

Background: The relationship between ion binding and conductance states of the dopamine transporter is poorly understood.
Results: The Li+ leak is mediated by the second sodium site (Na2) and is gated by Cl−.
Conclusion: Na2 controls transporter cation permeability and inner gate opening probability in a Cl−-dependent manner.
Significance: The data improve our mechanistic understanding of ion-coupling in neurotransmitter:sodium symporters.
Keywords: Dopamine Transporter, Electrophysiology, Membrane Transport, Monoamine Transporter, Neurotransmitter Transport, Site-directed Mutagenesis, Sodium Binding Sites, Structure-Function
Abstract
The dopamine transporter (DAT) belongs to the family of neurotransmitter:sodium symporters and controls dopamine (DA) homeostasis by mediating Na+- and Cl−-dependent reuptake of DA. Here we used two-electrode voltage clamp measurements in Xenopus oocytes together with targeted mutagenesis to investigate the mechanistic relationship between DAT ion binding sites and transporter conductances. In Li+, DAT displayed a cocaine-sensitive cation leak current ∼10-fold larger than the substrate-induced current in Na+. Mutation of Na+ coordinating residues in the first (Na1) and second (Na2) binding sites suggested that the Li+ leak depends on Li+ interaction with Na2 rather than Na1. DA caused a marked inhibition of the Li+ leak, consistent with the ability of the substrate to interact with the Li+-occupied state of the transporter. The leak current in Li+ was also potently inhibited by low millimolar concentrations of Na+, which according to our mutational data conceivably depended on high affinity binding to Na1. The Li+ leak was further regulated by Cl− that most likely increases Li+ permeation by allosterically lowering Na2 affinity. Interestingly, mutational lowering of Na2 affinity by substituting Asp-420 with asparagine dramatically increased cation permeability in Na+ to a level higher than seen in Li+. In addition to reveal a functional link between the bound Cl− and the cation bound in the Na2 site, the data support a key role of Na2 in determining cation permeability of the transporter and thereby possibly in regulating the opening probability of the inner gate.
Introduction
The dopamine transporter (DAT)4 mediates reuptake of the neurotransmitter dopamine (DA) from the extracellular space into the presynaptic dopaminergic neuron, thereby serving an important function in terminating dopaminergic signaling (1–3). The critical role of DAT in maintaining DA homeostasis is supported by recent data suggesting that altered or disrupted DAT function might be important in psychiatric diseases such as attention deficit hyperactivity disorder and autism (51, 52) as well as in infantile parkinsonism (4). Moreover, DAT is a target for multiple drugs of abuse including cocaine and amphetamine (1–3). These drugs act to increase the extracellular DA concentration; cocaine by binding and blocking the uptake of substrate by the transporter (5, 6) and amphetamine by competing for uptake of DA as well as by promoting reverse transport of DA via DAT (7, 8). Together with the closely related serotonin transporter (SERT) and the norepinephrine transporter, DAT belongs to the class of neurotransmitter:sodium symporters (NSSs), also referred to as the solute carrier family 6 (SLC6) (1, 3). NSS proteins operate by coupling transport of Na+ down its concentration gradient with “uphill” transport of substrate (1, 3). In addition, several of the eukaryotic NSS proteins display co-transport of Cl−. For DAT, this results in a transport associated stoichiometry of 1DA:1Cl−:2Na+ (9, 10).
High resolution crystal structures of the prokaryotic NSS member LeuT (11–14) and more recently of the Drosophila DAT (dDAT) (15) have provided critical insight into the tertiary structure of NSS proteins and the putative transport mechanism. The structures revealed a unique structural-fold consisting of 12 tightly packed transmembrane segments of which the first 10 segments are arranged in a pseudo-2-fold symmetric pattern (11–14). The structures substantiated that NSS proteins operate by an alternating access mechanism in which binding of substrate and ions from the extracellular side of the transporter induces conformational changes in the transporter leading to the translocation of substrate and ions to the intracellular side. Two Na+ binding sites, designated Na1 and Na2, were identified adjacent to the primary central substrate binding cavity (11, 13–15), and a Cl− binding site was predicted to lie immediately adjacent to the Na1 site in the eukaryotic Cl−-dependent transporters (15–18). Nonetheless, the mechanistic relationship between the two sodium ions, the chloride ion, and the substrate remains poorly understood. Specifically, the role of Cl− in the transport process is unsettled and might involve participation in several steps of the translocation cycle (16, 19).
Electrophysiological measurements in a number of biological systems, including Xenopus laevis oocytes, heterologous cells, and cultured dopaminergic neurons, have demonstrated several different conducting states for DAT (20–24). These include, in addition to the predicted coupled substrate-dependent current, a substrate-dependent uncoupled anion conductance and a small tonic cation leak that is blocked by cocaine (20–22). Interestingly, this tonic leak is not present in the Drosophila DAT and thus is a feature that might have appeared later in evolution (25). Importantly, uncoupled currents have been identified in other NSS proteins, such as the norepinephrine transporter, SERT, and the γ-aminobutyric acid transporter-1 (GAT-1) (26–30). A leak current in Li+ has also been identified in DAT as well as in GAT-1 and SERT (20, 26, 29, 31, 32). Although a physiological significance of the cation leak remains to be established, the uncoupled anion current has been reported to be of such a magnitude that it might have physiological relevance; that is, the uncoupled DAT-mediated anion current was sufficient to affect the firing rate of dopaminergic neurons in culture (21).
In the present study we employ the two-electrode voltage clamp technique in Xenopus oocytes in combination with targeted mutagenesis to obtain further insight into the molecular basis for how anion and cation binding may collaborate to control the different DAT-conducting states. Our data provide evidence that the Li+ leak in DAT is dependent on the Na2 site rather than the Na1 site, in good agreement with previous predictions for the Li+ leak in GAT-1 (31, 33, 34). Importantly, we also demonstrate that the Li+ leak is regulated by Cl−, suggesting a hitherto unknown functional link between the Cl− site and Li+ binding in the Na2 site. Moreover, our results support a key role of Na2 in determining cation permeability of the transporter. We speculate that binding in Na2 is tightly coupled to the intracellular gate and that during the transport process Cl− might facilitate transport by lowering Na2 affinity when the inner gate is open.
EXPERIMENTAL PROCEDURES
Molecular Biology
Mouse DAT, residing in pcDNA3, was kindly provided by Dr. Howard Gu, Ohio State University, Columbus, OH. Mutants were generated by Invitrogen in the pcDNA3 background and subcloned into the pFROG vector (35) using HindIII and XbaI (New England Biolabs). cRNA was generated as run-off transcripts from pFROG mouse DAT vector linearized with MluI using T7 mESSAGE-mMACHINE (Ambion) and purified using Megaclear (Ambion) following manufacturer's instructions. Stage V defolliculated X. laevis oocytes were ordered from EcoCyte BioScience. Oocytes were injected with ∼50 ng of cRNA and incubated in Kulori's medium (88 mm NaCl, 1 mm KCl, 1 mm MgCl2, 1 mm CaCl2, 5 mm Hepes-Tris) at 18 °C for ∼5 days or until use.
Electrophysiology
Xenopus oocytes were voltage-clamped similar to previously reported methods (22) using Dagan CA1-B (Dagan Corp.), connected to a PC with Digidata 1440A (Axon Instruments), and controlled with pClamp 9.2 (Axon Instruments). An Ag+/AgCl grounding electrode was connected to the recording chamber via a 2 m KCl, 2% agarose bridge to minimize liquid junction potential offsets. Current was acquired every 0.1 ms, and 50 Hz interference was off-line filtered using the build-in functions of Clampfit 9.2 (Axon Instruments). All oocytes were clamped in +20-mV steps between −100 mV (250 ms) and +40 mV (250 ms) and returned to the holding potential of −60 mV (250 ms) before each voltage step. All buffers were gravity-perfused, and current voltage relationships (I/V) and dose-response relations were analyzed on steady-state currents using Origin 8 (OriginLab). The substrate-induced current is defined as (INaCl,DA − INaCl wash). Inhibition of the Li+ leak current by Na+ is defined as (ILi − ILi+Na) and the inhibition by substrate as (ILi − ILi+DA). Cocaine-sensitive currents are defined as (I − ICOC). Unless indicated otherwise in the figure, all experiments were carried out in the 130 mm NaCl standard buffer or one of its isotonic substitutes. All recording buffers include 2.5 mm KCl, 1.8 mm CaCl2, 1 mm MgCl2, and 1 mm Hepes-Tris. For ion substitution experiments the standard buffer was isotonically substituted with the indicated buffers. Lithium gluconate was made by mixing LiOH with gluconic acid. Choline gluconate was made by mixing choline bicarbonate with gluconic acid.
Modeling
A homology human DAT model was built based on the crystal structure of dDAT (PDB code 4M48) (15) using Modeler 9v13 (36). The EL2 loop of human DAT, which does not have a complete template from dDAT, was further refined by the ab initio loop modeling module of Modeler (37). The bound Na+ (Na1 and Na2) and Cl+ ions in human DAT were modeled at their aligned positions in dDAT.
Calculations
Data were analyzed by nonlinear regression analysis using Prism 4.0 (GraphPad Software, San Diego, CA). An unpaired t test was used for statistical calculations.
RESULTS
DAT Mediates a Li+-induced Leak Current
The mouse DAT was expressed in X. laevis oocytes, and two-electrode voltage clamp was used to evaluate DAT-mediated currents. We chose to use the mouse DAT because we consistently observed larger DAT-mediated currents when expressing this transporter as compared with the rat and human DAT (data not shown). In agreement with previous reports on rat and human DAT (20–22), we observed for mouse DAT that perfusion with DA (20 μm) induced an inward current at negative potentials in 130 mm NaCl. This current is believed to represent the sum of the stoichiometric-coupled current and an uncoupled flux of anions (Fig. 1A) (20–22). Assessing the current at different membrane potentials yielded the I/V plot in Fig. 1B showing the current during DA perfusion minus the control current during perfusion with buffer. As previously observed for human DAT (20), the I/V curve showed an inwardly rectifying current at negative potentials, whereas at positive potentials the horizontal asymptote and off-set from the zero current level is thought to reflect inhibition by DA of a tonic cation leak (20). This tonic leak conductance has previously been characterized as a non-selective cation conductance (20), but the molecular mechanisms underlying the conductance remain poorly understood. To further characterize the tonic leak, we performed ion substitution experiments. Substitution of 130 mm NaCl with 130 mm lithium chloride (LiCl) resulted in a tonic leak that was ∼10-fold larger than the substrate-induced current (Fig. 1, A and C). The current observed when substituting Na+ with Li+ was indeed DAT-mediated, as the current was blocked by perfusion with 10 μm cocaine (Fig. 1D). Li+ does not, however, support transport in DAT (38), but importantly, DA can still bind to the transporter as perfusion with 20 μm DA markedly decreased the current in Li+ (Fig. 1, A and C).
FIGURE 1.

DAT mediates a Li+-induced leak current. A, trace recording of mouse DAT expressing oocytes voltage-clamped at −60 mV. Buffer changes were made as indicated. The concentration of DA (+DA) was 20 μm and of Na+ (+Na) was 3 mm. The figure shows a representative trace. B, I/V plot of steady-state currents induced by DA (20 μm) with control (no DA) subtracted (means ± S.E., n = 6). C, I/V plot of steady-state currents in NaCl plus 20 μm DA (open squares), LiCl (open blue triangles), LiCl plus DA (open red triangle), LiCl plus 3 mm Na+ (open green triangle), and Ligluc (open circles). Data are normalized to the maximum response in LiCl (means ± S.E., n = 4–6). D, I/V plot of steady-state current showing responses in NaCl and LiCl in the absence and presence of cocaine (COC, 10 μm) (means ± S.E., n = 4–6). The I/V plots were generated in 20-mV steps from −100 to +40mV (B–D).
It has been shown for GAT-1 that low millimolar concentrations of Na+ blocks the Li+ leak, and it was suggested that this inhibition reflects restoration of the conformational equilibrium between different functional states in the transport cycle (31, 32). Similar to these findings, perfusion with 3 mm Na+ caused a prominent reduction in the DAT Li+ leak without changing the reversal potential (Fig. 1, A and C). Finally, we tested the importance of anions on the Li+ leak current by substituting Cl− with gluconate. As shown in the current trace in Fig. 1A and in the I/V diagram in Fig. 1B, substitution of Cl− with gluconate also caused a prominent reduction in the current as compared with the current in LiCl. There was no change in the reversal potential of the current upon Cl− substitution, supporting that the current in Li+ is not carried by anions (Fig. 1C). Note that perfusion with DA also did not change the reversal potential (Fig. 1C). Summarizing, in the presence of Li+, DAT mediates a major tonic leak that is sensitive to substrate as well as to Na+ and Cl−.
The Dopamine Affinity Is Reduced in Li+
We estimated the apparent affinity of DA for the transporter in Na+ by measuring the DA-induced current at increasing DA concentrations. From this experiment we obtained an EC50 for DA of 1.19 ± 0.11 μm (mean ± S.E., n = 5) (Fig. 2A) in good agreement with the Km value for DA uptake obtained in heterologous expression systems (6, 9, 38). To assess the apparent affinity of DA in Li+, we took advantage of the inhibition by DA of the Li+ current and determined the magnitude of the Li+ leak inhibition at increasing DA concentrations. A half-maximal effect (EC50) was observed at 23.5 ± 1.9 μm (mean ± S.E., n = 5) (Fig. 2B). This suggests that the apparent affinity of DA is lower for the Li+-bound state than for the Na+-bound state, supporting that Li+ promotes a distinct conformation of the transporter, as has been suggested before for GAT-1 and SERT (31, 39, 40). Of interest, according to recent data obtained for SERT, the Li+-bound state of the transporter might represent an inward facing conformation with the inner gate open (40).
FIGURE 2.
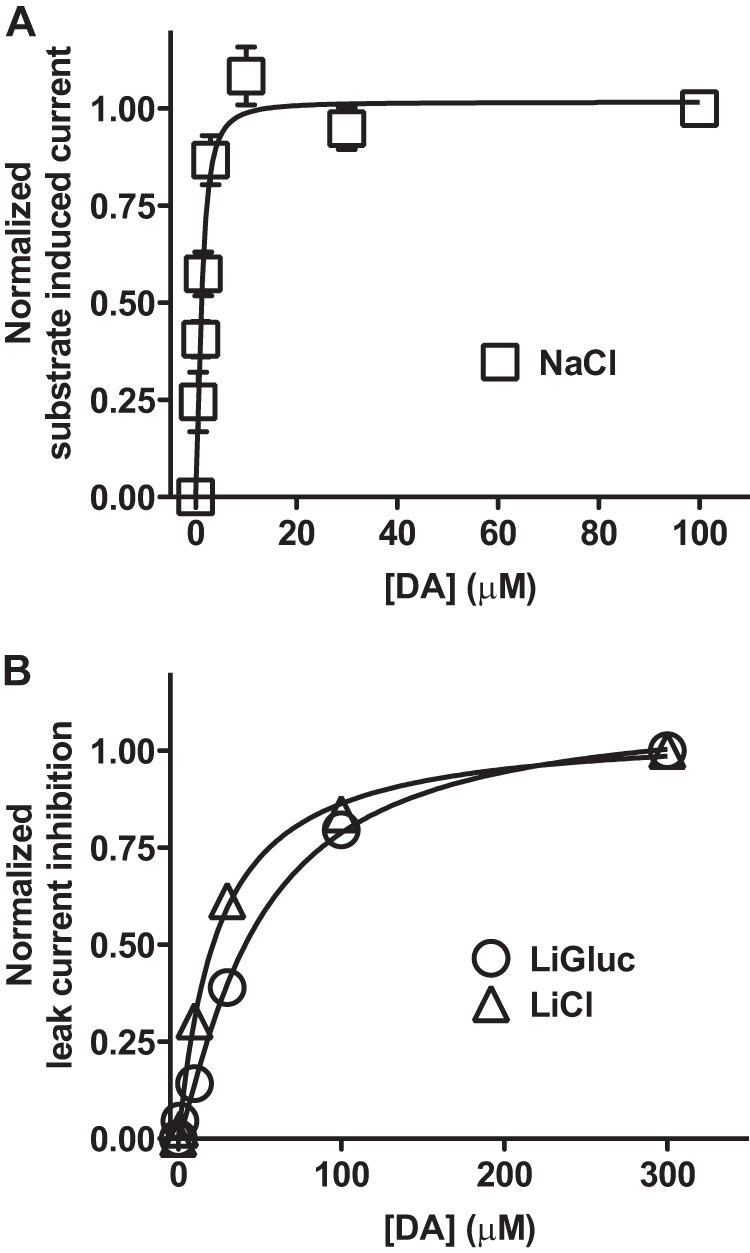
The DA affinity is dramatically changed in the Li+-bound state of the transporter. A, substrate-induced current at −100 mV plotted as a function of increasing DA concentrations and normalized to the maximum response. EC50 = 1.19 ± 0.11 μm (mean ± S.E., n = 5). B, inhibition of the leak current by DA at −100 mV plotted as a function of increasing DA concentrations after normalization to the maximum inhibition (Ligluc). IC50LiCl = 23.5 ± 1.9 μm; mean ± S.E., n = 7. IC50Ligluconate = 48 ± 5 μm; mean ± S.E., n = 3).
We also assessed the apparent DA affinity upon substitution of Cl− with gluconate and found only a rather small change (EC50 = 48 ± 5 μm (mean ± S.E., n = 5) (Fig. 2B), indicating little coupling between the Cl− and the DA binding sites in the Li+-bound state of the transporter. Notably, a tighter coupling is likely in the Na+-bound state as uptake experiments on DAT expressed in COS-7 cells suggested a >3-fold increase in Km for dopamine uptake when decreasing the Cl− concentration from 200 to 30 mm.5
Cl− Decreases the Affinity of Li+
By isotonically substituting Na+ with Li+, we determined the potency of Na+ for inhibition of the Li+ leak. Half-maximal effect (IC50) was observed at a Na+ concentration of 2.72 ± 0.09 mm (mean ± S.E., n = 5) (Fig. 3A), and thus the potency was similar to that observed for Na+ inhibition of the Li+ leak in GAT-1 (31, 32). The potency of Na+ appeared not to be affected by the presence of Cl−; that is, the potency of Na+ was unaltered when substituting Cl− with gluconate (IC50 = 3.7 ± 0.2 mm; mean ± S.E., n = 5) (Fig. 3A). Nonetheless, Cl− had a major effect on the apparent affinity for Li+ itself. This was determined by titrating Li+ against choline (which does not permeate the transporter) in Cl− or in gluconate. At a membrane potential of −100 mV, the EC50 for Li+ was 81 ± 18 mm in Cl− and was reduced to 23 ± 4 mm in gluconate (means ± S.E., n = 5) (Fig. 3B). This suggests that Cl− might increase Li+ permeation by lowering the Li+ affinity for the transporter.
FIGURE 3.
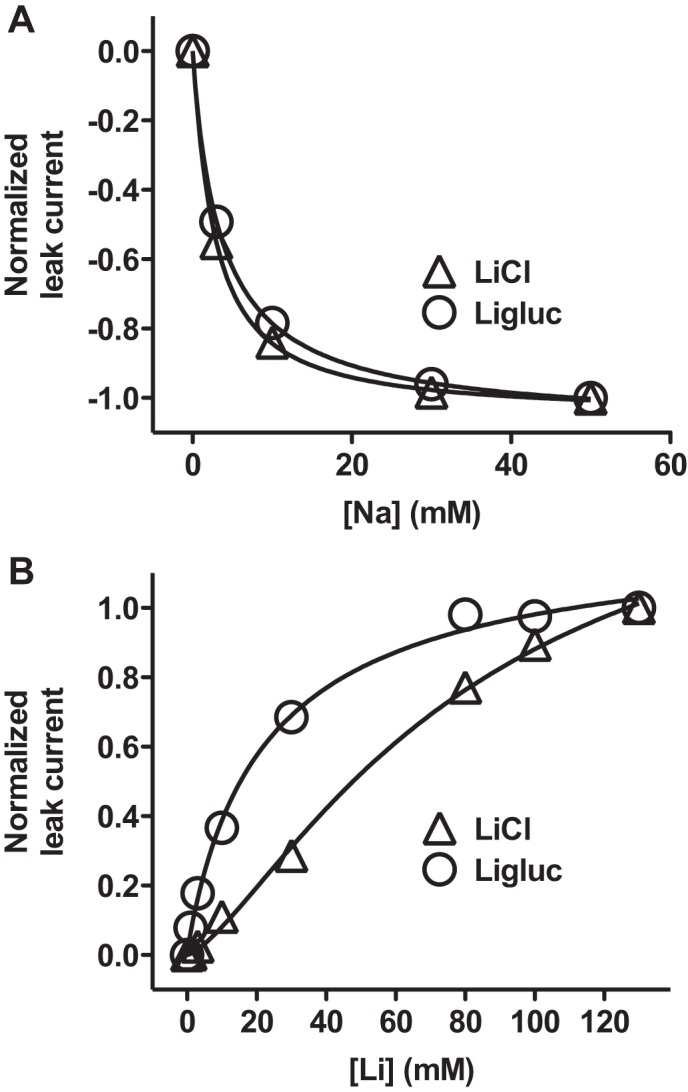
Chloride decreases the Li+ affinity but not Na+ affinity. A, inhibition of the Li+ leak by Na+. The currents were normalized to the maximum leak current observed at −100 mV and plotted against increasing Na+ concentrations. In 80 mm LiCl, 50 mm ChCl the ChCl was isotonically substituted with increasing concentrations of NaCl. IC50,LiCl = 2.72 ± 0.09 mm (mean ± S.E., n = 3). In 80 mm Ligluc, 50 mm choline gluconate, the choline gluconate was isotonically substituted with increasing concentrations of sodium gluconate. IC50,Ligluc = 3.7 ± 0.2 mm; mean ± S.E., n = 5. B, lithium dose-response curve. The currents were normalized to the maximum Li+ current and plotted against increasing Li+ concentrations. ChCl or choline gluconate was isotonically substituted with LiCl or Ligluc. The apparent Li+ affinity was 81 ± 18 mm in LiCl and 23 ± 4 mm in Ligluc, respectively (means ± S.E., n = 3) (B).
Mutation of the Na1 Site Does Not Affect the Affinity and Selectivity of the Cation Leak Current
To further evaluate the molecular basis of the leak current, we first mutated Ser-320 that according to a DAT model based on the Drosophila DAT structure (11, 15) is involved in direct coordination of Na+ binding to Na1 (Fig. 4A). Mutation of Ser-320 to Ala eliminated all currents and thus resulted in a transporter that was not amenable to further electrophysiological analysis in the oocytes (data not shown). Instead, we generated the more conservative substitution S320T, which displayed a cocaine-sensitive Li+ leak, as seen in the wild type (WT) transporter (Fig. 4B and Table 1). No cocaine-sensitive current was seen in uninjected oocytes (Fig. 4D). Strikingly, Na+ inhibited the tonic Li+ leak more potently in S320T than in the WT (Fig. 5A). In fact, the inhibition was so potent that it was not possible to obtain a reliable IC50 value (Fig. 5A). This might suggest that the S320T mutation increases the interaction of Na+ with the Na1 site. A possible explanation for this finding would be that because threonine is β-branched and less flexible it will have less entropy to loose when it binds Na+.
FIGURE 4.

Mutation of the second sodium site changes the selectivity of the leak current. A, model of DAT based on the structure of Drosophila DAT illustrating the involvement of Ser-320 in coordinating Na+ binding to Na1 and of Asp-420 in coordinating Na+ binding to Na2. Note that Ser-320 also is involved in coordination of Cl−. B, cocaine-sensitive leak currents in S320T shown as I/V plot of steady-state currents in NaCl (squares) and in LiCl (triangles) (means ± S.E. n = 5). The properties are similar to WT. C, cocaine-sensitive leak currents in D420N shown as I/V plot of steady-state currents in NaCl (squares) and in LiCl (triangles) (means ± S.E., n = 5). D, cocaine-sensitive leak currents in uninjected oocytes shown as I/V plot of steady-state currents in NaCl (squares) and in LiCl (triangles) (means ± S.E., n = 5). The mutation (D420N) preserved the leak in LiCl but dramatically increased the leak observed in NaCl. E, quantified cocaine-sensitive leak currents in WT, S320T, and D420N at a holding potential of −100 mV. Data are normalized to the current in Na+ (means ± S.E., n = 4–5).
TABLE 1.
Leak currents in NaCl or LiCl for WT and mutants
Data are unsubstracted in oocytes expressing the indicated constructs.
| Buffer |
Vhold = −100 mV (n = 5–8) |
||
|---|---|---|---|
| WT | S320T | D420N | |
| nA ± S.E | |||
| NaCl | −173 ± 31 | −114 ± 7 | −752 ± 91 |
| LiCl | −728 ± 67 | −389 ± 37 | −503 ± 76 |
FIGURE 5.
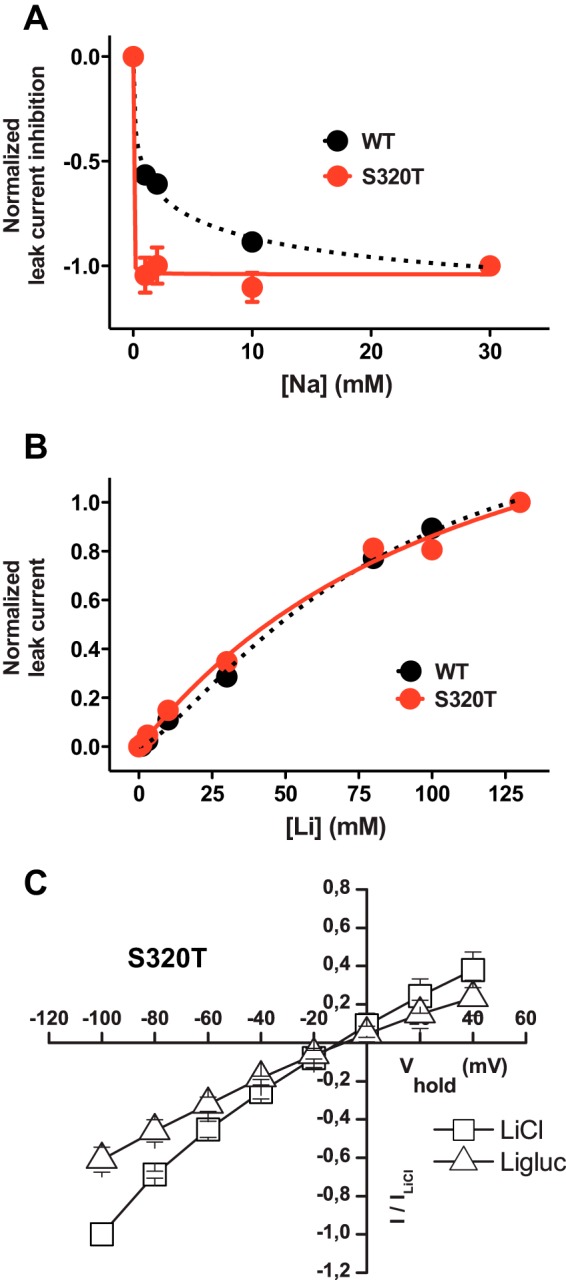
Mutation of the first sodium site changes affinity for Na+ but not Li+. A, inhibition in WT (black circles/dotted line) and S320T (red circles/solid line) of the leak current by Na+ at −100 mV plotted against increasing Na+ concentrations after normalization to the maximum inhibition (means ± S.E., n = 3–4). Mutation of the Na1 site (S320T) dramatically increased the affinity for Na+ as evaluated by the ability of Na+ to inhibit the Li+ leak with full inhibition observed at concentrations ∼1 mm. B, Li+ dose-response curves for WT (black circles/dotted line) and S320T (red circles/solid line). The currents were normalized to the maximum Li+ current and plotted against increasing Li+ concentrations. ChCl was isotonically substituted with LiCl. EC50 was 111 ± 36 mm for WT and 81 ± 13 mm for S320T (means ± S.E., n = 3–4). C, I/V plot of steady-state leak currents in S320T in LiCl and Ligluc (means ± S.E., n = 4) showing that Cl− gates the Li+ current in S320T similar to our findings for the WT. The I/V plot was generated in 20-mV steps from −100 to +40 mV.
We also determined the apparent affinity of Li+ for S320T by isotonically substituting choline with Li+. As illustrated in Fig. 5B, the apparent affinity of Li+ was largely unchanged compared with WT (EC50 = 111 ± 36 mm for WT versus 81 ± 13 mm for S320T; means ± S.E., n = 4) (Fig. 5B). This suggests that Ser-320, and thereby possibly Na1, is not critical for the DAT-Li+ interaction and thus for the tonic Li+ leak.
Of note, we were unable to measure a substrate-induced current for S320T. This could relate to the increased Na+ affinity that perhaps could slow down transporter turnover rate and thereby diminish the substrate-induced current. Because Ser-320 also is involved in coordination of Cl− (Fig. 4A), it is possible that altered Cl− binding might affect transporter function as well. However, the ability of Cl− to stimulate the Li+ leak and thus to bind to the transporter was fully preserved in S320T, i.e. the leak current observed in LiCl was larger than the leak in lithium gluconate (Ligluc), and no change was seen in the reversal potential (Fig. 5C).
Mutation of the Na2 Site Affects the Affinity and Selectivity of the Cation Leak Current
To investigate the importance of Na2, we mutated Asp-420 to asparagine (D420N). Asp-420 coordinates Na+ binding to Na2 according to our DAT model based on the Drosophila DAT structure (11, 15) (Fig. 4A). Similar to observations for S320T, a large Li+ leak current was seen in D420N (Fig. 4C and Table 1). However, whereas we hardly were able to measure a cocaine-sensitive leak in the WT and S320T, we observed in D420N a cocaine-sensitive leak current in Na+ that was larger than that the cocaine-sensitive current in Li+ but with the same reversal potential (Fig. 4, C and E, and Table 1). Furthermore, the apparent Li+ affinity was, in contrast to our findings for S320T, markedly decreased in D420N. As shown in Fig. 6A, the Li+ dose-response curve showed no signs of saturation even up to a concentration of 125 mm Li+, and thus, it was not even possible to estimate the affinity. Together, these results suggest a critical role of Asp-420 and thereby likely Na2 in mediating the Li+ leak and possibly in controlling cation permeation of the transporter.
FIGURE 6.
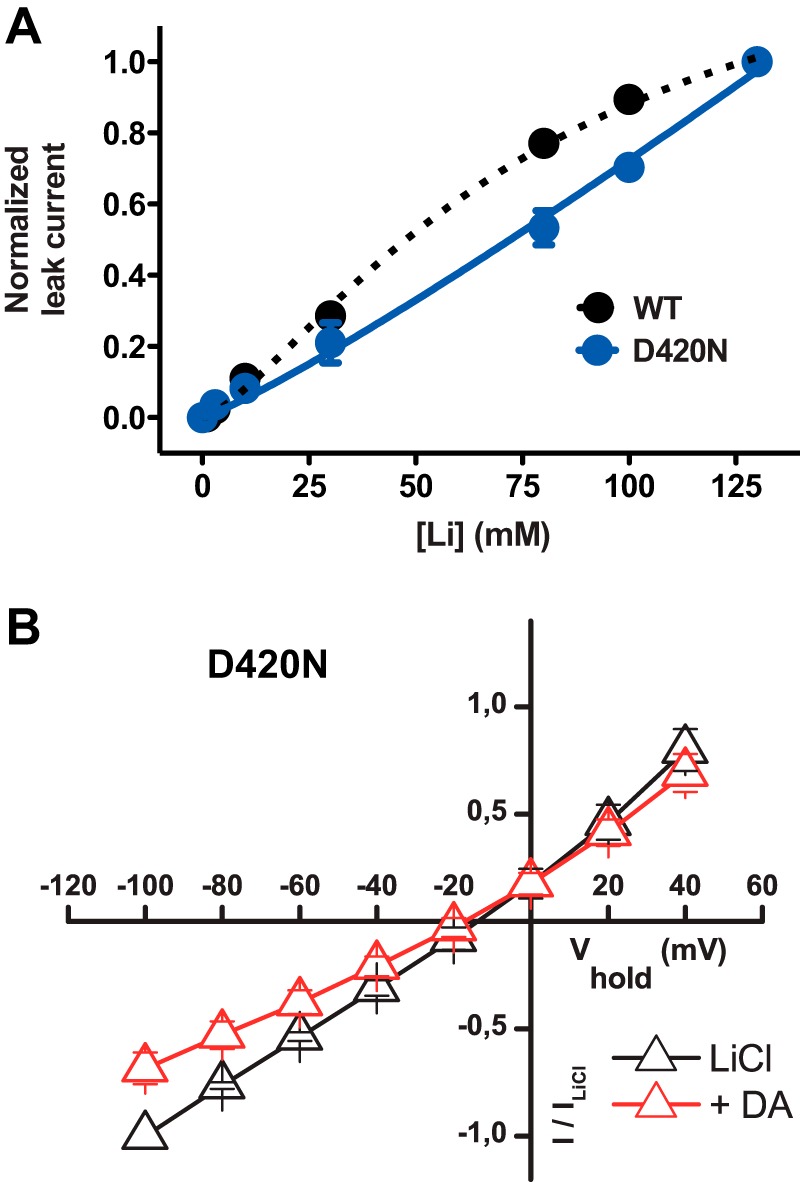
Mutation of the Na2 site lowers apparent Li+ affinity but preserves DA binding. A, lithium dose-response curve for D420N (blue circles/solid line) compared with WT (black circles/dotted line). The currents were normalized to the maximum Li+ current and plotted against increasing Li+ concentrations. ChCl was isotonically substituted with LiCl. An EC50 value could not be determined for D420N because no saturation was observed. B, I/V plot of steady-state leak currents in D420N in LiCl with and without perfusion of DA (100 μm) (means ± S.E., n = 5), showing that DA can bind to D420N.
To further characterize the properties of D420N, we assessed the effect of DA perfusion on the leak current in Na+. This caused an inhibition of the current similar to what we observed for the WT (Fig. 6B), substantiating that DA can bind to the mutant transporter.
Finally, we performed a series of ion substitution experiments comparing WT and D420N (Fig. 7, A–D). Changing from NaCl to choline chloride (ChCl) caused a dramatic left shift in the reversal potential of ∼40 mV, consistent with the leak in D420N being carried at least in part by Na+ ions. The D420N leak current in Na+ is most likely not carried by anions because we observed no change in reversal potential by changing NaCl to sodium gluconate. Nonetheless, the D420N leak is influenced by the anion present: substitution of Cl− with the more permeable anion thiocyanate (SCN−) dramatically increased the magnitude of the current without changing the reversal potential (Fig. 7, D–F).
FIGURE 7.

Mutation of the Na2 site changes cation selectivity of the leak. A and C, I/V plot of steady-state cocaine-sensitive leak currents in WT (A) and D420N (C) assessed in NaCl (open black squares), LiCl (open blue triangles), and ChCl (open green triangles) (means ± S.E., n = 3–4). A large leak current was seen in NaCl for D420N but not for the WT. When Na+ was removed from the extracellular side by changing from NaCl to ChCl, a change in reversal potential was observed for D420N in agreement with Na+ being the principal permeating ion. B and D, I/V plot of steady-state cocaine-sensitive leak currents in WT (B) and D420N (D) assessed in NaCl (open black squares), sodium gluconate (Nagluc; open red circles) and NaSCN (open brown diamonds) (means ± S.E., n = 3–4). E and F, trace recordings of WT and D420N clamped at −60 mV, illustrating the effect of substituting with thiocyanate (SCN). Buffer changes were made as indicated. The figure shows representative traces.
DISCUSSION
NSS proteins constitute a major family of Na+-coupled transport proteins conserved from prokaryotic organisms to man (1). Application of x-ray crystallography to the prokaryotic NSS protein LeuT and more recently to the Drosophila DAT has provided insight into the structure of these proteins at atomic resolution (11, 18). The structures have aided our understanding of the transport mechanism and revealed the structural context of ion binding to the transporters. Nevertheless, we continue to lack a mechanistic understanding of the coupling between Na+, Cl−, and substrate in the translocation cycle. Due to its slow turnover rates, LeuT has not been used much in conventional functional assays to study ion binding dynamics of NSS proteins (12). Rather, studies of ion binding dynamics in LeuT have been mostly computational (41–44). In contrast, DAT has been intensively studied in radioactive uptake assays and with electrophysiological measurements both in heterologous and in native systems (1–3). These studies have led to the identification of several ion-conducting states of the transporter and have predicted the ion coupling stoichiometry (9, 20–23). Studies have also been carried out on homologous mammalian transporters (for review, see Refs. 1–3). In SERT, for example, it was recently shown how mutation of an asparagine in TM1 (Asn-101) rendered Ca2+ capable of replacing Na+ at Na1. The data indicate that Na+ bound to Na1 is not co-transported and that Na+ bound to Na2 is co-transported, and thus that Na2 is critical for determining cation permeation in SERT. The study accordingly provided a mechanism by which SERT, in contrast to e.g. DAT, co-transports only one Na+ despite the presence of two Na+ binding sites (45). In DAT, however, the individual roles of the distinct ion binding sites have not been discerned.
Here we have applied electrophysiological analyses in X. laevis oocytes combined with targeted mutagenesis to investigate the ion binding dynamics in DAT. The findings substantiate that DAT, like GAT-1 and SERT (20, 26, 29, 31, 34), possesses a large inwardly rectifying leak current in Li+. The current is ∼10-fold larger than the substrate-induced current and is thus by far the largest DAT-mediated current described. A leak in Li+ has been described before for DAT, but because the previous measurements were done with 2 mm Na+ present in the solution (20), the actual magnitude of the current was not revealed due to the inhibitory action of Na+ demonstrated here (Fig. 1 and below).
Similar to previous observations for the Li+ leak in GAT-1 (31, 32), the DAT Li+ leak was inhibited by low concentrations of Na+. This inhibition is likely the result of high affinity Na+ binding to Na1, as suggested by the findings for the S320T mutant showing increased potency of Na+ in inhibiting the Li+ leak. This would be consistent with an increased affinity of Na+ for Na1 that may result from the less flexible nature of the β-branched threonine as compared with serine. The result supports that high affinity Na+ binding to Na1 restores the conformational equilibrium of the transporter and thereby impairs Li+ permeation. Our data also suggest that the Na1 coordinating residue Ser-320 is unlikely to be important for mediating the Li+ leak as the S320T mutation changed neither the Li+ current nor the apparent affinity of Li+ for the transporter.
According to the structure of dDAT, Ser-320 is also involved in the Cl− binding site, and thus the phenotype resulting from mutating this residue could in part be a consequence of altered Cl− binding. However, the enhanced potency of Na+ for inhibition of the Li+ leak in S320T is less likely the result of altered Cl− coordination, and Cl− is indeed able to interact with the mutant transporter as removal of Cl− caused a marked reduction in the Li+ leak of S320T (Fig. 5C). Interestingly, it was shown by Henry et al. (46) that mutation of the corresponding residue in SERT (Ser-336) resulted in transport activity largely independent on Cl−, indicating a change in Cl− interaction. In DAT, however, we were unable to measure reliable uptake when mutating Ser-320 to Thr or Ala in DAT (data not shown). This strong functional impairment did not only prevent us from assessing Cl− dependence in the mutants but also suggests that the residue might serve different roles in the two transporters. Of further interest, the study by Henry et al. (46) also suggested a tight link between Cl− and Na1 in SERT, i.e. their data indicated that Asn-101, which is involved in Na1 coordination but does not coordinate to the bound Cl−, was critical for coupling Cl− binding to concentrative serotonin uptake. Future studies should assess whether the corresponding residue in DAT serves a similar function.
In contrast to our findings for S320T, we observed for the D420N mutation in Na2 a major shift in the apparent Li+ affinity, as reflected in an unsaturable Li+ dose-response curve. This suggests that the Li+ leak depends on the interaction of Li+ with Na2. Notably, Li+ interaction with Na2 has also been suggested to be critical for the Li+ leak observed in GAT-1 (34). A remarkable additional phenotype of the D420N mutation was a large cocaine-sensitive leak conductance in Na+ that was even larger than the current observed in Li+. This is in stark contrast to the WT DAT, which in Na+ displays a barely detectable cocaine-sensitive cation leak (20). The current observed in Na+ for D420N is at least in part carried by Na+ because substituting Na+ with choline caused a dramatic leftward shift in the reversal potential (Fig. 7C). We observed no change in the reversal potential upon anion substitution, arguing that the current is not carried by anions. It is nonetheless important to note that the reversal potential observed in Na+ was markedly more negative (−10 mV) than the Nernst potential for Na+ alone determined from the Goldman-Hodgkin-Katz equation (+61 mV) (47), indicating that the net current is not carried solely by Na+. Hence, although the majority of the current probably is carried by Na+, the current is also partially carried by other cations such as potassium (as suggested by left-shift toward the reversal potential for K+ in response to substitution of Na+ with choline). Of further interest, our findings for D420N might have pathophysiological perspectives. In a patient diagnosed with early-onset parkinsonism and attention deficit hyperactivity disorder we recently identified two DAT missense mutations (48). One of the mutants (D421N) was equivalent to the Na2 mutation we have studied here in mouse DAT (D420N). Indeed, an initial electrophysiological assessment of D421N in human DAT revealed a phenotype similar to that for mouse D420N showing that the phenotype of the mutant is conserved across different species.
The Li+ leak current in WT DAT was not merely regulated by DA and Na+ but also by Cl−. A regulation of the Li+ leak by Cl− has also been suggested for GAT-1 (28). It is important to point out that Cl− is likely to serve several roles at different steps in the transport cycle. First, it has been proposed that Cl− plays a role in counterbalancing the charge of the co-transported cations (DA and Na+) during binding and translocation (16). During binding this might involve electrostatic facilitation of binding of the positively charged substrate to the substrate binding site, predicted to lie in close proximity to the Cl− site. The putative importance of the charge counterbalancing effect of Cl− has been supported by data suggesting that Cl−-independent prokaryotic NSS proteins, such as LeuT, Tyt1, and TnaT, contain a negatively charged aspartate/glutamate corresponding to the predicted Cl− site and that deprotonation/protonation of this residue might serve a similar role as Cl− in the transport cycle (11, 16, 18, 49). Secondly, it has been suggested that the transport process is dependent not only on extracellular Cl− but also on intracellular Cl− based on whole-cell patch clamp and use of a rapid solution exchange protocol in DAT-expressing HEK293 cells where intracellular Cl− application was found to positively regulate DAT turn-over rate (19). It was accordingly proposed that that Cl− occupancy from the inside played a yet unappreciated role in the transporter return step.
Finally, the present data suggest a coupling between the bound Cl− and the cation bound in Na2. This conclusion is based on our observations for D420N that link the Li+ current to Na2 and on the finding that removal of Cl− caused a prominent reduction in the Li+ current while at the same time causing a surprising decrease in the apparent Li+ affinity. Because Cl− does not permeate in the presence of Li+ according to the electrophysiological analyses, we suggest that binding of Cl− weakens the interaction of Li+ with the transporter by allosterically lowering the Li+ affinity for Na2 and that this leads to enhanced permeation. Our results suggest accordingly an interesting link between Cl− and the cation bound in Na2 in the Li+ leak mode of the transporter.
The link between Cl− and Na2 becomes particularly interesting in light of recent results obtained in the homologous SERT, providing strong evidence that Li+ promotes a state equivalent to an open inner gate configuration (40). A preferred inward-facing configuration of the transporter would also fit with our finding that substrate (DA) can bind to the transporter in Li+ but with lowered affinity; hence, we have evidence from studies on the homologous LeuT that the inward-facing configuration of this transporter displays lowered substrate affinity.6 In further agreement with a switch to the inward facing configuration in Li+, previous application of noise analysis on SERT currents reported that changing from Na+ to Li+ increased the frequency of channels openings (50), which could reflect an increased inner gate opening probability in the presence of Li+. We speculate moreover that when pushing the conformational equilibrium toward an inward-facing configuration with increased inner gate opening probability, lithium increases the likelihood that both gates are simultaneously open, thereby permitting lithium permeation in putative “channel”-like mode of the transporter. Importantly, molecular dynamics studies have further strengthened the link between inner gate opening and the Na2 site by showing how unbinding of Na+ from the Na2 site leads to destabilization of the thin inner gate (41, 44). According to our data, Cl− would be expected in this scenario to increase Li+ permeation by lowering its affinity for Na2 and thereby possible increase inner gate opening probability. Moreover, the large cation leak seen for D420N in Na+ suggests that the notion may apply to Na+ as well. The lower cation affinity of Na2 might weaken the interaction of Na+ with Na2, thereby leading to increased inner gate opening probability and augmentation of spontaneous Na+ permeation. In line with this, it could be argued that D420N promotes a conformational equilibrium in Na+ that mimics the equilibrium seen in Li+ for the WT. The potential change in conformational equilibrium induced by the D420N mutation might further explain the lack of inhibition of the leak by sodium binding to the Na1 site as D420N might promote a change in the conformational equilibrium that leads to a structural perturbation preventing sodium from inhibiting the leak by interacting with Na1. It is also interesting to note the dramatic increase in the cation leak of D420N when substituting Cl− with the thiocyanate (SCN−), adding further support to a functional link between the anion binding site and Na2.
Summarized, the present study assigns for the first time discrete functional properties to the two presumed Na+ binding sites in DAT. Specifically, we unravel a conceivable key role of Na2 in controlling cation permeation of the transporter and inner gate opening. Moreover, we provide evidence for regulation of the Na2 site by Cl−, and our results thereby add further important insight into the mechanistic role of Cl− in the transport cycle of NSS proteins.
This work was supported, in whole or in part, by National Institutes of Health Grant P01 DA 12408 (to U. G. and H. W.). This work was also supported by the Danish Medical Research Council (to U. G.), University of Copenhagen BioScaRT Program of Excellence (to U. G.), Lundbeck Foundation Center for Biomembranes in Nanomedicine (to U. G.), the Novo Nordisk Foundation (to U. G.), Fabrikant Vilhelm Pedersen og Hustrus Mindelegat (to U. G.), and The Lundbeck Foundation (to L. B.).
L. Borre, T. F. Andreassen, and U. Gether, unpublished observation.
K. Gotfryd, C. J. Loland, and U. Gether, unpublished observation.
- DAT
- dopamine (DA) transporter
- dDAT
- Drosophila DAT
- SERT
- serotonin transporter
- NSS
- neurotransmitter:sodium symporter
- GAT-1
- γ-aminobutyric acid transporter-1
- Ligluc
- lithium gluconate
- ChCl
- choline chloride
- NSS
- neurotransmitter:sodium symporter.
REFERENCES
- 1. Kristensen A. S., Andersen J., Jørgensen T. N., Sørensen L., Eriksen J., Loland C. J., Strømgaard K., Gether U. (2011) SLC6 neurotransmitter transporters: structure, function, and regulation. Pharmacol. Rev. 63, 585–640 [DOI] [PubMed] [Google Scholar]
- 2. Chen N., Reith M. E. (2000) Structure and function of the dopamine transporter. Eur J. Pharmacol. 405, 329–339 [DOI] [PubMed] [Google Scholar]
- 3. Torres G. E., Gainetdinov R. R., Caron M. G. (2003) Plasma membrane monoamine transporters: structure, regulation, and function. Nat. Rev. Neurosci. 4, 13–25 [DOI] [PubMed] [Google Scholar]
- 4. Kurian M. A., Gissen P., Smith M., Heales S., Jr., Clayton P. T. (2011) The monoamine neurotransmitter disorders: an expanding range of neurological syndromes. Lancet Neurol. 10, 721–733 [DOI] [PubMed] [Google Scholar]
- 5. Schmitt K. C., Rothman R. B., Reith M. E. (2013) Nonclassical pharmacology of the dopamine transporter: atypical inhibitors, allosteric modulators, and partial substrates. J. Pharmacol. Exp. Ther. 346, 2–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Beuming T., Kniazeff J., Bergmann M. L., Shi L., Gracia L., Raniszewska K., Newman A. H., Javitch J. A., Weinstein H., Gether U., Loland C. J. (2008) The binding sites for cocaine and dopamine in the dopamine transporter overlap. Nat. Neurosci. 11, 780–789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sulzer D. (2011) How addictive drugs disrupt presynaptic dopamine neurotransmission. Neuron 69, 628–649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wall S. C., Gu H., Rudnick G. (1995) Biogenic amine flux mediated by cloned transporters stably expressed in cultured cell lines: amphetamine specificity for inhibition and efflux. Mol. Pharmacol. 47, 544–550 [PubMed] [Google Scholar]
- 9. Gu H., Wall S. C., Rudnick G. (1994) Stable expression of biogenic amine transporters reveals differences in inhibitor sensitivity, kinetics, and ion dependence. J. Biol. Chem. 269, 7124–7130 [PubMed] [Google Scholar]
- 10. Krueger B. K. (1990) Kinetics and block of dopamine uptake in synaptosomes from rat caudate nucleus. J. Neurochem. 55, 260–267 [DOI] [PubMed] [Google Scholar]
- 11. Yamashita A., Singh S. K., Kawate T., Jin Y., Gouaux E. (2005) Crystal structure of a bacterial homologue of Na+/Cl−-dependent neurotransmitter transporters. Nature 437, 215–223 [DOI] [PubMed] [Google Scholar]
- 12. Singh S. K., Piscitelli C. L., Yamashita A., Gouaux E. (2008) A competitive inhibitor traps LeuT in an open-to-out conformation. Science 322, 1655–1661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zhou Z., Zhen J., Karpowich N. K., Goetz R. M., Law C. J., Reith M. E., Wang D. N. (2007) LeuT-desipramine structure reveals how antidepressants block neurotransmitter reuptake. Science 317, 1390–1393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Krishnamurthy H., Gouaux E. (2012) X-ray structures of LeuT in substrate-free outward-open and apo inward-open states. Nature 481, 469–474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Penmatsa A., Wang K. H., Gouaux E. (2013) X-ray structure of dopamine transporter elucidates antidepressant mechanism. Nature 503, 85–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zomot E., Bendahan A., Quick M., Zhao Y., Javitch J. A., Kanner B. I. (2007) Mechanism of chloride interaction with neurotransmitter:sodium symporters. Nature 449, 726–730 [DOI] [PubMed] [Google Scholar]
- 17. Forrest L. R., Tavoulari S., Zhang Y. W., Rudnick G., Honig B. (2007) Identification of a chloride ion binding site in Na+/Cl−-dependent transporters. Proc. Natl. Acad. Sci. U.S.A. 104, 12761–12766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kantcheva A. K., Quick M., Shi L., Winther A. M., Stolzenberg S., Weinstein H., Javitch J. A., Nissen P. (2013) Chloride binding site of neurotransmitter sodium symporters. Proc. Natl. Acad. Sci. U.S.A. 110, 8489–8494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Erreger K., Grewer C., Javitch J. A., Galli A. (2008) Currents in response to rapid concentration jumps of amphetamine uncover novel aspects of human dopamine transporter function. J. Neurosci. 28, 976–989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sonders M. S., Zhu S. J., Zahniser N. R., Kavanaugh M. P., Amara S. G. (1997) Multiple ionic conductances of the human dopamine transporter: the actions of dopamine and psychostimulants. J. Neurosci. 17, 960–974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ingram S. L., Prasad B. M., Amara S. G. (2002) Dopamine transporter-mediated conductances increase excitability of midbrain dopamine neurons. Nat. Neurosci. 5, 971–978 [DOI] [PubMed] [Google Scholar]
- 22. Meinild A. K., Sitte H. H., Gether U. (2004) Zinc potentiates an uncoupled anion conductance associated with the dopamine transporter. J. Biol. Chem. 279, 49671–49679 [DOI] [PubMed] [Google Scholar]
- 23. Sitte H. H., Huck S., Reither H., Boehm S., Singer E. A., Pifl C. (1998) Carrier-mediated release, transport rates, and charge transfer induced by amphetamine, tyramine, and dopamine in mammalian cells transfected with the human dopamine transporter. J. Neurochem. 71, 1289–1297 [DOI] [PubMed] [Google Scholar]
- 24. Carvelli L., McDonald P. W., Blakely R. D., Defelice L. J. (2004) Dopamine transporters depolarize neurons by a channel mechanism. Proc. Natl. Acad. Sci. U.S.A. 101, 16046–16051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Pörzgen P., Park S. K., Hirsh J., Sonders M. S., Amara S. G. (2001) The antidepressant-sensitive dopamine transporter in Drosophila melanogaster: a primordial carrier for catecholamines. Mol. Pharmacol. 59, 83–95 [DOI] [PubMed] [Google Scholar]
- 26. Mager S., Min C., Henry D. J., Chavkin C., Hoffman B. J., Davidson N., Lester H. A. (1994) Conducting states of a mammalian serotonin transporter. Neuron 12, 845–859 [DOI] [PubMed] [Google Scholar]
- 27. Galli A., Petersen C. I., deBlaquiere M., Blakely R. D., DeFelice L. J. (1997) Drosophila serotonin transporters have voltage-dependent uptake coupled to a serotonin-gated ion channel. J. Neurosci. 17, 3401–3411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Mager S., Kleinberger-Doron N., Keshet G. I., Davidson N., Kanner B. I., Lester H. A. (1996) Ion binding and permeation at the GABA transporter GAT1. J. Neurosci. 16, 5405–5414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Mager S., Naeve J., Quick M., Labarca C., Davidson N., Lester H. A. (1993) Steady states, charge movements, and rates for a cloned GABA transporter expressed in Xenopus oocytes. Neuron 10, 177–188 [DOI] [PubMed] [Google Scholar]
- 30. Galli A., Blakely R. D., DeFelice L. J. (1996) Norepinephrine transporters have channel modes of conduction. Proc. Natl. Acad. Sci. U.S.A. 93, 8671–8676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. MacAulay N., Zeuthen T., Gether U. (2002) Conformational basis for the Li+-induced leak current in the rat γ-aminobutyric acid (GABA) transporter-1. J. Physiol. 544, 447–458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kanner B. I. (2003) Transmembrane domain I of the γ-aminobutyric acid transporter GAT-1 plays a crucial role in the transition between cation leak and transport modes. J. Biol. Chem. 278, 3705–3712 [DOI] [PubMed] [Google Scholar]
- 33. Meinild A. K., Forster I. C. (2012) Using lithium to probe sequential cation interactions with GAT1. Am. J. Physiol. Cell Physiol 302, C1661–C1675 [DOI] [PubMed] [Google Scholar]
- 34. Zhou Y., Zomot E., Kanner B. I. (2006) Identification of a lithium interaction site in the γ-aminobutyric acid (GABA) transporter GAT-1. J. Biol. Chem. 281, 22092–22099 [DOI] [PubMed] [Google Scholar]
- 35. Günther W., Lüchow A., Cluzeaud F., Vandewalle A., Jentsch T. J. (1998) ClC-5, the chloride channel mutated in Dent's disease, colocalizes with the proton pump in endocytotically active kidney cells. Proc. Natl. Acad. Sci. U.S.A. 95, 8075–8080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sali A., Blundell T. L. (1993) Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 234, 779–815 [DOI] [PubMed] [Google Scholar]
- 37. Fiser A., Do R. K., Sali A. (2000) Modeling of loops in protein structures. Protein Sci. 9, 1753–1773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Giros B., el Mestikawy S., Godinot N., Zheng K., Han H., Yang-Feng T., Caron M. G. (1992) Cloning, pharmacological characterization, and chromosome assignment of the human dopamine transporter. Mol. Pharmacol. 42, 383–390 [PubMed] [Google Scholar]
- 39. Ni Y. G., Chen J. G., Androutsellis-Theotokis A., Huang C. J., Moczydlowski E., Rudnick G. (2001) A lithium-induced conformational change in serotonin transporter alters cocaine binding, ion conductance, and reactivity of Cys-109. J. Biol. Chem. 276, 30942–30947 [DOI] [PubMed] [Google Scholar]
- 40. Schicker K., Uzelac Z., Gesmonde J., Bulling S., Stockner T., Freissmuth M., Boehm S., Rudnick G., Sitte H. H., Sandtner W. (2012) Unifying concept of serotonin transporter-associated currents. J. Biol. Chem. 287, 438–445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Zhao C., Noskov S. Y. (2011) The role of local hydration and hydrogen-bonding dynamics in ion and solute release from ion-coupled secondary transporters. Biochemistry 50, 1848–1856 [DOI] [PubMed] [Google Scholar]
- 42. Caplan D. A., Subbotina J. O., Noskov S. Y. (2008) Molecular mechanism of ion-ion and ion-substrate coupling in the Na+-dependent leucine transporter LeuT. Biophys. J. 95, 4613–4621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Noskov S. Y., Roux B. (2008) Control of ion selectivity in LeuT: two Na+ binding sites with two different mechanisms. J. Mol. Biol. 377, 804–818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Shi L., Quick M., Zhao Y., Weinstein H., Javitch J. A. (2008) The mechanism of a neurotransmitter:sodium symporter-inward release of Na+ and substrate is triggered by substrate in a second binding site. Mol. Cell 30, 667–677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Felts B., Pramod A. B., Sandtner W., Burbach N., Bulling S., Sitte H. H., Henry L. K. (2014) The two Na+ sites in the human serotonin transporter play distinct roles in the ion coupling and electrogenicity of transport. J. Biol. Chem. 289, 1825–1840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Henry L. K., Iwamoto H., Field J. R., Kaufmann K., Dawson E. S., Jacobs M. T., Adams C., Felts B., Zdravkovic I., Armstrong V., Combs S., Solis E., Rudnick G., Noskov S. Y., DeFelice L. J., Meiler J., Blakely R. D. (2011) A conserved asparagine residue in transmembrane segment 1 (TM1) of serotonin transporter dictates chloride-coupled neurotransmitter transport. J. Biol. Chem. 286, 30823–30836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Costa P. F., Emilio M. G., Fernandes P. L., Ferreira H. G., Ferreira K. G. (1989) Determination of ionic permeability coefficients of the plasma membrane of Xenopus laevis oocytes under voltage clamp. J. Physiol. 413, 199–211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Hansen F. H., Skjørringe T., Yasmeen S., Arends N. V., Sahai M. A., Erreger K., Andreassen T. F., Holy M., Hamilton P. J., Neergheen V., Karlsborg M., Newman A. H., Pope S., Heales S. J., Friberg L., Law I., Pinborg L. H., Sitte H. H., Loland C., Shi L., Weinstein H., Galli A., Hjermind L. E., Møller L. B, Gether U. (2014) Missense mutations in the dopamine transporter gene associate with adult parkinsonism and ADHD. J. Clin. Invest. 124, 3107–3120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Zhao Y., Quick M., Shi L., Mehler E. L., Weinstein H., Javitch J. A. (2010) Substrate-dependent proton antiport in neurotransmitter:sodium symporters. Nat. Chem. Biol. 6, 109–116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Lin F., Lester H. A., Mager S. (1996) Single-channel currents produced by the serotonin transporter and analysis of a mutation affecting ion permeation. Biophys. J. 71, 3126–3135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Mazei-Robison M. S., Bowton E., Holy M., Schmudermaier M., Freissmuth M., Sitte H. H., Galli A., Blakely R. D. (2008) Anomalous dopamine release associated with a human dopamine transporter coding variant. J. Neurosci. 28, 7040–7046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Hamilton P. J., Campbell N. G., Sharma S., Erreger K., Herborg Hansen F., Saunders C., Belovich A. N., Consortium N. A. A. S., Sahai M. A., Cook E. H., Gether U., McHaourab H. S., Matthies H. J., Sutcliffe J. S., Galli A. (2013) De novo mutation in the dopamine transporter gene associates dopamine dysfunction with autism spectrum disorder. Mol. Psychiatry 18, 1315–1323 [DOI] [PMC free article] [PubMed] [Google Scholar]