

Background: Overexpression of MERTK tyrosine kinase is observed in many human cancers.
Results: MERTK has a potent gain-of-function capacity to stimulate efferocytosis in epithelial cells.
Conclusion: When overexpressed, MERTK acts as a preeminent efferocytosis receptor that is targetable by soluble Ig-domain containing TAM receptors.
Significance: Our findings provide new mechanistic insight into how MERTK may impinge on tumor progression by enhancing efferocytosis.
Keywords: Apoptosis, Breast Cancer, Epithelial Cell, Phagocytosis, Receptor-tyrosine Kinase, AXL, Efferocytosis, GAS6, MERTK, Protein S
Abstract
MERTK, a member of the TAM (TYRO3, AXL, and MERTK) receptor tyrosine kinases, has complex and diverse roles in cell biology. On the one hand, knock-out of MERTK results in age-dependent autoimmunity characterized by failure of apoptotic cell clearance, while on the other, MERTK overexpression in cancer drives classical oncogene pathways leading to cell transformation. To better understand the interplay between cell transformation and efferocytosis, we stably expressed MERTK in human MCF10A cells, a non-tumorigenic breast epithelial cell line devoid of endogenous MERTK. While stable expression of MERTK in MCF10A resulted in enhanced motility and AKT-mediated chemoprotection, MERTK-10A cells did not form stable colonies in soft agar, or enhance proliferation compared with parental MCF10A cells. Concomitant to chemoresistance, MERTK also stimulated efferocytosis in a gain-of-function capacity. However, unlike AXL, MERTK activation was highly dependent on apoptotic cells, suggesting MERTK may preferentially interface with phosphatidylserine. Consistent with this idea, knockdown of MERTK in breast cancer cells MDA-MB 231 reduced efferocytosis, while transient or stable expression of MERTK stimulated apoptotic cell clearance in all cell lines tested. Moreover, human breast cancer cells with higher endogenous MERTK showed higher levels of efferocytosis that could be blocked by soluble TAM receptors. Finally, through MERTK, apoptotic cells induced PD-L1 expression, an immune checkpoint blockade, suggesting that cancer cells may adopt MERTK-driven efferocytosis as an immune suppression mechanism for their advantage. These data collectively identify MERTK as a significant link between cancer progression and efferocytosis, and a potentially unrealized tumor-promoting event when MERTK is overexpressed in epithelial cells.
Introduction
TYRO3, AXL, and M ERTK receptors (abbreviated TAMs)3 are a family of three homologous receptor-tyrosine kinases expressed predominantly on myeloid-derived hematopoietic cells that function as inhibitory receptors to dampen inflammatory responses and control tissue tolerance (1–6). All three TAM members belong to the same subfamily of receptor tyrosine kinases based on their unique features that include a conserved sequence KW(I/L)A(I/L)ES in the catalytic kinase domain and similar organization of the extracellular region, i.e. two immunoglobulin-like IG domains (Ig1 and Ig2) and two fibronectin type III domains (7, 8).
The best-characterized ligands for TAMs are secreted glycoproteins, Growth Arrest Specific Factor-6 (GAS6), and Protein S (PROS1), which bind via their C-terminal regions to the Ig1 and Ig2 domains of the TAMs (9). Both GAS6 and PROS1 are γ-carboxylated on glutamic acid residues in their N-terminal Gla (γ-carboxyglutamic acid-rich) domains by a vitamin K-dependent γ-carboxylase and, in doing so, enable the Gla domain to bind Ca2+ and attain a calcium-dependent conformation which promotes the interaction of PROS1 and GAS6 with anionic phospholipid surfaces, that include externalized phosphatidylserine (PS) on apoptotic cells (ACs) (7, 9–14). The γ-carboxylation enables TAMs to act as indirect receptors for ACs and hence contributes to their roles as immunoregulatory receptors promoting tolerance (2, 10, 15, 16).
Targeted disruption of all three TAMs (TYRO3/AXL/MERTK triple knock-out) have surprisingly unremarkable phenotypes in development, but adult mice develop age-dependent lymphoproliferative disease reminiscent of systemic lupus erythematosis and show impaired ability to clear ACs in multiple tissues (6, 7, 17–20). Although the single knock-out of mouse Mertk(−/−) has a milder phenotype, it recapitulates much of the biology of the triple knock-out with respect to autoimmunity, as tingible body macrophages in germinal centers from Mertk(−/−) mice are defective to clear ACs leading to auto-antibody production (21–23). Mertk(−/−) mice also develop age-dependent blindness, abnormal spermatogenesis and infertility in males, and impaired clearance of ACs in the post-partum involuting mammary glands (24–26). The latter effect results from the inability of epithelial cells to engulf ACs, a process termed efferocytosis in recent years, demonstrating that Mertk also acts as an important efferocytosis receptor in the involuting mammary gland.
GAS6 or Protein S binding to TAM receptors induces classic ligand-inducible dimerization, causing receptor autophosphorylation, recruitment of signaling proteins with SH2 or PTB domains, and activation of downstream signaling (7, 27, 28). In AXL and MERTK transformed cells, several conventional pathways are activated that include the PI3-kinase/AKT/Bcl-2 axis to stimulate survival, as well as activation of SRC, ERK, and BRAF that stimulate cell proliferation (7, 29–32). In addition to their role in cell proliferation and survival, TAMs also have non-canonical roles as “dampening receptors” that suppress TLR signaling to NF-κβ and down-regulate pro-inflammatory cytokine production (1, 2, 16, 32). Investigating signaling from the MERTK receptor, we previously reported that the effects on efferocytosis versus suppression of NF-κβ were separable and dissociable based on mutation of tyrosine Y867 in the kinase domain that blocks efferocytosis but still permits inhibition of NF-κβ (33). However, TAMs can also serve as co-receptors, such as for β5 integrin and interferon receptor (34). In this latter pathway, TAMs have been reported to stimulate phosphorylation of STAT1 acting as negative regulators of pro-inflammatory TLR-signaling and promoting the induction of suppressors of cytokine signaling SOCS-1 and SOCS-3 expression, which may in part explain why TAM(−/−) mice have elevated inflammatory cytokines such as IL-6 and TNF-α especially upon the activation of TLRs (35–37).
MERTK up-regulation induced by ACs is limited to early involution while expression is negatively regulated in the mammary gland under physiological conditions (26). However, each of the three TAMs has been implicated in human cancers by virtue of their pathological overexpression and their ability to activate oncogenic and survival signaling pathways (4, 7, 8, 38–42). Previous studies have shown negative regulation of AXL and MERTK expression by miR-34, miR-199a/b, and miR-126, which are down-regulated in a variety of cancers (43, 44). Moreover, in a majority of primary breast carcinomas from patients who relapse, the decreased expression of miR-335, an inhibitor of cancer metastasis predicted to target the 3′-UTR of MERTK, is potentially explaining the up-regulation of MERTK in these predisposed tumors (45). Clinically, in independent studies, AXL and MERTK overexpression in solid cancers is associated with more aggressive and invasive malignant behavior, and associated with poorer patient survival outcomes (7, 16, 42, 46–50). In addition, GAS6 is also concomitantly overexpressed in human cancers, implying that, besides receptor overexpression, TAMs participate in autocrine circuits by overexpressing both receptors and ligands (46, 51, 52).
Given the versatility of MERTK signaling in the mammary gland, and the seemingly contradictory roles that MERTK play in oncogenic and efferocytic signaling pathways, we set out to explore the biology of MERTK overexpression in mammary epithelial cells. Here we show that MERTK has pleiotropic biological functions in mammary cells. Overexpression of MERTK promotes chemoresistance of epithelial cells and migration, as well as increases their efferocytic activity leading to the induction of Programmed death-ligand 1 (PD-L1), a known blockade of immune checkpoints in cancer. Overall, these data suggest that MERTK is sufficient to be a stand-alone receptor driving epithelial efferocytosis in mammary cells, and, when overexpressed, it acts as a preeminent engulfment receptor that is targetable by soluble TAM receptors.
EXPERIMENTAL PROCEDURES
Cell Culture
MCF10A cells were maintained in DMEM/F-12 medium containing 5% horse serum (HS), and MCF10A supplements consisting of 10 μg/ml insulin (Sigma), 20 ng/ml EGF (Invitrogen), 500 ng/ml hydrocortisone (Sigma), 100 ng/ml cholera toxin (Sigma). MCF10A-pMSCV and MCF10A-MERTK stable cell lines were cultured in similar medium in addition of 0.5 μg/ml puromycin. Human T cell lymphoblast-like cell lines CEM, 293TN, HeLa, MDA-MB 231, MCF-7, and THP-1 cells were cultured in RPMI medium plus 5% FBS, 100 units/liter penicillin, and 100 units/liter streptomycin. THP-1 monocytes were differentiated into macrophages by addition of 50 ng/ml phorbol 12-myristate 13-acetate (Sigma) for 48 h followed by incubation in fresh RPMI medium plus 5% FBS for additional 48 h. Immortalized mouse primary mammary epithelial cells 21 (53), a kind gift from Dr. Vassiliki Karantza-Wadsworth, were maintained in DMEM/F-12 medium containing 5% FBS, 10 μg/ml insulin (Sigma), 20 ng/ml EGF (Invitrogen), 500 ng/ml hydrocortisone (Sigma), and 100 ng/ml cholera toxin (Sigma).
Transfection and Infection
Bicistronic plasmids pIRES-EGFP-Mertk (mouse) were generated as previously described (33). HeLa or immortalized mouse primary mammary epithelial cells 21 were transfected with control pIRES2-EGFP or pIRES-EGFP-Mertk using Lipofectamine 2000 (Invitrogen) at a DNA:liposome ratio of 1:4. Efferocytosis was performed 48 h post-transfection. For retroviral infection, human MERTK (NM_006343.2) was cloned into pMSCV puro retroviral vector (Addgene) between EcoRI and XhoI by Genewiz. 5 × 106 BOSC23 cells were transfected with 3 μg of pMSCV empty vector or pMSCV-MERTK plasmids, and 1.5 μg of pCL-Eco (Addgene), 1.5 μg of pMD2.G (Addgene), and 12 μl of X-tremeGENE (Roche). Forty-eight hours post-transfection, about 5 ml of conditioned medium containing viral particles was collected and filter sterilized. 500 μl of viral particles was used to infect 4 × 105 MCF-10A cells for 12 h after which viral conditioned medium was replaced with fresh medium. For transient expression of MERTK or pMSCV empty vector, efferocytosis assay was started 72 h post-infection. For stable expression, 0.5 μg/ml puromycin was used to select MCF10A stably expressing MERTK or pMSCV 72 h post-infection. For MERTK knockdown of MDA-MB 231 cells, 5 × 106 of 293TN cells were first transfected with 4 μg of lentiviral vectors (pLKO.1) containing shRNA sequences targeting MERTK (Sigma, oligo ID: TRCN0000000862) or scramble shRNA (Sigma), 4 μg of psPAX2, 4 μg of pMD2.G, and 36 μl of X-tremeGENE transfection reagent. 72 h after transfection, conditioned medium containing lentiviral particles was collected, filter sterilized, and used to infect 5 × 105 cells of MDA-MB-231. Efferocytosis assay was started 72 h post-infection.
Human GAS6 Conditioned Medium
3 to 5 μg of pSecTag2-human GAS6 plasmid (a kind gift from Dr. Peter Carmeliet) and 12 to 15 μl of X-tremeGENE mixture was used to transfect into a 10-cm plate of 293TN cells at 70% confluence in the presence of 10 μg/ml vitamin K1 (Phytonadione injectable emulsion from Hospira). After overnight incubation, the same 293TN cells were transfected again in the same manner. 6 h post-second transfection, the complete medium with 10% FBS was then changed into 0% serum in the presence of vitamin K1. About 36 h later, the GAS6 conditioned medium was collected. For mock-conditioned medium, GFP was used in place of GAS6 plasmid.
Immunoblotting
Cells were washed with cold PBS, lysed in 1% HNTG buffer (20 mm HEPES, pH 7.5, 150 mm NaCl; 1% Triton X-100; 10% glycerol; 1 mm phenylmethylsulfonyl fluoride; 1 mm sodium vanadate; 0.1 mm sodium molybdate, and 20 μg/ml aprotinin), and protein concentrations were determined by Bradford method (Bio-Rad). Proteins were then resolved by SDS-PAGE and transferred to polyvinylidene difluoride membranes after which blots were incubated with appropriate primary antibodies overnight and horseradish peroxidase-conjugated secondary antibodies for 1 h followed by detection by chemiluminescence. Antibodies used were human MERTK (Cell Signaling), human phosphor MERTK (FabGennix), human AXL (Santa Cruz Biotechnology), human phosphor AXL (R&D), human GAS6 (R&D), human Protein S (Santa Cruz Biotechnology), human AKT (Cell Signaling), human phosphor AKT (Cell Signaling), and His-tagged antibodies (Thomas Scientific).
Proliferation Assay
MCF10A-pMSCV and MCF10A-MERTK stable cell lines were starved overnight in medium containing 0.5% HS and full MCF10A supplements. Cells were collected in starvation serum medium (0.5% HS and full MCF10A supplements), after which 50 μl of cell suspension containing 2.5 × 103 cells was added to 100 μl of GAS6 conditioned medium or normal growth medium containing 5% HS and full MCF10A supplements. Real-time proliferation was monitored every 5 h for 160 h by xCELLigence system (ACEA Biosciences).
Colony Formation Assay
In each 35-mm plate, 1.5 ml of 0.5% base agar layer in human GAS6 conditioned medium plus 1% serum (and MCF10A supplements described above for all MCF10A cells). About 5000 cells (MCF10A-pMSCV, MCF10A-MERTK, or MDA-MB 231) were then embedded in 1.5 ml of 0.35% top agar layer with GAS6-conditioned medium plus 1% serum (and other MCF10A supplements as needed). Approximately 0.75 ml of GAS6 conditioned medium plus 1% serum (plus other MCF10A supplements when necessary) was added to each plate, and medium was changed every 3 days. After 21 days, each plate was stained with 0.75 ml of 0.005% crystal violet for 4 h, and plates were photographed, and colonies were counted.
Cell Death Assay
MCF10A-MERTK and MCF10A-pMSCV were first starved for 6 h and then treated with 50 μm of camptothecin (Sigma) or 20 μm of 5-Fluorouracil (5-FU) (Sigma) in the human GAS6 conditioned medium with or without 50 μm of LY-294002 (Cell Signaling). After 24 h, cells were stained with FITC-conjugated Annexin V and propidium iodine (PI) according to the manufacturer's protocol (BioLegend), and cell death (Annexin V and/or PI positive) was evaluated by flow cytometry.
Acini Formation Assay
MCF10A-pMSV and MCF10A-MERTK cells were grown in three-dimensional cell cultures containing growth factor reduced matrigel (BD Biosciences) as previously described (54). Briefly, about 5000 cells were overlaid in each well of an eight-chambered slide that had been pre-coated with 40 μl of matrigel, and the three-dimensional cell cultures were cultured for 12 days, with assay medium changed every 4 days. The cells were then fixed in 3.7% paraformaldehyde, permeabilized with 0.5% Triton X-100 in PBS, and stained with Alexa Fluor 594 phalloidin (Invitrogen), DAPI (Sigma), and cleaved caspase-3 (Cell Signaling). Confocal analyses were performed with Nikon A1R confocal microscope.
Migration Assay
MCF10A-MERTK and MCF10A-pMSCV were starved for 6 h and then 5 × 104 cells were added to the top chamber of the CIM-Plate 16 pre-coated with 10 μg/ml fibronectin. Medium in the top chamber containing 3% HS and one-fifth of the MCF10A supplements described above. On the bottom chamber, human GAS6 conditioned medium in 1% serum or mock-conditioned medium in 1% serum was used to induce the migration of those cells. Real time migration was monitored by xCELLigence system (ACEA Biosciences) every 15 min for 10 h. Each experiment was done in four replicates.
Primary Mammary Epithelial Cell (PMEC) Isolation
Six to eight-week-old female C57BL/6 mice, wild type or knock-out Mertk(−/−), were euthanized after which mammary glands were collected into PBS with penicillin and streptomycin on ice. Mammary tissues were then transferred into sterilized 10-cm glass Petri dish and minced with two scalpels. Minced tissues were digested with 2 mg/ml collagenase A (Roche) and 0.02g/ml hyaluronidase (Sigma) in DMEM medium with 5% FBS and penicillin and streptomycin for 6 to 8 h at 37 °C on a shaker. Dissociated tissue was then washed three times with DMEM plus 5% FBS, and pellets highly enriched in epithelial organoids were collected into 5 ml of 0.25% trypsin-EDTA. Pipetting was done to break up the cells, and then cells were washed with DMEM medium plus 5% FBS for three times. The pellet was resuspended in 2 ml of 5 mg/ml Dispase (Roche) and 200 μl of 1 mg/ml DNase I (Roche), washed with DMEM plus 5% FBS, and filtered through a 40-μm cell strainer. Collected cells were culture in DMEM/F12 medium containing 5% FBS, 5 μg/ml insulin (Sigma), 1 μg/ml hydrocortisone (Sigma), and 5 ng/ml EGF (Invitrogen). Cells were allowed to recover for at least 2 days before any further experiment.
Efferocytosis
CEM or MCF10A were used as apoptotic cells (ACs). When phagocytes were labeled green with PKH67 (Sigma) dye or GFP from mouse Mertk bicistronic vectors or CellTracker Green CFDA (Invitrogen), ACs were labeled red with PKH26 dye (Sigma) according to the manufacturer's protocol. However, if phagocytes were labeled PKH26-red, ACs would be labeled PKH67-green. Apoptosis was induced by irradiation of CEM cells with 25mJ/cm2 ultraviolet B (cells were then incubated at 37 °C for 6–8 h before co-incubation with phagocytes) or by starvation of MCF10A in medium without HS and other supplements for 24 to 48 h. Apoptosis was verified by Annexin V conjugated FITC and PI staining according to the manufacturer's protocol (BioLegend). Phagocytes were starved for 6 h and co-cultured with ACs at the ratio of 1:5 or 1:10 for 1 to 5 h depending on the experiment in the presence of 10% serum or GAS6 conditioned medium. After co-incubation, cells were washed three times with 10 mm EDTA to minimize binding of ACs to phagocytes, fixed with 1% paraformaldehyde, and analyzed by flow cytometry, Nikon A1R Confocal Microscope System, or Amnis Image Stream. For efferocytosis inhibition assay using TAM soluble receptors, efferocytosis was carried on as described above and ACs were mixed with soluble receptors for 6 h prior to co-incubation of ACs and phagocytes.
Immunohistochemistry
12 breast tumor adjacent normal tissue samples (normal) and 34 Grade 2 moderately differentiated breast tumor samples were collected. Paraffin-embedded breast cancer tissue sections (4 mm) were deparaffinized in xylene, and rehydrated in graded solutions of ethanol. Antigen retrieval was done by heating the sections in 0.01 m sodium citrate buffer (pH 6.0) at 100 °C for 15 min. The sections were then incubated in 3% hydrogen peroxide to block the endogenous peroxidase activity at room temperature for 10 min. After blocking with 5% BSA for 20 min at room temperature, the sections were incubated with the rabbit monoclonal anti-MERTK (1:100, Cell Signaling Technology, #4319) overnight at 4 °C. The slides were then incubated with biotin labeled goat anti-rabbit IgG for 20 min at room temperature and further incubated with streptavidin peroxidase solution (SABC, SA1022, Boster Biotechnology, Wuhan, China). The staining was visualized by reaction with DAB (AR1022, Boster Biotechnology, Wuhan, China). Slides were counterstained with hematoxylin, washed, dehydrated, and mounted. Five random microscopic fields per slide were photographed using a Nikon Light Microscope. Positive staining was shown in brown color.
Soluble TAM Receptor Production
First, cDNAs corresponding to human Ig1-Ig2 domains, of sTYRO3 (residues 42–223), sAXL (residues 33–220), and sMERTK (residues 95–285) were each cloned into baculovirus transfer vector pIEx (EMD Millipore). Transfection and virus amplification was then performed with Bacmagic-2 (EMD Millipore) in suspension cultures of Sf9 insect cells, after which suspension cultures of insect High Five cells grown in Express Five SFM (Invitrogen) to a density of 2 × 106 cells/ml were infected at a multiplicity of infection of ∼10. About 72 h post-infection, cells were collected in 25 mm Hepes pH 7.5, 250 mm NaCl, 500 mm l-Arg, 10% glycerol, and lysed by sonication (Sonic Dismembrator 500, Fisher Scientific). Soluble TAM were then recovered from cell lysate by affinity chromatography using Ni-IDA resin (Clontech), and further purified by ion-exchange chromatography over a Mono Q 5/50 GL column (GE Healthcare) followed by size exclusion chromatography over a HiLoad 16/60 Superdex 200 prep grade column (GE Healthcare) into a final buffer condition of 25 mm HEPES pH 7.5 with 200 mm NaCl.
Soluble TAM Receptor Binding Assay
Human GAS6-conditioned medium or 100 nm human Protein S (Hematologic Technologies) was incubated with 200 nm of individual purified soluble receptors sTYRO3, sAXL, or sMERTK overnight at 4 °C. Those complexes of individual soluble receptors and the GAS6 or Protein S were then pulled down using Ni-NTA-agarose according to the manufacturer's protocol (Qiagen). GAS6 or Protein S alone without any soluble receptors was used as a control (mock). Those pulled down complexes were then resolved in SDS-PAGE and immunoblotted with GAS6 and His-Tag antibodies.
Generation of Chimeric Fc-Mertk Receptor Construct
DNA fragments encoded intracellular domain (ICDs) of mouse Mertk (500–995) were amplified by PCR using sequence specific primers flanked with restriction enzyme sites at the ends. DNA fragments were cut and cloned into responding site of pEF2-FL-Fc/IFN-γR1 to replace the intracellular domain of IFN-γ receptor. The ligation resulted in pEF2-FL-Fc-Mertk, which contain mouse Fc region of immunoglobulin and transmembrane and intracellular domain of mouse Mertk.
Real-time PCR Detection of PD-L1 and PD-L2
293TN cells were transfected with pEF2-FL-Fc/truncated-IFN-γR1 control or pEF2-FL-Fc-Mertk expression plasmids. Cells were replenished with fresh DMEM containing 10% FBS after 6 h and cells were collected 12 or 24 h post-transfection by adding lysis buffer. RNA isolation was performed according to the Quick-RNA MicroPrep manufacturer's protocol (ZymoResearch). Total RNA was used for the first strand cDNA synthesis by iScript reverse transcription supermix (Bio-Rad). PCR reactions contained cDNA, forward and reverse primers for PD-L1 or PD-L2 (Qiagen) and iTaq Universal SYBR Green Supermix (Bio-Rad) in 20 μl per reaction. Real-Time PCRs were performed by CFX96 Touch Real-Time PCR Detection System (Bio-Rad). Data were analyzed by CFX Manager Software (Bio-Rad). Gene expression analysis was carried by normalized expression ΔΔCq, relative quantity of genes of interest is normalized to relative quantity of the reference gene (β-actin) across samples.
Surface PD-L1 and PD-L2 Expression
293TN cells were transfected with pEF2-FL-Fc/truncated-IFN-γR1 control or pEF2-FL-Fc-Mertk expression plasmids. 48 h post-transfection, cells were collected and stained with FITC-conjugated anti-FLAG (Sigma) and PE-conjugated anti-PD-L1 (BioLegend) or APC-conjugated anti-PD-L2 (Biolegend) according to the manufacturer's protocol, and analyzed by flow cytometry. MDA-MB 231 cells were transfected with either scramble shRNAs (pLKO.1 from Sigma) or MERTK targeting shRNAs (Sigma, oligo ID: TRCN0000000862) using XtremeGene (Roche) at the ratio of 1:3. 30 h post-transfection, cells were starved for 5 h in serum-free medium and then treated with or without apoptotic MCF10A plus GAS6-conditioned medium. Six hours later, apoptotic MCF10A cells were washed away, and MDA-MB 231 cells were incubated in growth medium with 0.5% serum for another 30 h. Cells were then collected, stained with PE-conjugated anti-PD-L1 (BioLegend) according to the manufacturer's protocol, and analyzed by flow cytometry.
Statistical Analysis
Statistical analysis was done by GraphPad Prism. Descriptive statistics for quantitative variables were summarized using mean ± S.D. Differences between groups were tested by t test or one-way ANOVA followed by Tukey post-hoc test. p values by t test or Tukey post-hoc test are shown, and p < 0.05 is considered as significant.
RESULTS
Homozygous deletion of MERTK results in systemic autoimmunity from the failure to clear ACs by myeloid cells in peripheral tissues as well as defects in the post-partum mammary gland involution by failure to clear ACs by mammary epithelial cells (5, 7, 21, 23, 26). Given the strong phenotype of MERTK deficiency in primary mammary epithelial cells, in combination with the fact that MERTK is frequently overexpressed in mammary carcinoma (44–46), we set out to study the effects of MERTK when overexpressed in mammary epithelial cells. First, MERTK was stably expressed by retroviral transduction in MCF10A cells, a non-transformed immortalized mammary epithelial cell lines devoid of endogenous MERTK (Fig. 1A). Despite the fact that MERTK was stably expressed on the cell surface of MCF10A, these cells did not form colonies in soft agar as a marker of cell transformation by primary oncogenes (Fig. 1B), nor did they proliferate faster than the control MCF10A-pMSCV cells (Fig. 1C). Also, MCF10A-MERTK and parental cells both formed hollow luminal structures in a three-dimensional acini forming assay where the luminal cells died by factor-deprivation induced anoikis (Fig. 1D). In contrast, MCF10A-MERTK cells were measured to have enhanced motility compared with control MCF10-pMSCV cells toward a serum gradient, and this motility was increased by the addition of GAS6 into the chemotaxis gradient (Fig. 2A). In addition, MERTK protected MCF10A from camptothecin and 5-flurouracil (5-FU) induced cell death. Annexin V and propidium iodine staining revealed that ∼45 and 50% of control MCF-10A-pMSCV cells was dying while only 23 and 24% of MCF-10A-MERTK cells underwent cell death when treated with 50 μm camptothecin and 20 μm 5-FU, respectively (Fig. 2, B and C). Since chemoresistance has been linked to the alteration of AKT signaling, we interrogated AKT activation downstream of MERTK signaling. When MERTK was activated by its classical ligand GAS6, AKT phosphorylation was strongly elevated (Fig. 2D) while ERK1/2 phosphorylation was slightly increased (data not shown). To confirm that AKT mediated the chemoresistance in MCF10A-MERTK, PI3K/AKT inhibitor, LY-294002, was employed and the chemoprotective effect of MERTK on MCF10A was indeed reversible (Fig. 2, B and C). Taken together, these data suggest that overexpression of MERTK in mammary epithelial cells could be an advantage for cancer cells by enhancing cell motility and promoting chemoresistance through AKT.
FIGURE 1.
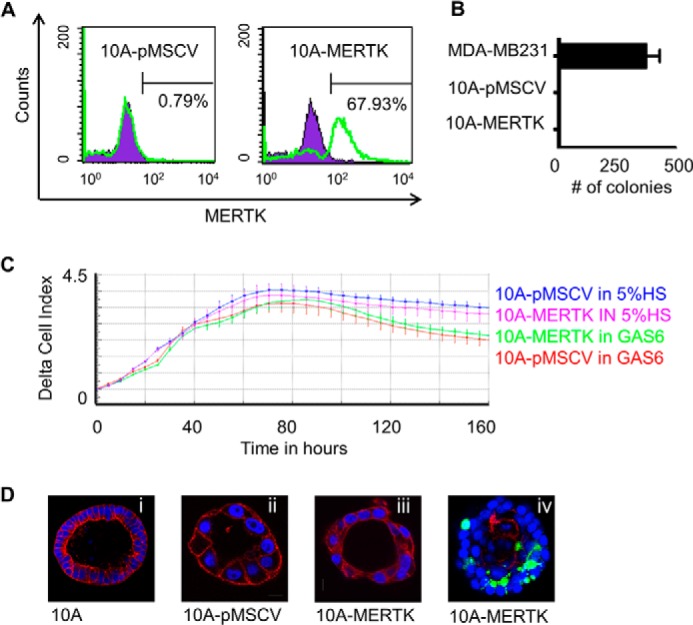
Stable MERTK expression in non-transformed MCF10A cells. A, MCF10A cells were infected with either pMSCV empty vector (left panel) or pMSCV-MERTK (right panel) retroviral particles, and after selection with puromycin, MERTK was detected with anti-MERTK antibodies by flow cytometry. B, MCF10A-pMSCV empty vector control, MCF10A-MERTK, or highly invasive MDA-MB-231 cells were grown in soft agar in the presence of human GAS6, and colonies (where applicable) were stained with crystal violet after 21 days. C, MCF10A-pMSCV and MCF10A-MERTK stable cell lines were starved overnight in medium containing 0.5% HS and full MCF10A supplements. Cells were collected in starvation serum (0.5% HS plus full MCF10A supplements), after which 50 μl of cell suspension containing 2.5 × 103 cells was added to 100 μl of GAS6 conditioned medium or normal growth medium containing 5% HS and full MCF10A supplements. Real-time proliferation was monitored every 5 h for 160 h by xCELLigence (ACEA Biosciences). D, representative confocal imaging of acini by MCF10A (20× magnification), MCF10A-MERTK (40× magnification), and MCF10A-pMSCV (40× magnification) cells grown in Matrigel for 7 (iv) or 12 (i, ii, and iii) days. Acini were fixed and stained for actin conjugated with Alexa Fluor® 568 phalloidin (red), nuclei with DAPI (blue), and cleaved caspase-3 (green) to identify apoptotic cells (iv).
FIGURE 2.

MERTK promotes migration and chemoprotection. A, real-time monitoring of cell migration in the MCF10A-MERTK and MCF10A-pMSCV stable cell lines was analyzed by xCELLigence. Fibronectin (10 μg/ml) was coated in the top chamber of the CIM-Plate 16, after which 5 × 104 MCF10A-MERTK or MCF10A-pMSCV cells were cultured in 3% serum and one fifth of the complete MCF10A supplements described under “Experimental Procedures.” MCF10A-MERTK and MCF10A-pMSCV cells were allowed to migrate toward either human GAS6 conditioned medium in 1% serum or control conditioned medium in 1% serum located in the bottom chamber of the CIM-Plate 16. The experiment was done in four replicates, and the representative of two independent experiments is shown (n = 2). B and C, MCF10A-MERTK and MCF10A-pMSCV (in the presence of human GAS6) were treated with 50 μm of camptothecin (B) or 20 μm of 5-FU (C) with or without PI3K/AKT inhibitor, LY-294002 (50 μm), for 24 h and stained with FITC-conjugated Annexin V and propidium iodine (PI). Total cell death (Annexin V and/or PI positive) was evaluated by flow cytometry. The experiment was done in triplicate, and the representative of two independent experiments is shown (n = 2). D, MCF10A-MERTK and MCF10A-pMSCV were starved for 6 h and treated with either human GAS6-conditioned medium or control conditioned medium for 30 min, after which cell lysates were collected, and MERTK and AKT phosphorylation were determined by immunoblotting with their respective antibodies.
We next explored the capacity of MCF10A-MERTK cells to promote the clearance of ACs, a process termed efferocytosis in recent years. Previous results have shown that MERTK deficiency results in the inability of mammary epithelial cells to clear ACs in the involuting mammary glands, suggesting that MERTK has a preeminent role in efferocytosis during involution (26). Consistent with these findings, primary mammary epithelial cells (PMECs) from Mertk(−/−) mice had only ∼50% of the efferocytic capacity as normal PMECs (Fig. 3A), while PMECs from Axl(−/−) mice were spared such an effect (data not shown). In addition, stable MERTK expression in MCF10A-MERTK cells described above promoted over 2-fold difference in its efferocytosis capacity when compared with control MCF10A-pMSCV cells by flow cytometry analysis, demonstrating that MERTK could act as a gain-of-function receptor for efferocytosis when overexpressed in epithelial cells (Fig. 3, B and C). Indeed, confocal imaging showed that internalized green fragments of apoptotic CEM cells were readily engulfed by red MCF10A-MERTK while, in contrast, it was quite rare to find efferocytosis by MCF10A-pMSCV cells (Fig. 3D).
FIGURE 3.

MERTK drives efferocytosis in PMECs and when stably expressed in MCF10A cells. A, primary mammary epithelial cells (PMECs) isolated from wild type (WT) or knock-out Mertk(−/−) mice were labeled with CFDA-green. Cells were starved for 6 h and then co-cultured with PKH26-red apoptotic CEM for 5 h and efferocytosis was analyzed by flow cytometry. The experiment was done in triplicate and the representative of 2 independent experiments is shown (n = 2). B, PKH26-red MCF10A-MERTK and MCF10A-pMSCV stable cells were starved for 6 h and then co-cultured with PKH67-green apoptotic CEM in 10% FBS medium for 3 h, and efferocytosis was determined by flow cytometry. C, efferocytosis was carried out as described in B with human GAS6 conditioned medium, GAS6 (1) or (0.5), instead of 10% serum. GAS6 (0.5) was generated by mixing equal volumes of GAS6 (1) with fresh RPMI. NS = not significant. D, representative confocal imaging of PKH26-red MCF10A-MERTK or MCF10A-pMSCV phagocytes and PKH67-green apoptotic CEM co-cultures. The experiment was performed as described in B in an 8-chambered slide.
To ascertain whether enhanced MERTK-inducible efferocytosis in stable MCF10A-MERTK cells could be recapitulated by transient overexpression to minimize compensatory up-regulation of co-receptors, MERTK was introduced into MCF10A by means of pMSCV retroviral vectors and efferocytosis was again increased 3-fold as compared with pMSCV control cells (Fig. 4A). To further demonstrate the role of MERTK as a preeminent efferocytic receptor, primary immortalized mouse mammary epithelial cells 21 or cervical cancer cells HeLa were transiently transfected with a bi-cistronic vector expressing Mertk and GFP. When 21 and HeLa cells were gated for GFP as a marker for Mertk co-expression by bi-cistronic vectors, efferocytosis was increased 3- and 5-fold, respectively, as compared with GFP control 21 and HeLa cells (Fig. 4, B and C). These data suggested that MERTK was capable of immediately driving efferocytosis as a stand-alone receptor when ectopically overexpressed in different epithelial cells, including non-transformed mammary cells and epithelial cells transformed by other oncogenes.
FIGURE 4.
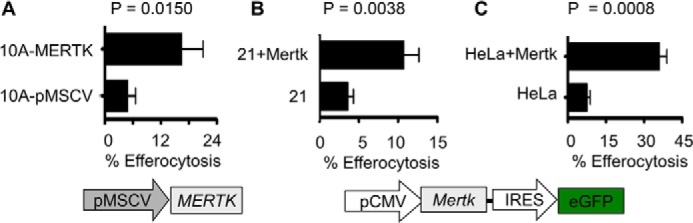
Transient expression of MERTK accelerates efferocytosis. A, MCF10A was infected with either control pMSCV or human MERTK retroviral vectors and labeled with PKH26-red. 72 h post-infection, efferocytosis was carried out as described in Fig. 3B and analyzed by flow cytometry. B, immortalized mouse primary mammary epithelial cells 21 or C, HeLa cells were transfected with either bi-cistronic plasmids pIRES-EGFP-Mertk (mouse) or control pIRES2-EGFP. 48 h post transfection, these phagocytes were co-cultured with PKH26-labeled (red) apoptotic CEM cells. Efferocytosis was assayed 5 h (21 cells) or 2 h (HeLa) post co-culture, whereby double positive cells (red + green) were scored from GFP-expressing cells and analyzed by flow cytometry.
To further implicate the relevance of MERTK directed efferocytosis in mammary epithelial cancer, we investigated the expression status of MERTK in breast cancer. Of 34 breast carcinoma and 12 normal breast samples that we analyzed, 24 of the cancer samples (70%) had highly elevated level of MERTK while all normal samples were below detection. Fig. 5A shows representative samples of breast tumor and normal adjacent tissues immunohistochemically stained with MERTK antibodies. Consistent with the tumor expression data, in several breast cancer cell lines that we surveyed including MCF-7 and MDA-MB-231, MERTK was overexpressed as compared with the non-transformed breast cells MCF10A reported above (Fig. 5B). In addition, while examining the association between endogenous MERTK expression and efferocytic capacity, we found that both breast cancer cell lines MCF-7 and MDA-MB-231 had 2 or 4-fold higher than MCF10A in their efferocytic capacity (Fig. 5C), while the most invasive cancer cell line MDA-MB-231 (with the highest level of endogenous MERTK) had the highest level of efferocytosis, which approached that observed in THP-1 macrophages, a professional phagocyte cell line (Fig. 5C). To confirm that MERTK indeed drove efferocytosis in MDA-MB 231 cells that expressed the highest level of MERTK among tested epithelial cells, we employed shRNA viral transduction to knockdown MERTK in these cells. As a result, the efferocytosis capacity of MERTK knockdown cells was reduced indicating that MERTK facilitated efferocytosis in highly invasive breast cancer cells (Fig. 5D).
FIGURE 5.

MERTK overexpression in human breast cancer. A, representatives of 34 breast tumor tissues and 12 normal (tumor adjacent) tissues were immunohistochemical stained with MERTK antibodies. Brown color denotes positive staining. Scale bar is 10 μm. Magnification 200×. All normal tissues were below detection for MERTK, while 24 out of 34 breast cancer tissues (70%) were positive for MERTK. B, human non-transformed breast cells MCF10A and breast cancer cells MDA-MB 231 and MCF-7 were characterized for MERTK expression by immunoblotting. Macrophage THP-1 cells were used as a positive control for MERTK. C, efferocytosis of PKH67-green labeled apoptotic CEM by PKH26-red labeled MCF10A, MCF-7, MDA-MB 231, and THP-1 macrophages was determined by flow cytometry after co-incubation for 3 h. D, MDA-MB 231 cells were infected with MERTK shRNA or scramble shRNA control. MERTK knockdown was verified by immunoblotting. 72 h post-infection, efferocytosis (for 3 h) of apoptotic CEM (PKH67 green) by MDA-MB 231 cells (PKH26 red) was analyzed by flow cytometry.
To explore whether the efferocytosis activity in high-MERTK expressing breast carcinoma cells could be targeted by inhibitors, we employed purified soluble human TAM receptors containing Ig1-Ig2 domain to selectively inhibit MERTK. As shown in Fig. 6A, we used soluble recombinant TAM ecto-domains (sTYRO3, sAXL, and sMERTK) comprising the tandem extracellular Ig1-Ig2 repeats as decoys for ligands of MERTK. Initially, when we assessed the ability of the sTAMs to bind GAS6 in vitro, sAXL bound much better to recombinant GAS6, while sMERTK and sTYRO3 bound much weaker (Fig. 6B). However, the related TAM ligand PROS1 preferentially bound sTYRO3 and had much weaker binding to sAXL and sMERTK (Fig. 6C), in agreement with observations by Tsou et al. (68) that PROS1 preferentially activates TYRO-3 and GAS6 favors AXL over the other TAM members. When testing the ability of soluble receptors to inhibit MERTK activation, we found that at the concentration of 0.1 μm, sAXL effectively inhibited GAS6-induced MERTK phosphorylation while sMERTK could not (Fig. 6D).
FIGURE 6.
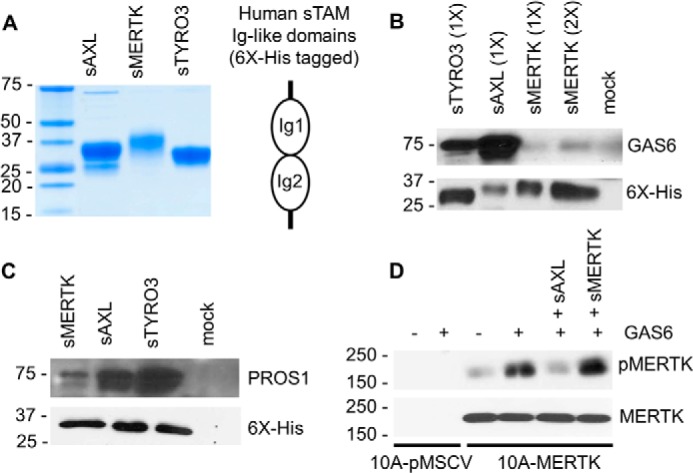
AXL soluble receptors block MERTK activation. A, Coomassie Blue staining of 6×-His-tagged human soluble receptors sAXL, sMERTK, and sTYRO3 used in this study. B, human GAS6 conditioned medium was incubated with 200 nm of each individual human soluble receptor at 4 °C overnight. Complexes were then “pulled down” using Ni-NTA-agarose and the GAS6 alone without any soluble receptor was used as a control (mock). The pulled down complexes were resolved in SDS-PAGE and immunoblotted with GAS6 or His-Tag antibodies. 1× = 10 μl and 2× = 20 μl of eluted protein complex loaded onto SDS gel. C, an experiment was performed similarly to B with 100 nm of human Protein S (PROS1) instead of GAS6. D, MCF10A-MERTK and MCF10A-pMSCV were starved for 6 h and treated with either GAS6 conditioned medium or control conditioned medium for 15 min. Soluble receptors sAXL (0.1 μm) or sMERTK (0.1 μm) was used as indicated, and MERTK phosphorylation was verified by immunoblotting.
To directly examine the role of ACs in MERTK activation, we pre-mixed ACs with GAS6 or PROS1 before treatment of MCF10A-MERTK or MCF10A-pMSCV cells in the presence or absence of soluble receptors. Consistent with the data of Tsou et al. (68) obtained on TAM reporter cell lines, ACs strongly enhanced GAS6 and PROS1 inducible MERTK phosphorylation but not GAS6 or PROS1 inducible AXL phosphorylation (Fig. 7, A and B). This indicated that ACs enhanced the activity of GAS6 and PROS1 toward MERTK but not AXL suggesting the modes of ligand-dependent interaction by AXL and MERTK were different. However, despite the dramatic effects of ACs on MERTK activation, sAXL and sTYRO3 saturated apoptotic cell surface receptors and successfully blocked GAS6 and PROS1 inducible activation of MERTK as evidence by examining the level of MERTK autophosphorylation (Fig. 7, A and B). Moreover, not only was sAXL highly effective as a receptor decoy to prevent MERTK activation, but it also completely prevented the efferocytosis enhancing effects of MERTK in MCF10A cells, suggesting it acted as a functional trap to block MERTK-mediated efferocytosis independent of the fibronectin type III domains (Fig. 8A).
FIGURE 7.

Apoptotic cells enhance MERTK activation by GAS6 or PROS1. MCF10A-MERTK and MCF10A-pMSCV were starved for 6 h and treated with either (A) human GAS6-conditioned medium or control conditioned medium or (B) 0.5 μm human PROS1 for 15 min in the presence or absence of MCF10A apoptotic cells (ACs). Human sTYRO3 (1 μm), sAXL (1 μm), or sMERTK (5 μm) was pre-incubated in lanes 7–9 (A) or lanes 3 and 4 (B). MERTK and AXL phosphorylation was verified by immunoblotting. Densitometry of immunoblots was quantified by ImageJ where auto-phosphorylation status of MERTK or AXL was normalized by total MERTK or AXL expression level, respectively.
FIGURE 8.

AXL soluble receptors inhibit MERTK-driven efferocytosis. A, MCF10A-MERTK and MCF10A-pMSCV labeled with PKH26-red were co-cultured with PKH67-green apoptotic CEM for 3 h in the presence or absence of 1 μm human sAXL and efferocytosis was analyzed by flow cytometry. B, a similar experiment was performed as described in A with MDA-MB 231 instead of MCF10A phagocytes. C, efferocytosis was carried as in panel A for 4 h and analyzed by Amnis Imagestream. However, instead of apoptotic CEM cells, apoptotic MCF10A cells were used in this experiment.
To translate these findings into the complex biology of epithelial efferocytosis in MERTK-expressing breast cancer cells, we explored the effects of adding sAXL when co-incubated ACs with MDA-MB-231 cells that overexpressed endogenous MERTK. As shown in Fig. 8B, sAXL blocked efferocytosis by over 50%, suggesting that MERTK is a preeminent efferocytosis receptor in cancer mammary epithelial cells that up-regulate MERTK. Finally, Amnis Image Stream which had the capacity to both quantify and capture fluorescent imaging of internalization of ACs by phagocytes strongly confirmed that MERTK indeed enhanced efferocytosis in MCF-10A, which could be blocked by sAXL (Fig. 8C). Also, since apoptotic MCF10A cells were used in our Amnis experiments instead of apoptotic CEM cells, it better mimicked the cancer microenvironment where MERTK-overexpressing cancer cells engulfed surrounding dead cells highly abundant in the core of tumor.
While the above findings indicate that MERTK can enhance efferocytosis in epithelial cells, the full consequences of epithelial efferocytosis are not yet realized. To begin addressing this issue, we examined the effect of MERTK on the expression of the immune checkpoint blockades, Programmed death-ligand 1 (PD-L1) and 2 (PD-L2). PD-L1 and PD-L2 are often overexpressed in poorly immunogenic tumors, and allow cancers to evade host immune responses and promote cancer invasiveness (55–58). In our study, when Mertk was constitutively activated in 293T cells by means of Fc-Mertk transfection, mRNA transcripts and surface expression of PD-L1 and PD-L2 were increased (Fig. 9, A and B). Furthermore, consistent with the current literatures that highly invasive cancer cells often express PD-L1, we found that the aggressive breast cancer cell line MDA-MB 231 highly expressed PD-L1 and knockdown of MERTK strongly reduced PD-L1 level as compared with scramble knockdown control (Fig. 9C). More importantly, AC treatment further induces PD-L1 level in MDA-MB 231 suggesting that MERTK driven efferocytosis has the potential to inhibit immune checkpoints, an event that may advance tumor progression (Fig. 9C).
FIGURE 9.

MERTK activation induces PD-L1 and PD-L2 expression. 293TN cells were transfected with pEF2-FL-Fc control or pEF2-FL-Fc-Mertk plasmids. A, 12 and 24 h post-transfection, mRNA transcripts of PD-L1 and PD-L2 in transfected cells were analyzed by real time PCR. B, 48 h post transfection, cells were collected and stained with FITC-conjugated anti-FLAG and PE-conjugated anti-PD-L1 or APC-conjugated anti-PD-L2 and analyzed by flow cytometry. Cells were gated on FLAG positive expression and then analyzed for percent cells that expressed PD-L1 or PD-L2. C, MDA-MB 231cells were transfected with either scramble shRNAs or MERTK targeting shRNAs. 30 h post-transfection, cells were starved for 5 h in serum-free medium and then treated with or without apoptotic MCF10A plus GAS6 conditioned medium. Six hours later, apoptotic MCF10A cells were washed away, and MDA-MB 231 cells were incubated in growth medium with 0.5% serum for another 30 h. Cells were then collected, stained with PE-conjugated anti PD-L1, and analyzed by flow cytometry.
DISCUSSION
In the present study, we provide evidence that overexpression of MERTK in epithelial cells results in increased motility and chemoresistance, as well as endowed epithelial cells with a robust gain-of-function capacity for efferocytosis. We show that MERTK alters the chemoresistance/efferocytosis axis, and acts as a double-edged sword in epithelial cells; first to spare tumor cells from chemotherapy drug induced cell death by promoting AKT survival pathway and second to drive phosphatidylserine (PS)-dependent interactions and processing of bystander ACs by MERTK-expressing tumor phagocytes in the tumor environment. In addition, epithelial cells with MERTK induce PD-L1 and PD-L2 expression, which could alter the immune balance for tumor tolerance and progression. Finally, MERTK directed efferocytosis may be an attractive target in cancers where soluble TAMs as therapeutics can block receptor activation and prevent PS-mediated efferocytosis and tolerance.
While the best understood roles for MERTK lies in their ability to regulate efferocytosis in myeloid-derived cells, such as macrophages and dendritic cells, where systemic MERTK knock-out leads to age-dependent autoimmunity and a systemic elevation in pro-inflammatory cytokines (1, 10, 21–23, 35, 59), MERTK has equally important roles in clearance in solid tissues that include (i) the clearance of shed rod outer segments by RPEs (24), (ii) the clearance of immature spermatogonia by Sertoli cells (25), and (iii) the efferocytosis of dying mammary epithelial by neighboring epithelial cells during involution (26). In this latter scenario during post-lactational involution, there occurs apoptosis of epithelial cells that require rapid clearance to prevent inflammation and fibrosis. Interestingly, within the mammary gland, ACs are cleared within hours of removing pups from a nursing dam by neighboring epithelial cells prior to the infiltration of macrophages to the mammary gland (26). Recent studies indicate that efferocytic epithelial cells are exquisitely dependent on Mertk, as mouse Mertk(−/−) fail to clear ACs in the mammary gland, while transplantation of Mertk (+/+) mammary epithelial cells into the mammary fat pads rescues this phenotype (26, 37). It is noteworthy that the loss of Mertk in mammary epithelial cells not only caused impaired clearance, but also leads to fibrosis, inflammation, and impaired mammary regeneration (26, 37). This suggests that physiological clearance is associated with the production of homeostatic and regeneration factors within the mammary microenvironment.
While the loss of Mertk function in the mouse mammary gland have strong physiological outcomes that affect post-parturition involution, the consequences of Mertk overexpression and/or its native ligand GAS6 overexpression in epithelial cell efferocytosis especially at the tumor micro-environment is not well understood. In the studies described here, while human MERTK was not detectable in normal breast tissue, about 70% of breast cancer tissues expressed high levels of MERTK (Fig. 5A). Moreover, the MCF10A cells lacking endogenous MERTK have limited efferocytic capacity even when saturated with ACs. However, when MERTK was overexpressed, either ectopically or endogenously, this receptor strongly supported efferocytosis as a “stand-alone” receptor in all cases, even after transient expression where compensatory up-regulation of other receptors was not likely (Figs. 3 and 4). In addition, unpublished data by Tsou et al. (manuscript JBC/2014/569020 submitted to JBC) demonstrated that ACs imposed a strong effect on MERTK but not AXL chimeric receptor activation and this observation was also confirmed in our MCF10A-MERTK stable cell line (Fig. 7). These data suggest that up-regulation of MERTK on epithelial cancer cells, as an isolated entity, promotes a favorable environment whereby efferocytosis and receptor activation will be enhanced. Our studies raise the provocative question as to whether tumor cells have evolved to adopt efferocytosis to promote tumor activities especially since classical efferocytic outcomes involve immune tolerance.
The tumor microenvironment consists of epithelial cells, fibroblast, infiltrating immune cells, soluble, and signaling factors such as chemokines and cytokines, and extracellular matrix, which all support neoplastic transformation, proliferation, invasion, and immune tolerance. Recently, the important role of infiltrating immune cells in cancer has become a central focus of cancer biology. Tumor-associated macrophages have been found to enhance tumor initiation, progression and metastasis, while infiltration of CD8+ lymphocytes is associated with tumor regression and favorable survival outcomes (37, 60, 61). Moreover, M2-polarized tumor-associated macrophages are capable of up-regulating mesenchymal markers, and increasing proliferation and migration of cancer cells (62). While the interaction of apoptotic tumor cells with macrophages and dendritic cells in the tumor microenvironment has been greatly studied in recent years, it will be equally important to study the role of tumor cells themselves as a phagocytic entity.
A future goal will be to explore the consequences of MERTK up-regulation and increased efferocytosis on skewing immune responses. Recently, Cook et al. reported that inhibition of Mertk (knock-out) in tumor leukocytes reduced tumor growth and metastasis (37). In mouse models of breast cancer, melanoma, and colon cancer, all tumors proliferated and metastasized poorly in the context of Mertk(−/−) myeloid cells (37, 63). At the mechanistic level, Mertk(−/−) leukocytes, in response to tumor cells, produced lower levels of IL-10 and GAS6 and higher levels of inflammatory cytokines such as IL-12 and IL-6 as compared with wild-type mice. Intratumoral CD8+ T lymphocytes were more abundant in the Mertk(−/−) tumor microenvironment and the elimination of them resulted in tumor progression. While this study elegantly demonstrated the importance of Mertk receptor in tumor innate immunity and progression in the context of myeloid cells, the contribution of Mertk expression in epithelial tumors themselves was not investigated, particularly whether epithelial cells have the potential to skew the balance of cytokines and chemokines to influence tumor tolerance and progression.
In our pursuit of identifying human MERTK as a suppressor of anti-tumor immunity, we found that breast cancer cell line MDA-MB 231 highly expressed PD-L1, a critical blockade of immune checkpoint that is well known for regulating the balance between T cell activation, tolerance, and immunopathology (55–58). Binding of PD-L1 to PD1 expressed on immune cells has been reported to induce IL-10 production and inhibitory effect on effector T cell responses, which might explain the association of PD-L1 expression in cancer with poorer prognosis (64). In our study, we found that, upon treatment of apoptotic cells, PD-L1 expression was further induced while knocking down of MERTK significantly reduced PD-L1 level in highly invasive breast cancer cells MDA-MB 231 (Fig. 9). In addition, constitutive activation of mouse Mertk in 293TN cells promoted both mRNA transcription and surface expression of PD-L1 and a related molecule, PD-L2. This suggests the potential of MERTK inducible efferocytosis in altering immune checkpoint regulation, which will be investigated more extensively in future studies.
Finally, at the functional level, it is attractive to predict that inhibition of MERTK mediated efferocytosis in the tumor microenvironment will have therapeutic value, perhaps in an analogous manner as the PS targeting antibodies Bavituxamab and 2aG4 (65–67). These latter antibodies target PS on the tumor vasculature and inhibit their signaling, with the net result being the repolarization of tumor-associated macrophages from the M2 to the M1-like phenotype. Collectively, our data demonstrate that MERTK may act as a tumor-promoting receptor tyrosine kinase, which conventionally promotes AKT activation, chemoprotection, and migration while non-canonically drives PS-dependent efferocytosis to enhance its tumor-promoting potential. Since MERTK-driven efferocytosis traditionally tends to release factors that promote tissue remodeling and wound healing as well as immune suppression, it is possible that cancer cells adopt MERTK enhanced efferocytosis as a strategy for tumor tolerance.
Acknowledgments
We thank members of the Birge laboratory for helpful comments and discussions and the Society of Research Scholars Foundation for supporting Khanh-Quynh Nguyen.
This work was supported in part by National Institutes of Health Grant R01 165077 (to R. B. B.), NIH R01 AI104669 (to S. V. K.), the Rutgers-NJMS Translational Research Award and Bridge Research Grant (to R. B. B. and S. V. K.), NIH Grants GM094662 and GM094665 (to S. C. A.), the Albert Einstein College of Medicine Cancer Center P30 CA013330 Grant, the National Natural Science Foundation of China Grant (No. 31070710) and Zhejiang Qianjiang Talent Project B Grant (No. 2010R10045) (to B. L.), and the Key Science and Technology Innovation Team of Zhejiang Province Grant (2010R50048).
- TAM
- TYRO3, AXL, and MERTK receptors
- MERTK
- human c-MERTK proto-oncogene tyrosine kinase
- Mertk
- mouse c-Mertk proto-oncogene tyrosine kinase
- ACs
- apoptotic cells
- KO
- knockout
- PS
- phosphatidylserine
- sTYRO3
- soluble form of TYRO3 corresponding to Ig1-Ig2 domain
- sAXL
- soluble form of AXL corresponding to Ig1-Ig2 domain
- sMERTK
- soluble form of MERTK corresponding to Ig1-Ig2 domain
- GAS6
- growth arrest specific factor-6
- PROS1
- Protein S
- ERK
- extracellular signal-regulated kinase
- AKT
- serine/threonine-protein kinase B
- SH2
- Src homology 2
- PTB
- phosphotyrosine-binding domain
- PI3-kinase
- phosphatidylinositol 3-kinase
- Bcl-2
- B-cell lymphoma 2
- BRAF
- serine/threonine-protein kinase B-Raf murine sarcoma viral oncogene homolog B1
- TLR
- Toll-like receptor
- NF-κβ
- nuclear factor kappa-light-chain-enhancer of activated B cells
- STAT1
- signal transducer and activator of transcription 1
- SOCS
- suppressors of cytokine signaling
- miR
- microRNA
- PD-L
- programmed death-ligand.
REFERENCES
- 1. Rothlin C. V., Ghosh S., Zuniga E. I., Oldstone M. B., Lemke G. (2007) TAM receptors are pleiotropic inhibitors of the innate immune response. Cell 131, 1124–1136 [DOI] [PubMed] [Google Scholar]
- 2. Carrera Silva E. A., Chan P. Y., Joannas L., Errasti A. E., Gagliani N., Bosurgi L., Jabbour M., Perry A., Smith-Chakmakova F., Mucida D., Cheroutre H., Burstyn-Cohen T., Leighton J. A., Lemke G., Ghosh S., Rothlin C. V. (2013) T cell-derived protein S engages TAM receptor signaling in dendritic cells to control the magnitude of the immune response. Immunity 39, 160–170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Camenisch T. D., Koller B. H., Earp H. S., Matsushima G. K. (1999) A novel receptor tyrosine kinase, Mer, inhibits TNF-α production and lipopolysaccharide-induced endotoxic shock. J. Immunol. 162, 3498–3503 [PubMed] [Google Scholar]
- 4. Graham D. K., Bowman G. W., Dawson T. L., Stanford W. L., Earp H. S., Snodgrass H. R. (1995) Cloning and developmental expression analysis of the murine c-mer tyrosine kinase. Oncogene 10, 2349–2359 [PubMed] [Google Scholar]
- 5. Seitz H. M., Camenisch T. D., Lemke G., Earp H. S., Matsushima G. K. (2007) Macrophages and dendritic cells use different Axl/Mertk/Tyro3 receptors in clearance of apoptotic cells. J. Immunol. 178, 5635–5642 [DOI] [PubMed] [Google Scholar]
- 6. Behrens E. M., Gadue P., Gong S. Y., Garrett S., Stein P. L., Cohen P. L. (2003) The mer receptor tyrosine kinase: expression and function suggest a role in innate immunity. Eur. J. Immunol. 33, 2160–2167 [DOI] [PubMed] [Google Scholar]
- 7. Linger R. M., Keating A. K., Earp H. S., Graham D. K. (2008) TAM receptor tyrosine kinases: biologic functions, signaling, and potential therapeutic targeting in human cancer. Adv. Cancer Res. 100, 35–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Graham D. K., Dawson T. L., Mullaney D. L., Snodgrass H. R., Earp H. S. (1994) Cloning and mRNA expression analysis of a novel human protooncogene, c-mer. Cell Growth Differ. 5, 647–657 [PubMed] [Google Scholar]
- 9. Mark M. R., Chen J., Hammonds R. G., Sadick M., Godowsk P. J. (1996) Characterization of Gas6, a member of the superfamily of G domain-containing proteins, as a ligand for Rse and Axl. J. Biol. Chem. 271, 9785–9789 [DOI] [PubMed] [Google Scholar]
- 10. Anderson H. A., Maylock C. A., Williams J. A., Paweletz C. P., Shu H., Shacter E. (2003) Serum-derived protein S binds to phosphatidylserine and stimulates the phagocytosis of apoptotic cells. Nat. Immunol. 4, 87–91 [DOI] [PubMed] [Google Scholar]
- 11. Stenhoff J., Dahlbäck B., Hafizi S. (2004) Vitamin K-dependent Gas6 activates ERK kinase and stimulates growth of cardiac fibroblasts. Biochem. Biophys. Res. Commun. 319, 871–878 [DOI] [PubMed] [Google Scholar]
- 12. Fisher P. W., Brigham-Burke M., Wu S. J., Luo J., Carton J., Staquet K., Gao W., Jackson S., Bethea D., Chen C., Hu B., Giles-Komar J., Yang J. (2005) A novel site contributing to growth-arrest-specific gene 6 binding to its receptors as revealed by a human monoclonal antibody. Biochem. J. 387, 727–735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sasaki T., Knyazev P. G., Cheburkin Y., Göhring W., Tisi D., Ullrich A., Timpl R., Hohenester E. (2002) Crystal structure of a C-terminal fragment of growth arrest-specific protein Gas6. Receptor tyrosine kinase activation by laminin G-like domains. J. Biol. Chem. 277, 44164–44170 [DOI] [PubMed] [Google Scholar]
- 14. Sasaki T., Knyazev P. G., Clout N. J., Cheburkin Y., Göhring W., Ullrich A., Timpl R., Hohenester E. (2006) Structural basis for Gas6-Axl signalling. EMBO J. 25, 80–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lemke G., Rothlin C. V. (2008) Immunobiology of the TAM receptors. Nat. Rev. Immunol. 8, 327–336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Nguyen K. Q., Tsou W. I., Kotenko S., Birge R. B. (2013) TAM receptors in apoptotic cell clearance, autoimmunity, and cancer. Autoimmunity 46, 294–297 [DOI] [PubMed] [Google Scholar]
- 17. Rothlin C. V., Lemke G. (2010) TAM receptor signaling and autoimmune disease. Curr. Opin. Immunol. 22, 740–746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lu Q., Lemke G. (2001) Homeostatic regulation of the immune system by receptor tyrosine kinases of the Tyro 3 family. Science 293, 306–311 [DOI] [PubMed] [Google Scholar]
- 19. Lu Q., Gore M., Zhang Q., Camenisch T., Boast S., Casagranda F., Lai C., Skinner M. K., Klein R., Matsushima G. K., Earp H. S., Goff S. P., Lemke G. (1999) Tyro-3 family receptors are essential regulators of mammalian spermatogenesis. Nature 398, 723–728 [DOI] [PubMed] [Google Scholar]
- 20. Duncan J. L., LaVail M. M., Yasumura D., Matthes M. T., Yang H., Trautmann N., Chappelow A. V., Feng W., Earp H. S., Matsushima G. K., Vollrath D. (2003) An RCS-like retinal dystrophy phenotype in mer knockout mice. Invest. Ophthalmol. Vis. Sci. 44, 826–838 [DOI] [PubMed] [Google Scholar]
- 21. Rahman Z. S., Shao W. H., Khan T. N., Zhen Y., Cohen P. L. (2010) Impaired apoptotic cell clearance in the germinal center by Mer-deficient tingible body macrophages leads to enhanced antibody-forming cell and germinal center responses. J. Immunol. 185, 5859–5868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Cohen P. L., Caricchio R., Abraham V., Camenisch T. D., Jennette J. C., Roubey R. A., Earp H. S., Matsushima G., Reap E. A. (2002) Delayed apoptotic cell clearance and lupus-like autoimmunity in mice lacking the c-mer membrane tyrosine kinase. J. Exp. Med. 196, 135–140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Scott R. S., McMahon E. J., Pop S. M., Reap E. A., Caricchio R., Cohen P. L., Earp H. S., Matsushima G. K. (2001) Phagocytosis and clearance of apoptotic cells is mediated by MER. Nature 411, 207–211 [DOI] [PubMed] [Google Scholar]
- 24. Prasad D., Rothlin C. V., Burrola P., Burstyn-Cohen T., Lu Q., Garcia de Frutos P., Lemke G. (2006) TAM receptor function in the retinal pigment epithelium. Mol. Cell Neurosci. 33, 96–108 [DOI] [PubMed] [Google Scholar]
- 25. Chen Y., Wang H., Qi N., Wu H., Xiong W., Ma J., Lu Q., Han D. (2009) Functions of TAM RTKs in regulating spermatogenesis and male fertility in mice. Reproduction 138, 655–666 [DOI] [PubMed] [Google Scholar]
- 26. Sandahl M., Hunter D. M., Strunk K. E., Earp H. S., Cook R. S. (2010) Epithelial cell-directed efferocytosis in the post-partum mammary gland is necessary for tissue homeostasis and future lactation. BMC Dev. Biol. 10, 122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ling L., Templeton D., Kung H. J. (1996) Identification of the major autophosphorylation sites of Nyk/Mer, an NCAM-related receptor tyrosine kinase. J. Biol. Chem. 271, 18355–18362 [DOI] [PubMed] [Google Scholar]
- 28. Mahajan N. P., Earp H. S. (2003) An SH2 domain-dependent, phosphotyrosine-independent interaction between Vav1 and the Mer receptor tyrosine kinase: a mechanism for localizing guanine nucleotide-exchange factor action. J. Biol. Chem. 278, 42596–42603 [DOI] [PubMed] [Google Scholar]
- 29. Braunger J., Schleithoff L., Schulz A. S., Kessler H., Lammers R., Ullrich A., Bartram C. R., Janssen J. W. (1997) Intracellular signaling of the Ufo/Axl receptor tyrosine kinase is mediated mainly by a multi-substrate docking-site. Oncogene 14, 2619–2631 [DOI] [PubMed] [Google Scholar]
- 30. Toshima J., Ohashi K., Iwashita S., Mizuno K. (1995) Autophosphorylation activity and association with Src family kinase of Sky receptor tyrosine kinase. Biochem. Biophys. Res. Commun. 209, 656–663 [DOI] [PubMed] [Google Scholar]
- 31. Fridell Y. W., Jin Y., Quilliam L. A., Burchert A., McCloskey P., Spizz G., Varnum B., Der C., Liu E. T. (1996) Differential activation of the Ras/extracellular-signal-regulated protein kinase pathway is responsible for the biological consequences induced by the Axl receptor tyrosine kinase. Mol. Cell Biol. 16, 135–145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sen P., Wallet M. A., Yi Z., Huang Y., Henderson M., Mathews C. E., Earp H. S., Matsushima G., Baldwin A. S., Jr., Tisch R. M. (2007) Apoptotic cells induce Mer tyrosine kinase-dependent blockade of NF-κB activation in dendritic cells. Blood 109, 653–660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Tibrewal N., Wu Y., D'Mello V., Akakura R., George T. C., Varnum B., Birge R. B. (2008) Autophosphorylation docking site Tyr-867 in Mer receptor tyrosine kinase allows for dissociation of multiple signaling pathways for phagocytosis of apoptotic cells and down-modulation of lipopolysaccharide-inducible NF-κB transcriptional activation. J. Biol. Chem. 283, 3618–3627 [DOI] [PubMed] [Google Scholar]
- 34. Wu Y., Singh S., Georgescu M. M., Birge R. B. (2005) A role for Mer tyrosine kinase in αvβ5 integrin-mediated phagocytosis of apoptotic cells. J. Cell Sci. 118, 539–553 [DOI] [PubMed] [Google Scholar]
- 35. Li Q., Lu Q., Lu H., Tian S., Lu Q. (2013) Systemic autoimmunity in TAM triple knockout mice causes inflammatory brain damage and cell death. PLoS One 8, e64812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ji R., Tian S., Lu H. J., Lu Q., Zheng Y., Wang X., Ding J., Li Q., Lu Q. (2013) TAM receptors affect adult brain neurogenesis by negative regulation of microglial cell activation. J. Immunol. 191, 6165–6177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Cook R. S., Jacobsen K. M., Wofford A. M., DeRyckere D., Stanford J., Prieto A. L., Redente E., Sandahl M., Hunter D. M., Strunk K. E., Graham D. K., Earp H. S., 3rd. (2013) MerTK inhibition in tumor leukocytes decreases tumor growth and metastasis. J. Clin. Invest. 123, 3231–3242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Burchert A., Attar E. C., McCloskey P., Fridell Y. W., Liu E. T. (1998) Determinants for transformation induced by the Axl receptor tyrosine kinase. Oncogene 16, 3177–3187 [DOI] [PubMed] [Google Scholar]
- 39. Lan Z., Wu H., Li W., Wu S., Lu L., Xu M., Dai W. (2000) Transforming activity of receptor tyrosine kinase tyro3 is mediated, at least in part, by the PI3 kinase-signaling pathway. Blood 95, 633–638 [PubMed] [Google Scholar]
- 40. Keating A. K., Salzberg D. B., Sather S., Liang X., Nickoloff S., Anwar A., Deryckere D., Hill K., Joung D., Sawczyn K. K., Park J., Curran-Everett D., McGavran L., Meltesen L., Gore L., Johnson G. L., Graham D. K. (2006) Lymphoblastic leukemia/lymphoma in mice overexpressing the Mer (MerTK) receptor tyrosine kinase. Oncogene 25, 6092–6100 [DOI] [PubMed] [Google Scholar]
- 41. Guttridge K. L., Luft J. C., Dawson T. L., Kozlowska E., Mahajan N. P., Varnum B., Earp H. S. (2002) Mer receptor tyrosine kinase signaling: prevention of apoptosis and alteration of cytoskeletal architecture without stimulation or proliferation. J. Biol. Chem. 277, 24057–24066 [DOI] [PubMed] [Google Scholar]
- 42. Wu Y. M., Robinson D. R., Kung H. J. (2004) Signal pathways in up-regulation of chemokines by tyrosine kinase MER/NYK in prostate cancer cells. Cancer Res. 64, 7311–7320 [DOI] [PubMed] [Google Scholar]
- 43. Mudduluru G., Ceppi P., Kumarswamy R., Scagliotti G. V., Papotti M., Allgayer H. (2011) Regulation of Axl receptor tyrosine kinase expression by miR-34a and miR-199a/b in solid cancer. Oncogene 30, 2888–2899 [DOI] [PubMed] [Google Scholar]
- 44. Png K. J., Halberg N., Yoshida M., Tavazoie S. F. (2012) A microRNA regulon that mediates endothelial recruitment and metastasis by cancer cells. Nature 481, 190–194 [DOI] [PubMed] [Google Scholar]
- 45. Tavazoie S. F., Alarcón C., Oskarsson T., Padua D., Wang Q., Bos P. D., Gerald W. L., Massagué J. (2008) Endogenous human microRNAs that suppress breast cancer metastasis. Nature 451, 147–152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Verma A., Warner S. L., Vankayalapati H., Bearss D. J., Sharma S. (2011) Targeting Axl and Mer kinases in cancer. Mol. Cancer Ther. 10, 1763–1773 [DOI] [PubMed] [Google Scholar]
- 47. Graham D. K., Salzberg D. B., Kurtzberg J., Sather S., Matsushima G. K., Keating A. K., Liang X., Lovell M. A., Williams S. A., Dawson T. L., Schell M. J., Anwar A. A., Snodgrass H. R., Earp H. S. (2006) Ectopic expression of the proto-oncogene Mer in pediatric T-cell acute lymphoblastic leukemia. Clin. Cancer Res. 12, 2662–2669 [DOI] [PubMed] [Google Scholar]
- 48. Nakano T., Tani M., Ishibashi Y., Kimura K., Park Y. B., Imaizumi N., Tsuda H., Aoyagi K., Sasaki H., Ohwada S., Yokota J. (2003) Biological properties and gene expression associated with metastatic potential of human osteosarcoma. Clin. Exp. Metastasis 20, 665–674 [DOI] [PubMed] [Google Scholar]
- 49. Wu C. W., Li A. F., Chi C. W., Lai C. H., Huang C. L., Lo S. S., Lui W. Y., Lin W. C. (2002) Clinical significance of AXL kinase family in gastric cancer. Anticancer Res. 22, 1071–1078 [PubMed] [Google Scholar]
- 50. Shieh Y. S., Lai C. Y., Kao Y. R., Shiah S. G., Chu Y. W., Lee H. S., Wu C. W. (2005) Expression of axl in lung adenocarcinoma and correlation with tumor progression. Neoplasia 7, 1058–1064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Vajkoczy P., Knyazev P., Kunkel A., Capelle H. H., Behrndt S., von Tengg-Kobligk H., Kiessling F., Eichelsbacher U., Essig M., Read T. A., Erber R., Ullrich A. (2006) Dominant-negative inhibition of the Axl receptor tyrosine kinase suppresses brain tumor cell growth and invasion and prolongs survival. Proc. Natl. Acad. Sci. U.S.A. 103, 5799–5804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Holland S. J., Powell M. J., Franci C., Chan E. W., Friera A. M., Atchison R. E., McLaughlin J., Swift S. E., Pali E. S., Yam G., Wong S., Lasaga J., Shen M. R., Yu S., Xu W., Hitoshi Y., Bogenberger J., Nör J. E., Payan D. G., Lorens J. B. (2005) Multiple roles for the receptor tyrosine kinase axl in tumor formation. Cancer Res. 65, 9294–9303 [DOI] [PubMed] [Google Scholar]
- 53. Karantza-Wadsworth V., White E. (2008) A mouse mammary epithelial cell model to identify molecular mechanisms regulating breast cancer progression. Methods Enzymol. 446, 61–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Debnath J., Muthuswamy S. K., Brugge J. S. (2003) Morphogenesis and oncogenesis of MCF-10A mammary epithelial acini grown in three-dimensional basement membrane cultures. Methods 30, 256–268 [DOI] [PubMed] [Google Scholar]
- 55. Soliman H., Khalil F., Antonia S. (2014) PD-L1 expression is increased in a subset of basal type breast cancer cells. PLoS One 9, e88557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Haile S. T., Dalal S. P., Clements V., Tamada K., Ostrand-Rosenberg S. (2013) Soluble CD80 restores T cell activation and overcomes tumor cell programmed death ligand 1-mediated immune suppression. J. Immunol. 191, 2829–2836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Berghoff A. S., Ricken G., Widhalm G., Rajky O., Hainfellner J. A., Birner P., Raderer M., Preusser M. (2014) PD1 (CD279) and PD-L1 (CD274, B7H1) expression in primary central nervous system lymphomas (PCNSL). Clin. Neuropathol. 33, 42–49 [DOI] [PubMed] [Google Scholar]
- 58. Basso D., Fogar P., Falconi M., Fadi E., Sperti C., Frasson C., Greco E., Tamburrino D., Teolato S., Moz S., Bozzato D., Pelloso M., Padoan A., De Franchis G., Gnatta E., Facco M., Zambon C. F., Navaglia F., Pasquali C., Basso G., Semenzato G., Pedrazzoli S., Pederzoli P., Plebani M. (2013) Pancreatic tumors and immature immunosuppressive myeloid cells in blood and spleen: role of inhibitory co-stimulatory molecules PDL1 and CTLA4. An in vivo and in vitro study. PLoS One 8, e54824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Caberoy N. B., Alvarado G., Bigcas J. L., Li W. (2012) Galectin-3 is a new MerTK-specific eat-me signal. J. Cell Physiol. 227, 401–407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Helm O., Held-Feindt J., Grage-Griebenow E., Reiling N., Ungefroren H., Vogel I., Kruger U., Becker T., Ebsen M., Rocken C., Kabelitz D., Schafer H., Sebens S. (2014) Tumor-associated macrophages exhibit pro- and anti-inflammatory properties by which they impact on pancreatic tumorigenesis. Int. J. Cancer 135, 843–861 [DOI] [PubMed] [Google Scholar]
- 61. Maeda R., Ishii G., Neri S., Aoyagi K., Haga H., Sasaki H., Nagai K., Ochiai A. (2014) Circulating CD14+CD204+ Cells Predict Postoperative Recurrence in Non-Small-Cell Lung Cancer Patients. J. Thorac. Oncol. 9, 179–188 [DOI] [PubMed] [Google Scholar]
- 62. Liu C. Y., Xu J. Y., Shi X. Y., Huang W., Ruan T. Y., Xie P., Ding J. L. (2013) M2-polarized tumor-associated macrophages promoted epithelial-mesenchymal transition in pancreatic cancer cells, partially through TLR4/IL-10 signaling pathway. Lab. Invest. 93, 844–854 [DOI] [PubMed] [Google Scholar]
- 63. Schlegel J., Sambade M. J., Sather S., Moschos S. J., Tan A. C., Winges A., DeRyckere D., Carson C. C., Trembath D. G., Tentler J. J., Eckhardt S. G., Kuan P. F., Hamilton R. L., Duncan L. M., Miller C. R., Nikolaishvili-Feinberg N., Midkiff B. R., Liu J., Zhang W., Yang C., Wang X., Frye S. V., Earp H. S., Shields J. M., Graham D. K. (2013) MERTK receptor tyrosine kinase is a therapeutic target in melanoma. J. Clin. Invest. 123, 2257–2267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Said E. A., Dupuy F. P., Trautmann L., Zhang Y., Shi Y., El-Far M., Hill B. J., Noto A., Ancuta P., Peretz Y., Fonseca S. G., Van Grevenynghe J., Boulassel M. R., Bruneau J., Shoukry N. H., Routy J. P., Douek D. C., Haddad E. K., Sekaly R. P. (2010) Programmed death-1-induced interleukin-10 production by monocytes impairs CD4+ T cell activation during HIV infection. Nat. Med. 16, 452–459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. DeRose P., Thorpe P. E., Gerber D. E. (2011) Development of bavituximab, a vascular targeting agent with immune-modulating properties, for lung cancer treatment. Immunotherapy 3, 933–944 [DOI] [PubMed] [Google Scholar]
- 66. Gerber D. E., Stopeck A. T., Wong L., Rosen L. S., Thorpe P. E., Shan J. S., Ibrahim N. K. (2011) Phase I safety and pharmacokinetic study of bavituximab, a chimeric phosphatidylserine-targeting monoclonal antibody, in patients with advanced solid tumors. Clin. Cancer Res. 17, 6888–6896 [DOI] [PubMed] [Google Scholar]
- 67. He J., Luster T. A., Thorpe P. E. (2007) Radiation-enhanced vascular targeting of human lung cancers in mice with a monoclonal antibody that binds anionic phospholipids. Clin. Cancer Res. 13, 5211–5218 [DOI] [PubMed] [Google Scholar]
- 68. Tsou W.-I., Nguyen K.-Q. N., Calarese D. A., Garforth S. J., Antes A. L., Smirnov S. V., Almo S. C., Birge R. B., Kotenko S. V. (2014) Receptor tyrosine kinases, TYRO3, AXL, and MER, demonstrate distinct patterns and complex regulation of ligand-induced activation. J. Biol. Chem. 289, 25750–25763 [DOI] [PMC free article] [PubMed] [Google Scholar]