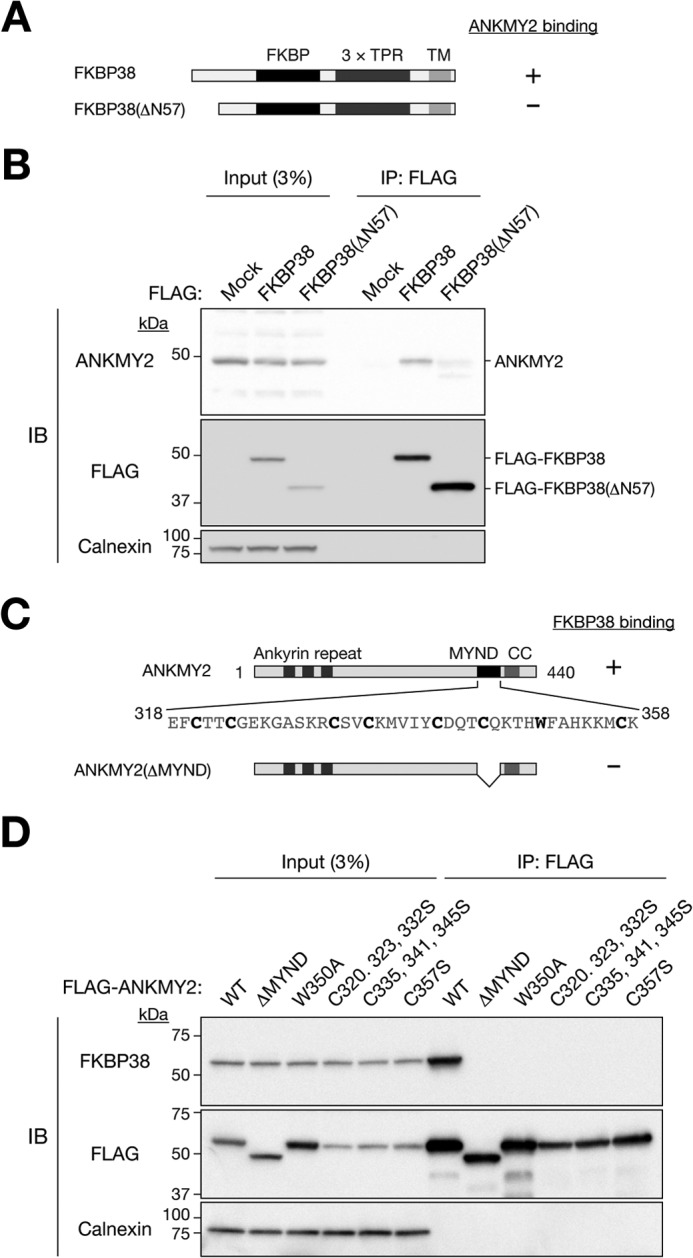
Delineation of the regions of FKBP38 and ANKMY2 responsible for their interaction. A, domain organization of human FKBP38 and structure of the FKBP38(ΔN57) mutant. TPR, tetratricopeptide repeat; TM, transmembrane. A summary of the ability of the WT and mutant proteins to bind ANKMY2 as determined in B is shown on the right. B, extracts of HEK293T cells transiently transfected with an expression vector for 3×FLAG-tagged human FKBP38 or FKBP38(ΔN57) were subjected to immunoprecipitation with anti-FLAG. The resulting precipitates, as well as a portion (3% of the input for immunoprecipitation (IP)) of the cell extracts, were subjected to immunoblot analysis with anti-ANKMY2, anti-FLAG, and anti-calnexin. C, domain organization of mouse ANKMY2 and structure of the ANKMY2(ΔMYND) mutant. CC, coiled-coil domain. A summary of the ability of the WT and mutant proteins to bind FKBP38 as determined in D is shown on the right. Individual residues of the MYND domain mutated in D are also indicated in boldface type. D, extracts of HEK293T cells transiently transfected with expression vectors for 3×FLAG-tagged WT or mutant forms of mouse ANKMY2 were subjected to immunoprecipitation with anti-FLAG. The resulting precipitates, as well as a portion (3% of the input for immunoprecipitation) of the cell extracts, were subjected to immunoblot analysis with anti-FKBP38, anti-FLAG, and anti-calnexin.
ANKMY2 contains three ankyrin repeats, a MYND domain, and a coiled-coil domain (Fig. 3C). Given that FKBP38 interacted with not only ANKMY2 but also other related proteins harboring the MYND domain, we hypothesized that the MYND domain of ANKMY2 is required for interaction with FKBP38. To test this hypothesis, we generated a deletion mutant of mouse ANKMY2 that lacks the MYND domain (ΔMYND). Co-immunoprecipitation analysis revealed that this mutant was defective with regard to the ability to interact with FKBP38 (Fig. 3D). Given that the MYND domain contains two predicted Zn2+ finger motifs, we replaced amino acids required for the formation of these structures to generate W350A, C320S/C323S/C332S, C335S/C341S/C345S, and C357S mutants (Fig. 3C). None of these mutants associated with FKBP38 (Fig. 3D), suggesting that the integrity of the Zn2+ finger motifs of ANKMY2 is required for binding to FKBP38.
FKBP38 Inhibits Shh Signaling by Suppressing the Activity of ANKMY2
We next examined Shh signaling in MEFs derived from Fkbp38+/+ or Fkbp38−/− embryos, given that FKBP38 has been implicated as a negative regulator of Shh signaling during development (21). The genes for Gli1 and Ptch1 are two early targets of Shh signaling, with the levels of the corresponding mRNAs and proteins increasing in a time-dependent manner (34). Confluent cultures of MEFs were deprived of serum for 24 h by incubation in Opti-MEM reduced serum medium and were then exposed to mouse Shh for assay of Shh signaling. Under this condition, we confirmed that both Fkbp38+/+ and Fkbp38−/− MEFs possessed cilia with a basal body containing γ-tubulin and an axoneme containing acetylated α-tubulin (Fig. 4A). Treatment of Fkbp38+/+ MEFs with Shh would be expected to activate the Shh pathway at Smo and thereby trigger downstream events that lead to up-regulation of Gli1 expression. We found that the up-regulation of Gli1 protein by Shh was markedly more pronounced in Fkbp38−/− MEFs than in Fkbp38+/+ MEFs (Fig. 4B). RT and real-time PCR analysis also showed that the amounts of Gli1 and Ptch1 mRNAs were increased by Shh to a significantly greater extent in Fkbp38−/− MEFs than in the WT cells (Fig. 4C). Together, these results were consistent with the notion that FKBP38 is a negative regulator of Shh signaling in mammalian cells.
FIGURE 4.

FKBP38 is a negative regulator of Shh signal transduction. A, confluent cultures of Fkbp38+/+ or Fkbp38−/− MEFs that had been deprived of serum for 24 h by incubation in Opti-MEM reduced serum medium were fixed and processed for immunofluorescence analysis with antibodies to γ-tubulin and to acetylated α-tubulin. Nuclei were also stained with Hoechst 33258, and the cells were then examined with a confocal microscope. Merged images are also shown; scale bars, 20 μm. Higher magnification views are boxed; scale bars, 5 μm. B, Fkbp38+/+ and Fkbp38−/− MEFs were incubated in the absence or presence of Shh for 48 h, after which whole cell homogenates were prepared and subjected to immunoblot analysis (IB) with anti-Gli1, anti-FKBP38, anti-ANKMY2, and anti-HSP90. C, Fkbp38+/+ and Fkbp38−/− MEFs were incubated in the absence or presence of Shh for 24 h, after which total RNA was extracted from the cells and subjected to RT and real-time PCR analysis of Gli1 and Ptch1 mRNAs. Data are presented as relative -fold induction by Shh and are means ± S.D. from five independent experiments. **, p < 0.01 versus the corresponding value for Fkbp38+/+ cells (Student's t test).
ANKMY2 localizes to sensory cilia in Caenorhabditis elegans, with such localization being required for the proper ciliary localization of guanylyl cyclase (35, 36). Given that primary cilia are essential for signal transduction by the vertebrate Shh pathway (8, 9), we examined whether ANKMY2 functions as a regulator of Shh signaling in MEFs. Depletion of endogenous ANKMY2 mediated by two independent siRNAs attenuated the stimulatory effects of Shh on Gli1 expression at the protein (Fig. 5A) and mRNA (Fig. 5B) levels as well as on Ptch1 mRNA abundance (Fig. 5B). Conversely, forced expression of ANKMY2 in MEFs enhanced these effects of Shh on Gli1 and Ptch1 expression (Fig. 5, C and D). Given that ANKMY2(ΔMYND) also up-regulated the expression of Gli1 and Ptch1 to an extent similar to that observed with the WT protein (Fig. 5D), the MYND domain of ANKMY2 does not appear to be required for the activation of Shh signaling per se. We also examined the effects of forced expression of either WT or ΔN57 mutant forms of FKBP38 on Gli1 abundance in FKBP38-deficient MEFs. The up-regulation of Gli1 expression apparent in Fkbp38−/− MEFs compared with Fkbp38+/+ cells was suppressed by forced expression of WT FKBP38 but not by that of FKBP38(ΔN57) (Fig. 5E), suggesting that the interaction of FKBP38 with ANKMY2 via the NH2-terminal domain of the former protein is essential for the suppression of ANKMY2 function with regard to activation of Shh signaling. Together, these results indicated that FKBP38 and ANKMY2 are negative and positive regulators of mammalian Shh signaling, respectively.
FIGURE 5.

ANKMY2 is a positive regulator of Shh signal transduction. A, MEFs were transfected for 48 h with control or Ankmy2 siRNAs and then incubated in the absence or presence of Shh for 48 h, after which whole cell homogenates were prepared and subjected to immunoblot analysis (IB) with anti-Gli1, anti-ANKMY2, and anti-HSP90. The asterisk indicates a nonspecific band. B, MEFs transfected with siRNAs as in A were incubated in the absence or presence of Shh for 24 h, after which total RNA was extracted from the cells and subjected to RT and real-time PCR analysis of Gli1 and Ptch1 mRNAs. Data are means ± S.D. (error bars) from three independent experiments. **, p < 0.01 versus the corresponding control value (Student's t test). C, MEFs stably expressing FLAG-tagged mouse ANKMY2 were incubated in the absence or presence of Shh for 48 h, after which whole cell homogenates were subjected to immunoblot analysis with anti-Gli1, anti-ANKMY2, and anti-HSP90. D, MEFs stably expressing FLAG-tagged ANKMY2 or ANKMY2(ΔMYND) were incubated in the absence or presence of Shh for 24 h and then subjected to RT and real-time PCR analysis of Gli1 and Ptch1 mRNAs. Data are means ± S.D. from three independent experiments. **, p < 0.01 versus the corresponding value for cells infected with the empty retrovirus (Student's t test). E, Fkbp38+/+ or Fkbp38−/− MEFs stably expressing EGFP-tagged human FKBP38 or FKBP38(ΔN57) were incubated in the absence or presence of Shh for 48 h, after which whole cell homogenates were subjected to immunoblot analysis with anti-Gli1, anti-FKBP38, and anti-HSP90.
We next examined how FKBP38 and ANKMY2 regulate Shh signaling by knockdown of ANKMY2 in FKBP38-deficient MEFs. Whereas FKBP38 deficiency enhanced the Shh-induced up-regulation of Gli1 expression (Fig. 4), depletion of ANKMY2 in Fkbp38−/− MEFs reversed this effect at both the protein (Fig. 6A) and mRNA (Fig. 6B) levels. Similarly, whereas Shh-induced Ptch1 transcription was enhanced in Fkbp38−/− MEFs compared with Fkbp38+/+ MEFs (Fig. 4C), this difference was abolished by additional depletion of ANKMY2 (Fig. 6B). This genetic evidence indicates that ANKMY2 is a downstream effector of FKBP38 (see Fig. 10). Specifically, our results suggested that FKBP38 is an upstream regulator that inhibits Shh signaling via suppression of the stimulatory activity of ANKMY2.
FIGURE 6.

FKBP38 inhibits Shh signaling via suppression of the stimulatory activity of ANKMY2. A, Fkbp38+/+ or Fkbp38−/− MEFs were transfected for 48 h with control or Ankmy2 siRNAs and then incubated in the absence or presence of Shh for 48 h, after which whole cell homogenates were prepared and subjected to immunoblot analysis with anti-Gli1, anti-FKBP38, anti-ANKMY2, and anti-HSP90. B, Fkbp38+/+ or Fkbp38−/− MEFs transfected with siRNAs as in A were incubated in the absence or presence of Shh for 24 h, after which total RNA was extracted from the cells and subjected to RT and real-time PCR analysis of Gli1 (left) and Ptch1 (right) mRNAs. C, MEFs stably expressing EGFP-tagged human FKBP38 and FLAG-tagged mouse ANKMY2, as indicated, were incubated in the absence or presence of Shh for 24 h, after which total RNA was extracted from the cells and subjected to RT and real-time PCR analysis of Gli1 (left) and Ptch1 (right) mRNAs. D, Fkbp38+/+ or Fkbp38−/− MEFs stably expressing FLAG-tagged ANKMY2 were incubated in the absence or presence of Shh for 24 h, after which total RNA was extracted from the cells and subjected to RT and real-time PCR analysis of Gli1 (left) and Ptch1 (right) mRNAs. Data in B–D are means ± S.D. (error bars) from three independent experiments. **, p < 0.01 (Student's t test). NS, not significant.
FIGURE 10.
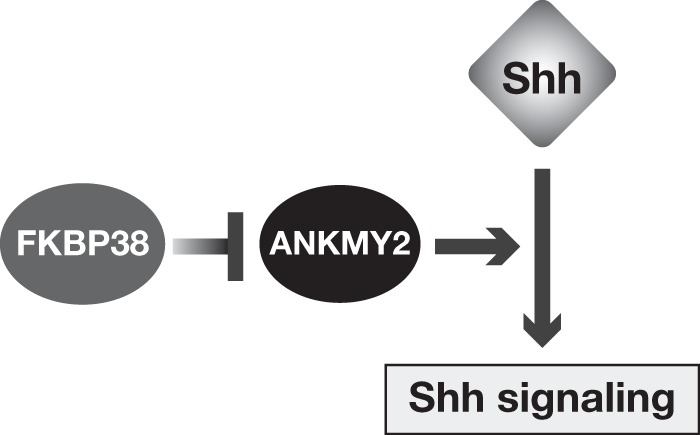
Model for FKBP38-mediated regulation of Shh signaling. FKBP38 regulates Shh signaling by suppression of ANKMY2 function. Loss of FKBP38 results in the activation of ANKMY2 and enhanced Shh signaling, leading to aberrant neural tube development.
We also examined whether ANKMY2 overexpression might reverse the effect of FKBP38 overexpression on Shh target gene transcription in MEFs. The down-regulation of Gli1 and Ptch1 expression apparent in cells overexpressing FKBP38 was prevented by overexpression of ANKMY2 (Fig. 6C), again suggesting that FKBP38 antagonizes ANKMY2 function in the enhancement of Shh signaling. We also examined the effect of overexpression of ANKMY2 on Gli1 and Ptch1 expression in WT and FKBP38-deficient MEFs (Fig. 6D). Forced expression of ANKMY2 increased Gli1 or Ptch1 expression to similar levels in cells of both genotypes, suggesting that the observed levels of gene expression were maximal.
ANKMY2 Functions as a Positive Regulator of Shh Signaling in Vivo
We next sought to determine whether ANKMY2 is required for Shh signal transduction during embryogenesis in a living organism. Zebrafish possesses two paralogous ankmy2 genes (ankmy2a, ankmy2b), probably as a result of a gene duplication event that occurred after its divergence from mammals. However, we were unable to detect ankmy2b mRNA in zebrafish by sensitive RT-PCR analysis (data not shown), suggesting that the gene is expressed at only a low level or not at all. To confirm the interaction between zebrafish FKBP38 and ANKMY2, we performed a co-immunoprecipitation assay. Immunoprecipitates were prepared with anti-HA from lysates of HEK293T cells transfected with expression vectors both for FLAG-tagged Danio rerio ANKMY2a (dANKMY2a) and for HA-tagged dFKBP38, and the resulting precipitates were subjected to immunoblot analysis with anti-HA and anti-FLAG (Fig. 7A). FLAG-dANKMY2a was detected in the immunoprecipitates prepared from the cells expressing HA-dFKBP38, suggesting that the interaction of FKBP38 with ANKMY2 is conserved in zebrafish.
FIGURE 7.

Zebrafish ankmy2a expression is required for Shh signaling. A, extracts of HEK293T cells transiently transfected with expression vectors for 3×FLAG-tagged dANKMY2a and for 2×HA-tagged dFKBP38 were subjected to immunoprecipitation (IP) with anti-HA. The resulting precipitates, as well as a portion (3% of the input for IP) of the cell extracts, were subjected to immunoblot analysis (IB) with anti-FLAG, anti-HA, and anti-calnexin. B, in situ hybridization of whole-mount zebrafish embryos at the indicated stages (hpf, hours postfertilization) with an antisense ankmy2a RNA probe. C, in situ hybridization of whole-mount zebrafish embryos at 24 hpf with antisense gli1 or myoD RNA probes. The embryos had been injected with ankmy2a and p53 antisense MOs at the one-cell stage or treated with cyclopamine, as indicated. D, percentages of embryos exhibiting normal or reduced gli1 or myoD expression as determined in C. The n values correspond to the total numbers of embryos examined. E, in situ hybridization of whole-mount zebrafish embryos at 24 hpf with an antisense gli1 RNA probe. The embryos had been injected with ankmy2a and p53 antisense MOs in the absence or presence of mouse Shh (mShh) mRNA at the one-cell stage. F, percentages of embryos exhibiting normal or reduced versus increased gli1 expression as determined in E. Scale bars (B, C, and E), 200 μm.
In situ hybridization revealed that ankmy2a is expressed in the neural tube of zebrafish embryos as early as the postsomitogenesis stage (Fig. 7B). We therefore investigated the functional role of dANKMY2a in Shh signaling. To this end, we inhibited the function of dANKMY2a with the use of antisense MOs, ankmy2a MO (for translational inhibition) or ankmy2a spl MO (for splicing inhibition). The Gli1 gene (gli1) is a target of the Shh pathway in fish and exhibits adaxial expression in normal embryos at 24 h postfertilization (hpf) (Fig. 7C). As a control, treatment of fish with cyclopamine, an inhibitor of the hedgehog signaling pathway, attenuated the expression of gli1 (Fig. 7, C and D). Suppression of ankmy2a expression with antisense MOs resulted in down-regulation of the adaxial expression of gli1 (Fig. 7, C and D), suggestive of reduced Shh signaling in the receiving cells. In situ hybridization analysis also revealed that the expression of myoD, a marker for muscle development, was attenuated by injection of ankmy2a antisense MOs or cyclopamine treatment (Fig. 7, C and D). We confirmed that the expression of gli1 was enhanced by injection of embryos with mouse Shh mRNA (Fig. 7, E and F). Furthermore, this effect was reversed by co-injection of either ankmy2a MO or ankmy2a spl MO. Collectively, these results suggested that dANKMY2a is required for full activation of Shh signaling in zebrafish embryos.
Given that attenuation of Shh signaling was previously shown to impair the development of slow muscle in fish (37), we examined the effect of injection of ankmy2a antisense MOs on this process. Immunohistochemical staining for myosin heavy chain (expressed in slow muscle) and Engrailed (expressed in muscle pioneer cells, the anlagen of slow muscle), both of which are targets of Shh signaling, was indeed reduced by injection of either ankmy2a MO or ankmy2a spl MO (Fig. 8, A–C). This down-regulation of myosin heavy chain and Engrailed expression was prevented by co-injection of human ANKMY2 mRNA (Fig. 8, A–C), excluding the possibility of off-target effects of these antisense MOs and suggesting that the function of ANKMY2 is conserved from fish to humans. Collectively, these results suggested that dANKMY2a indeed contributes to slow muscle development and plays an integral role in activation of Shh signaling in vivo.
FIGURE 8.

Zebrafish ankmy2a expression is required for muscle development. A and B, immunohistochemical staining of zebrafish embryos at 24 hpf with antibodies to myosin heavy chain (A) or to Engrailed (B). The embryos had been injected with ankmy2a and p53 antisense MOs in the absence or presence of human ANKMY2 (hANKMY2) mRNA at the one-cell stage or treated with cyclopamine, as indicated. C, percentages of embryos exhibiting normal or reduced myosin heavy chain or Engrailed expression, as determined in A and B. The n values correspond to the total numbers of embryos examined. D, in situ hybridization of whole-mount zebrafish embryos at 16 or 24 hpf with antisense gli1, fox2a, or nkx2.2 RNA probes. The embryos had been injected with ankmy2a and p53 antisense MOs at the one-cell stage. Scale bars (A, B, D), 200 μm.
We also examined the effect of injection of ankmy2a MOs on dorsoventral patterning of the neural tube. We expected that the area expressing ventral markers in the neural tube might be reduced by suppression of ankmy2a expression. Unexpectedly, however, the expression patterns of fox2a and nkx2.2 were unaffected by injection of either ankmy2a MO or ankmy2a spl MO (Fig. 8D). One possible explanation for these results is that dANKMY2a might be a context-dependent regulator rather than a universal activator of Shh signaling; it might thus influence gli1 expression and slow muscle development but not dorsoventral patterning of the neural tube. Alternatively, given that muscle development begins at a relatively late stage of embryogenesis, whereas dorsoventral patterning of the neural tube occurs earlier at ∼18 hpf, dANKMY2a might regulate Shh signaling only in the late phase of development in zebrafish Consistent with this notion, injection of ankmy2a MOs did not reduce the expression of gli1 at 16 hpf (Fig. 8D) but did so at 24 hpf (Fig. 7, C and D).
FKBP38 Regulates ANKMY2 Localization
We next examined whether FKBP38 might affect the subcellular localization of ANKMY2, given that FKBP38 contributes to the mitochondrial localization of Bcl-2 and Bcl-xL (15). We thus determined the subcellular localization of endogenous FKBP38 and EGFP-tagged ANKMY2 in MEFs with a primary cilium, a sensor for Shh. We found that FKBP38 was localized predominantly to mitochondria, with no apparent localization to cilia (Fig. 9A). Whereas ANKMY2 was previously shown to localize to cilia in C. elegans (35, 36), we found that EGFP-ANKMY2 was distributed throughout the cytoplasm of MEFs, with no apparent localization to cilia (Fig. 9B). This discrepancy is probably attributable to the difference in cell type or species studied. We speculate that a small but undetectable amount of ANKMY2 is present at cilia and that this fraction is reduced by sequestration of the protein from cilia to mitochondria mediated by the interaction with FKBP38. Consistent with this hypothesis, forced expression of HA-FKBP38 resulted in the apparent redistribution of FLAG-ANKMY2 from the cytoplasm to mitochondria, whereas it did not affect the localization of FLAG-ANKMY2(ΔMYND), which is not able to interact with FKBP38 (Fig. 9C). We also found that HA-FKBP38(ΔN57), which is not able to interact with ANKMY2 or to suppress its function, also did not affect the localization of FLAG-ANKMY2 (Fig. 9D). Together, these results revealed a correlation between the localization of ANKMY2 and its function in activation of Shh signaling, suggesting that FKBP38 probably regulates the function of ANKMY2 by controlling its localization through interaction with this protein.
FIGURE 9.

FKBP38 regulates ANKMY2 localization. A, confluent cultures of Fkbp38+/+ or Fkbp38−/− MEFs that had been deprived of serum for 24 h by incubation in Opti-MEM reduced serum medium were fixed and processed for immunofluorescence analysis with antibodies to acetylated α-tubulin and to FKBP38. Nuclei were stained with Hoechst 33258, and the cells were then examined with a confocal microscope. Merged images are also shown; scale bars, 20 μm. Higher magnification views are boxed; scale bar, 5 μm. B, confluent cultures of MEFs stably expressing EGFP-tagged human ANKMY2 were deprived of serum for 24 h, fixed, and processed for immunofluorescence analysis with antibodies to acetylated α-tubulin. Nuclei were stained with Hoechst 33258, and the cells were then examined with a confocal microscope. The fluorescence of EGFP-ANKMY2 was monitored directly. A merged image is also shown; scale bar, 20 μm. Higher magnification views are boxed; scale bar, 5 μm. C, HeLa cells expressing 2×HA-tagged human FKBP38 and 3×FLAG-tagged mouse ANKMY2 or ANKMY2(ΔMYND) were fixed and processed for immunofluorescence staining with anti-FLAG and anti-HA. Nuclei were stained with Hoechst 33258, and the cells were then examined with a confocal microscope. Merged images are also shown; scale bars, 20 μm. Higher magnification views are boxed; scale bars, 10 μm. An arrowhead indicates colocalization of ANKMY2 and FKBP38 signals; an arrow indicates that ANKMY2(ΔMYND) and FKBP38 signals are not colocalized. D, HeLa cells expressing 3×FLAG-tagged ANKMY2 and 2×HA-tagged FKBP38 or FKBP38(ΔN57) were fixed and processed for immunofluorescence staining with anti-HA and anti-FLAG. Nuclei were stained with Hoechst 33258, and the cells were then examined with a confocal microscope. Merged images are also shown; scale bars, 20 μm. Higher magnification views are boxed; scale bars, 10 μm. An arrowhead indicates colocalization of ANKMY2 and FKBP38 signals; an arrow indicates that ANKMY2 and FKBP38(ΔN57) signals are not colocalized.
DISCUSSION
Although the phenotypes associated with FKBP38 ablation in mice suggested that FKBP38 suppresses Shh signaling during development, the detailed mechanism by which FKBP38 inhibits such signaling had remained unclear. To explore this mechanism, we applied a dual affinity purification system to FKBP38 tagged with His6 and FLAG epitopes and expressed in the brain of transgenic mice. This system allowed us to identify physiological targets of FKBP38 in vivo, with ANKMY2 being the target with the highest sequence coverage. Co-immunoprecipitation analysis further revealed that the MYND domain of ANKMY2 interacts with the NH2-terminal domain of FKBP38. Although PHD2 (ZMYND6), another MYND-type Zn2+ finger protein, was previously shown to interact with FKBP38 (18), we found that the extent of this interaction was minimal compared with that between FKBP38 and ANKMY2 (ZMYND20). Our finding that the MYND-type Zn2+ finger domain of ANKMY2 is responsible for binding to FKBP38 suggests that other proteins with this type of domain might also associate with FKBP38. Indeed, we found that ZMYND7, -10, and -14 also interacted with FKBP38, although the physiological roles of these interactions remain unknown.
We have shown that ANKMY2 is a positive regulator of Shh signaling and that FKBP38 is an upstream regulator that inhibits Shh signaling by abrogation of the function of ANKMY2 (Fig. 10). The mechanisms by which ANKMY2 enhances Shh signaling and by which FKBP38 antagonizes this action of ANKMY2 remain unclear, however. ANKMY2 localizes to cilia and contributes to the intraflagellar transport-independent translocation of guanylyl cyclase in C. elegans (35, 36). We confirmed the interaction between ANKMY2 and guanylyl cyclase in mammalian cells (data not shown). In this regard, it is of note that cGMP activates Shh signaling in mammals (38). Given that protein kinase G (PKG) activated by cGMP phosphorylates Gli3, resulting in activation of Shh signaling (39), it is possible that the FKBP38-ANKMY2 axis controls Shh signaling via the cGMP-PKG pathway.
One of the molecular functions of FKBP38 is recruitment of its interacting proteins, such as Bcl-2 and Bcl-xL, to the mitochondrial outer membrane. Overexpression of FKBP38 blocks apoptosis as a result of enhanced recruitment of Bcl-2 and Bcl-xL to mitochondria, whereas depletion of FKBP38 results in mislocalization of these proteins (15). Similarly, we found that overexpression of FKBP38 in mammalian cells triggered translocation of ANKMY2 to mitochondria. Such translocation of ANKMY2 induced by FKBP38 might be responsible for the abrogation of putative ANKMY2 function that contributes to the activation of guanylyl cyclase in cilia. This hypothesis remains to be tested in the future.
Acknowledgments
We thank Y. Minamishima, S. S. Wing, S. Nagata, T. Kitamura, and T. Akagi for providing vectors and DNA; K. Tsunematsu and other laboratory members for technical assistance; and A. Ohta for help in preparation of the manuscript.
This work was supported in part by a Japan Society for the Promotion of Science (JSPS) fellowship (to S. S.).
- FKBP
- FK506-binding protein
- MEF
- mouse embryonic fibroblast
- MO
- morpholino oligonucleotide
- NTA
- nitrilotriacetic acid
- E11.5
- embryonic day 11.5
- hpf
- hours postfertilization
- EGFP
- enhanced green fluorescent protein.
REFERENCES
- 1. Huangfu D., Anderson K. V. (2005) Cilia and Hedgehog responsiveness in the mouse. Proc. Natl. Acad. Sci. U.S.A. 102, 11325–11330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lum L., Beachy P. A. (2004) The Hedgehog response network: sensors, switches, and routers. Science 304, 1755–1759 [DOI] [PubMed] [Google Scholar]
- 3. Rohatgi R., Scott M. P. (2007) Patching the gaps in Hedgehog signalling. Nat. Cell Biol. 9, 1005–1009 [DOI] [PubMed] [Google Scholar]
- 4. Alcedo J., Ayzenzon M., Von Ohlen T., Noll M., Hooper J. E. (1996) The Drosophila smoothened gene encodes a seven-pass membrane protein, a putative receptor for the hedgehog signal. Cell 86, 221–232 [DOI] [PubMed] [Google Scholar]
- 5. Marigo V., Davey R. A., Zuo Y., Cunningham J. M., Tabin C. J. (1996) Biochemical evidence that patched is the Hedgehog receptor. Nature 384, 176–179 [DOI] [PubMed] [Google Scholar]
- 6. Stone D. M., Hynes M., Armanini M., Swanson T. A., Gu Q., Johnson R. L., Scott M. P., Pennica D., Goddard A., Phillips H., Noll M., Hooper J. E., de Sauvage F., Rosenthal A. (1996) The tumour-suppressor gene patched encodes a candidate receptor for Sonic hedgehog. Nature 384, 129–134 [DOI] [PubMed] [Google Scholar]
- 7. Hui C. C., Angers S. (2011) Gli proteins in development and disease. Annu. Rev. Cell Dev. Biol. 27, 513–537 [DOI] [PubMed] [Google Scholar]
- 8. Murdoch J. N., Copp A. J. (2010) The relationship between sonic Hedgehog signaling, cilia, and neural tube defects. Birth Defects Res. A Clin. Mol. Teratol. 88, 633–652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Fliegauf M., Benzing T., Omran H. (2007) When cilia go bad: cilia defects and ciliopathies. Nat. Rev. Mol. Cell Biol. 8, 880–893 [DOI] [PubMed] [Google Scholar]
- 10. Rohatgi R., Milenkovic L., Scott M. P. (2007) Patched1 regulates hedgehog signaling at the primary cilium. Science 317, 372–376 [DOI] [PubMed] [Google Scholar]
- 11. Corbit K. C., Aanstad P., Singla V., Norman A. R., Stainier D. Y., Reiter J. F. (2005) Vertebrate Smoothened functions at the primary cilium. Nature 437, 1018–1021 [DOI] [PubMed] [Google Scholar]
- 12. Barik S. (2006) Immunophilins: for the love of proteins. Cell. Mol. Life Sci. 63, 2889–2900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hamilton G. S., Steiner J. P. (1998) Immunophilins: beyond immunosuppression. J. Med. Chem. 41, 5119–5143 [DOI] [PubMed] [Google Scholar]
- 14. Lam E., Martin M., Wiederrecht G. (1995) Isolation of a cDNA encoding a novel human FK506-binding protein homolog containing leucine zipper and tetratricopeptide repeat motifs. Gene 160, 297–302 [DOI] [PubMed] [Google Scholar]
- 15. Shirane M., Nakayama K. I. (2003) Inherent calcineurin inhibitor FKBP38 targets Bcl-2 to mitochondria and inhibits apoptosis. Nat. Cell Biol. 5, 28–37 [DOI] [PubMed] [Google Scholar]
- 16. Saita S., Shirane M., Nakayama K. I. (2013) Selective escape of proteins from the mitochondria during mitophagy. Nat. Commun. 4, 1410. [DOI] [PubMed] [Google Scholar]
- 17. Nakagawa T., Shirane M., Iemura S., Natsume T., Nakayama K. I. (2007) Anchoring of the 26S proteasome to the organellar membrane by FKBP38. Genes Cells 12, 709–719 [DOI] [PubMed] [Google Scholar]
- 18. Barth S., Edlich F., Berchner-Pfannschmidt U., Gneuss S., Jahreis G., Hasgall P. A., Fandrey J., Wenger R. H., Camenisch G. (2009) Hypoxia-inducible factor prolyl-4-hydroxylase PHD2 protein abundance depends on integral membrane anchoring of FKBP38. J. Biol. Chem. 284, 23046–23058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Nielsen J. V., Mitchelmore C., Pedersen K. M., Kjaerulff K. M., Finsen B., Jensen N. A. (2004) Fkbp8: novel isoforms, genomic organization, and characterization of a forebrain promoter in transgenic mice. Genomics 83, 181–192 [DOI] [PubMed] [Google Scholar]
- 20. Shirane M., Ogawa M., Motoyama J., Nakayama K. I. (2008) Regulation of apoptosis and neurite extension by FKBP38 is required for neural tube formation in the mouse. Genes Cells 13, 635–651 [DOI] [PubMed] [Google Scholar]
- 21. Cho A., Ko H. W., Eggenschwiler J. T. (2008) FKBP8 cell-autonomously controls neural tube patterning through a Gli2- and Kif3a-dependent mechanism. Dev. Biol. 321, 27–39 [DOI] [PubMed] [Google Scholar]
- 22. Nakayama K., Ishida N., Shirane M., Inomata A., Inoue T., Shishido N., Horii I., Loh D. Y., Nakayama K. I. (1996) Mice lacking p27Kip1 display increased body size, multiple organ hyperplasia, retinal dysplasia, and pituitary tumors. Cell 85, 707–720 [DOI] [PubMed] [Google Scholar]
- 23. Nakayama K., Nagahama H., Minamishima Y. A., Matsumoto M., Nakamichi I., Kitagawa K., Shirane M., Tsunematsu R., Tsukiyama T., Ishida N., Kitagawa M., Nakayama K., Hatakeyama S. (2000) Targeted disruption of Skp2 results in accumulation of cyclin E and p27Kip1, polyploidy and centrosome overduplication. EMBO J. 19, 2069–2081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kamura T., Hara T., Matsumoto M., Ishida N., Okumura F., Hatakeyama S., Yoshida M., Nakayama K., Nakayama K. I. (2004) Cytoplasmic ubiquitin ligase KPC regulates proteolysis of p27Kip1 at G1 phase. Nat. Cell Biol. 6, 1229–1235 [DOI] [PubMed] [Google Scholar]
- 25. Matsuzaki F., Shirane M., Matsumoto M., Nakayama K. I. (2011) Protrudin serves as an adaptor molecule that connects KIF5 and its cargoes in vesicular transport during process formation. Mol. Biol. Cell 22, 4602–4620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Borchelt D. R., Davis J., Fischer M., Lee M. K., Slunt H. H., Ratovitsky T., Regard J., Copeland N. G., Jenkins N. A., Sisodia S. S., Price D. L. (1996) A vector for expressing foreign genes in the brains and hearts of transgenic mice. Genet. Anal. 13, 159–163 [DOI] [PubMed] [Google Scholar]
- 27. Susaki E., Kaneko-Oshikawa C., Miyata K., Tabata M., Yamada T., Oike Y., Katagiri H., Nakayama K. I. (2010) Increased E4 activity in mice leads to ubiquitin-containing aggregates and degeneration of hypothalamic neurons resulting in obesity. J. Biol. Chem. 285, 15538–15547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Shirane M., Nakayama K. I. (2006) Protrudin induces neurite formation by directional membrane trafficking. Science 314, 818–821 [DOI] [PubMed] [Google Scholar]
- 29. Ota S., Ishitani S., Shimizu N., Matsumoto K., Itoh M., Ishitani T. (2012) NLK positively regulates Wnt/β-catenin signalling by phosphorylating LEF1 in neural progenitor cells. EMBO J. 31, 1904–1915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ishitani T., Matsumoto K., Chitnis A. B., Itoh M. (2005) Nrarp functions to modulate neural-crest-cell differentiation by regulating LEF1 protein stability. Nat. Cell Biol. 7, 1106–1112 [DOI] [PubMed] [Google Scholar]
- 31. Miller J. B., Teal S. B., Stockdale F. E. (1989) Evolutionarily conserved sequences of striated muscle myosin heavy chain isoforms: epitope mapping by cDNA expression. J. Biol. Chem. 264, 13122–13130 [PubMed] [Google Scholar]
- 32. Patel N. H., Martin-Blanco E., Coleman K. G., Poole S. J., Ellis M. C., Kornberg T. B., Goodman C. S. (1989) Expression of engrailed proteins in arthropods, annelids, and chordates. Cell 58, 955–968 [DOI] [PubMed] [Google Scholar]
- 33. Saita S., Shirane M., Natume T., Iemura S., Nakayama K. I. (2009) Promotion of neurite extension by protrudin requires its interaction with vesicle-associated membrane protein-associated protein. J. Biol. Chem. 284, 13766–13777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Humke E. W., Dorn K. V., Milenkovic L., Scott M. P., Rohatgi R. (2010) The output of Hedgehog signaling is controlled by the dynamic association between Suppressor of Fused and the Gli proteins. Genes Dev. 24, 670–682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Fujiwara M., Teramoto T., Ishihara T., Ohshima Y., McIntire S. L. (2010) A novel zf-MYND protein, CHB-3, mediates guanylyl cyclase localization to sensory cilia and controls body size of Caenorhabditis elegans. PLoS Genet. 6, e1001211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Jensen V. L., Bialas N. J., Bishop-Hurley S. L., Molday L. L., Kida K., Nguyen P. A., Blacque O. E., Molday R. S., Leroux M. R., Riddle D. L. (2010) Localization of a guanylyl cyclase to chemosensory cilia requires the novel ciliary MYND domain protein DAF-25. PLoS Genet. 6, e1001199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Blagden C. S., Currie P. D., Ingham P. W., Hughes S. M. (1997) Notochord induction of zebrafish slow muscle mediated by Sonic hedgehog. Genes Dev. 11, 2163–2175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Robertson C. P., Gibbs S. M., Roelink H. (2001) cGMP enhances the sonic hedgehog response in neural plate cells. Dev. Biol. 238, 157–167 [DOI] [PubMed] [Google Scholar]
- 39. Yamamoto T., Suzuki N. (2005) Expression and function of cGMP-dependent protein kinase type I during medaka fish embryogenesis. J. Biol. Chem. 280, 16979–16986 [DOI] [PubMed] [Google Scholar]