

Background: Magnesium protoporphyrin IX methyltransferase (ChlM) catalyzes the second step in the magnesium branch of tetrapyrrole biosynthesis.
Results: The SAM- and SAH-bound ChlM structures were obtained, and the ChlM active site was characterized.
Conclusion: Flexibility of two regions could be a key modulator for methyltransferase activity.
Significance: This work provides first structural insights into the catalytic mechanism of ChlM.
Keywords: Chlorophyll, Chloroplast, Enzyme Mechanism, Enzyme Structure, Photosynthesis, Protein Structure
Abstract
Magnesium protoporphyrin IX O-methyltransferase (ChlM) catalyzes transfer of the methyl group from S-adenosylmethionine to the carboxyl group of the C13 propionate side chain of magnesium protoporphyrin IX. This reaction is the second committed step in chlorophyll biosynthesis from protoporphyrin IX. Here we report the crystal structures of ChlM from the cyanobacterium Synechocystis sp. PCC 6803 in complex with S-adenosylmethionine and S-adenosylhomocysteine at resolutions of 1.6 and 1.7 Å, respectively. The structures illustrate the molecular basis for cofactor and substrate binding and suggest that conformational changes of the two “arm” regions may modulate binding and release of substrates/products to and from the active site. Tyr-28 and His-139 were identified to play essential roles for methyl transfer reaction but are not indispensable for cofactor/substrate binding. Based on these structural and functional findings, a catalytic model is proposed.
Introduction
Chlorophylls are the primary pigments that harvest solar energy for the light reaction of photosynthesis. The biosynthesis of chlorophylls is a vital process in photosynthetic organisms and consists of multiple enzymatic steps (1, 2). Protoporphyrin IX is the last common tetrapyrrole precursor located at the branch point of heme and chlorophyll biosynthesis. The initial steps of the chlorophyll branch are tightly regulated by interplays of proteins involved in sequential conversions of protoporphyrin IX to magnesium protoporphyrin IX (MgP)2 and Mg-protoporphyrin IX monomethyl ester (MgPME). These interacting proteins include magnesium chelatase, a complex enzyme comprising three subunits (ChlH, ChlI, and ChlD), magnesium protoporphyrin IX O-methyltransferase (ChlM), genomes uncoupled 4 protein (Gun4), and redox regulators such as thioredoxin and the NADPH-dependent thioredoxin reductase C (3–5). In addition, the intermediates during the process, MgP and MgPME, are potential retrograde signaling molecules in plastid-containing photosynthetic organisms (6–8). The structural characterization of some these proteins, including magnesium chelatase (9–12) and Gun4 (13, 14), has provided a basis to elucidate their regulatory mechanisms and the signaling issues. However, details about the catalytic mechanism are not well understood due to the lack of high resolution structural information, particularly for ChlH and ChlM.
ChlM (E.C. 2.1.1.11) catalyzes methyl transfer from the common methyl donor SAM to the carboxyl group of the C13 propionate side chain of MgP, resulting in the formation of MgPME and SAH. Genetic studies have shown that ChlM is essential for chlorophyll biosynthesis and chloroplast development (15, 16). The enzymatic kinetics of the cyanobacterium Synechocystis ChlM (SyChlM) have been elaborately studied (17, 18). These kinetic studies have established a random sequential mechanism in which SAM and MgP bind to SyChlM in either order. Sequence analysis revealed that SyChlM is homologous to small molecule methyltransferases, but structural information about the active site was not available. Furthermore, it has been shown that Synechocystis ChlH can stimulate SyChlM activity (19). Such a regulatory coupling of ChlH and ChlM is conserved from the purple bacteria to higher plants (20–23). An efficient intermediate channeling between ChlH and ChlM has been proposed to facilitate the flow of protoporphyrin IX to the chlorophyll branch (2, 3). To understand the molecular basis underlying the catalytic and regulatory mechanisms of ChlM, we set out to solve the structure of SyChlM.
In the present work we report the SAM- and SAH-bound SyChlM structures at resolutions of 1.6 and 1.7 Å, respectively. The structures demonstrate that the N-terminal region and a 21-residue insertion protruding from the core of the protein are flexible. Based on the SyChlM structure and supported by biochemical experiments on site-directed mutants, we identified two residues that are required for the methyl transfer reaction. In addition, these findings suggest a possible molecular basis for ChlM catalytic and regulatory mechanism.
EXPERIMENTAL PROCEDURES
Expression, Purification, and Crystallization
The chlM gene from Synechocystis sp. PCC 6803 was inserted into pET-22b(+) (Novagen) to generate the pET-22b(+)-SyChlM-His6 plasmid. The recombinant His-tagged SyChlM was expressed in Escherichia coli BL21(DE3) cells and purified by nickel-nitrilotriacetic acid column (Qiagen) followed by size-exclusion chromatography using a HiLoad 16/60 Superdex 75 column (GE Healthcare). The purified protein was concentrated by ultrafiltration in a buffer containing 20 mm citrate sodium, pH 5.8, 150 mm NaCl, and 100 mm glycerol, shock-frozen in liquid nitrogen, and stored at −80 °C. For crystallization, the purified protein was diluted to a concentration of 10 mg/ml and then incubated with 2 mm SAM or SAH (Sigma) at 4 °C for 30 min. Both native and selenomethionine-substituted crystals of the SyChlM-SAM complex were obtained using the hanging-drop vapor diffusion method at 4 °C in a mixed drop containing 1–2 μl of protein sample and an equal volume of well solution (0.1 m MES, pH 6.7, 300 mm NaCl, and 10% PEG 3350) taken from a 1-ml reservoir. Crystals qualified for x-ray data collection were optimized by the seeding method. The SyChlM-SAH crystals were obtained using sitting-drop vapor diffusion at 4 °C by mixing 1–2 μl of protein sample with an equal volume of crystallization solution (0.1 m citrate sodium, pH 5.8, 10% PEG 4000, and 20% isopropyl alcohol) taken from a 200-μl reservoir.
Data Collection and Structure Determination
The harvested crystals were cryo-protected in crystallization solution supplemented with 20% (v/v) glycol and then shock-frozen in liquid nitrogen. X-ray diffraction data were collected at beamline BL17U of the Shanghai Synchrotron Radiation Facility at a wavelength of 0.9793 Å at 100 K. The data were indexed, integrated, and scaled using DENZO and SCALEPACK as implemented in HKL2000 (24). Selenium positions in the SyChlM-SAM crystal and preliminary model-building were resolved using the program AutoSol in the PHENIX suite (25). Additional missing residues in the auto-built model were manually added according to the 2Fo − Fc and Fo − Fc electron density maps in Coot (26). The native SyChlM-SAM and SyChlM-SAH structures were determined by molecular replacement using the selenomethionine-substituted SyChlM-SAM structure as the searching model. Automatic model building was performed using ARP/wARP (27), and manual model correction was done in Coot. The models were refined further in PHENIX, and the overall quality of the final structural models was assessed by PROCHECK (28). Statistics for data collection and structure refinement are summarized in Table 1. The protein structure figures were prepared using the program PyMOL.
TABLE 1.
Data collection and structure refinement statistics
Numbers in parentheses represent the value for the highest resolution shell.
| SyChlM-SAM | SyChlM-SAH | SeMet SyChlM-SAM | |
|---|---|---|---|
| Data collection | |||
| Space group | P212121 | P21 | P212121 |
| Resolution range (Å) | 50-1.6 (1.66-1.60) | 50-1.7 (1.76-1.70) | 50-2.5 (2.59-2.50) |
| Unit cell parameters | |||
| a, b, c (Å) | 49.3, 61.0, 68.8 | 49.0, 98.8, 52.9 | 49.3, 60.3, 70.9 |
| α, β, γ (°) | 90, 90, 90 | 90, 95.8, 90 | 90, 90, 90 |
| Wavelength (Å) | 0.9793 | 0.9793 | 0.9793 |
| Total reflections | 342,952 (17,979) | 200,818 (19,645) | 212,291 (21,588) |
| Unique reflections | 2,7436 (2,305) | 54,275 (5,457) | 7,776 (747) |
| No. of protein molecules in asymmetric unit | 1 | 2 | 1 |
| Completeness (%) | 97.9 (84.3) | 99.5 (98.9) | 100 (100) |
| I/σ(I) | 40.7 (5.2) | 9.3 (2.4) | 56.7 (18.6) |
| Rmergea | 0.096 (0.317) | 0.125 (0.547) | 0.146 (0.452) |
| Refinement statistics | |||
| Resolution range (Å) | 50-1.6 | 50-1.7 | |
| Rwork/Rfreeb | 0.14/0.17 | 0.18/0.22 | |
| r.m.s.d. bond length (Å) | 0.003 | 0.003 | |
| r.m.s.d. bond angle (°) | 0.840 | 0.745 | |
| Ramachandran plot | |||
| Most favored (%) | 96.7 | 97.0 | |
| Additional allowed (%) | 3.3 | 3.0 | |
| Disallowed (%) | 0.0 | 0.0 | |
a Rmerge = Σ|Ii − Im|/ΣIi, where Ii is the intensity of the measured reflection, and Im is the mean intensity of all symmetry related reflections.
b Rwork = Σ‖Fobs| − |Fcal‖/Σ|Fobs|, where Fobs and Fcal are observed and calculated structure factors. Rfree = ΣT‖Fobs| − |Fcal‖/ΣT|Fobs|, where T is a test data set of ∼5% of the total reflections randomly chosen and set aside before refinement.
MgP Docking
In silico docking of the substrate MgP to SyChlM was performed with AutoDock Vina 1.1.2 (29). The MgP coordinates were extracted from a high resolution x-ray crystal structure (Protein Data Bank code 1QSI (30)). Protein coordinates were taken from chain A of the SyChlM-SAH structure. Before simulation, the cofactor SAH in the SyChlM-SAH structure was replaced by SAM. Hydrogen atoms, Gasteiger partial charges, and ligand torsions were added using the program AutoDockTool in the MGLtools software (31). During simulation, the ChlM and SAM structures were kept rigid, and grid maps were calculated using 60 × 40 × 45 grid points with a spacing of 0.55 Å. Grids were centered such that they covered the whole substrate binding site, and the most plausible model was selected based on the steric accessibilities of the C13 propionate side chain of MgP and the calculated binding affinity energy.
Site-directed Mutagenesis
SyChlM mutants were generated with the Fast Mutagenesis System kit (TransGen Biotech, Beijing) using pET-22b(+)-SyChlM-His6 as template. All mutant plasmids were sequenced to confirm the desired mutations. The procedure for purification of the SyChlM mutant proteins was the same as that of the wild type.
Methyltransferase Assay
SyChlM activity was measured using an enzyme-coupled continuous spectrophotometric assay (32, 33). The reaction mixtures contained 100 mm Tris-HCl, pH 7.5, 100 mm NaCl, 5% polypropylene glycol P 400, 500 μm MnCl2, 50 μm SAM, 50 μm MgP (Frontier Scientific), 10 μm SAH nucleosidase, 0.2 μm adenine deaminase, and 2 μm SyChlM. The recombinant SAH nucleosidase and adenine deaminase were expressed and purified following the established procedures (32, 34). The assay was performed in a thermostatted 1-cm quartz cuvette at 30 °C. The coupled reaction was measured by continuously recording the absorbance at 265 nm for 5 min, which indicates the conversion of adenine to hypoxanthine that correlates to the production of SAH.
Isothermal Titration Calorimetry
ITC experiments were performed on a MicroCal iTC200 calorimeter (GE Healthcare) at 20 °C. The purified SyChlM protein was changed into the buffer of 100 mm Tris-HCl, pH 7.4, 100 mm NaCl, and 300 mm glycerol. The ligand SAH was directly dissolved in the above buffer, and MgP was dissolved in the buffer supplemented with 0.1% Tween 80. The concentrations of SAH and MgP were determined spectrophotometrically using the molar absorption coefficients of ϵ260 nm = 15,400 m−1cm−1 and ϵ408 nm = 278,000 m−1cm−1, respectively (35, 36). Each titration series consisted of 20 injections (2 μl) of ligand in the syringe into the protein sample (200 μl). Control experiments were carried out by injecting each ligand into the buffer, and the resulting heat of dilution was subtracted from the binding isotherm data. The first injection was ignored in the final analysis. The raw ITC data were processed using the Origin software and fitted to a single-site binding model.
RESULTS
Overall Structure of SyChlM
We obtained crystals of SyChlM in the apo-, SAM-bound, and SAH-bound forms. However, attempts to solve the apo structure were unsuccessful due to poor diffraction quality of the crystals. The structures of SyChlM bound to SAM and SAH were determined at resolutions of 1.6 and 1.7 Å, respectively (Table 1). The secondary structural elements of SyChlM are defined by DSSP (37), and the topology is depicted in Fig. 1A. An α/β/α sandwich forms the core of fold with the central seven-stranded β-sheet having a strand order of 3-2-1-4-5-7-6, which is a characteristic feature of SAM-dependent methyltransferase (38, 39). The eight flanking α-helices are named sequentially in alphabetical order (αA to αH). In the SyChlM-SAM structure (Fig. 1B), electron density is missing for residues 168–188. In the SyChlM-SAH structure (Fig. 1C), residues 169–178 are observed as an α-helix (αG), which protrudes from the core of the protein. It is of note that the packing arrangements of SyChlM-SAM and SyChlM-SAH are in different space groups. Whereas SyChlM-SAM is a monomer in one asymmetric unit (ASU), there are two SyChlM-SAH monomers per ASU related by noncrystallographic symmetry. A superimposition of SyChlM in these three monomeric forms reveals that their major differences lie in the orientation of αG and the N-terminal sequence (including αA and the loop between αB and αC) (Fig. 1D). These differences correlate with the increased B factors for these regions, indicating their conformational flexibility.
FIGURE 1.

Overall structure of SyChlM. A, topology diagram of SynChlM. Helices and β-strands are represented by cylinders and arrows. The starting and ending residue numbers for each secondary structural element are labeled. B, ribbon representation of the monomeric SyChlM-SAM. The SAM cofactor is in stick model. The dotted lines indicate the missing sequences. The starting and ending residue numbers for sequences not observed in the electron density are labeled. C, ribbon representation of the two SyChlM-SAH monomers with SAH shown in stick model. D, superimposition of the monomeric forms of SyChlM (as in B and C). A dashed line is drawn between the αG region and the core of SyChlM. The radius of the tubes corresponds to the B factor.
SAM/SAH Binding Pocket
Sequence alignment of ChlMs from different species reveals a DXGCGXG motif after β1 and a conserved aspartic acid at the end of β2 (Fig. 2A). These are the two conserved structural elements that are required for cofactor SAM binding among the Class I methyltransferases (39). In both SyChlM-SAM and SyChlM-SAH structures, the electron density for the cofactor SAM/SAH is well defined, and no significant differences were observed between SAM and SAH except the S-methyl group (Fig. 2B). Due to its higher resolution, the SyChlM-SAM structure is used to describe the SAM/SAH binding pocket. This pocket is covered by an N-terminal arm consisting of αA and αB (Figs. 1B and 2C) and is shaped by residues from helices αA and αC and loops β1-αD, β2-αE, β3-β4, and β4-αF. The SAM binding in this pocket is stabilized by interactions as follows (Fig. 2D). The adenine ring is surrounded by hydrophobic residues Val-12, Phe-16, Ile-92, Leu-121, and Val-136. Three ring nitrogen atoms, N1, N6, and N7, form a hydrogen-bond network with the carboxylic group of Asp-120, the backbone amine of Leu-121, and the phenolic hydroxyl group of Tyr-140. The ribose 2′- and 3′-hydroxyl groups form hydrogen bonds with the side-chain carboxyl group of Asp-91. The amine group of the SAM methionine moiety forms a hydrogen bond with the backbone carbonyl group of Leu-134, and the carboxylic group interacts directly with the Nϵ2 nitrogen of His-44. The water-mediated hydrogen bonds are observed between the methionine moiety and residues Asp-68, Ala-69, Cys-71, Val-73, Ser-75, and Leu-134.
FIGURE 2.

Multiple ChlM sequences alignment and the cofactor binding pocket of SyChlM. A, multiple sequence alignment of ChlMs. Identical amino acids are in white on a red background, with the DXGCGXG motifs on a cyan background. The similar residues are in red and boxed. Dots indicate gaps introduced during alignment. Pink ellipses and green stars denote the residues involved in binding to the cofactor and to MgP, respectively. The two catalytically essential residues are marked with blue triangles. B, electron density maps of SAM and SAH. The 2Fo − Fc maps at 0.5 σ for SAM (top) and SAH (bottom) are shown. C, clipped view of the SAM binding pocket. SyChlM is in gray. D, residues interacting with SAM in the pocket. Residues involved in hydrophobic interaction with SAM are in green; residues involved in hydrogen bond formation are in yellow. The backbone of the DXGCGXG motif is in blue. Two waters forming hydrogen bonds with SAM are shown as red spheres. Hydrogen bonds are depicted as dotted dashes.
Substrate Binding Site
We further tried to obtain crystals of SyChlM complexed with MgP by co-crystallization and soaking methods, but the solved structure revealed no bound MgP irrespective of the presence or absence of SAM/SAH. Based on the structure of SyChlM bound to SAM/SAH, we analyzed the substrate binding site of SyChlM by in silico docking using the program AutoDock Vina (29). The protein coordinates of the SyChlM-SAH structure were used for the docking model because the SyChlM-SAM structure misses αG, which protrudes from the protein core in the SyChlM-SAH structure and leaves the substrate binding site open (Fig. 3A). This site is a distinct deep hydrophobic cleft with a dimension of ∼20 × 11 × 15 Å3. The MgP-docked model showed that the substrate MgP is bound by helices αA, αB, αC, and αG and loops β4-αF, β5-αG, αG-αH, and β6-β7 (Fig. 3B). The predicted interactions between MgP and SyChlM are summarized as follows: hydrophobic interactions between the tetrapyrrole rings of MgP and the side chains of residues Tyr-15, Phe-16, Trp-24, Ile-27, Val-36, Ile-40, Ile-138, Leu-174, Phe-219, Tyr-220, and Tyr-221; hydrogen bonds between the propionate groups of MgP and the hydroxyl groups of residues Tyr-28 and Tyr-221; electrostatic interaction between the C13 propionate group of MgP and the Nϵ2 nitrogen of His-139. The aforementioned residues are conserved in ChlMs from different species with one exception, the purple bacterium Rhodobacter, suggesting a conserved mode of MgP binding (Fig. 2A). The active site of Rhodobacter ChlM (hereafter referred to as RhBchM) is addressed under “Discussion.”
FIGURE 3.
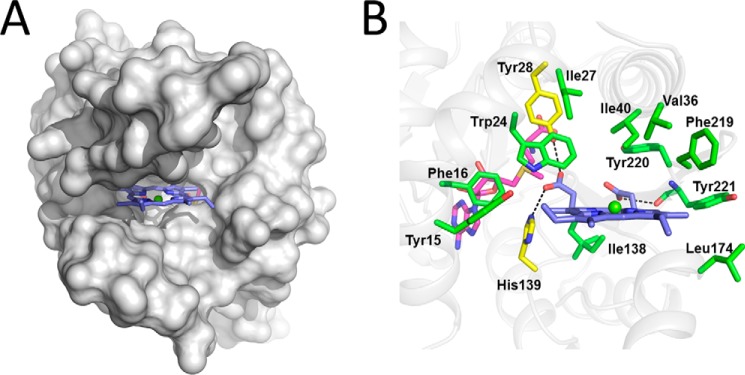
Model of SyChlM-SAM-MgP. A, MgP docked into SyChlM. SyChlM is presented in gray, and MgP is in indigo with the magnesium colored green. B, the substrate binding site. Residues predicted to interact with MgP are in green with Tyr-28 and His-139 colored in yellow. Putative hydrogen bonds are depicted as dotted dashes.
Tyr-28 and His-139 Are Essential for Catalysis
The proximity of the C13 propionate group of MgP to the hydroxyl group of Tyr-28, the Nϵ2 nitrogen of His-139, and the S-methyl group of SAM (Fig. 3B) suggests that Tyr-28 and His-139 participate in the methyl transfer reaction. To address their potential involvement in catalysis, we mutated these two residues individually. Mutation of Tyr-28 to phenylalanine led to inclusion body formation during bacterial expression, and therefore, the activity of the Y28F mutant could not be assayed. The Y28A, H139N, H139Q, and H139A mutants lost >80% methyltransferase activity as measured in the described in vitro assay (Fig. 4). Because these two residues are also involved in MgP binding as suggested by in silico docking, the lost methyltransferase activity of the mutants could be a result of an impaired substrate affinity. To test this possibility, we measured MgP binding by ITC (Fig. 5A and Table 2). The apparent Kd value between the wild type SyChlM and MgP is 6.0 μm, and all mutants have comparable Kd values as the wild type protein. These results indicate that the Y28A, H139N, H139Q, and H139A mutants largely retain the MgP binding affinity. Another explanation for the lost methyltransferase activity is that these mutations might disrupt the integrity of the cofactor binding pocket. Due to the instability of SAM (40), SAH was used for ITC analysis to evaluate the interaction between SyChlM and its cofactor (Fig. 5B and Table 2). The apparent Kd value between the wild type SyChlM and SAH is 1.4 μm, and all mutants have increased Kd values of ∼2–22-fold. Whereas these mutations resulted in reduced cofactor binding affinity to various extents, no significant difference was observed in the methyltransferase assay in which the activity of each mutant was reduced to 0 ∼ 20% that of the wild type (Fig. 4). These results indicate that Tyr-28 and particularly His-139 are implicated in cofactor binding, but neither residue is indispensable. Taken together, we conclude that their essential role is in methyl transfer.
FIGURE 4.
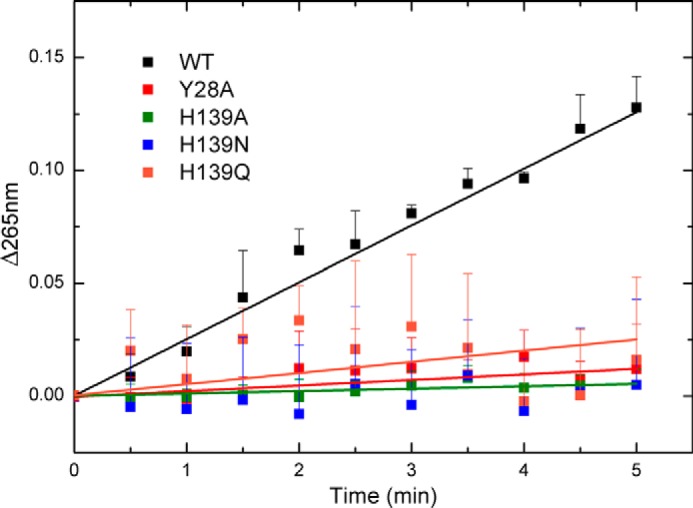
Spectrophotometric methyltransferase assay. The reaction mixture was measured by recording absorbance at 265 nm for 5 min. Data are presented as the mean ± S.E. of at least three independent experiments.
FIGURE 5.

ITC analysis of ligands binding. A, titration of SyChlMWT with MgP. The top panel shows the heat response to injections, and the bottom panel shows the integrated heats of each injection (■) and the fit (–) to a single-site binding model. B, titration of SyChlMWT with SAH. Top and bottom panels are the same as in A.
TABLE 2.
ITC analysis of MgP and SAH binding
Titrations were carried out as described under “Experimental Procedures.”
| Protein-Ligand | Kd | ΔG | ΔH | ΔS |
|---|---|---|---|---|
| μm | kcal mol−1 | kcal mol−1 | cal mol−1 K−1 | |
| SyChlMWT-MgP | 6.0 ± 0.4 | −7.0 ± 0.4 | −10.4 ± 0.1 | −11.5 |
| SyChlMY28A-MgP | 4.9 ± 0.7 | −7.1 ± 1.1 | −14.0 ± 0.6 | −23.1 |
| SyChlMH139A-MgP | 8.1 ± 0.3 | −6.8 ± 0.3 | −11.8 ± 0.1 | −17.0 |
| SyChlMH139N-MgP | 3.6 ± 0.3 | −7.3 ± 0.6 | −14.7 ± 0.3 | −25.2 |
| SyChlMH139Q-MgP | 4.1 ± 0.2 | −7.2 ± 0.4 | −12.1 ± 0.1 | −17.0 |
| SyChlMWT-SAH | 1.4 ± 0.1 | −7.8 ± 0.3 | −16.1 ± 0.0 | −28.0 |
| SyChlMY28A-SAH | 2.5 ± 0.2 | −7.5 ± 0.5 | −16.1 ± 0.2 | −29.3 |
| SyChlMH139A-SAH | 15.5 ± 1.8 | −6.4 ± 0.8 | −8.5 ± 0.3 | −7.0 |
| SyChlMH139N-SAH | 8.1 ± 0.2 | −6.8 ± 0.2 | −13.5 ± 0.1 | −22.9 |
| SyChlMH139Q-SAH | 31.0 ± 1.7 | −6.0 ± 0.3 | −12.0 ± 0.4 | −20.4 |
| SyChlM-SAH-MgPa | 2.7 ± 0.4 | −7.5 ± 1.1 | −7.6 ± 0.2 | −0.5 |
| SyChlM-MgP-SAHb | 1.9 ± 0.1 | −7.7 ± 0.3 | −16.0 ± 0.1 | −28.3 |
a MgP titration to the SAH-saturated SyChlM; 50 μm SyChlM was mixed with 150 μm SAH and then titrated with 0.40 mm MgP. See “Discussion.”
b SAH titration to the MgP-saturated SyChlM; 50 μm SyChlM was mixed with 100 μm MgP and then titrated with 0.60 mm SAH.
DISCUSSION
Structural Comparison
Although the structure of SyChlM is highly similar to a few members of the Class I SAM-dependent methyltransferases (Table 3), none of them catalyzes carboxyl methylation. The structures identified by Dali search (41) with Z > 18 include: Nods N-methyltransferase (42), tRNA (cmo5U34)-methyltransferase (43), YecO (44), phosphonate O-methyltransferase DhpI (45), a putative RNA methyltransferase (46), aclacinomycin-10-hydroxylase (47), nicotinamide N-methyltransferase (48), cyclopropane synthetase 1 (49), geranyl diphosphate 2-C-methyltransferase (50), glycine N-methyltransferase (51), and phosphoethanolamine N-methyltransferase (52). These 11 structures can be superimposed to SyChlM with r.m.s.d. values ≤3 Å (Fig. 6A). Among them five catalyze N-methylation (42, 46, 48, 51, 52), two catalyze C-methylation (49, 50), two catalyze the conversion of SAM to carboxy-SAM (43, 44, 53), one is a hydroxylase (47), and the only O-methyltransferase DhpI methylates the phosphonic acid group (45). Interestingly, like SyChlM, DhpI has an unusual insertion that encompasses a helix called the “capping helix” (topologically equivalent to αG in SyChlM) (Fig. 6B). This insertion between β5 and α6 (equivalent to αH in SyChlM) of DhpI plays a critical role in modulating substrate binding (45). It is likely that a similar binding mechanism can be applied to account for the formation of SyChlM-MgP complex in which the motion of αG allows conformational change during MgP recognition. This also suggests that the flexibility of the αG region together with the N-terminal region and the DXGCGXG motif may reduce the homogeneity of apoSyChlM molecules in the crystals and cause poor diffraction. Thermodynamic measurements using ITC reveal that both MgP and SAH can result in decreased motion of SyChlM as reflected by the decreased entropy values (Table 2). Thus, flexibility (as inferred from Fig. 1D) may be an intrinsic property required for binding the cofactor SAM or the substrate MgP.
TABLE 3.
Structural homologs of SyChlM identified by DALI with Z score above 18
| Protein | PDB | Za | r.m.s.d.b | Lalic | Identityd | Methyl acceptor | Reference |
|---|---|---|---|---|---|---|---|
| Å | n | % | |||||
| NodS N-methyltransferase (Bradyrhizobium japonicum) | 3OFK | 20.8 | 2.3 | 184 | 11 |
 NH2 NH2
|
42 |
| tRNA (cmo5U34)-methyltransferase, CmoA (E. coli) | 4GEK | 19 | 2.9 | 193 | 15 | NAe | 43 |
| YecO (Haemophilus influenzae) | 1IM8 | 19 | 2.7 | 189 | 16 | NA | 44, 53 |
| Phosphonate O-methyltransferase, DhpI (Streptomyces luridus) | 3OU2 | 18.9 | 2.6 | 179 | 16 |
OH |
45 |
| Putative RNA methyltransferase PH1948 (Pyrococcus horikoshii) | 1WY7 | 18.8 | 2.1 | 161 | 16 |
NH2
|
46 |
| Aclacinomycin-10-hydroxylase, RdmB (Streptomyces purpurascens) | 1QZZ | 18.3 | 2.6 | 182 | 15 | NA | 47 |
| Nicotinamide N-methyltransferase (Homo sapiens) | 3ROD | 18.3 | 2.8 | 196 | 17 |
N
|
48 |
| cyclopropane synthetase 1 (Mycobacterium tuberculosis) | 1KPG | 18.2 | 2.7 | 195 | 14 |
CH
|
49 |
| Geranyl diphosphate 2-C-methyltransferase (Streptomyces lasaliensis) | 4F86 | 18.2 | 2.8 | 191 | 13 |
CH
|
50 |
| Glycine N-methyltransferase (Rattus norvegicus) | 1NBH | 18.2 | 2.9 | 192 | 19 |
NH2
|
51 |
| Phosphoethanolamine N-methyltransferase (Plasmodium falciparum) | 3UJ8 | 18.1 | 3 | 194 | 13 |
NH2
|
52 |
a Strength of structural similarity in S.D. above expected.
b r.m.s.d. of superimposed Cα atoms.
c Lali, number of equivalent residues.
d Percentage of sequence identity over equivalent position.
e NA, not applicable.
FIGURE 6.
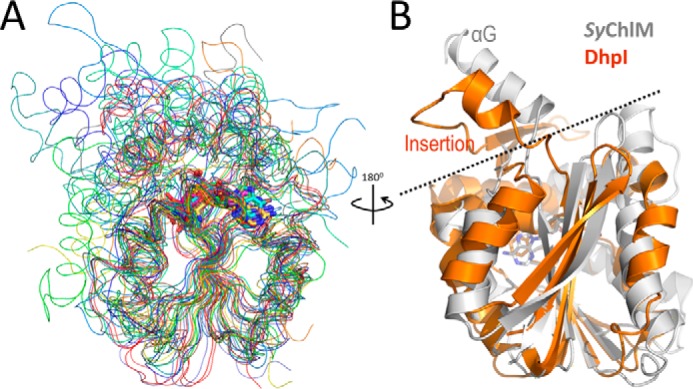
Comparison of SyChlM and its structural homologs. A, backbone superimposition of SyChlM-SAH (gray) and its structural homologs (red to blue) with a Z score above 18. Cooler color corresponds to lower Z score listed in Table 3. B, ribbon superimposition of SyChlM (gray) with DhpI (orange). A dashed line is drawn between the insertion regions and the protein cores. The coordinates of DhpI are from PDB entry 3OU6 (44) to show the capping helix that is not observed in 3OU2.
Catalytic and Regulatory Mechanism
The SAM-dependent methyltransferases utilize an SN2 replacement mechanism in which the nucleophile (methyl acceptor) directly attacks the S-methyl of SAM (methyl donor) (39). Based on structures of SyChlM, the nucleophile, the carboxyl oxygen of the C13 propionate group, is likely to be properly positioned by Tyr-28 and His-139. Such an arrangement of the active site might facilitate a direct methyl transfer from the methyl donor to the negatively charged acceptor. It is likely that His-139 serves as the base responsible for deprotonation of the carboxyl of MgP; thus, the negatively charged carboxyl attacks the high energy S-CH3 bond. During this process Tyr-28 helps to position the carboxyl in the proximity for efficient nucleophile attack. The catalytic role of the Tyr-His pair is reflected by the loss of methyltransferase activity if either residue is mutated. This pair is conserved across the species with the exception of RhBchM in which the corresponding residues are Arg-43 and Tyr-135, which compensates for these structural changes compared with other ChlMs (Fig. 7A). Upon the completion of the methyl transfer, the resulting monomethyl ester of the product MgPME might cause a steric interference with the surrounding residues, thus pushing MgPME away from the substrate binding site.
FIGURE 7.

Model of ChlM catalysis. A, predicted active site models of SyChlM and RhBchM. The homology model of RhBchM was built by SWISS-MODEL using the SyChlM coordinates as template. B, surface model of SyChlM (gray) with the two flexible regions shown as yellow ribbons. Before substrate binding, the N-terminal arm and the αG arm are intrinsically flexible (shown in transparent mode). Upon SAM and MgP binding, these two arms become stable. The right panel shows surface presentation of the whole protein, with these two arms in yellow to illustrate their independent status.
A thiol-based redox regulation of the chlorophyll branch has been identified (5). The redox status of the cysteine residues in Arabidopsis ChlM has been demonstrated to be controlled by the NADPH-dependent thioredoxin reductase C (54). Whereas the three cysteines are buried within the protein interior in SyChlM-SAM and SyChlM-SAH structures, we suggest that the conserved cysteine of the DXGCGXG motif (Cys-71 in SyChlM) on the loop β1-αD (Fig. 2D) may undergo a large conformational change when no cofactor is present and that oxidation of this cysteine may disturb the cofactor binding so as to negatively regulate the methyltransferase activity.
The cofactor binding and the substrate binding sites of SyChlM are possibly controlled by two flexible regions (the N-terminal arm and the αG arm), respectively (Fig. 7B). Each arm may independently undergo a conformational change after binding one ligand without obstructing the binding of the other one. This model would explain the random binding of the cofactor and the substrate to SyChlM as shown previously (17) and also by our ITC experiments, in which saturation of SyChlM with one ligand did not prevent binding to the other one (Table 2). Notably, SyChlM saturated with SAH appears less disordered during MgP binding as indicated by the least decreased entropy. We suggest that this is due to stabilization of the N-terminal arm of SyChlM in the presence of SAH. Furthermore, the stimulatory effect of ChlH (the protoporphyrin binding subunit of magnesium chelatase) on ChlM could possibly be achieved through the interaction between ChlH and these two flexible arms of ChlM, as channeling of the tetrapyrrole intermediates (2, 3) needs direct protein-protein communications. The αG arm is of particular interest considering a distinct protruding helix from the core of the protein.
The residues in the N-terminal arm and the αG arm are conserved with a few exceptions for RhBchM (Fig. 2A). Because all known photosynthetic organisms contain only a single bchM/chlM gene in the chlorophyll branch from MgP to chlorophyllide a, these differences might reflect the uniqueness of the anoxygenic photosynthetic bacteria in which the step subsequent to MgPME production (formation of the fifth ring) depends on the oxic/anoxic environments (2). The components of the Synechocystis MgPME cyclase that catalyzes the fifth ring formation are being identified, and their coupling with SyChlM has been demonstrated (55, 56). Further structural and functional studies with the preceding magnesium chelatase, Gun4, and the subsequent MgPME cyclase are required to understand how ChlM activity is regulated by the protein-protein interactions within the initial steps of the chlorophyll branch.
This work was supported by National Natural Science Foundation of China Grant 31340044, National Basic Research Program of China Grant 2011CBA00901, and the Hundred Talents Program of Chinese Academy of Sciences.
The atomic coordinates and structure factors (codes 4QDJ and 4QDK) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- MgP
- magnesium protoporphyrin IX
- MgPME
- magnesium protoporphyrin IX monomethylester
- ChlM
- magnesium protoporphyrin IX O-methyltransferase
- SyChlM
- Synechocystis ChlM
- SAM
- S-adenosylmethionine
- SAH
- S-adenosylhomocysteine
- ITC
- isothermal titration calorimetry
- r.m.s.d.
- root mean square deviation.
REFERENCES
- 1. Von Wettstein D., Gough S., Kannangara C. G. (1995) Chlorophyll biosynthesis. Plant Cell 7, 1039–1057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Chew A. G., Bryant D. A. (2007) Chlorophyll biosynthesis in bacteria: the origins of structural and functional diversity. Annu. Rev. Microbiol. 61, 113–129 [DOI] [PubMed] [Google Scholar]
- 3. Tanaka R., Tanaka A. (2007) Tetrapyrrole biosynthesis in higher plants. Annu. Rev. Plant Biol. 58, 321–346 [DOI] [PubMed] [Google Scholar]
- 4. Mochizuki N., Tanaka R., Grimm B., Masuda T., Moulin M., Smith A. G., Tanaka A., Terry M. J. (2010) The cell biology of tetrapyrroles: a life and death struggle. Trends Plant Sci. 15, 488–498 [DOI] [PubMed] [Google Scholar]
- 5. Richter A. S., Grimm B. (2013) Thiol-based redox control of enzymes involved in the tetrapyrrole biosynthesis pathway in plants. Front. Plant Sci. 4, 371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Nott A., Jung H. S., Koussevitzky S., Chory J. (2006) Plastid-to-nucleus retrograde signaling. Annu. Rev. Plant Biol. 57, 739–759 [DOI] [PubMed] [Google Scholar]
- 7. Chi W., Sun X., Zhang L. (2013) Intracellular signaling from plastid to nucleus. Annu. Rev. Plant Biol. 64, 559–582 [DOI] [PubMed] [Google Scholar]
- 8. Schlicke H., Hartwig A. S., Firtzlaff V., Richter A. S., Glässer C., Maier K., Finkemeier I., Grimm B. (2014) Induced deactivation of genes encoding chlorophyll biosynthesis enzymes disentangles tetrapyrrole-mediated retrograde signaling. Mol. Plant 7, 1211–1227 [DOI] [PubMed] [Google Scholar]
- 9. Fodje M. N., Hansson A., Hansson M., Olsen J. G., Gough S., Willows R. D., Al-Karadaghi S. (2001) Interplay between an AAA module and an integrin I domain may regulate the function of magnesium chelatase. J. Mol. Biol. 311, 111–122 [DOI] [PubMed] [Google Scholar]
- 10. Sirijovski N., Lundqvist J., Rosenbäck M., Elmlund H., Al-Karadaghi S., Willows R. D., Hansson M. (2008) Substrate-binding model of the chlorophyll biosynthetic magnesium chelatase BchH subunit. J. Biol. Chem. 283, 11652–11660 [DOI] [PubMed] [Google Scholar]
- 11. Lundqvist J., Elmlund H., Wulff R. P., Berglund L., Elmlund D., Emanuelsson C., Hebert H., Willows R. D., Hansson M., Lindahl M., Al-Karadaghi S. (2010) ATP-induced conformational dynamics in the AAA+ motor unit of magnesium chelatase. Structure 18, 354–365 [DOI] [PubMed] [Google Scholar]
- 12. Qian P., Marklew C. J., Viney J., Davison P. A., Brindley A. A., Söderberg C., Al-Karadaghi S., Bullough P. A., Grossmann J. G., Hunter C. N. (2012) Structure of the cyanobacterial magnesium chelatase H subunit determined by single particle reconstruction and small-angle x-ray scattering. J. Biol. Chem. 287, 4946–4956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Verdecia M. A., Larkin R. M., Ferrer J. L., Riek R., Chory J., Noel J. P. (2005) Structure of the Mg-chelatase cofactor GUN4 reveals a novel hand-shaped fold for porphyrin binding. PLoS Biol. 3, e151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Davison P. A., Schubert H. L., Reid J. D., Iorg C. D., Heroux A., Hill C. P., Hunter C. N. (2005) Structural and biochemical characterization of Gun4 suggests a mechanism for its role in chlorophyll biosynthesis. Biochemistry 44, 7603–7612 [DOI] [PubMed] [Google Scholar]
- 15. Alawady A. E., Grimm B. (2005) Tobacco Mg protoporphyrin IX methyltransferase is involved in inverse activation of Mg porphyrin and protoheme synthesis. Plant J. 41, 282–290 [DOI] [PubMed] [Google Scholar]
- 16. Pontier D., Albrieux C., Joyard J., Lagrange T., Block M. A. (2007) Knock-out of the magnesium protoporphyrin IX methyltransferase gene in Arabidopsis: effect on chloroplast development and on chloroplast-to-nucleus signaling. J. Biol. Chem. 282, 2297–2304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Shepherd M., Reid J. D., Hunter C. N. (2003) Purification and kinetic characterization of the magnesium protoporphyrin IX methyltransferase from Synechocystis PCC6803. Biochem. J. 371, 351–360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Shepherd M., Hunter C. N. (2004) Transient kinetics of the reaction catalysed by magnesium protoporphyrin IX methyltransferase. Biochem. J. 382, 1009–1013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Shepherd M., McLean S., Hunter C. N. (2005) Kinetic basis for linking the first two enzymes of chlorophyll biosynthesis. FEBS J. 272, 4532–4539 [DOI] [PubMed] [Google Scholar]
- 20. Hinchigeri S. B., Hundle B., Richards W. R. (1997) Demonstration that the BchH protein of Rhodobacter capsulatus activates S-adenosyl-l-methionine:magnesium protoporphyrin IX methyltransferase. FEBS Lett. 407, 337–342 [DOI] [PubMed] [Google Scholar]
- 21. Sawicki A., Willows R. D. (2010) BchJ and BchM interact in a 1:1 ratio with the magnesium chelatase BchH subunit of Rhodobacter capsulatus. FEBS J. 277, 4709–4721 [DOI] [PubMed] [Google Scholar]
- 22. Johnson E. T., Schmidt-Dannert C. (2008) Characterization of three homologs of the large subunit of the magnesium chelatase from Chlorobaculum tepidum and interaction with the magnesium protoporphyrin IX methyltransferase. J. Biol. Chem. 283, 27776–27784 [DOI] [PubMed] [Google Scholar]
- 23. Alawady A., Reski R., Yaronskaya E., Grimm B. (2005) Cloning and expression of the tobacco CHLM sequence encoding Mg protoporphyrin IX methyltransferase and its interaction with Mg chelatase. Plant Mol. Biol. 57, 679–691 [DOI] [PubMed] [Google Scholar]
- 24. Otwinowski Z., Minor W. (1997) Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326 [DOI] [PubMed] [Google Scholar]
- 25. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., Zwart P. H. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Emsley P., Lohkamp B., Scott W. G., Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D. Biol. Crystallogr. 66, 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Perrakis A., Morris R., Lamzin V. S. (1999) Automated protein model building combined with iterative structure refinement. Nat. Struct. Biol. 6, 458–463 [DOI] [PubMed] [Google Scholar]
- 28. Laskowski R. A., MacArthur M. W., Moss D. S., Thornton J. M. (1993) PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 26, 283–291 [Google Scholar]
- 29. Trott O., Olson A. J. (2010) AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 31, 455–461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Miyazaki G., Morimoto H., Yun K. M., Park S. Y., Nakagawa A., Minagawa H., Shibayama N. (1999) Magnesium(II) and zinc(II)-protoporphyrin IX's stabilize the lowest oxygen affinity state of human hemoglobin even more strongly than deoxyheme. J. Mol. Biol. 292, 1121–1136 [DOI] [PubMed] [Google Scholar]
- 31. Morris G. M., Huey R., Lindstrom W., Sanner M. F., Belew R. K., Goodsell D. S., Olson A. J. (2009) AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. J. Comput. Chem. 30, 2785–2791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Dorgan K. M., Wooderchak W. L., Wynn D. P., Karschner E. L., Alfaro J. F., Cui Y., Zhou Z. S., Hevel J. M. (2006) An enzyme-coupled continuous spectrophotometric assay for S-adenosylmethionine-dependent methyltransferases. Anal. Biochem. 350, 249–255 [DOI] [PubMed] [Google Scholar]
- 33. McLean S., Hunter C. N. (2009) An enzyme-coupled continuous spectrophotometric assay for magnesium protoporphyrin IX methyltransferases. Anal. Biochem. 394, 223–228 [DOI] [PubMed] [Google Scholar]
- 34. Lee J. E., Cornell K. A., Riscoe M. K., Howell P. L. (2001) Expression, purification, crystallization and preliminary X-ray analysis of Escherichia coli 5′-methylthioadenosine/S-adenosylhomocysteine nucleosidase. Acta Crystallogr. D. Biol. Crystallogr. 57, 150–152 [DOI] [PubMed] [Google Scholar]
- 35. Montange R. K., Mondragón E., van Tyne D., Garst A. D., Ceres P., Batey R. T. (2010) Discrimination between closely related cellular metabolites by the SAM-I riboswitch. J. Mol. Biol. 396, 761–772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Karger G. A., Reid J. D., Hunter C. N. (2001) Characterization of the binding of deuteroporphyrin IX to the magnesium chelatase H subunit and spectroscopic properties of the complex. Biochemistry 40, 9291–9299 [DOI] [PubMed] [Google Scholar]
- 37. Kabsch W., Sander C. (1983) Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 22, 2577–2637 [DOI] [PubMed] [Google Scholar]
- 38. Martin J. L., McMillan F. M. (2002) SAM (dependent) I AM: the S-adenosylmethionine-dependent methyltransferase fold. Curr. Opin. Struct. Biol. 12, 783–793 [DOI] [PubMed] [Google Scholar]
- 39. Schubert H. L., Blumenthal R. M., Cheng X. (2003) Many paths to methyltransfer: a chronicle of convergence. Trends Biochem. Sci. 28, 329–335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hoffman J. L. (1986) Chromatographic analysis of the chiral and covalent instability of S-adenosyl-l-methionine. Biochemistry 25, 4444–4449 [DOI] [PubMed] [Google Scholar]
- 41. Holm L., Rosenström P. (2010) Dali server: conservation mapping in 3D. Nucleic Acids Res. 38, W545–W549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Cakici O., Sikorski M., Stepkowski T., Bujacz G., Jaskolski M. (2010) Crystal structures of NodS N-methyltransferase from Bradyrhizobium japonicum in ligand-free form and as SAH complex. J. Mol. Biol. 404, 874–889 [DOI] [PubMed] [Google Scholar]
- 43. Kim J., Xiao H., Bonanno J. B., Kalyanaraman C., Brown S., Tang X., Al-Obaidi N. F., Patskovsky Y., Babbitt P. C., Jacobson M. P., Lee Y. S., Almo S. C. (2013) Structure-guided discovery of the metabolite carboxy-SAM that modulates tRNA function. Nature 498, 123–126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lim K., Zhang H., Tempczyk A., Bonander N., Toedt J., Howard A., Eisenstein E., Herzberg O. (2001) Crystal structure of YecO from Haemophilus influenzae (HI0319) reveals a methyltransferase fold and a bound S-adenosylhomocysteine. Proteins 45, 397–407 [DOI] [PubMed] [Google Scholar]
- 45. Lee J. H., Bae B., Kuemin M., Circello B. T., Metcalf W. W., Nair S. K., van der Donk W. A. (2010) Characterization and structure of DhpI, a phosphonate O-methyltransferase involved in dehydrophos biosynthesis. Proc. Natl. Acad. Sci. U.S.A. 107, 17557–17562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Gao Y. G., Yao M., Yong Z., Tanaka I. (2005) Crystal structure of the putative RNA methyltransferase PH1948 from Pyrococcus horikoshii, in complex with the copurified S-adenosyl-l-homocysteine. Proteins 61, 1141–1145 [DOI] [PubMed] [Google Scholar]
- 47. Jansson A., Niemi J., Lindqvist Y., Mäntsälä P., Schneider G. (2003) Crystal structure of aclacinomycin-10-hydroxylase, a S-adenosyl-l-methionine-dependent methyltransferase homolog involved in anthracycline biosynthesis in Streptomyces purpurascens. J. Mol. Biol. 334, 269–280 [DOI] [PubMed] [Google Scholar]
- 48. Peng Y., Sartini D., Pozzi V., Wilk D., Emanuelli M., Yee V. C. (2011) Structural basis of substrate recognition in human nicotinamide N-methyltransferase. Biochemistry 50, 7800–7808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Huang C. C., Smith C. V., Glickman M. S., Jacobs W. R., Jr., Sacchettini J. C. (2002) Crystal structures of mycolic acid cyclopropane synthases from Mycobacterium tuberculosis. J. Biol. Chem. 277, 11559–11569 [DOI] [PubMed] [Google Scholar]
- 50. Ariyawutthiphan O., Ose T., Minami A., Shinde S., Sinde S., Tsuda M., Gao Y. G., Yao M., Oikawa H., Tanaka I. (2012) Structure analysis of geranyl pyrophosphate methyltransferase and the proposed reaction mechanism of SAM-dependent C-methylation. Acta Crystallogr. D. Biol. Crystallogr. 68, 1558–1569 [DOI] [PubMed] [Google Scholar]
- 51. Takata Y., Huang Y., Komoto J., Yamada T., Konishi K., Ogawa H., Gomi T., Fujioka M., Takusagawa F. (2003) Catalytic mechanism of glycine N-methyltransferase. Biochemistry 42, 8394–8402 [DOI] [PubMed] [Google Scholar]
- 52. Lee S. G., Kim Y., Alpert T. D., Nagata A., Jez J. M. (2012) Structure and reaction mechanism of phosphoethanolamine methyltransferase from the malaria parasite Plasmodium falciparum: an antiparasitic drug target. J. Biol. Chem. 287, 1426–1434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Byrne R. T., Whelan F., Aller P., Bird L. E., Dowle A., Lobley C. M., Reddivari Y., Nettleship J. E., Owens R. J., Antson A. A., Waterman D. G. (2013) S-Adenosyl-S-carboxymethyl-l-homocysteine: a novel cofactor found in the putative tRNA-modifying enzyme CmoA. Acta Crystallogr. D. Biol. Crystallogr. 69, 1090–1098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Richter A. S., Peter E., Rothbart M., Schlicke H., Toivola J., Rintamäki E., Grimm B. (2013) Posttranslational influence of NADPH-dependent thioredoxin reductase C on enzymes in tetrapyrrole synthesis. Plant Physiol. 162, 63–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Minamizaki K., Mizoguchi T., Goto T., Tamiaki H., Fujita Y. (2008) Identification of two homologous genes, chlAI and chlAII, that are differentially involved in isocyclic ring formation of chlorophyll a in the cyanobacterium Synechocystis sp. PCC 6803. J. Biol. Chem. 283, 2684–2692 [DOI] [PubMed] [Google Scholar]
- 56. Hollingshead S., Kopecná J., Jackson P. J., Canniffe D. P., Davison P. A., Dickman M. J., Sobotka R., Hunter C. N. (2012) Conserved chloroplast open-reading frame ycf54 is required for activity of the magnesium protoporphyrin monomethylester oxidative cyclase in Synechocystis PCC 6803. J. Biol. Chem. 287, 27823–27833 [DOI] [PMC free article] [PubMed] [Google Scholar]