

Background: The mechanisms by which ligands activate TAM receptors (TYRO3, AXL, and MER) are not well understood.
Results: We created a series of TAM reporter cell lines and interrogated ligand-inducible TAM activation.
Conclusion: TAMs are differentially activated by GAS6 and protein S and have distinct requirements for phosphatidylserine.
Significance: Results reveal molecular mechanisms and rationale for non-overlapping functions of TAMs.
Keywords: Apoptosis, Phospholipid, Receptor-tyrosine Kinase, Signal Transduction, Virus, AXL, GAS6, MER, TYRO3, Protein S
Abstract
TYRO3, AXL, and MER receptors (TAMs) are three homologous type I receptor-tyrosine kinases that are activated by endogenous ligands, protein S (PROS1) and growth arrest-specific gene 6 (GAS6). These ligands can either activate TAMs as soluble factors, or, in turn, opsonize phosphatidylserine (PS) on apoptotic cells (ACs) and serve as bridging molecules between ACs and TAMs. Abnormal expression and activation of TAMs have been implicated in promoting proliferation and survival of cancer cells, as well as in suppressing anti-tumor immunity. Despite the fact that TAM receptors share significant similarity, little is known about the specificity of interaction between TAM receptors and their ligands, particularly in the context of ACs, and about the functional diversity of TAM receptors. To study ligand-mediated activation of TAMs, we generated a series of reporter cell lines expressing chimeric TAM receptors. Using this system, we found that each TAM receptor has a unique pattern of interaction with and activation by GAS6 and PROS1, which is also differentially affected by the presence of ACs, PS-containing lipid vesicles and enveloped virus. We also demonstrated that γ-carboxylation of ligands is essential for the full activation of TAMs and that soluble immunoglobulin-like TAM domains act as specific ligand antagonists. These studies demonstrate that, despite their similarity, TYRO3, AXL, and MER are likely to perform distinct functions in both immunoregulation and the recognition and removal of ACs.
Introduction
Receptor-tyrosine kinases (RTKs)3 TYRO3, AXL, and MER share similar structural organization of their extracellular domains that are composed of two tandem N-terminal immunoglobulin-like domains (Igs) followed by two tandem membrane-proximal fibronectin type III-like (FNIII) domains (1–3). The intracellular regions of TAMs contain a tyrosine kinase domain that is highly conserved and includes a TAM-specific sequence KW(I/L)A(I/L)ES in the catalytic domain. Based on these unique structural features, TYRO3, AXL, and MER form a subfamily of RTKs abbreviated as TAMs.
Mice lacking all three TAMs (a TYRO3/AXL/MER triple knock-out) develop normally but demonstrate impaired ability to clear apoptotic cells (ACs) in multiple tissues, elevated levels of pro-inflammatory cytokines such as TNF-α and IL-6, and auto-antibody production. With age, these mice develop symptoms reminiscent of systemic lupus erythematosus (4). Unlike other RTK knock-out mice that are often embryonically lethal, single TAM mutant mice are viable and have mild abnormalities; TYRO3 knock-out mice show neurological disorders, and AXL knock-out mice have increased vascular permeability and impaired vascular remodeling (5–7). Interestingly, only MER knock-out mice recapitulate much of the biology of the triple knock-out with respect to autoimmunity (5, 8–10). Thus, TAMs appear to contribute to the clearance of ACs and control of inflammatory responses (11).
While the loss of TAM receptors in adult tissues causes defects in cellular processes that regulate homeostasis, remodeling, and inflammation, all three TAMs have been also implicated in human cancers (12). Although TAMs are expressed predominantly in myeloid-derived hematopoietic cells, they are also found in other cell types including normal epithelial and endothelial cells (2, 11, 13). Notably, TAMs are overexpressed in a variety of cancers and possess a gain-of-function ability to activate oncogenic and survival signaling pathways (14). In many of these cancers, the level of overexpression positively correlates with chemo-resistance, metastasis, and poor survival outcomes (15).
TAMs are activated by the interaction with growth arrest-specific gene 6 (GAS6) and protein S (PROS1), the two best characterized ligands for TAMs (16). These secreted glycoproteins share about 40% sequence identity and both contain an N-terminal glutamic acid-rich Gla domain, followed by four tandem EGF-like domains, and a C-terminal sex hormone-binding globulin (SHBG) domain comprised of two Laminin G (LG) domains (17). The LG domains are required for TAM binding and the activation of post-receptor signaling pathways that include PI3-kinase/AKT, ERK, and PLC-γ (18–20). The functional importance of other domains of GAS6 and PROS1 for TAM activation remains to be elucidated.
During secretion from producing cells, both GAS6 and PROS1 are constitutively γ-carboxylated on glutamic acid residues in their N-terminal Gla domains by a vitamin K-dependent γ-carboxylase. The γ-carboxylated Gla domain binds Ca2+ and enables PROS1 and GAS6 to regulate coagulation and clotting (21, 22). In addition, the γ-carboxylation of GAS6 and PROS1 facilitates their Ca2+-dependent interaction with anionic phospholipids including externalized phosphatidylserine (PS) on ACs (23) and enveloped viruses such as Ebola, HIV, and Dengue (24–26). Therefore, GAS6 and PROS1 act as bridging factors: their γ-carboxylated Gla domains bind to PS and opsonize ACs, whereas their LG domains interact with TAMs. This enables TAMs to bind indirectly to ACs and act as efferocytosis receptors (27). Moreover, activation of TAMs leads to the suppression of NF-κβ signaling; thus, TAMs act as immuno-regulatory receptors that dampen inflammation (28). In recent years, several additional TAM ligands, including TUBBY, TULP-1, and Galectin-3, have been reported; however the functions of these new ligands are still emerging (29, 30).
While a general paradigm for TAM signaling has emerged in recent years, it is important to investigate whether TAMs have unique or overlapping modes of ligand-triggered receptor activation. While previous studies have shown that different TAMs have various affinities toward their ligands (31), these studies did not investigate receptor activation status or whether the nature of the signaling is altered in the presence of ACs.
To compare ligand-receptor interaction of all three TAM receptors, we generated a series of reporter cell lines expressing chimeric TAM receptors. Our studies revealed that each TAM receptor demonstrates a unique pattern of activation by GAS6 and PROS1 that is also differentially affected by the presence of ACs or PS-containing vesicles. These studies suggest that, despite their similarity, TYRO3, AXL, and MER perform distinct functions in the recognition and removal of ACs.
EXPERIMENTAL PROCEDURES
Cells, Culture Condition, and Apoptosis Induction
CHO-derived 16–9 cells (32) were cultured in HAM's F12 media. Jurkat cells were cultured in RPMI media. HEK293TN cells were cultured in DMEM media. All media were supplemented with 10% FBS, 100 IU/ml ampicillin, and 100 μg/ml streptomycin. All cells were cultured in a humidified chamber with 5% CO2. For apoptosis induction, Jurkat cells were washed with PBS twice and treated with 10 μm camptothecin (CPT) for 5 h in serum-free media or UV irradiated for 1 min (25 mJ/cm2) and left in serum-free media for 5 h. The percentage of cell death was evaluated by propidium iodide (PI; BioLegend) and FITC-conjugated Annexin V (BioLegend) staining followed by flow cytometry.
Generation of Chimeric Receptor Constructs and Reporter Cell Lines
cDNA fragments encoding the extracellular domains (ECDs) of mTYRO3 (1–418), mAXL (20–443), mMER (21–496), hTRYO3 (42–428), hAXL (33–451), and hMER (21–500) were amplified by PCR using sequence specific primers flanked with restriction enzyme sites at the ends. PCR-generated DNA fragments were digested with restriction enzymes and cloned into corresponding sites of pEF2-FL-CRF2–12/IFN-γR1 plasmid (33) to replace the extracellular domain of IFN-λR1. The cloning generated pEF2-mTyro3/IFN-γR1, pEF2-FL-mAxl/IFN-γR1, pEF2-FL-mMer/IFN-γR1, pEF2-FL-hTYRO3/IFN-γR1, pEF2-FL-hAXL/IFN-γR1, and pEF2-FL-hMER/IFN-γR1 plasmids, which contain mouse or human TAM extracellular domains and transmembrane and intracellular domains of human IFN-γR1. The constructs were transfected into CHO-derived 16-9 cells and stable cells were selected in 400 μg/ml G418. Single stable clones were selected by limiting dilution and responsiveness of individual clones to GAS6 and PROS1 was determined by ligand-induced pSTAT1 activation. The expression of chimeric receptors was determined by immunoblotting with antibody (Ab) against human IFN-γR1 C terminus (Santa Cruz Biotechnology).
Detection of Activation of Chimeric TAM Receptors
Stable cell lines expressing chimeric TAM receptors were starved for 5 h in serum-free HAM's F12 media. Purified GAS6 (Amgen, R&D Systems or Abnova) or hPROS1 (Hematologic Technologies Inc.), or HEK293TN cell-conditioned media containing GAS6 or PROS1 were added, and cells were incubated at 37 °C for 30 min. For AC, liposome, and viral assays, protein ligands were premixed with 2 × 106 apoptosis-stimulated or untreated Jurkat cells, or 0.5 mm phospholipid vesicles, or viral particles (1 to 10 × 106 CFU) for 30 min at 22 °C and then added to the reporter cells for 30 min at 37 °C. Jurkat cells, liposomes, or viral particles were washed away by PBS without Ca2+ and Mg2+. Whole cell lysates of reporter cells were prepared by lysing cell pellets in lysis buffer containing 100 mm Tris-HCl (pH 8), 150 mm NaCl, 1% Nonidet P-40 (Nonidet P-40), 20% glycerol, 0.2 mm EDTA, 1 mm DTT, 0.2 mm PMSF, 1 mm Na3VO4, protease inhibitor mixture (Sigma). Protein concentration was determined by Bradford assay using BSA as a standard (Bio-Rad). Equal amounts of proteins were subjected to SDS-PAGE followed by immunoblotting with Ab against pSTAT1-Y701 (BD Bioscience) to assess the activation of chimeric TAM receptors.
Generation of Soluble TAM Receptor Construct and PNGaseF Treatment
cDNA fragments encoding mTYRO3 (26–418), mAXL (20–443), mMER (21–496) were cloned into a pEF2-SPFL vector (34) that provided a signal peptide followed by the FLAG epitope tag fused in-frame with mTAM ECDs. HEK293TN cells were transfected with the resulting expression plasmids (pEF2-FL-sol-mTyro3, pEF2-FL-sol-mAxl, and pEF2-FL-sol-mMer). Cells were refed with serum-free DMEM after 16 h, and the conditioned media were collected at 72 h post-transfection. Soluble mTAM receptors containing only Ig-like domains were produced in insect cells. cDNA corresponding to amino acid sequences of mTYRO3 (31–212), mAXL (26–221), and mMER (89–280) were cloned into pIEx baculovirus vector with a His-tag (EMD Millipore). Transfection and virus amplification were performed with Bacmagic-2 (EMD Millipore) in suspension cultures of Sf9 insect cells. For protein production, insect High Five cells were cultured in suspension and grown to a density of 2 × 106 cells/ml in Express Five SFM (Invitrogen). High Five cells were then infected at a multiplicity of infection (MOI) of 10 and harvested by centrifugation (800 × g, 20 min) 72 h postinfection. Cell pellet were resuspended in 25 mm Hepes (pH 7.5), 250 mm NaCl, 500 mm l-Arg, 10% glycerol, and lysed by sonication. Soluble TAM-Igs were purified from cell lysate by using Ni-IDA affinity column (Clontech). Further protein purification was achieved by ion-exchange chromatography over a Mono Q 5/50 GL column (GE Healthcare) followed by size exclusion chromatography over a HiLoad 16/60 Superdex 200 prep grade column (GE Healthcare) into a buffer containing 25 mm HEPES (pH 7.5), 200 mm NaCl.
To investigate the N-linked glycosylation, PNGase F (New England Biolabs) treatment was performed according to the manufacturer's protocol. In brief, 50 ng of soluble receptors were boiled in denaturing buffer (0.5% SDS, 40 mm DTT) for 10 min. 50 units of PNGase F, 1% Nonidet P-40, and 50 mm sodium phosphate buffer (pH 7.5) were added, and reactions were incubated for 2 h at 37 °C. Products were separated by SDS-PAGE followed by immunoblotting with Abs against FLAG (M2, Sigma) or His-tag (Thomas Scientific).
Production of γ-Carboxylated and Non-carboxylated GAS6 and PROS1
HEK293TN cells were transfected with pSecTag2-hGAS6, pcDNA6-mPROS1-myc-His, or pCMV-SPORT6-mGAS6. After 16 h transfection, media were replaced with serum-free DMEM supplemented with 10 μg/ml vitamin K1 (Phytonadione injectable emulsion; Hospira) for γ-carboxylated protein production. Conditioned media were collected 72 h post-transfection. For non-carboxylated protein production, cells were initially grown in complete media containing 2 μm warfarin (Sigma) for 24 h before transfection, and warfarin was also added after transfection to inhibit γ-carboxylation until conditioned media collection. Protein expression and γ-carboxylation were checked by immunoblotting with Abs against GAS6 (R&D Systems), His-tag and γ-carboxylglutamic acid (Sekisui Diagnostics). hGAS6 concentration in conditioned media was estimated by comparison to purified hGAS6 (Amgen) by immunoblotting with GAS6 Ab.
Preparation of Phospholipid Vesicles
Large multilamellar vesicles (LMV) were either prepared by mixing l-α-phosphatidylcholine (PC; chicken egg, Avanti Polar Lipids) and l-α-phosphatidylserine (PS; porcine brain, sodium salt, Avanti Polar Lipids) in chloroform at 7:3 molar ratios or by using pure phospholipids. Lipids were stored in chloroform and dried under constant nitrogen gas to evaporate organic solvent in chemical hood for 2 h. The dry lipid films were rehydrated by adding PBS at a concentration of 5 mm and vigorous votexing for 5 min to form a milky liposome solution. All liposomes were used the same day of preparation at 0.5 mm final concentration in all experiments.
GAS6 and Soluble TAM-Igs Co-precipitation
Each TAM-Igs-His tag (10 nm) was mixed with γ-carboxylated or non-carboxylated hGAS6-conditioned media (∼50 nm) in 0.2 ml binding buffer containing 10 mm HEPES (pH 7.4), 140 mm NaCl, 5 mm KCl, 2.5 mm CaCl2, 1 mm MgCl2, and the mixtures were incubated at 4 °C for 1 h. One-twentieth of volume was saved for input loading control. TALON metal affinity resin (15 μl, Clontech) and 0.5 ml binding buffer were added to the mixture and kept at 4 °C overnight with gentle rotation. The beads were washed with binding buffer containing 0.1% Triton X-100 and Nonidet P-40 three times. Protein complexes were eluted by non-reducing protein loading dye at 100 °C for 10 min. A third of the eluted protein mixture was reduced by the addition of 2-mercaptoethnol (5% final concentration) for reducing condition. Proteins were separated by SDS-PAGE and immunoblotted with Abs against hGAS6 (non-reducing condition) and His-tag (reducing condition).
Virus Production
Vesicular stomatitis virus (VSV) was prepared as described previously (35). In brief, ARPE-19 cells were inoculated with VSV, and virus containing media were collected at 24 h postinfection. The media were centrifuged at 3000 × g for 10 min to remove large cellular debris followed by 71,000 × g at 4 °C for 1 h to pellet the viral particles. The virus pellet was resuspended in TE buffer (1 mm Tris-HCl (pH 7.5), 1 mm EDTA) with 10% DMSO and centrifuged through a 7–60% discontinuous sucrose gradient composed of steps of 2 ml of 60% (w/w) sucrose, 3 ml of 45% sucrose, 4.5 ml of 25% sucrose and 1.5 ml of 7% sucrose. Sucrose solutions were prepared in HEN buffer (10 mm HEPES (pH 7.4), 1 mm EDTA, 100 mm NaCl). The virus containing band was collected from the gradient after overnight centrifugation at 130,000 × g at 4 °C and diluted with TE buffer. The virus was pelleted again by centrifugation at 130,000 × g at 4 °C for 1 h to remove sucrose and resuspended in TE/DMSO buffer. Viral titers were determined by standard plaque assay on ARPE-19 cells.
Quantification of Immunoblot Intensities
Immunoblot data were obtained within a linear range of exposure and intensities were quantified by Image Studio Lite software (LI-COR). The levels of TAM/γR1 activation were measured by pSTAT1 signal intensities normalized to intensities of actin protein loading controls. For the combination treatments, signal intensities of pSTAT1 activation induced by lipid-containing liposomes or ACs alone were subtracted from intensities of signals induced by the combination treatments; and the levels of pSTAT1 activation induced by ligand combined with other treatments were plotted as a fold of enhancement or reduction over intensities induced by ligand alone (set as 1).
RESULTS
γ-Carboxylation of GAS6 and PROS1 Is Required for TAM Receptor Activation
To determine whether TAM RTKs have distinct or overlapping mechanisms of ligand-induced activation, we created a series of CHO-based reporter cell lines expressing human or mouse chimeric TAM receptors in which the extracellular domain of each TAM was fused in-frame with the transmembrane and intracellular domains of the human IFN-γR1 chain (Fig. 1A). RTK activation is assumed to be triggered by ligand-induced receptor dimerization or oligomerization. Homo-dimerization of the IFN-γR1 intracellular domains is sufficient to trigger downstream IFN-γ specific signaling and biological activities (36). Binding of TAM ligands, GAS6 and PROS1, results in the dimerization of TAM extracellular domains (37, 38) that should cause dimerization of the IFN-γR1 intracellular domains and induction of the downstream JAK-STAT signaling pathway in the reporter cells. Therefore, the activation of chimeric TAM receptors can be detected by TAM ligand-induced STAT1 phosphorylation as a common readout for all chimeric TAM receptors (Fig. 1A). Immunoblotting with an antibody against the intracellular domain of human IFN-γR1 demonstrated comparable levels of chimeric receptor expression in all stable cell lines (Fig. 1B). Therefore, levels of STAT1 activation were expected to reflect the intensity of ligand-induced activation of chimeric TAM receptors.
FIGURE 1.

Ligand specificity of TAM receptor activation. A, structures of intact TAM and chimeric TAM/IFN-γR1 receptors are schematically shown. Gla: γ-carboxyglutamic acid domain; EGF-like: epidermal growth factor-like domain; SHBG: sex hormone-binding globulin domain; LG: Laminin G domain; Ig-like: immunoglobulin domain; FNIII: fibronectin type III domain; RTK: receptor tyrosine kinase domain; TAM-EC: TAM receptor extracellular domain; IFN-γR1(TM+IC): interferon γ receptor 1, transmembrane domain and intracellular domain. B, expression levels of chimeric TAM receptors were assessed by immunoblotting with Ab against the intracellular domain of human IFN-γR1. C, reporter cells expressing chimeric mAXL/γR1 receptors were starved for 5 h in serum-free media and treated with GAS6 obtained from different sources (Amgen (50, 10, or 2 nm hGAS6), R&D Systems (100 nm hGAS6 or mGAS6), Abnova (100, 10 nm hGAS6)] for 30 min at 37 °C. The activation of chimeric mAXL/γR1 receptors was assessed by pSTAT1 immunoblotting. D, HEK293TN cells were transiently transfected with expression plasmids encoding human GAS6 (hGAS6) and His-tagged mouse PROS1 (mPROS1) and grown in serum-free media containing 10 μg/ml vitamin K1 (marked as K) or 2 μm warfarin (marked as W) for 72 h. The conditioned media were collected and resolved by SDS-PAGE followed by immunoblotting with Abs against hGAS6, His-tag and γ-carboxyglutamic acid. Sample preparation for the detection of hGAS6 and His-tag were done under non-reducing conditions. E—G, serum-starved reporter cells expressing chimeric mTAM/γR1 receptors were treated with either conditioned media containing TAM ligands produced in the presence of either vitamin K1 (K) or warfarin (W) in E, or with 50 nm of recombinant hGAS6 (Amgen) or hPROS1, or 20% fetal bovine serum (FBS) in F or with 50 nm of hGAS6 (Amgen), hPROS1 or hamster IFN-γ (10 ng/ml) in the absence or presence of 0.5 mm EDTA in G for 30 min at 37 °C. Warfarin was also added to the conditioned media with vitamin K-derived ligands (pW) immediately before mMER/γR1 cell stimulation (E, most right panel). The activation of chimeric TAM receptors was assessed by pSTAT1 immunoblotting. Parental cells stably transfected with plasmid expressing red fluorescent protein (RFP) were used as a negative control; hamster IFN-γ (10 ng/ml) was used as a positive control to trigger STAT1 activation through the endogenous hamster IFN-γ receptor (F, most right panel). Immunoblotting results are representative results of three independent experiments.
Initially, we found that recombinant GAS6 proteins from different sources were not equally potent for triggering STAT1 phosphorylation in AXL/γR1 reporter cells (Fig. 1C). Since both GAS6 and PROS1 have been reported to undergo vitamin K-dependent γ-carboxylation within the Gla domain, we investigated whether the γ-carboxylation of ligands affected their abilities to trigger TAM receptor activation. To produce non-carboxylated ligands, GAS6 or PROS1-expressing HEK293TN cells were treated with warfarin, which blocks endogenous vitamin K epoxide reductase and prevents γ-carboxylation biosynthesis. In the presence of warfarin, GAS6 and PROS1 were still secreted into the conditioned media, but γ-carboxylation of the Gla domain was completely abolished (Fig. 1D). Interestingly, although both γ-carboxylated or non-carboxylated hGAS6 and mPROS1 were predominantly present as monomers in solution, they also formed dimers and γ-carboxylation appeared to increase the ratio between monomers and dimers of both ligands.
While it is well known that γ-carboxylation within Gla domains of coagulation factors are essential for their clotting functions (21), the importance of γ-carboxylation of PROS1 and GAS6 in TAM receptor activation is still unclear. To determine the relationship between GAS6 and PROS1 γ-carboxylation and the ability of ligands to activate TAMs, we used HEK293TN cell culture supernatants containing γ-carboxylated or non-carboxylated GAS6 and PROS1 to stimulate TAM reporter cells. STAT1 activation was strongly induced only by γ-carboxylated ligands produced in the presence of vitamin K, whereas the inhibition of γ-carboxylation by warfarin dramatically reduced the ability of GAS6 and PROS1 to activate chimeric TAM receptors (Fig. 1E). To eliminate the possibility that warfarin directly affected the chimeric receptor activation, we added warfarin to vitamin K derived ligands immediately before cell stimulation (labeled pW in the right panel in Fig. 1E). Under these conditions, the addition of warfarin failed to block ligand induced activation of STAT1, confirming that γ-carboxylation, a post-translational modification following protein synthesis, is required for both GAS6 and PROS1-induced TAM activation. Therefore, commercial available recombinant GAS6 proteins which were lacking Gla domain or generated with insufficient supplement of vitamin K were not as potent as those purified by affinity chromatography specifically for γ-carboxylated proteins (Amgen) to induce TAM receptor activation (Fig. 1C).
Chimeric TAM Receptors Show Distinct Ligand Inducible-activation Patterns
When we analyzed the activation of the AXL/γR1 cells with γ-carboxylated GAS6 and PROS1, only the former activated the receptor (Fig. 1, C, right panel and E). Therefore, to further investigate ligand specificity in relation to receptor activation, reporter cells were treated with recombinant ligands at a 50 nm concentration (Fig. 1F). We observed that each of the TAM receptors responded to ligands in a distinct manner. PROS1 was more potent in triggering TYRO3 activation than equimolar amounts of GAS6, whereas MER responded to both ligands with relatively similar activation patterns. In stark contrast to TYRO3 and MER, AXL was exclusively activated by GAS6 and failed to respond to PROS1, either purified protein from pooled human plasma or fetal bovine serum that contains ∼60 nm of bovine PROS1 in 20% FBS. There was no pSTAT1 detected in parental cells expressing red fluorescent protein (RFP) following treatment with either GAS6 or PROS1, whereas hamster IFN-γ triggered strong STAT1 phosphorylation through the endogenous hamster IFN-γ receptor, showing the integrity of the JAK-STAT pathway in the cells (Fig. 1F). These results demonstrate that STAT1 activation was mediated by chimeric TAM/γR1 receptors expressed in reporter cell lines, and therefore, STAT1 activation in reporter cell lines can be utilized as a standardized readout to evaluate and compare biological potencies of GAS6 and PROS1 toward specific TAM receptors.
Metal ions may participate in the regulation of protein functions, protein folding and protein-protein interactions. There are several calcium-binding sites within the Gla, EGF-like, and SHBG domains of PROS1 and GAS6 that may contribute to the ligand activity (39, 40). Crystal structures of TAM domains also revealed the interaction between Ni2+, Zn2+, Ca2+, and receptor residues (37, 38). Therefore, we tested if metal ions may be involved in ligand-induced TAM receptor activation. PROS1 or GAS6 failed to activate TYRO3 or AXL, respectively, in the presence of chelating agent EDTA, whereas hamster IFN-γ signaling was unaffected by EDTA in the reporter cells (Fig. 1G). These results indicate that the metal ions present in the culture media are essential for both GAS6 and PROS1 induced TAM receptor activation, and ion binding may be required to maintain proper ligand structures, ligand-receptor interactions or conformational rearrangements.
Soluble TAM Receptor Ig Domains Can Serve as Specific Ligand Antagonists
Three-dimensional crystal structures of TYRO3 and AXL:GAS6 complexes reveal that the GAS6 binding pockets are located close to the interface of N-terminal Ig domains (37, 38), although the contribution of the FNIII domains to ligand binding has not been investigated. To test if the FNIII domains of TAM receptors are involved in ligand binding, we compared the abilities of soluble mTAM receptors containing either entire TAM extracellular domains (2 Ig domains + 2 FNIII domains; sTAM) or only N-terminal Ig domains (TAM-Igs) to act as ligand antagonists (Fig. 2). Since each of the TAM receptors have multiple NX(S/T) glycosylation sites predicted by protein sequences (data not shown), we first treated soluble receptors with PNGaseF, an amidase that cleaves complex N-linked oligosaccharides from glycoproteins. Notably, both types of soluble TAM receptors produced from either HEK293TN cells (sTAM) or insect cells (TAM-Igs), showed reduction of molecular weight compared with untreated soluble receptors (Fig. 2, A and B), demonstrating N-linked glycosylation of TAM receptors.
FIGURE 2.

Inhibition of GAS6 and PROS1 activities by soluble TAM receptors. A and B, soluble mTAM receptors composed of either two FNIII and two Ig domains (marked as sTAM) or two Ig domains only (marked as TAM-Igs) are schematically depicted and were produced in mammalian or insect cells, respectively. Conditioned media containing sTAMs (5 μl) or 50 ng of TAM-Igs were treated with PNGaseF for 2 h and then subjected to SDS-PAGE. Soluble mTAM receptors were immunoblotted with Abs against FLAG-tag (sTAM, A) or against His-Tag (TAM-Igs, B). C, mTAM-Igs (10 nm) were incubated with conditioned media containing 50 nm γ-carboxylated hGAS6 (K) or non-carboxylated hGAS6 (W). The hGAS6:mTAM-Igs complexes were pulled down by cobalt resin, resolved by SDS-PAGE and immunoblotted with Abs against GAS6 (non-reducing condition, top panels) or His-tag (reducing condition, bottom panels). Beads-alone lane showed non-carboxylated GAS6 weakly bound nonspecifically to cobalt resin (W lane on the GAS6 pull down, long exposure panel). One-twentieth of total reaction volume was used for input control (left two panels). Right two panels show results of pull down experiments. The short and long exposure films were both displayed for anti-GAS6 results. D–F, conditioned media containing soluble mTAMs (50 μl, D) or mTAM-Igs (500 nm, D–F) were mixed with 50 nm of hGAS6 (D) or hPROS1 (E and F), or 20% FBS (F) and incubated at 22 °C for 30 min. Serum-starved mAXL/γR1 (D), mTYRO3/γR1 (E), and mMER/γR1 (F) reporter cells were then treated with the mixtures as indicated on the figure for 30 min at 37 °C, and the activation of STAT1 was detected by pSTAT1 immunoblotting. Immunoblotting results are representative results of three independent experiments.
Next, we assessed the ability of mTAM-Igs to bind γ-carboxylated or non-carboxylated hGAS6 by performing co-precipitation assay (Fig. 2C). Among TAM-Igs, AXL-Igs were capable of binding the highest amount of GAS6, and both monomeric and dimeric forms of GAS6 were co-precipitated in approximately the same ratio as present in loading control. Because the majority of either γ-carboxylated or non-carboxylated GAS6 were detected as monomers in solution (Figs. 1D and 2C, left panel), TYRO3-Igs demonstrated preference in binding GAS6 dimers irrespectively of γ-carboxylation, whereas MER-Igs seemed to prefer binding monomers of GAS6 (when weak nonspecific binding of non-carboxylated GAS6 dimers to beads alone is subtracted; Fig. 2C). In addition, both AXL-Igs and TYRO3-Igs preferentially bound to carboxylated GAS6, whereas MER-Igs showed better binding toward non-carboxylated GAS6. However, despite the fact that binding between non-carboxylated GAS6 and TAM-Igs was observed, none of TAMs was activated by non-carboxylated ligands (Fig. 1E), suggesting that γ-carboxylation may be required for a specific structural conformation.
To investigate whether TAM soluble receptors can serve as ligand antagonists, GAS6 was first mixed and incubated with equimolar amounts of soluble TAM receptors, and the mixture was then added to the AXL/γR1 reporter cells because they selectively respond to GAS6 and not PROS1. Both sAXL and AXL-Igs blocked GAS6 induced STAT1 activation in the reporter cells, while at these concentrations none of the forms of soluble TYRO3 or MER were able to fully antagonize GAS6 activity (Fig. 2D). These experiments support the notion that, in comparison to TYRO3 and MER, AXL has highest binding affinity to GAS6. Moreover, because the soluble Ig1 and Ig2 domains were sufficient to inhibit GAS6 activity, these data suggest that the FNIII domains do not play a major role in ligand binding.
Since PROS1 showed a greater specificity for TYRO3 (Fig. 1F), we next examined if the soluble TYRO3 can act as a PROS1 antagonist (Fig. 2, E and F). Indeed, in contrast to the AXL-Ig domains that were specific for GAS6, soluble TYRO3-Ig domains showed a strong inhibitory effect against PROS1 and were highly effective in blocking FBS, which contains ∼60 nm endogenous bovine PROS1 (Fig. 2F). Finally, soluble MER-Ig domains had minimal blocking effects toward either GAS6 or PROS1, and could not neutralize FBS induced activation. These results suggest that MER binds with the lowest affinity to GAS6 and PROS1.
To further analyze blocking abilities of TAM-Igs toward each ligand, we pre-incubated different concentrations of soluble TAM-Igs with GAS6 or PROS1. AXL-Igs achieved 50% inhibitory effect toward GAS6 at 10 nm concentration, which was hundreds of times lower than TYRO3-Igs (1.5 μm) and MER-Igs (6 μm) (Fig. 3A). In contrast, because PROS1 has the highest binding affinity to TYRO3, only TYRO3-Igs could achieve 50% of inhibition of PROS1 induced TAM receptor activation in the nanomolar concentration range (Fig. 3B). Taken together, these data are consistent with the fact that individual TAM receptors have distinct ligand-dependent activation, and the Ig domains of TYRO3 and AXL are sufficient to block PROS1- and GAS6-mediated activities, respectively.
FIGURE 3.
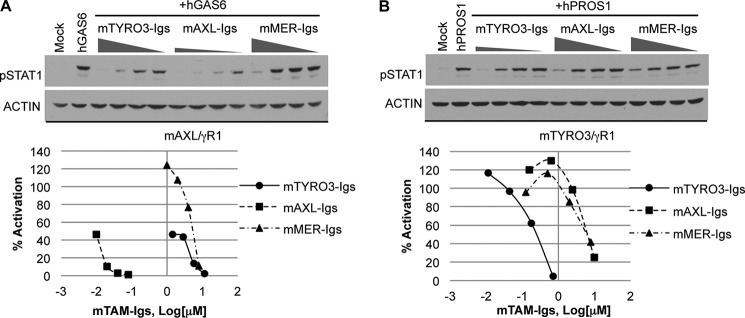
Blocking GAS6 and PROS1 activities by soluble TAM-Igs. mTYRO3/γR1 and mAXL/γR1 reporter cells were starved for 5 h in serum-free media and incubated with serial dilution of soluble mTAM-Igs for 30 min at 22 °C. The size of triangles represent the concentration of TAM-Igs: mTYRO3-Igs (1.43–11.43 μm), mAXL-Igs (10–80 nm), mMER-Igs (1–8 μm) were premixted with AXL/γR1 cells (A); and mTYRO3-Igs (11–714 nm), mAXL-Igs (0.16–10 μm), mMER-Igs (0.13–8 μm) were premixed with mTYRO3/γR1 cells (B). hGAS6 (10 nm, Amgen) and hPROS1 (20 nm) were then added to the cells:TAM-Igs mixture for additional 30 min at 37 °C. The activation of chimeric receptors was assessed by measuring levels of pSTAT1 by immunoblotting, and the levels of pSTAT1 activation induced by ligands alone was set as 100% after normalization to actin loading control. The curve was plotted according to the blocking effects and the concentration of mTAM-Igs. Immunoblotting results are representative results of at least three independent experiments.
Phosphatidylserine (PS) Liposomes and Apoptotic Cells Differentially Modulate Ligand-induced TAM Activation
Because ligand-inducible activation of TAMs depends on γ-carboxylation, a biochemical modification predicted to mediate binding to anionic lipids such as PS, we investigated the effect of PS liposomes with the respect to the activation of each TAM receptor. The reporter cell lines were treated with either GAS6 or PROS1 in the presence of phospholipid liposomes containing either PS or phosphatidylcholine (PC), a neutral lipid (Fig. 4). PC liposomes failed to enhance GAS6 or PROS1 induced activation of any of the three chimeric TAM receptors. In contrast, purified reconstituted PS liposomes or liposomes containing mixtures of PC and PS (PC/PS; 7:3 molar ratio) promoted both GAS6 and PROS1 induced activation of TYRO3 (Fig. 4, A and B). Although it was previously reported that hPROS1 did not activate hTYRO3 (16, 41), we observed that hPROS1 induced signaling through hTYRO3/γR1 at 100 nm concentration by which the activation of hMER/γR1 was also achieved. The signaling was further enhanced by PS liposomes indicating that hPROS1 became a more potent ligand toward hTYRO3 in the presence of anionic lipid (Fig. 4B). In addition, PS liposomes also strongly enhanced the activation of MER by either PROS1 or GAS6 (Fig. 4, C and D). The effects of liposomes containing a PC/PS mixture on GAS6 and PROS1 activities were quite different, in that the presence of PC in liposomes strongly inhibited the enhancement of PROS1 stimulated MER activation by liposomes containing pure PS (Fig. 4, C and D). PC was able to attenuate PS-mediated enhancement of PROS1-dependent MER activation only when PC and PS were present in the same liposome, because this inhibitory effect of PC was not observed when PC- and PS-containing liposomes were prepared separately and then mixed in 7:3 molar ratios (PC+PS; Fig. 4C). These results suggest that GAS6 and PROS1 interact with phospholipids differently, and that PROS1 induced MER activation is highly dependent on the presence and the local concentration of PS. Finally, unlike TYRO3 and MER, the activation of AXL by GAS6 was not enhanced in the presence of PS liposomes (Fig. 4, E and F), and we even observed the reduction of activation in hAXL (Fig. 4F), suggesting that AXL mainly responds to the soluble GAS6, and that AXL signaling is not enhanced and possibly even attenuated by anionic lipids.
FIGURE 4.

Modulation of ligand induced TAM activation by lipids. Conditioned media containing hGAS6 (50 nm in A–D, 10 nm in E–F) or hPROS1 (20 nm in A, 100 nm in B--F) were premixed with or without 0.5 mm of phosphatylcholine (PC), phosphatylserine (PS), or 7:3 molar ratios of phospholipids (PC/PS indicates the presence of both lipids in the same multilamellar liposomes; PC+PS represents a mixture of PC- and PS-containing individual liposomes) for 30 min at 22 °C. The mixture was added to the serum-starved reporter cells for 30 min at 37 °C. The activation of chimeric TAM receptors was assessed by measuring levels of pSTAT1 in mTYRO3/γR1 (A), hTYRO3/γR1 (B), mMER/γR1 (C), hMER/γR1 (D), mAXL/γR1 (E), or hAXL/γR1 (F) reporter cells by immunoblotting. G and H, densitometric analysis was done as described under “Experimental Procedures” and plots represent immunoblots shown in A--F. I, 50 nm hGAS6 or hPROS1 were pre-incubated with or without 0.5 μm mTRYO3-Igs, mAXL-Igs or Annexin V in the absence or presence of 0.5 mm PS liposomes at 22 °C for 30 min. The mixture was added to mMER/γR1 reporter cells for 30 min at 37 °C; and STAT1 activation in cells was measured by immunoblotting as described. Immunoblotting results are representative results of at least three independent experiments.
Since MER was observed to have the greatest PS dependence on ligand-induced activation, we further investigated whether this enhancement can be affected by the presence of ligand antagonists. Consistent with our results (Fig. 2), AXL-Igs and TYRO3-Igs acted as effective ligand antagonists of GAS6 and PROS1, respectively, even in the presence of PS liposomes (Fig. 4I). In addition, because Annexin V binds to PS, we evaluated whether the presence of Annexin V may alter the effect of PS liposomes on GAS6 and PROS1 activities. We observed that PS-mediated enhancement of ligand-induced MER activation was greatly reduced, suggesting that Annexin V may act as a competitor of PROS1 and GAS6 for PS binding sites on liposomes.
To further evaluate the relevance of the PS enhancement on TAM-mediated signaling, we tested whether apoptotic cells (ACs) also impinge on the ligand-dependent activation of TAM receptors (Fig. 5). In these assays, apoptosis was induced in Jurkat cells by either camptothecin (CPT) or UV irradiation in serum free media. The Annexin V positive population was 7% in untreated, 45% in CPT-treated, and 80% in UV-treated cells (Fig. 5A). ACs were pre-incubated with ligands to coat ACs with either GAS6 or PROS1. To avoid the saturation of signal, we used lower concentrations of ligands. Activation of TYRO3 was moderately up-regulated after GAS6 or PROS1 was pre-mixed with live untreated Jurkat cells (Fig. 5, B and C). The addition of ACs with ligands did not further enhance TYRO3 activation; even though the percentage of PS positive cell is higher in ACs than in live (untreated) cells (45 and 80% versus 7%; Fig. 5A). The observation that the addition of Jurkat cells (untreated or ACs) alone can trigger weak activation of TYRO3 suggests that PS positive Jurkat cells are likely to be coated with bovine PROS1 from serum-containing media during cell culture before apoptosis induction. Thus, the residual bovine PROS1 bound to PS-containing Jurkat cells stimulated TYRO3 activation. In contrast, the activation of MER was moderately enhanced by untreated Jurkat cells pre-incubated with ligands, and the signal was further boosted by GAS6 and PROS1 coated ACs (Fig. 5, D and E). These results imply that MER receptor is fully activated only in the presence of ACs. Unlike TYRO3 and MER receptors, ligand induced activation of AXL was only moderately affected by ACs (Fig. 5, F and G). Altogether, these results indicate that all three TAM receptors possess very distinct activation patterns in the context of ACs and the proportion of PS significantly alters the ligand-induced activation in a TAM-specific manner.
FIGURE 5.

Effects of apoptotic cells on ligand-mediated TAM activation. A, Jurkat cells were incubated in serum-free media containing 10 μm camptothecin (CPT) for 5 h or treated by UV irradiation (25 mJ/cm2, 1 min). The percentage of cell death was evaluated by PI and Annexin V-FITC staining followed by flow cytometry. B–G, conditioned media containing hGAS6 (50 nm in B–E, 10 nm in F–G), mGAS6-conditioned media (1:1 dilution in F and G) or hPROS1 (20 nm in B, 100 nm in C–G) were premixed with untreated Jurkat (Live) or CPT or UV-treated Jurkat cells for 30 min at 22 °C. The mixtures were added to the indicated serum-starved reporter cells for 30 min at 37 °C. Jurkat cells were washed away by PBS without Ca2+ and Mg2+ six times. The activation of chimeric TAM receptors was assessed by measuring levels of pSTAT1 in mTYRO3/γR1 (B), hTYRO3/γR1 (C), mMER/γR1 (D), hMER/γR1 (E), mAXL/γR1 (F), or hAXL/γR1 (G) reporter cells by immunoblotting. H and I, densitometric analysis was done as described under “Experimental Procedures” and plots represent immunoblots shown in B–G. Immunoblotting results are representative results of at least three independent experiments.
Enveloped Viruses Potentiate Ligand-dependent Activation of TAM Receptors
Several studies have shown that TAM receptors and their ligands are involved in the entry of enveloped viruses into host cells (24–26, 42, 43). GAS6 and PROS1 bind to PS on enveloped viruses, and these GAS6/PROS1-coated viruses may mimic ACs and utilize efferocytosis machinery to facilitate infection (44). Although the kinase domain is dispensable for the viral entry (25, 43), it is possible that ligand-coated enveloped viruses can still modulate TAM receptor signaling. To investigate whether enveloped viruses may potentiate ligand-induced TAM activation, we pre-incubated enveloped vesicular stomatitis virus (VSV) with either GAS6 or PROS1 for 30 min before TAM stimulation. The presence of VSV particles along with GAS6 or PROS1 potentiated ligand-inducible activation of TYRO3 (Fig. 6, A and B) and MER receptors (Fig. 6, C and D), but did not modulate AXL activation (Fig. 6, E and F). We then tested whether higher concentrations of VSV particles would further enhance TAM activation. Consistent with the AC and PS liposome results (Figs. 4 and 5), MER activation by both ligands was enhanced in a VSV dose dependent manner (Fig. 6, C and D). TYRO3 activation particularly by PROS1 was also strongly increased in the presence of VSV (Fig. 6, A and B).
FIGURE 6.

Effects of enveloped viruses on ligand-dependent TAM activation. Conditioned media containing hGAS6 (50 nm in A–D, 10 nm in E–F or hPROS1 (20 nm in A or 100 nm in B–F were premixed with or without purified VSV particles (1, 3 or 10 × 106 CFU) for 30 min at 37 °C. The ligand-coated viral particles were added to the serum-starved TAM/γR1 reporter cells for 30 min at 37 °C. The activation of chimeric TAM receptors was assessed by measuring levels of pSTAT1 in mTYRO3/γR1 (A), hTYRO3/γR1 (B), mMER/γR1 (C), hMER/γR1 (D), mAXL/γR1 (E), or hAXL/γR1 (F) reporter cells by pSTAT1 immunoblotting. G and H, densitometric analysis was done as described under “Experimental Procedures” and plots represent immunoblots shown in A–F. Immunoblotting results are representative results of at least three independent experiments.
DISCUSSION
While the TAM receptors (TYRO3, AXL, and MER) are defined by high sequence conservation in their catalytic kinase domain (∼75% amino acid identity), and a common organization (two Ig-like domains and two FNIII-like domains; Fig. 1A) in their extracellular regions, it remains unclear at the functional level whether TAMs possess specific or overlapping modes of ligand-inducible activation. To systematically evaluate TAM activation by conventional ligands, GAS6 and PROS1, we created chimeric TAM/IFN-γR1 (TAM/γR1) receptors containing human or mouse TAM extracellular domains fused in frame to the transmembrane and intracellular domains of the human IFN-γR1 chain. In this system whereby receptor dimerization triggers the well-characterized IFN-γ receptor signaling, TAM receptor activation is assessed and normalized by the tyrosine phosphorylation of STAT1 in cells expressing TAM/γR1. Using this reporter system, we show that TAMs have distinct and dynamic patterns of activation by PROS1 and GAS6 (Fig. 7, A–C). In particular, we observed that AXL is activated exclusively by GAS6 but not PROS1 (Figs. 1, 4, and 6). In contrast, TYRO3 and MER are activated by both GAS6 and PROS1, although TYRO3 respond preferentially to PROS1, while MER shows weak activation toward both ligands (Figs. 1, 4, 5, and 6).
FIGURE 7.

Interaction of TAM RTKs with their ligands. A model depicts the interaction of TAM receptors with their ligands (GAS6 and PROS1). The thickness of the arrows reflects the activation strength of each ligand to each receptor (A). Phosphatidylserine lipid vesicles, apoptotic cells, and enveloped viruses significantly potentiate the ligand-induced activation of MER and to a lesser extent TYRO3, but not AXL (B). Soluble TYRO3-Igs and AXL-Igs act as effective ligand antagonists by blocking PROS1- and GAS6-induced TAM receptor activation, respectively. Soluble MER-Igs possess weak inhibitory activities toward both ligands (C). D and E, binding of GAS6 alone causes homodimerization of AXL leading to receptor auto-phosphorylation and induction of strong downstream signaling (D). Only weak signaling is induced upon binding of GAS6 alone to either TYRO3 or MER, while PS-containing vesicles provide nucleation force to facilitate receptor oligomerization and strongly enhance downstream signaling (E).
Previous studies demonstrated that the concentration of GAS6 and PROS1 could be used as biomarkers for certain diseases. Many cancers, including breast cancer, demonstrate elevated GAS6 production along with AXL and MER overexpression (15), whereas lupus patients show different profiles of soluble TAM receptors and free PROS1 and GAS6 in plasma, which are also associated with common genetic variants and stages of disease (45, 46). Therefore, one of the important utilities of these TAM reporter cell lines is to assess the activity of functional TAM ligands, as current conventional ELISA technology only provides information on the ligand concentrations, independent of whether the ligand is active or inactive. These cell lines can be also used to determine whether recently identified non-conventional TAM ligands (29, 30) can signal through TAM homodimers. In addition, these cell lines can be applied for screening potential agonists or receptor specific inhibitors in the form of neutralizing antibodies or drugs. Because TYRO3 and AXL respond preferentially to PROS1 and GAS6, respectively, TYRO3/γR1 and AXL/γR1 reporter cells can be used to detect and quantify levels of biologically active PROS1 and GAS6 in biological samples. Indeed, we observed that FBS and human plasma triggered pSTAT1 activation in TYRO3, but not AXL, reporter cells (Fig. 1, C and F and data not shown). The concentration of the free form of PROS1 is ∼100 nm, which is a hundred times higher than GAS6 concentration that is lower than 0.5 nm in human plasma (46, 47). The fact that we did not observe activation of AXL by human plasma or FBS might also be explained by studies reporting that serum GAS6 exists in a complex with naturally occurring sAXL (48). It is likely that GAS6 is functionally inert in the sAXL:GAS6 complex, because AXL-Igs act as a potent GAS6 antagonist (Figs. 2 and 3).
Importantly, we discovered that both GAS6 and PROS1 required vitamin K dependent γ-carboxylation of their N-terminal Gla domain for activity. We found that when GAS6 and PROS1 were prepared in the producer cells treated with warfarin, an inhibitor of cellular vitamin K epoxide reductase, the γ-carboxylation of both ligands was blocked (Fig. 1D) and neither ligand had measurable activity (Fig. 1E). While one interpretation is that γ-carboxylation of GAS6 and PROS1 is required for the binding of the proteins to anionic lipids (i.e. PS) in a manner analogous to coagulation factors, the fact that γ-carboxylation is also required for GAS6-inducible activation of AXL (where PS does not enhance AXL activation; Figs. 4–6) suggests a more complex scenario whereby γ-carboxylation might be required for a specific structural conformation, or that this post-translational modification promotes assembly of a functional ligand:receptor complexes. However, despite the fact that non-carboxylated GAS6 had undetectable activity toward AXL, curiously it still retained binding activity to recombinant AXL-Igs (Fig. 2C), albeit at a lower level, suggesting that receptor binding can be dissociated from functional activation. We are currently testing GAS6 truncation mutants in the Gla and EGF-like domains to better understand this relationship.
Interestingly, in addition to differences in the activation patterns by native ligands, we also found significant differences in the PS-dependence for the different TAMs. While PS enhanced activation of MER and TYRO3 by their respective ligands, it did not promote but rather attenuated the activity of GAS6 toward AXL (Figs. 4–6). The crystal structure of GAS6-LG domains bound to AXL-Ig domains reveals that the first LG domain has two distinct epitopes that interact with and crosslink two molecules of AXL-Igs whereby each of them also possesses two independent spatially separate GAS6 binding sites (38). Therefore, GAS6 alone is sufficient to induce AXL homodimerization in 2:2 stoichiometry. Interestingly, only one minor GAS6 binding site is highly conserved in all TAMs, whereas the second major GAS6 binding site in MER and TYRO3 is not preserved. This opens a possibility that whereas GAS6 alone can cause AXL homodimerization, and perhaps heterodimerization of AXL with MER or TYRO3, GAS6 alone is unable to induce MER or TYRO3 homodimerization (Fig. 7D). Therefore, additional factors are required to promote dimerization or oligomerization of MER or TYRO3 bound to GAS6 in 1:1 stoichiometry. In support of this idea, PS exposed on the surface of ACs or enveloped viruses can provide crosslinking force sufficient to cause oligomerization of GAS6-bound MER or TYRO3 (Fig. 7E). Indeed, we observed that only MER and TYRO3, but not AXL, demonstrated strong enhancement of GAS6 mediated activation in PS concentration dependent manner (Figs. 4–6). In this scenario, the local concentration of PS can regulate the intensity of MER and TYRO3 signaling and their functions. Since only crystal structures of TYRO3-Igs and AXL-Igs:GAS6-LGs (37, 38), but not MER, have been solved, a completion of the three-dimensional structures of all TAM receptors with their ligands is needed to further advance our understanding of the functional architecture of ligand:TAM complexes.
Consistent with previous affinity studies demonstrating that GAS6 has a considerably higher affinity for AXL compared with MER (49), we also observed that, although all TAM receptors can be activated by GAS6, this ligand alone triggers stronger activation of AXL (Fig. 1F) that was not further enhanced by the presence of PS (Figs. 4–6). In addition, in both co-precipitation studies (Fig. 2) and in competition studies with soluble TAMs (Fig. 3), the GAS6:AXL interaction was much more prominent compared with GAS6:MER and GAS6:TYRO3 interactions, suggesting that if AXL is co-expressed with either MER or TYRO3 in a single cell, AXL would be the preferred receptor for native GAS6 and will be preferentially engaged in ligand binding. However, in the presence of PS, either in the form of PS liposomes, apoptotic cells, or enveloped viruses (Figs. 4–6), GAS6 acquires the capability to induce stronger activation of MER; therefore, MER may act as a preferred receptor for GAS6 opsoninzed to PS. This hypothesis is also supported by studies of Nguyen et al. (50) demonstrating that ACs only enhance GAS6 induced receptor autophosphorylation in cells expressing MER, but not AXL. This raises an interesting possibility that following PS opsonization, GAS6 switches from preferential AXL binding to MER binding, and, taken further, that MER will function mainly as an efferocytosis receptor that interacts with the surface of ACs, while AXL interacts with un-opsonized GAS6 to stimulate conventional RTK signaling. In accordance with this scenario, we even observed that PS liposomes and high concentration of VSV virion particles reduced GAS6-mediated hAXL activation (Figs. 4F and 6F). Such distinctions might indeed explain why MER(−/−) mice display a strong defect in the clearance of apoptotic cells, while AXL(−/−) mice are spared in the phenotype of abnormal clearance of ACs. We also observed that either ACs or PS-containing liposomes most strongly promoted GAS6 or PROS1 mediated activation of MER among all TAM receptors (Figs. 4 and 5), further supporting the unique functional importance of MER in efferocytosis. The presence of PC within the PS-containing liposomes strongly inhibited enhancing effects of PS toward PROS1-induced MER activation (Fig. 4, C and D). These results suggest that a certain threshold in local concentration of PS should be achieved on the surface of dying cells to mark them as apoptotic and appropriate for removal by MER-mediated efferocytosis. Presence of PC on the cell surface may prevent reaching levels of MER activation by PROS1 sufficient for efferocytosis and therefore spare cells from efferocytosis by allowing phagocytes to discriminate between ACs and non-ACs.
Recent studies showed that enveloped viruses could mimic ACs to enter cells by efferocytosis via PS recognizing TAM and TIM receptors (25, 51). It is well established that viruses can hijack cellular machinery by many different ways from cell entry, replication to spreading (52). Although the intracellular domain of TAM receptors seems not to be necessary for viral entry, the kinase activity is required for potentiation of virus infectivity (25, 53). A recent study demonstrated that enveloped viruses can activate AXL and MER to attenuate type I IFN signaling in dendritic cells (43). Here, we showed that GAS6 and PROS1 coated VSV potentiated ligand-dependent activation of TYRO3 and MER (Fig. 6). These results indicate the PS coated GAS6 or PROS1 not only facilitate viral entry, but they also serve as “super ligands” to induce anti-inflammatory status by activating TAM receptors.
In addition to enveloped viruses and ACs, many cells including polymorphonuclear leukocytes, erythrocytes and tumor cells, secrete ectosomes, PS containing vesicles (54), which act similarly to ACs and stimulate production of anti-inflammatory cytokines in MER dependent manner (55). There is a growing body of evidence supporting the involvement of TAMs at multiple levels of tumor development and progression including promotion of proliferation and survival of cancer cells and establishment of immunosuppression (14). Recent studies by Paolino et al. (56) demonstrated that suppression of TAM activities with warfarin, kinase inhibitors or by ablation of TAM-specific E3 ligase exerts anti-metastatic activity by eliminating TAM-mediated inhibition of NK cells. Therefore, TAM signaling axis represents an attractive target for cancer therapy. Indeed, PS blockade with a monoclonal antibody bavituximab in patients with solid tumors resulted in tumor vessel occlusion and enhanced antitumor immunity (57). Our studies reveal distinct regulation of TAM receptor activation by GAS6, PROS1, and PS, and suggest that TAMs may play specific non-overlapping activities in cancer.
While our system thoroughly examined the differences in the binding of known ligands to TAMs, an equally important question is whether TAMs have unique or overlapping modes of post-receptor signaling. It is known that TAM receptors are differentially expressed in mature immune, nervous, reproductive, and vascular systems (11), and have different patterns of transcriptional regulation. For example, mammary epithelial cells express MER only in the involution stage, whereas AXL is up-regulated by IFN-α in dentritic cells (13, 58). Moreover, it remains to be determined whether in cells that co-express more than one TAM receptor, the receptors can heterodimerize, and if so, how this affects ligand receptor interactions as well as post-receptor signaling.
In conclusion, we have developed a complete series of reporter cell lines for human and mouse TAMs that allowed us to comprehensively compare the modes of ligand-inducible TAM activation by conventional TAM ligands, GAS6 and PROS1. The studies demonstrate that TAMs interact with their ligands in unique ways that depend on the context in which the ligands are presented to the TAM extracellular domains, and therefore reveal a previously unappreciated functional diversity of TAM receptors.
Acknowledgments
We thank Dr. Peter Carmeliet for providing the pSecTag2-hGAS6 plasmid, Dr. Carla Rothlin for the gift of pcDNA6-mPROS1 plasmid and Dr. Philip Cohen for providing pCMV-SPORT6-mGAS6 plasmid, and Dr. Brian Varnum (Amgen) for the gift of recombinant human GAS6.
This work was supported in part by National Institutes of Health Grants R01AI104669 (to S. V. K.) and R01NCI165077 (to R. B. B.), Rutgers University, NJMS translational research award & bridge grant (to S. V. K. and R. B. B.), National Institutes of Health Grants GM094662 & GM094665 (to S. C. A.), and P30CA013330 to Cancer Center of the Albert Einstein College of Medicine.
- RTK
- receptor tyrosine kinase
- TAM
- TYRO3/AXL/MER
- GAS6
- growth arrest specific gene 6
- PROS1
- protein S
- PS
- phosphatidylserine
- PC
- phosphatidylcholine
- ACs
- apoptotic cells
- FN
- fibronectin
- Ig
- immunoglobulin
- IL-6
- interleukin-6
- Gla
- γ-carboxyglutamic acid
- EGF
- epidermal growth factor
- SHBG
- sex hormone-binding globulin
- LG
- laminin G
- PI3K
- phosphatidylinositol-4,5-bisphosphate 3-kinase
- AKT
- protein kinase B
- ERK
- extracellular-signal-regulated kinases
- PLC-γ
- phospholipase Cγ
- HIV
- human immunodeficiency virus
- IFN
- interferon
- LMV
- large multilamellar vesicle
- VSV
- vesicular stomatitis virus
- STAT1
- signal transducer and activator of transcription 1
- JAK
- Janus kinase
- CPT
- camptothecin
- LPS
- lipopolysaccharides.
REFERENCES
- 1. O'Bryan J. P., Frye R. A., Cogswell P. C., Neubauer A., Kitch B., Prokop C., Espinosa R., 3rd, Le Beau M. M., Earp H. S., Liu E. T. (1991) axl, a transforming gene isolated from primary human myeloid leukemia cells, encodes a novel receptor tyrosine kinase. Mol. Cell. Biol. 11, 5016–5031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Graham D. K., Dawson T. L., Mullaney D. L., Snodgrass H. R., Earp H. S. (1994) Cloning and mRNA expression analysis of a novel human protooncogene, c-mer. Cell Growth Differ. 5, 647–657 [PubMed] [Google Scholar]
- 3. Lai C., Gore M., Lemke G. (1994) Structure, expression, and activity of Tyro 3, a neural adhesion-related receptor tyrosine kinase. Oncogene 9, 2567–2578 [PubMed] [Google Scholar]
- 4. Lu Q., Lemke G. (2001) Homeostatic regulation of the immune system by receptor tyrosine kinases of the Tyro 3 family. Science 293, 306–311 [DOI] [PubMed] [Google Scholar]
- 5. Lu Q., Gore M., Zhang Q., Camenisch T., Boast S., Casagranda F., Lai C., Skinner M., Klein R., Matsushima G., Earp H., Goff S., Lemke G. (1999) Tyro-3 family receptors are essential regulators of mammalian spermatogenesis. Nature 398, 723–728 [DOI] [PubMed] [Google Scholar]
- 6. Burstyn-Cohen T., Heeb M., Lemke G. (2009) Lack of protein S in mice causes embryonic lethal coagulopathy and vascular dysgenesis. The Journal of clinical investigation 119, 2942–2953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Korshunov V. A., Mohan A. M., Georger M. A., Berk B. C. (2006) Axl, a receptor tyrosine kinase, mediates flow-induced vascular remodeling. Circ. Res. 98, 1446–1452 [DOI] [PubMed] [Google Scholar]
- 8. Camenisch T. D., Koller B. H., Earp H. S., Matsushima G. K. (1999) A novel receptor tyrosine kinase, Mer, inhibits TNF-α production and lipopolysaccharide-induced endotoxic shock. J. Immunol. 162, 3498–3503 [PubMed] [Google Scholar]
- 9. Scott R. S., McMahon E. J., Pop S. M., Reap E. A., Caricchio R., Cohen P. L., Earp H. S., Matsushima G. K. (2001) Phagocytosis and clearance of apoptotic cells is mediated by MER. Nature 411, 207–211 [DOI] [PubMed] [Google Scholar]
- 10. Rahman Z. S., Shao W. H., Khan T. N., Zhen Y., Cohen P. L. (2010) Impaired apoptotic cell clearance in the germinal center by Mer-deficient tingible body macrophages leads to enhanced antibody-forming cell and germinal center responses. J. Immunol. 185, 5859–5868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lemke G., Rothlin C. (2008) Immunobiology of the TAM receptors. Nat. Rev. Immunol. 8, 327–336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Nguyen K. Q., Tsou W. I., Kotenko S., Birge R. B. (2013) TAM receptors in apoptotic cell clearance, autoimmunity, and cancer. Autoimmunity 46, 294–297 [DOI] [PubMed] [Google Scholar]
- 13. Sandahl M., Hunter D. M., Strunk K. E., Earp H. S., Cook R. S. (2010) Epithelial cell-directed efferocytosis in the post-partum mammary gland is necessary for tissue homeostasis and future lactation. BMC Dev. Biol. 10, 122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Linger R. M., Keating A. K., Earp H. S., Graham D. K. (2008) TAM receptor tyrosine kinases: biologic functions, signaling, and potential therapeutic targeting in human cancer. Adv. Cancer Res. 100, 35–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Verma A., Warner S. L., Vankayalapati H., Bearss D. J., Sharma S. (2011) Targeting Axl and Mer kinases in cancer. Mol. Cancer Ther. 10, 1763–1773 [DOI] [PubMed] [Google Scholar]
- 16. Stitt T., Conn G., Gore M., Lai C., Bruno J., Radziejewski C., Mattsson K., Fisher J., Gies D., Jones P. (1995) The anticoagulation factor protein S and its relative, Gas6, are ligands for the Tyro 3/Axl family of receptor tyrosine kinases. Cell 80, 661–670 [DOI] [PubMed] [Google Scholar]
- 17. Manfioletti G., Brancolini C., Avanzi G., Schneider C. (1993) The protein encoded by a growth arrest-specific gene (gas6) is a new member of the vitamin K-dependent proteins related to protein S, a negative coregulator in the blood coagulation cascade. Mol. Cell. Biol. 13, 4976–4985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Fridell Y. W., Jin Y., Quilliam L. A., Burchert A., McCloskey P., Spizz G., Varnum B., Der C., Liu E. T. (1996) Differential activation of the Ras/extracellular-signal-regulated protein kinase pathway is responsible for the biological consequences induced by the Axl receptor tyrosine kinase. Mol. Cell. Biol. 16, 135–145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Todt J. C., Hu B., Curtis J. L. (2004) The receptor tyrosine kinase MerTK activates phospholipase C γ2 during recognition of apoptotic thymocytes by murine macrophages. J. Leukoc. Biol. 75, 705–713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Li R., Chen J., Hammonds G., Phillips H., Armanini M., Wood P., Bunge R., Godowski P. J., Sliwkowski M. X., Mather J. P. (1996) Identification of Gas6 as a growth factor for human Schwann cells. J. Neurosci. 16, 2012–2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bandyopadhyay P. K. (2008) Vitamin K-dependent gamma-glutamylcarboxylation: an ancient posttranslational modification. Vitam. Horm. 78, 157–184 [DOI] [PubMed] [Google Scholar]
- 22. Robins R. S., Lemarié C. A., Laurance S., Aghourian M. N., Wu J., Blostein M. D. (2013) Vascular Gas6 contributes to thrombogenesis and promotes tissue factor up-regulation after vessel injury in mice. Blood 121, 692–699 [DOI] [PubMed] [Google Scholar]
- 23. Wu Y., Tibrewal N., Birge R. B. (2006) Phosphatidylserine recognition by phagocytes: a view to a kill. Trends Cell Biol. 16, 189–197 [DOI] [PubMed] [Google Scholar]
- 24. Shimojima M., Takada A., Ebihara H., Neumann G., Fujioka K., Irimura T., Jones S., Feldmann H., Kawaoka Y. (2006) Tyro3 family-mediated cell entry of Ebola and Marburg viruses. J. Virol. 80, 10109–10116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Meertens L., Carnec X., Lecoin M., Ramdasi R., Guivel-Benhassine F., Lew E., Lemke G., Schwartz O., Amara A. (2012) The TIM and TAM families of phosphatidylserine receptors mediate dengue virus entry. Cell Host Microbe 12, 544–557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Morizono K., Xie Y., Olafsen T., Lee B., Dasgupta A., Wu A., Chen I. (2011) The soluble serum protein Gas6 bridges virion envelope phosphatidylserine to the TAM receptor tyrosine kinase Axl to mediate viral entry. Cell Host Microbe 9, 286–298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Nakano T., Ishimoto Y., Kishino J., Umeda M., Inoue K., Nagata K., Ohashi K., Mizuno K., Arita H. (1997) Cell adhesion to phosphatidylserine mediated by a product of growth arrest-specific gene 6. J. Biol. Chem. 272, 29411–29414 [DOI] [PubMed] [Google Scholar]
- 28. Sen P., Wallet M., Yi Z., Huang Y., Henderson M., Mathews C., Earp H., Matsushima G., Baldwin A., Tisch R. (2007) Apoptotic cells induce Mer tyrosine kinase-dependent blockade of NF-kappaB activation in dendritic cells. Blood 109, 653–660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Caberoy N., Zhou Y., Li W. (2010) Tubby and tubby-like protein 1 are new MerTK ligands for phagocytosis. EMBO J. 29, 3898–3910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Caberoy N., Alvarado G., Bigcas J.-L., Li W. (2012) Galectin-3 is a new MerTK-specific eat-me signal. J. Cell. Physiol. 227, 401–407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hafizi S., Dahlbäck B. (2006) Gas6 and protein S. Vitamin K-dependent ligands for the Axl receptor tyrosine kinase subfamily. FEBS J. 273, 5231–5244 [DOI] [PubMed] [Google Scholar]
- 32. Soh J., Donnelly R. J., Mariano T. M., Cook J. R., Schwartz B., Pestka S. (1993) Identification of a yeast artificial chromosome clone encoding an accessory factor for the human interferon γ receptor: evidence for multiple accessory factors. Proc. Natl. Acad. Sci. U.S.A. 90, 8737–8741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kotenko S. V., Gallagher G., Baurin V. V., Lewis-Antes A., Shen M., Shah N. K., Langer J. A., Sheikh F., Dickensheets H., Donnelly R. P. (2003) IFN-lambdas mediate antiviral protection through a distinct class II cytokine receptor complex. Nat. Immunol. 4, 69–77 [DOI] [PubMed] [Google Scholar]
- 34. Kotenko S. V., Saccani S., Izotova L. S., Mirochnitchenko O. V., Pestka S. (2000) Human cytomegalovirus harbors its own unique IL-10 homolog (cmvIL-10). Proc. Natl. Acad. Sci. U.S.A. 97, 1695–1700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Moerdyk-Schauwecker M., Hwang S. I., Grdzelishvili V. Z. (2009) Analysis of virion associated host proteins in vesicular stomatitis virus using a proteomics approach. Virol J. 6, 166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Muthukumaran G., Kotenko S., Donnelly R., Ihle J. N., Pestka S. (1997) Chimeric erythropoietin-interferon γ receptors reveal differences in functional architecture of intracellular domains for signal transduction. J. Biol. Chem. 272, 4993–4999 [DOI] [PubMed] [Google Scholar]
- 37. Heiring C., Dahlbäck B., Muller Y. (2004) Ligand recognition and homophilic interactions in Tyro3: structural insights into the Axl/Tyro3 receptor tyrosine kinase family. J. Biol. Chem. 279, 6952–6958 [DOI] [PubMed] [Google Scholar]
- 38. Sasaki T., Knyazev P., Clout N., Cheburkin Y., Göhring W., Ullrich A., Timpl R., Hohenester E. (2006) Structural basis for Gas6-Axl signalling. EMBO J. 25, 80–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Sasaki T., Knyazev P. G., Cheburkin Y., Göhring W., Tisi D., Ullrich A., Timpl R., Hohenester E. (2002) Crystal structure of a C-terminal fragment of growth arrest-specific protein Gas6. Receptor tyrosine kinase activation by laminin G-like domains. J. Biol. Chem. 277, 44164–44170 [DOI] [PubMed] [Google Scholar]
- 40. Dahlbäck B., Hildebrand B., Linse S. (1990) Novel type of very high affinity calcium-binding sites in β-hydroxyasparagine-containing epidermal growth factor-like domains in vitamin K-dependent protein S. J. Biol. Chem. 265, 18481–18489 [PubMed] [Google Scholar]
- 41. Ohashi K., Nagata K., Toshima J., Nakano T., Arita H., Tsuda H., Suzuki K., Mizuno K. (1995) Stimulation of sky receptor tyrosine kinase by the product of growth arrest-specific gene 6. J. Biol. Chem. 270, 22681–22684 [DOI] [PubMed] [Google Scholar]
- 42. Hunt C., Kolokoltsov A., Davey R., Maury W. (2011) The Tyro3 receptor kinase Axl enhances macropinocytosis of Zaire ebolavirus. J. Virol. 85, 334–347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Bhattacharyya S., Zagórska A., Lew E., Shrestha B., Rothlin C., Naughton J., Diamond M., Lemke G., Young J. (2013) Enveloped viruses disable innate immune responses in dendritic cells by direct activation of TAM receptors. Cell Host Microbe 14, 136–147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Mercer J. (2011) Viral apoptotic mimicry party: P.S. Bring your own Gas6. Cell Host Microbe 9, 255–257 [DOI] [PubMed] [Google Scholar]
- 45. Recarte-Pelz P., Tàssies D., Espinosa G., Hurtado B., Sala N., Cervera R., Reverter J., de Frutos P. (2013) Vitamin K-dependent proteins GAS6 and Protein S and TAM receptors in patients of systemic lupus erythematosus: correlation with common genetic variants and disease activity. Arthritis Res. Ther. 15(2), R41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Suh C.-H., Hilliard B., Li S., Merrill J., Cohen P. (2010) TAM receptor ligands in lupus: protein S but not Gas6 levels reflect disease activity in systemic lupus erythematosus. Arthritis Res. Ther. 12(4), R146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Griffin J. H., Gruber A., Fernández J. A. (1992) Reevaluation of total, free, and bound protein S and C4b-binding protein levels in plasma anticoagulated with citrate or hirudin. Blood 79, 3203–3211 [PubMed] [Google Scholar]
- 48. Ekman C., Stenhoff J., Dahlbäck B. (2010) Gas6 is complexed to the soluble tyrosine kinase receptor Axl in human blood. J. Thromb. Haemost. 8, 838–844 [DOI] [PubMed] [Google Scholar]
- 49. Chen J., Carey K., Godowski P. (1997) Identification of Gas6 as a ligand for Mer, a neural cell adhesion molecule related receptor tyrosine kinase implicated in cellular transformation. Oncogene 14, 2033–2039 [DOI] [PubMed] [Google Scholar]
- 50. Nguyen K.-Q. N., Tsou W.-I., Calarese D. A., Kimani S. G., Singh S., Hsieh S., Liu Y., Lu B., Wu Y., Garforth S. J., Almo S. C., Kotenko S. V., Birge R. B. (2014) Overexpression of MERTK receptor tyrosine kinase in epithelial cancer cells drives efferocytosis in a gain-of-function capacity. J. Biol. Chem. 289, 25737–25749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Jemielity S., Wang J., Chan Y., Ahmed A., Li W., Monahan S., Bu X., Farzan M., Freeman G., Umetsu D., Dekruyff R., Choe H. (2013) TIM-family proteins promote infection of multiple enveloped viruses through virion-associated phosphatidylserine. PLoS Pathog. 9, e1003232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Giorda K. M., Hebert D. N. (2013) Viroporins customize host cells for efficient viral propagation. DNA Cell Biol. 32, 557–564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Shimojima M., Ikeda Y., Kawaoka Y. (2007) The mechanism of Axl-mediated Ebola virus infection. J. Infect. Dis. 196, S259–S263 [DOI] [PubMed] [Google Scholar]
- 54. Sadallah S., Eken C., Schifferli J. A. (2011) Ectosomes as modulators of inflammation and immunity. Clin. Exp. Immunol. 163, 26–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Eken C., Martin P. J., Sadallah S., Treves S., Schaller M., Schifferli J. A. (2010) Ectosomes released by polymorphonuclear neutrophils induce a MerTK-dependent anti-inflammatory pathway in macrophages. J. Biol. Chem. 285, 39914–39921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Paolino M., Choidas A., Wallner S., Pranjic B., Uribesalgo I., Loeser S., Jamieson A. M., Langdon W. Y., Ikeda F., Fededa J. P., Cronin S. J., Nitsch R., Schultz-Fademrecht C., Eickhoff J., Menninger S., Unger A., Torka R., Gruber T., Hinterleitner R., Baier G., Wolf D., Ullrich A., Klebl B. M., Penninger J. M. (2014) The E3 ligase Cbl-b and TAM receptors regulate cancer metastasis via natural killer cells. Nature 507, 508–512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Gerber D. E., Stopeck A. T., Wong L., Rosen L. S., Thorpe P. E., Shan J. S., Ibrahim N. K. (2011) Phase I safety and pharmacokinetic study of bavituximab, a chimeric phosphatidylserine-targeting monoclonal antibody, in patients with advanced solid tumors. Clin. Cancer Res. 17, 6888–6896 [DOI] [PubMed] [Google Scholar]
- 58. Scutera S., Fraone T., Musso T., Cappello P., Rossi S., Pierobon D., Orinska Z., Paus R., Bulfone-Paus S., Giovarelli M. (2009) Survival and migration of human dendritic cells are regulated by an IFN-α-inducible Axl/Gas6 pathway. J. Immunol. 183, 3004–3013 [DOI] [PubMed] [Google Scholar]