

Background: Cytoplasmic domains of Kv11.1 channels fine-tune their gating kinetics through unknown mechanisms.
Results: N-terminal positively charged residues form functional interactions with C-terminal negatively charged residues in Kv11.1 channels.
Conclusion: Cytoplasmic charge-charge interactions are critical for slow deactivation kinetics of Kv11.1 channels.
Significance: These results help clarify the molecular basis of slow Kv11.1 channel deactivation that is critical to opposing premature beats.
Keywords: Gating, Heart, hERG, Ion Channel, Mutagenesis, Potassium Channel, Protein Chimera, Protein Domain, Protein-Protein Interaction, Site-directed Mutagenesis
Abstract
The intracellular domains of many ion channels are important for fine-tuning their gating kinetics. In Kv11.1 channels, the slow kinetics of channel deactivation, which are critical for their function in the heart, are largely regulated by the N-terminal N-Cap and Per-Arnt-Sim (PAS) domains, as well as the C-terminal cyclic nucleotide-binding homology (cNBH) domain. Here, we use mutant cycle analysis to probe for functional interactions between the N-Cap/PAS domains and the cNBH domain. We identified a specific and stable charge-charge interaction between Arg56 of the PAS domain and Asp803 of the cNBH domain, as well an additional interaction between the cNBH domain and the N-Cap, both of which are critical for maintaining slow deactivation kinetics. Furthermore, we found that positively charged arginine residues within the disordered region of the N-Cap interact with negatively charged residues of the C-linker domain. Although this interaction is likely more transient than the PAS-cNBD interaction, it is strong enough to stabilize the open conformation of the channel and thus slow deactivation. These findings provide novel insights into the slow deactivation mechanism of Kv11.1 channels.
Introduction
The KCNH gene family encodes the Kv10.x (also known as the ether-a-go-go gene, EAG), Kv11.x (the ether-a-go-go related gene, ERG), and Kv12.x (the ether-a-go-go like gene, ELK) voltage-gated K+ channels (1). Members of the KCNH family share relatively close sequence homology but differ in the voltage dependence and kinetics of channel gating. The resulting diversity in current phenotype allows KCNH family members to perform a diverse range of physiological roles in the heart, during neuronal action potential firing (2), during phasic contraction in a range of smooth muscles (3, 4), in hormone secretion (5–7), and in cell proliferation (8).
The Kv11.1 K+ channel is the only member of the KCNH family to be expressed in the heart, where it forms the rapid component of the delayed rectifier K+ current (IKr), the major repolarizing current in ventricular myocytes and thus helps maintain the QT interval, a measure of ventricular action potential repolarization on an ECG, within the normal range (9, 10). Kv11.1 channels exhibit much slower kinetics of channel closure (also termed deactivation) than other members of the KCNH family, which is key to their role in ventricular action potential repolarization and in the prevention of premature beats. Dysfunction of Kv11.1 channels is implicated in the heart disease known as long QT syndrome type 2 (LQT2 syndrome), which is associated with an increased risk of cardiac arrhythmias and sudden cardiac arrest (11).
Kv11.1 channels share a similar protein structure to other voltage gated K+ channels. Within each subunit of the tetrameric channel, the first four transmembrane segments (S1–S4) form the voltage sensing domains, whereas the fifth and sixth transmembrane segments (S5 and S6) from each subunit come together to form the central pore domain. KCNH family members also have large N and C termini that can have profound effects on channel function. For example, the N-Cap/Per-Arnt-Sim (PAS)2 domain on the N termini plays a critical role in slowing deactivation gating in Kv11.1 channels (12–16), and this is likely mediated by an interaction with the cyclic nucleotide binding homology (cNBH) domain in the C terminus (17). A crystal structure complex of the PAS and cNBH domains from a closely related murine Kv10.1 channel provides the first atomic detail that these domains form a direct interaction (18). Interestingly the atomic structures of the PAS domain from the Kv10.1 and Kv11.1 channels are almost identical (19). However, the connection of the N-Cap domain to the PAS domain of these two channels appears to be different (18). Furthermore, in contrast to Kv11.1 channels, Kv10.1 channels do not exhibit slow deactivation kinetics (20, 21), suggesting that the functional interaction between N-Cap/PAS and cNBH domains may be different in Kv11.1 compared with other members of the KCNH family.
In this study, we have used a mutant cycle analysis approach to investigate the detailed interactions between the N-Cap/PAS domain and the cNBH domain that regulate slow deactivation gating in Kv11.1 channels. Our results show that the N-Cap/PAS domains of Kv10.1 and Kv11.1 channels are not interchangeable. Furthermore, we have identified specific interactions between the N-Cap/PAS domain and the cNBH domains, as well as between the N-Cap domain with the C-linker, that provide novel insights into the slow deactivation mechanism of Kv11.1 channels.
EXPERIMENTAL PROCEDURES
Molecular Biology
The cDNA of Kv11.1 (a gift from G. Robertson, University of Wisconsin, Madison, WI) was subcloned into a pBluescript vector containing the 5′-UTR and 3′-UTR of the Xenopus laevis β-globin gene (a gift from R. Vandenberg, University of Sydney). Mutagenesis of Kv11.1 cDNA was performed using the QuikChange method (Agilent Technologies, Santa Clara, CA) and confirmed by DNA sequencing.
To generate chimeric Kv11.1 channels with the N-Cap/PAS domains replaced with those of Kv10.1 channels (residues 1–135 of Kv10.1 + residues 136–1159 of Kv11.1), a mega-primer was generated using Kv10.1 cDNA as a template, with a forward primer containing a Bpu10I restriction site followed by residues 1–9 of Kv10.1 (5′-CCGCTCAGGATGACCATGGCTGGGGGCAGGAGGGGA-3′) and a reverse primer containing the overlap region at the end of the PAS domain (SDITAFK (Kv10.1)-DMVGSPA (Kv11.1)) (5′-AGCCGGGGACCCCACCATGTCTTTGAAAGCTGTTATGTCACT-3′). This mega forward primer was then used with a reverse primer that started from the BstEII restriction site of Kv11.1, and with Kv11.1 cDNA as the template, to generate a fragment containing a Bpu10I restriction site followed by Kv10.1 (residues 1–135) + Kv11.1 up to the BstEII restriction site. This fragment was then inserted into the Kv11.1 backbone using Bpu10I and BstEII restriction enzymes (New England Biolabs, Ipswich, MA). Similarly, to generate the Kv11.1 channel chimera with only the PAS domain of Kv10.1 (residues 1–25 of Kv11.1 + 26–135 of Kv101.1 + 136–1159 of Kv11.1), the above N-Cap/PAS Kv10.1/Kv11.1 chimera cDNA was used as the template, with a forward primer containing a Bpu10I restriction site followed by Kv11.1 sequence to the overlap region at the end of the N-Cap domain (MPV…EGQ(Kv11.1)-DTNFVL(Kv10.1)) (5′-CCGCTCAGGATGCCGGTGCGGAGGGGCCACGTCGCGCCGCAGAACACCTTCCTGGACACCATCATCCGCAAGTTTGAGGGCCAGGATACTAATTTTGTGTTG-3′) and a reverse primer that started from the BstEII restriction site of Kv11.1, to generate a fragment containing a Bpu10I restriction site followed by Kv11.1 (1–25) + Kv10.1 (26–135) + Kv11.1 up to the BstEII restriction site. This fragment was then inserted into the Kv11.1 backbone using Bpu10I and BstEII restriction enzymes (New England Biolabs). All constructs were confirmed by DNA sequencing. Linearization of DNA plasmids was performed using BamHI-HF (New England Biolabs), and the cRNA was transcribed in vitro using the mMessage mMachine kit (Ambion, Austin, TX).
Electrophysiology
Female X. laevis frogs were purchased from Nasco (Fort Atkinson, WI). All experiments carried out in this study were approved by the Garvan/St. Vincent's Animal Ethics Committee (approval identifier 11/37). The ovarian lobes were removed through a small abdominal incision following anaesthetization in 0.17% (w/v) tricaine. The follicular cell layer was removed by ∼1-h incubation with 1 mg/ml collagenase A (Roche Applied Science) in Ca2+-free ND96 solution containing 96 mm NaCl, 2 mm KCl, 1.0 mm MgCl2, and 5 mm Hepes (pH adjusted to 7.5 with NaOH). After rinsing with ND96 (as above, plus 1.8 mm CaCl2), stage V and VI oocytes were isolated and stored at 18 °C in tissue culture dishes containing ND96 supplemented with 2.5 mm pyruvic acid sodium salt, 0.5 mm theophylline, and 10 μg/ml gentamicin.
Isolated oocytes were injected with cRNA and incubated at 18 °C for 16–48 h prior to electrophysiological recordings. Two-electrode, voltage clamp experiments were performed at room temperature (20–22 °C) using a Geneclamp 500B amplifier (Molecular Devices, Sunnyvale, CA) with glass microelectrodes filled with 3 m KCl and tip resistances of 0.3–1.0 MΩ. Oocytes were perfused with ND96 solution during the recording. Data analysis was performed using pClamp software (version 10; Molecular Devices) and Prism 6 (GraphPad Software Inc., La Jolla, CA). All parameter values were calculated as means ± S.E. for n experiments, where n denotes the number of different oocytes studied for each construct.
The rates of deactivation were obtained by fitting a double exponential to the deactivation traces measured at voltages ranging from −150 to −50 mV. Both the fast and slow time constants at −120 mV, as well as the relative amplitude of the fast component compared with the slow component, are given in supplemental Table S1. For simplicity, in the figures we focus on the fast time constant of deactivation (τfast), which is the dominant component at −120 mV (i.e. relative amplitude ≥0.83 for WT and all mutants except D727R). A natural log of the fast time constant (ln·τfast,−120 mV) for each mutant was compared with that of WT channels to provide Δln·τfast,−120 mV.
Steady-state deactivation g-V curves were measured using tail current analysis, as previously described (22, 23). From a holding potential of −90 mV, cells were subjected to a 1-s depolarizing step to +40 mV and then a 3-s “test pulse” step to voltages in the range of −100 to +40 mV (the precise range depends on the mutant) in 10-mV increments, before measuring the tail current at −70 mV. Peak amplitude of the tail current were normalized to the maximum tail current value (Imax) and then fitted with a Boltzmann function,
where V0.5 is the half-maximal deactivation voltage, Vt is the test potential, and k is the slope factor. Alternatively, to calculate the changes in chemical potential energy (ΔG0) during the deactivation reaction, the data were fitted with a thermodynamic form of the Boltzmann function,
where ΔG0 is the work done at 0 mV, zg is the effective number of gating charges moving across the membrane electric field (E), F is the Faraday constant, R is the universal gas constant, and T is absolute temperature. The effect of a mutation (compared with WT) on changes in chemical potential energy was then calculated from Equation 3.
Double Mutant Cycle Analysis
The most commonly used technique for identifying pairs (24) or networks (25) of interacting residues is that of mutant cycle analysis, which can be performed in two main ways. For proposed charge-charge interactions between two oppositely charged side chains (from two residues in different domains), we initially used a charge reversal mutant cycle analysis approach. The theory behind this approach is relatively simple. Each member of a pair of residues is individually mutated to its opposite charge to disrupt the original charge-charge interaction. These individual mutations will produce quantifiable perturbations on an energetic property of the protein, measured kinetically by Δlnτfast or thermodynamically by ΔΔG0. If both residues are simultaneously mutated to their opposite charge, then the charge-charge interaction is restored, and perturbations to Δlnτfast or ΔΔG0 are reduced toward WT (i.e. a value close to 0), providing strong evidence for a native charge-charge interaction. It should be noted that in practice a complete restoration of WT-like function is difficult, as residues are often involved in multiple interactions that are not necessarily restored by charge swapping. In addition, this approach may not be appropriate for other, more complicated, forms of residue-residue interactions. For this reason, we examined a second form of thermodynamic mutant cycle analysis where an energetic coupling is considered to exist between two residues when the perturbations caused by the individual mutations (ΔGmut1 and ΔGmut2) are not additive when combined in a double mutant (i.e. ΔGmut1+mut2 < ΔGmut1 + ΔGmut2). Conversely, if perturbations caused by two single mutants were additive when combined in the double mutant (ΔGmut1+mut2 = ΔGmut1 + ΔGmut2), then the two residues are not energetically coupled. There are also limitations to this method of mutant cycle analysis that should be noted. First, mutations may introduce non-native interactions and give misleading results. Second, interacting residues may be “energetically silent” when mutated and will therefore not be identified (24). Lastly, residues that are energetically coupled are not necessarily interacting but may simply be part of the same mechanistic process.
Structure Generation
The structure of the PAS domain with the complete N-Cap domain bound was generated by aligning and fusing the N-Cap domain from one of the NMR structures (Protein Data Bank code 2L0W) to the crystal structure (Protein Data Bank code 4HP9) prior to removing the partial N-Cap helix. The structure of C-linker and cNBH domains was generated by modeling the mosquito agERG crystal structure (Protein Data Bank code 4L11) using the Swiss PdbViewer (26) and optimized using SWISS-MODEL Workspace (27, 28). The orientation of the C-linker of the resulting homology model was morphed to resemble the murine HCN2 C-linker conformation (Protein Data Bank code 1Q5O) using Chimera (29). The PAS and C-linker-cNBH domains were then superimposed to the murine Kv10.1 PAS-cNBH crystal structure (Protein Data Bank code 4LLO) to generate the model of Kv11.1 PAS-C-linker-cNBH domains. The final complex was energy-minimized and refined at 300 K using the protocol described previously (30) to remove any clashes between atoms using Amber12 (31).
RESULTS
N-Cap/PAS Domain of Kv11.1 Is Not Interchangeable with That of Kv10.1
The PAS domains from Kv10.1 and Kv11.1 channels show remarkable similarity in both their amino acid sequence and in their atomic structures (19). However, the kinetics of channel deactivation are much slower in Kv11.1 channels than in Kv10.1 channels (20), suggesting that the N-Cap/PAS domain and cNBH domains form different functional interactions in these two channels. To test this, we examined whether the N-Cap/PAS of Kv11.1 channels (shown in gray) was interchangeable with that of Kv10.1 (shown in green), using the kinetics of channel deactivation as a functional measure (Fig. 1, A–C). Deactivation kinetics, measured at voltages between −150 and −50 mV (Fig. 1A), were fitted with a double exponential. Exemplary fits of WT and Δ2–137 channel deactivation, measured at −120 mV, are shown in Fig. 1B. Both the fast and slow time constants for deactivation measured at −120 mV, as well as the relative amplitude of the fast and slow components, are given for WT and all mutant channels in supplemental Table S1. However, for simplicity, only the fast time constant of deactivation (τfast) is shown in Fig. 1C (and in all subsequent figures) because this represents the dominant component at −120 mV.
FIGURE 1.
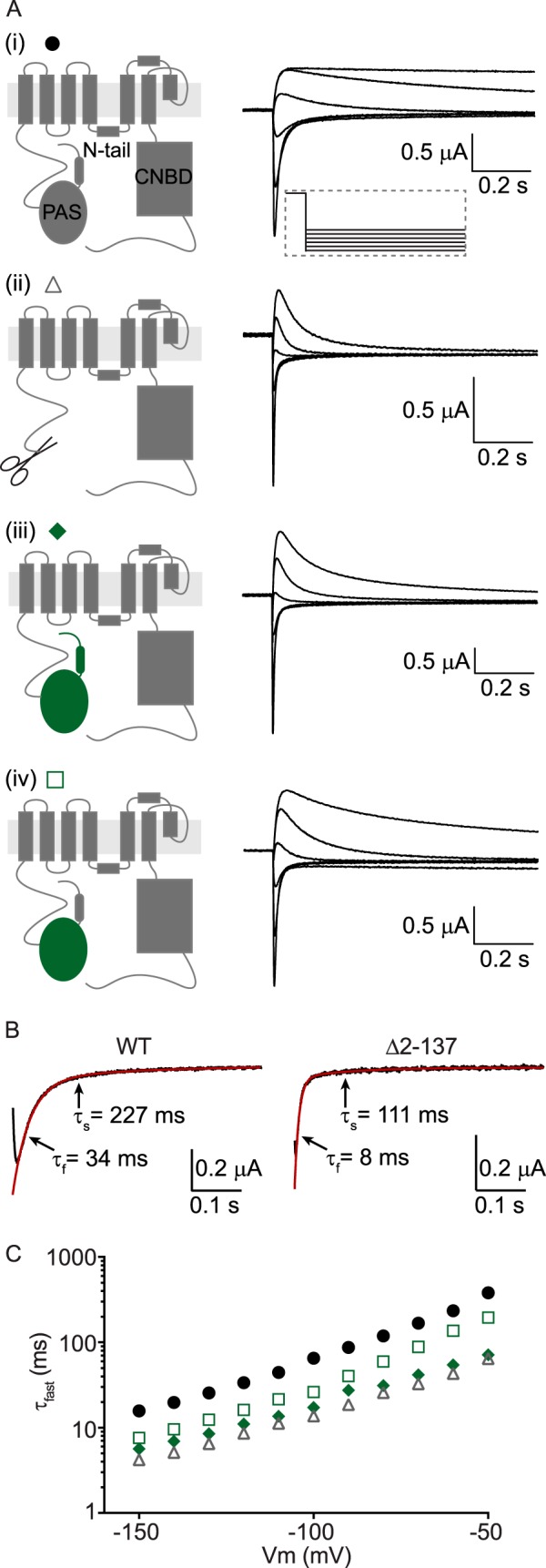
The N-Cap/PAS domain of Kv11. 1 is not interchangeable with that of Kv10.1. A, cartoon representation of a single subunit of either WT Kv11.1 channels (panel (i)), N-truncated (Δ2–137) Kv11.1 channels (panel (ii)), Kv11.1 channels with the N-Cap and PAS domains replaced by those of Kv10.1 (panel (iii)), or Kv11.1 channels with only the PAS domain of Kv10.1 (panel (iv)). Corresponding deactivation current traces are shown in the right hand panels for voltages between −50 and −150 mV in 20-mV increments. B, exemplary fits (red lines) of two exponential components to raw current traces measured at −120 mV (black lines) for WT and Δ2–137 channels. The fast (τfast) and slow (τslow) time constants are indicated. C, mean (± S.E.) rates of deactivation for WT Kv11.1 (black circles, n = 16), N-truncated Kv11.1 (gray triangles, n = 5), Kv11.1 with the N-Cap and PAS domain of Kv10.1 (green diamonds, n = 5), or Kv11.1 with only the PAS domain of Kv10.1 (open squares, n = 5). Note that error bars are included but are often within the symbols. Mean ± S.E. values for τfast, τslow, and the relative amplitudes of τfast to τslow for WT and all mutants are given in supplemental Table S1.
In contrast to WT Kv11.1 currents, which exhibit slow deactivation kinetics (Fig. 1, A, panel (i), and circles in C), Kv11.1 channels in which the N-Cap and PAS are replaced with the N-Cap and PAS of Kv10.1 channels (Fig. 1, A, panel (iii), and diamonds in C) show much faster deactivation gating kinetics that are comparable to removing the entire N-Cap and PAS domain of Kv11.1 (Fig. 1, A, panel (ii), and triangles in C). Interestingly, reintroducing just the N-Cap domain of Kv11.1 back into the N-Cap/PAS chimera (Fig. 1, A, panel (iv), and squares in C) is able to partially restore the rates of deactivation toward WT Kv11.1 channels. These data demonstrate that the N-Cap/PAS domains of Kv11.1 and Kv10.1 channels are not interchangeable and suggest that at least part of the channel-specific interaction arises from the N-terminal Cap.
Positive Charges in the N-Cap/PAS Are Important for Function
Several arginine residues in the N-Cap (Arg4, Arg5, and Arg20) and PAS (Arg56) domains (Fig. 2A) were previously found to be critical for slow deactivation of Kv11.1 channels (15, 32). To determine whether positive charge was the important factor in regulating slow deactivation of Kv11.1 channels, Arg4 (panel (i)), Arg5 (panel (ii)), Arg20 (panel (iii)), and Arg56 (panel (iv)) were individually mutated to a positively charged lysine side chain or negatively charged glutamic acid or aspartic acid side chains and then tested for their functionality (Fig. 2B). Our data strongly suggest that a positive charge at each position is critical for maintaining the slow deactivation, because mutation to either the glutamic acid (orange) or aspartic acid (red) residues produced channels that deactivate much faster than either the WT arginine or lysine mutant channels (as summarized in Fig. 2C). Similarly, an acceleration of the slow component of deactivation was also observed in the negatively charged side chain mutations but not in the positively charged lysine mutations at each position (see supplemental Table S1).
FIGURE 2.
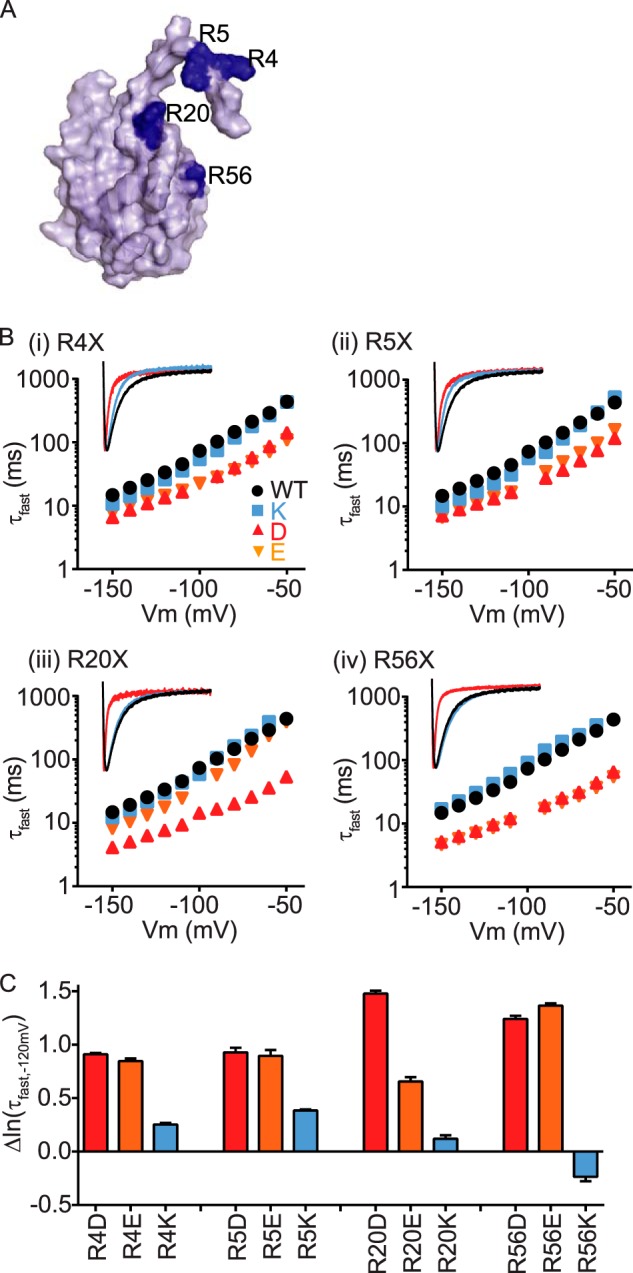
Positive charges in the N-Cap/PAS are important for function. A, surface representation of the N-Cap/PAS domains of Kv11.1 channels, showing the location of the positively charged arginine residues Arg4, Arg5, Arg20, and Arg56 (shown in dark blue). B, side chains of Arg4 (panel (i)), Arg5 (panel (ii)), Arg20 (panel (iii)), and Arg56 (panel (iv)) were mutated to either a positively charged lysine (blue squares), negatively charged aspartate (red triangles), or glutamate (orange inverted triangles) side chains, and mean (± S.E.) rates of deactivation were compared with the WT arginine (black circles). Note that error bars are included but are often within the symbols. Mean ± S.E. values for τfast, τslow, and the relative amplitudes of τfast to τslow for WT and all mutants are given in supplemental Table S1. For each mutated residue, the inset shows representative current traces at −120 mV for WT arginine (black), lysine (blue) or aspartate (red) side chains. C, mean (± S.E.) changes in natural log of the fast time constant of deactivation measured at −120 mV (ln·(τf,−120 mV) for mutant channels compared with WT (see also supplemental Table S1).
Surface-exposed Negative Charges on the cNBH Domain
To characterize the interaction between the N-Cap/PAS and the cNBH domains, we first sought to identify any critical negative charges on the cNBH domain of Kv11.1 channels by constructing a homology model of the Kv11.1 cNBH domain with an intact C-linker (Fig. 3A) (see “Structure Generation” under “Experimental Procedures”). We tested the functional importance of an initial 18 negatively charged residues, ranging from Asp712 to Glu857, by mutating to a positively charged arginine side chain and measuring kinetics of deactivation over a range of membrane potentials (Fig. 3B). Of the 18 negatively charged residues, only three: Asp774 (Fig. 3B, panel (i)), Glu788 (Fig. 3B, panel (ii)), and Asp803 (Fig. 3B, panel (iii)), exhibited clearly faster deactivation kinetics when mutated to arginine (Fig. 3, B and C). Of the remaining 15 mutations, two (D821R and E857R) exhibited small but significant accelerations of deactivation, whereas most had no effect, and two (D727R and D829R) exhibited significantly slowed deactivation kinetics (Fig. 3, B, panel (iv), and C). One mutant, E807R, did not form functional channels (Fig. 3C).
FIGURE 3.
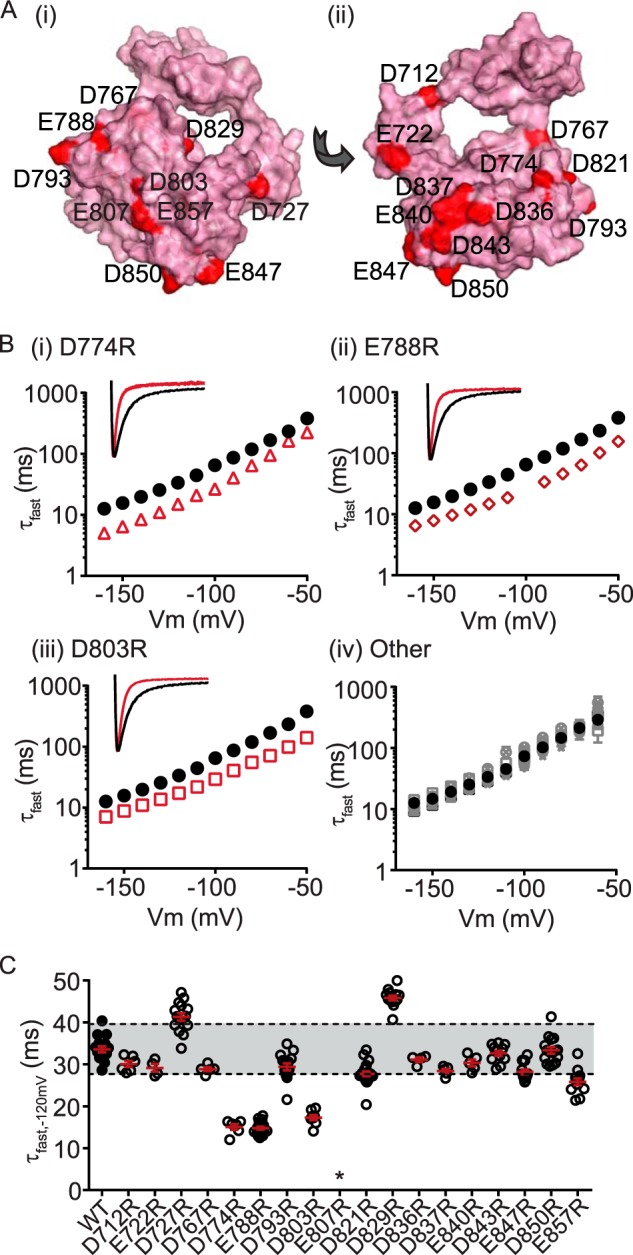
Several charged cNBH domain residues are critical for the slow deactivation kinetics of Kv11. 1 channels. A, surface representation homology model of the C-linker/cNBH domain of Kv11.1 channels showing the location of 18 negatively charged residues (red spheres). B, mean (± S.E.) rates of deactivation for the 18 negatively charged cNBH domain residues individually mutated to arginine. Three mutations exhibited substantially faster deactivation kinetics: D774R (panel (i), red triangles, n = 6), E788R (panel (ii), red diamonds, n = 21), and D803R (panel (iii), red squares, n = 8), whereas the remainder had deactivation kinetics similar to, or slower than, WT (panel (iv), gray symbols, n = 4–21). Note that error bars are included but are within the symbols. Mean ± S.E. values for τfast, τslow and the relative amplitudes of τfast to τslow for WT and all mutants are given in supplemental Table S1. Representative current traces at −120 mV for D774R, E788R, and D803R are shown in the inset. C, summary of all individual mutant effects on deactivation kinetics measured at −120 mV. Mutant perturbations were considered significant only when the mean was outside ± two standard deviations of the WT Kv11.1 (indicated by gray band). D774R, E788R, D803R, D821R, and E857R exhibited fast deactivation kinetics, whereas D727R and D829R were significantly slower than WT. * denotes that E807R did not express functional channels.
In our tetrameric homology model of the Kv11.1 cNBH domains, which is similar to the one generated by Haitin et al. (18), Asp774 is located within the interface of two subunits and is therefore likely to cause perturbation to the subunit-subunit interaction when mutated to arginine. In contrast, both Glu788 and Asp803 are surface-exposed negatively charged residues and could form charge-charge interactions with one or more of the critical N-Cap/PAS positively charged residues (Arg4, Arg5, Arg20, or Arg56).
Arg56 Interacts with Asp803
We performed charge reversal double mutant cycle analysis (see “Experimental Procedures”) to identify whether Asp803 interacts with any of the four positively charged residues from the N-Cap and PAS domains of Kv11.1 channels (Fig. 4A). The individual mutants, D803R (red squares) and RXD (blue inverted triangles) all exhibit fast deactivation (Fig. 4A, panels (i)–(iv)). However, when combined into double mutants (purple circles) only R56D (Fig. 4A, panel (iv)) was able to restore deactivation back to the WT in the background of D803R. In fact R56D/D803R restored both the fast (τfast) and slow (τslow) time constants of deactivation back to WT values (see supplemental Table S1). Changes in the Δlnτfast of deactivation compared with WT are summarized in Fig. 4B. Restoration of Δlnτfast toward WT (i.e. 0) in the R56D/D803R charge reversal mutant strongly indicates a native charge-charge interaction between Arg56 and Asp803. We sought to confirm the deactivation kinetics data by examining changes in chemical potential energy of individual and double mutants (versus WT, ΔΔG0) obtained from steady-state voltage dependence of deactivation g-V curves (Fig. 4C; see “Experimental Procedures” for details). Similar to the deactivation kinetics data, R56D/D803R restored ΔΔG0 values back toward WT (Fig. 4C). Furthermore, when we used a thermodynamic mutant cycle analysis approach where Arg56 and Asp803 are individually mutated to alanine, both produced a significant perturbation to ΔΔG0 (> 5 kj·mol−1), and these perturbations were not additive when combined in the double alanine mutant (Fig. 4D and supplemental Table S2). Together, these results clearly show that an interaction between Arg56 and Asp803 (depicted in Fig. 4E) is critical for slow deactivation of Kv11.1 channels.
FIGURE 4.
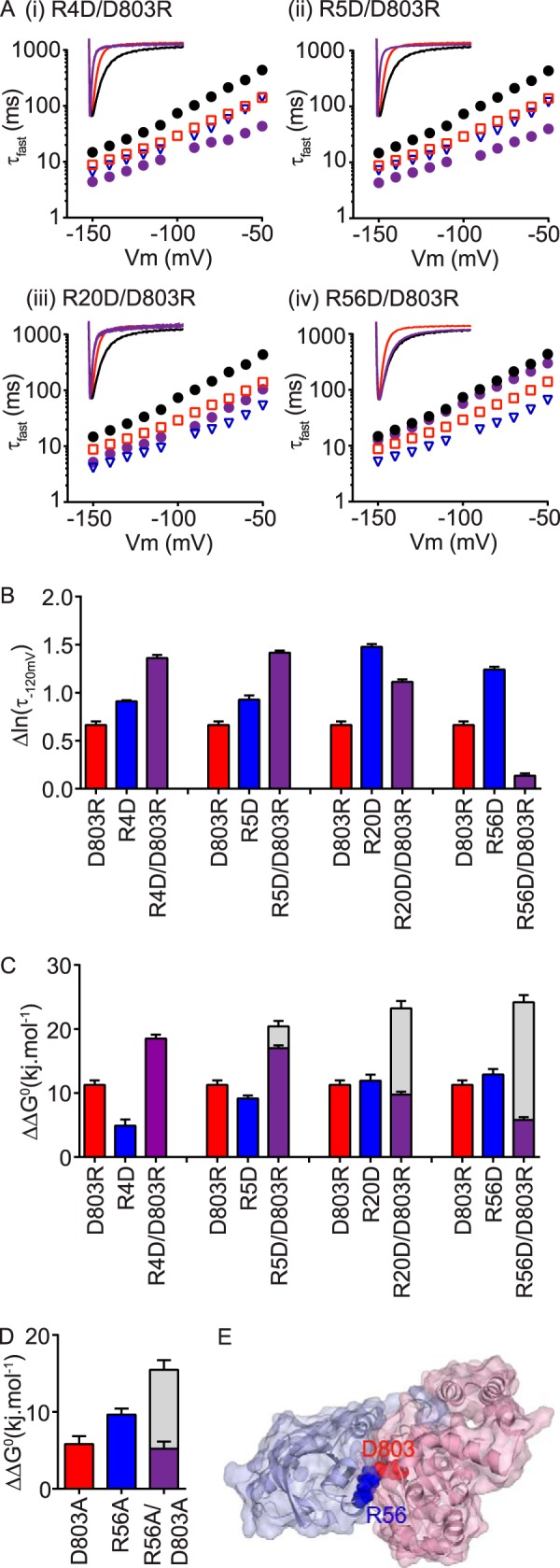
PAS domain residue Arg56 forms a charge-charge interaction with the cNBH domain residue Asp803. A, mean (± S.E.) rates of deactivation for N-Cap mutants R4D (panel (i)), R5D (panel (ii)), R20D (panel (iii)), or the PAS domain mutant R56D (panel (iv)), either alone (blue triangles, n = 5–14) or in the presence of the cNBH domain mutation D803R (purple circles, n = 7–16). WT Kv11.1 (black circles, n = 16) and D803R (red squares, n = 8) are shown for comparison. Note that error bars are included but are within the symbols. Mean ± S.E. values for τfast, τslow, and the relative amplitudes of τfast to τslow for WT and all mutants are given in supplemental Table S1. Representative current traces at −120 mV are shown within insets, with mutants color-coded as above. B, summary (means ± S.E.) showing complete restoration of slow deactivation kinetics, measured as Δln(τfast,−120 mV) compared with WT Kv11.1, only when R56D was combined with D803R, indicating a direct functional interaction between the native residues. C and D, summary (mean ± S.E.) ΔΔG0 values calculated from Boltzmann energy fits of steady-state g-V curves (see “Experimental Procedures” for details) for charge reversal (C) or alanine (D) mutant channels compared with the WT. Gray bars represent the theoretical additive values of the two individual mutant perturbations combined. Note that the double mutant R56D/D803R exhibits a reduced ΔΔG0 compared with the two single mutants alone. Mean ± S.E. values are given in supplemental Table S2. E, surface representation Kv11.1 homology model of the N-Cap/PAS domains (blue) interacting with the C-linker/cNBH domains (red), with the boxed region, expanded in the right panel, showing a charge-charge interaction between the side chains of the PAS domain residue Arg56 and Asp803 in the cNBH domain.
Glu788 Interacts with Asn12 but Not with the Four N-Cap/PAS Arginine Residues
Next we examined whether the cNBH domain surface-exposed negatively charged residue Glu788 interacted with any of the positively charged residues in the N-Cap/PAS domain, using a charge reversal double mutant cycle analysis approach (Fig. 5). Individual mutants, E788R (red squares), and RXE (blue triangles) all had fast deactivation kinetics compared with WT (Fig. 5A, panels (i)–(iv)). However, in the background of E788R, none of the arginine to glutamate mutations in the N-Cap/PAS (purple circles) slowed deactivation kinetics (summarized in Fig. 5B) or reversed thermodynamic ΔΔG0 values (summarized in Fig. 5C) back to that of WT Kv11.1 channels.
FIGURE 5.
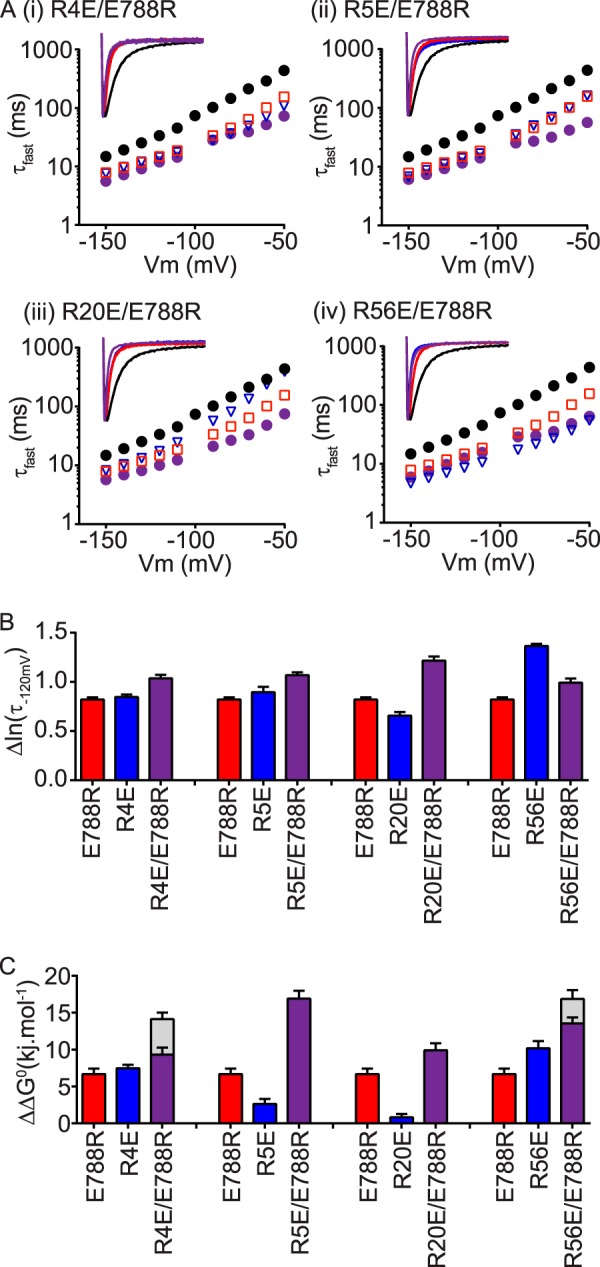
cNBH domain residue Glu788 does not appear to interact with the N-Cap/PAS domain arginine residues. A, mean (± S.E.) rates of deactivation for N-Cap mutants R4E (panel (i)), R5E (panel (ii)), R20E (panel (iii)), or the PAS domain mutant R56E (panel (iv)), either alone (blue triangles, n = 11–14) or in the presence of the cNBH domain mutation E788R (purple circles, n = 12–17). WT Kv11.1 (black circles, n = 16) and E788R (red squares, n = 21) are shown for comparison. Note that error bars are included but are within the symbols. Mean ± S.E. values for τfast, τslow, and the relative amplitudes of τfast to τslow for WT and all mutants are given in supplemental Table S1. Representative current traces at −120 mV are shown within the insets, with mutants color-coded as above. B and C, summary (mean ± S.E.) showing no restoration of slow deactivation kinetics (Δln(τfast, −120mV), B) or of chemical potential energy (ΔΔG0, C), compared with WT Kv11.1, with any of the combined N-Cap/PAS arginine mutants. Gray bars in C represent the theoretical additive values of the two individual mutant perturbations combined. Means ± S.E. for chemical potential energy parameters are given in supplemental Table S2.
To identify a possible interacting partner for Glu788, we returned back to the homology model of Kv11.1 N-Cap/PAS-C-linker/cNBH domain and identified a possible interaction with Asn12 of the N-Cap domain (Fig. 6A). We tested for an interaction between Asn12 and Glu788 (Fig. 6, B–C) and found that N12E is able to partially restore slow deactivation in the background of E788R (purple diamonds), compared with single mutants N12E (blue triangles) or E788R (red squares). In the thermodynamic analysis, N12E/E788R did not restore ΔΔG0 back toward WT, although the perturbation was similar to the least perturbing of the two single mutants (i.e. E788R rather than N12E, Fig. 6D). We were unable to confirm an interaction between Asn12 and Glu788 using alanine mutagenesis, because the N12A mutation did not significantly perturb ΔΔG0, even though the rate of channel deactivation was substantially faster for N12A (Fig. 6E and supplemental Tables S1 and S2). However, the partial restoration of slow deactivation in the double mutant N12E/E788R supports an interaction between Asn12 of the N-Cap and Glu788 of the cNBH domain.
FIGURE 6.
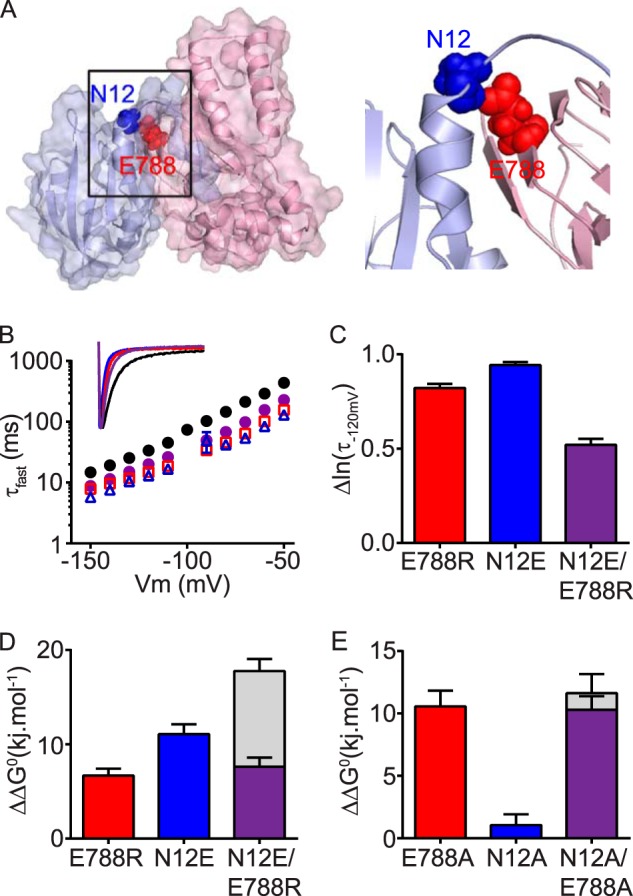
cNBH domain residue Glu788 interacts with the N-Cap residue Asn12. A, surface representation homology model of the N-Cap/PAS domain (blue) interacting with the C-linker/cNBH domain (red). The N-Cap residue Asn12 (blue spheres) interacts with Glu788 (red spheres) of the cNBH domain. B, mean (± S.E.) rates of deactivation for WT Kv11.1 (black circles, n = 16), N-Cap mutant N12E (blue triangles, n = 5), cNBH domain mutant E788R (red squares, n = 21), and combined mutant N12E/E788R (purple circles, n = 8). Note that error bars are included but are within the symbols. Mean ± S.E. values for τfast, τslow, and the relative amplitudes of τfast to τslow for WT and all mutants are given in supplemental Table S1. Inset, representative current traces at −120 mV for each mutant are color-coded as above. C, summary (mean ± S.E.) showing that the combined mutant N12E/E788R has slower deactivation kinetics, measured as smaller Δln(τ−120 mV) compared with WT Kv11.1, than either of the two single mutants alone. D and E, summary (mean ± S.E.) ΔΔG0 for charge reversal (D) or alanine (E) mutant channels compared with the WT. Note that the effects of the two single mutants are not additive when combined in the double mutant N12E/E788R. Gray bars represent the theoretical additive values of the two individual mutant perturbations combined. Means ± S.E. are given in supplemental Table S2.
Interaction Site for Arg4 and Arg5
As we have shown previously (15) and in this study (Fig. 2), arginine residues Arg4 and Arg5 in the N-Cap are critical for regulating slow deactivation. It is reasonable to assume that these two arginine residues will most likely interact with negatively charged residues, especially those that are in close proximity to one another. Guided by our homology model of the N-Cap/PAS and C-linker/cNBH domains, we identified two additional negatively charged glutamic acid residues, Glu698 and Glu699, that were not included in the initial scan of negatively charged residues. Both are located within the N-terminal portion of the C-linker and are in physical proximity to Arg4 and Arg5 from the N-Cap domain in our homology model (Fig. 7A). When mutated (Fig. 7B), both of the double mutants, R4E/R5E (blue triangles) and E698R/E699R (red squares), exhibited faster channel deactivation compared with WT Kv11.1. In comparison, a combination of the two double mutants (purple diamonds), that is R4E/R5E + E698R/E699R (EERR), produced channels with much slower deactivation kinetics than R4E/R5E but similar to E698R/E699R. Comparative changes to the kinetics of deactivation versus WT are summarized in Fig. 7C. Similar to the kinetic data, both double mutants produced significant perturbations to thermodynamic ΔΔG0 values that were not additive when the two double mutants were combined (Fig. 7D). Finally, we repeated the thermodynamic mutant cycle analysis using alanine mutants and observed significant perturbations to ΔΔG0 with the two double mutants (R4A/R5A and E698A/E699A). When combined (R4A/R5A + E698A/E699A, AAAA in Fig. 7E), the ΔΔG0 was reduced compared with R4A/R5A, but similar to E698A/E699A (i.e. the effects were not additive) consistent with an interaction (Fig. 6E). Based on these data, we suggest that Arg4 and Arg5 of the N-Cap can form a functional interaction with Glu698 and Glu699 of the C-linker region and that this interaction connects the N-Cap/PAS+cNBH complex to the S6 channel gate.
FIGURE 7.
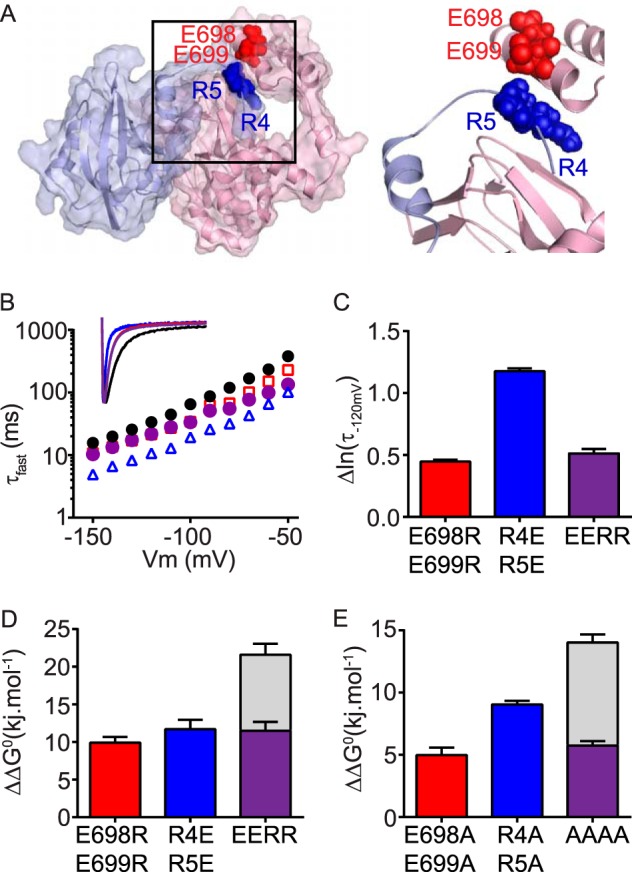
N-Cap residues Arg4/Arg5 may interact with the C-linker residues Glu698/Glu699. A, surface representation homology model showing that residues Arg4 and Arg5 (shown in “blue” spheres) of the N-Cap/PAS domains (blue) are in close proximity to residues Glu698 and Glu699 (shown in red spheres) of the C-linker/cNBH domains (red) in Kv11.1 channels. B, mean (± S.E.) rates of deactivation for WT Kv11.1 (black circles, n = 16), N-Cap double mutant R4E/R5E (blue triangles, n = 5), C-linker double mutant E698R/E699R (red squares, n = 5), and combined charge reversal mutant R4E/R5E+E698R/E699R (EERR, purple circles, n = 5). Note that error bars are included but are within the symbols. Mean ± S.E. values for τfast, τslow, and the relative amplitudes of τfast to τslow for WT and all mutants are given in supplemental Table S1. Inset, representative current traces at −120 mV for each mutant are color-coded as above. C, summary (means ± S.E.) showing that the charge reversal mutant R4E/R5E+E698R/E699R (EERR) exhibits altered deactivation kinetics, measured as Δln(τ−120 mV) compared with WT Kv11.1, that are similar to the C-linker double mutant E698R/E699R, but much slower (i.e. smaller shift) than the N-Cap double mutant R4E/R5E. D and E, summary (mean ± S.E.) ΔΔG0 for charge reversal (D) or alanine (E) mutant channels compared with the WT. Note that the effects of the N-Cap double mutant (R4E/R5E) and the C-linker/cNBH domain double mutant (E698R/E699R) are not additive when combined in the quadruple mutant. Gray bars represent the theoretical additive values of the two individual mutant perturbations combined. Means ± S.E. are given in supplemental Table S2.
To find additional evidence that the N-Cap and PAS domains share a common mechanism for the regulation of slow deactivation gating kinetics, i.e. via an interaction with the C-linker/cNBH domains, we combined an R4D/R5D N-Cap double mutant (orange triangles) with the PAS domain R56D mutant (red triangles) to generate a triple 4D5D56D N-Cap/PAS domain mutant (purple diamonds in Fig. 8A). All three mutant channels exhibited similar faster rates of deactivation than observed in WT (black circles). Importantly, the effect of the N-Cap double mutant (R4D/R5D) was not additive to the effect of the PAS domain mutant R56D, when examining both deactivation kinetics (Δlnτfast; Fig. 8B) and changes in chemical potential energy (ΔΔG0; Fig. 8C). These data provide further evidence that the N-Cap and PAS domain positively charged residue slow Kv11.1 channel deactivation through a common interaction with the C-linker/cNBH domains (Fig. 9).
FIGURE 8.
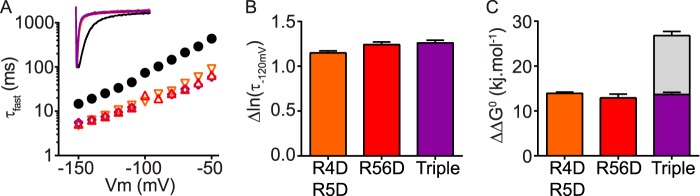
N-Cap and PAS domains share a common mechanism for the regulation of slow deactivation gating kinetics. A, mean (±S.E.) rates of deactivation for WT Kv11.1 (black circles, n = 16), the N-Cap domain double mutant R4D/R5D (orange inverted triangles, n = 5), the PAS domain mutant R56D (red triangles, n = 5), and the N-Cap/PAS triple mutant R4D/R5D+R56D (purple diamonds, n = 6). Note that error bars are included but are within the symbols. Mean ± S.E. values for τfast, τslow, and the relative amplitudes of τfast to τslow for WT and all mutants are given in supplemental Table S1. Inset, representative current traces at −120 mV for each mutant are color-coded as above. B and C, all mutants show similar accelerated deactivation kinetics (Δln(τ−120 mV), B) and similar changes in chemical potential energy (ΔΔG0, C) compared with WT Kv11.1, indicating a shared mechanism for slowing deactivation kinetics. Gray bars in C represent the theoretical additive values of the two individual mutant perturbations combined. Means ± S.E. for chemical potential energy parameters are given in supplemental Table S2.
FIGURE 9.
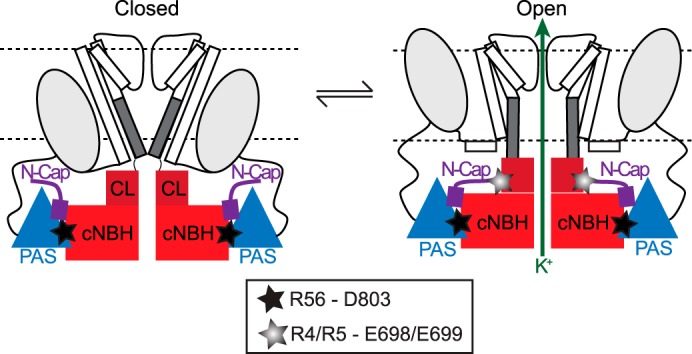
Proposed molecular mechanism for slow deactivation in Kv11. 1 channels. Cartoon of two opposing subunits of a tetrameric Kv11.1 channel shown in the closed (left) and open (right) conformations. In both conformations, Arg56 in the PAS domain (blue triangles) and Asp803 in the cNBH domain (red squares) form a highly stable interaction (denoted by black star). In the open conformation, Arg4 and Arg5 in the N-Cap domain (shown in purple) and Glu698 and Glu699 in the C-linker (shown in maroon) form an additional, but more transient, interaction (denoted by gray star) that stabilizes the open state, presumably by stabilizing the S6 activation gate (represented by dark gray bar) in the open conformation. The stable PAS-cNBH domain interaction is critical because it positions the N-Cap domain in the correct location to interact with the C-linker. These interactions underlie the slow deactivation gating observed in Kv11.1 channels.
DISCUSSION
The aim of this study was to identify the molecular basis of how the N-Cap/PAS domain slows deactivation of Kv11.1 channels. Our results show that positively charged residues in the N-terminal N-Cap and PAS domains form charge-charge interactions with surface-exposed negatively charged residues in the C-terminal C-linker and cNBH domains and that these interactions are critical for maintaining the slow deactivation kinetics of Kv11.1 channels. Specifically, we show that the PAS domain residue Arg56 forms a functional interaction with Asp803 of the cNBH domain, whereas Arg4 and Arg5 of the N-Cap domain most likely interact with Glu698 and Glu699 within the C-linker.
Kv10.1 and Kv11.1 are members of the same family of potassium channels and share remarkable sequence similarity, yet we found that the N-Cap/PAS domains of Kv10.1 are not interchangeable with those of Kv11.1 without dramatically accelerating the deactivation gating kinetics of Kv11.1 channels. Interestingly, restoring just the N-Cap domain of Kv11.1 can partially restore deactivation gating back toward the Kv11.1 WT. This observation is consistent with a previous study showing that injection of the N-Cap peptide is capable of partially restoring deactivation gating of a truncated Kv11.1 channel (33). These findings confirm that the N-Caps, as well as the PAS domains, are involved in slowing the deactivation kinetics of Kv11.1 channels.
R56Q is a clinically identified mutation in the PAS domain of Kv11.1 channels, which accelerates channel closure and predisposes patients to lethal arrhythmias (32). Although several studies proposed that the N-terminal PAS domains of Kv11.1 channels form interactions with the C-terminal cNBH domains (17, 34, 35), the exact nature of these interactions, including those of Arg56, were hitherto unclear. A recent crystal structure of isolated PAS and cNBH domains from mouse Kv10.1 channels showed that Arg57 in the PAS domain formed a specific salt bridge interaction with Asp642 in the cNBH domain (18). Consistent with this, we found a critical salt bridge interaction between Arg56 and Asp803, the corresponding positions in Kv11.1 channels. Our data suggest that a loss of interaction between Arg56 in the PAS domain and Asp803 in the cNBH domain is responsible for the fast deactivation gating phenotype, which underlies the pathophysiology of the R56Q mutation.
Several lines of evidence suggest that the PAS-cNBH domain interaction in Kv11.1 channels is relatively stable. For instance, the injection of isolated PAS domain protein into cells expressing N-terminal truncated Kv11.1 channels can restore slow deactivation, and this effect remains following excision of the membrane patch, suggesting that the exogenous PAS domain is stably bound to the Kv11.1 channel (16). In a different study, it was shown that FRET can be observed when CFP-tagged exogenous PAS domain protein is added to YFP-tagged R56Q mutant Kv11.1 channels but not when added to YFP-tagged WT Kv11.1 channels, indicating that the stably bound WT PAS domain cannot be supplanted by the exogenous CFP-tagged PAS domain protein (14). Thus, it appears that the Arg56–Asp803 salt bridge is one of several interactions that form a stable connection between the PAS and cNBH domains. Furthermore, this stable interaction is shared between at least two members of the EAG family of channels: Kv11.1 and Kv10.1. However, although in Kv11.1 channels this interaction is important for slowing the kinetics of deactivation (channel closure), in Kv10.1 channels the interaction appears to accelerate the kinetics of channel activation (channel opening) (18). The reason for this difference is unclear, but we postulated that it may reflect differences in the mechanism by which the N-Cap domains interact with the remainder of the channel.
The N-terminal N-Cap domains of Kv11.1 channels have been proposed to bind to various regions of the channel protein including the S4S5 linker (36, 37) or the cNBH domain (38). Recently, Haitin et al. (18) proposed that two charged residues, Arg7 and Arg8, in the N-Cap domain of Kv10.1 channels form a functional interaction with E627R of the cNBH domain. However, the structure of the co-crystallized isolated PAS and cNBH domains used in their study lacked the initial 15 residues of the N-Cap domain, including Arg7 and Arg8, as well as the majority of the C-linker (18). In our study, none of the positively charged residues within the N-Cap of Kv11.1 channels, including Arg4 and Arg5 (equivalent to Arg7 and Arg8 in Kv10.1), were able to restore the slow deactivation gating in the background of the cNBH domain mutation E788R (Glu627 in Kv10.1). Instead, we found that mutating Asn12 from the N-Cap domain to a glutamic acid partially restored the fast deactivation gating phenotype of E788R, suggesting that Asn12 is capable of forming a hydrogen bond with Glu788 that modulates gating (Fig. 5). Our homology model also supports the suggestion that Asn12 interacts with Glu788 (Fig. 6). That we get only partial restoration of the slow deactivation phenotype in the N12E/E788R mutant is not entirely surprising because this is not an exact exchange of residues (i.e. native Asn-Glu swapped with larger and more bulky Glu-Arg), so we should not necessarily expect complete restoration of the native interaction. Furthermore, any additional interactions between Asn12 or Glu788 with other nearby residues in the N-Cap/PAS and cNBH domains would likely not be restored. Finally, the introduction of a positively charged side chain at position 788 may introduce a non-native interaction with other nearby residues. These results reflect the limitations in the method of charge reversal mutant cycle analysis that are discussed under “Experimental Procedures.”
In the thermodynamic analysis, N12E/E788R did not restore ΔΔG0 back toward WT, although the perturbation was similar to the least perturbing of the two single mutants (i.e. E788R rather than N12E). From these data, we can at least conclude that the perturbations caused by the two single mutants are not additive when combined in the double mutant, consistent with Asn12 of the N-Cap and Glu788 of the cNBH domain being energetically coupled (i.e. part of the same mechanistic process). We also tested alanine mutants N12A and E788A, as well as the double alanine mutant N12A/E788A (supplemental Tables S1 and S2). Although N12A significantly accelerated the rate of channel deactivation, it did not produce a significant change in the chemical potential energy ΔΔG0, mainly because of a much steeper slope in the voltage dependence of deactivation for N12A compared with WT. Conversely, the slopes of the steady-state deactivation curves for E788A and E788A/N12A were similar to WT, and consequently the ΔΔG0 for E788A/N12A was not that different to the sum of the ΔΔG0 values for the individual mutants (see supplemental Table S2). Again, this may reflect the limitations of thermodynamic mutant cycle analysis, where mutations may introduce non-native interactions or mutation of interacting residues may be energetically silent (24), as discussed under “Experimental Procedures.” Despite this, the kinetic perturbations of N12A and E788A were not additive when combined in the double mutant, consistent with an energetic coupling between these residues during deactivation gating. Thus, although we cannot definitively conclude that Asn12 of the N-Cap and Glu788 of the cNBH domains interact, our data and the homology model indicate that this interaction is entirely plausible.
If Glu788 of the cNBH domain does not interact with either Arg4 or Arg5 from the N-Cap domain, then the Arg4 or Arg5 must be interacting with residues in close proximity, because the crystal structure of the Kv10.1 PAS-cNBH domain constrains the conformational space and distance to which the Arg4 and Arg5 may extend. Interestingly, our homology model shows that Arg4/Arg5 may be in close physical proximity to two glutamic acid residues, Glu698 and Glu699, within the C-linker. Although the quadruple mutant (R4E/R5E+E698R/E699R) exhibits no restoration of slow deactivation, the phenotype is clearly more similar to the slower deactivation profile of E698R/E699R rather than the much more rapidly deactivating R4E/R5E, suggesting that there is no additive effect. In other words, the quadruple mutant affects deactivation gating to the same extent as the E698R/E699R double mutant alone. The nonadditive perturbations of these two double mutants were confirmed using thermodynamic ΔΔG0 measurements of both the charge reversal mutants (Fig. 7D) and alanine mutants (Fig. 7E). One reason for the lack of restoration in the charge reversal mutants may be that we are simply introducing too much perturbation to both of the C-linker and N-Cap domain and thus are removing interactions additional to those directly between Arg4/Arg5 of the N-Cap and Glu698/Glu699 of the C-linker.
We (15, 23) and others (16) have previously shown that deletion of the N-terminal Cap residues (Δ2–25) results in a fast deactivation phenotype that is similar to that obtained by deletion of the entire PAS domain (16). Importantly, deletion of the N-Cap (Δ2–25) accelerates the kinetics of deactivation without substantially altering the kinetics of activation (i.e. has no effect on the forward transition), suggesting that the N-tail interaction occurs when channels are in the open conformation and dissociates upon channel closure (23). In support of a transient interaction for the N-tail, Wang et al. (33) have shown that the application of an exogenous peptide that mimics the first 16 residues of the N-Cap can restore a slow deactivation phenotype to N-Cap truncated Kv11.1 channels (Δ2–16) and that this restoration is reversible on washout (31). Thus, in contrast to the relatively stable interaction between Arg56 of the PAS domain and Asp803 of the cNBH domain, which occurs both in Kv10.1 channels as well as Kv11.1 channels, it is likely that the N-Cap (Arg4/Arg5) interaction with the C-linker (Glu698/Glu699) is much more transient. Furthermore, this interaction is likely very specific to Kv11.1 channels because the equivalent residues in Kv10.1 (Asp/Met) would not provide the same negatively charged surface for binding of positively charged Arg7/Arg8 residues. This Kv11.1 specific interaction therefore allows the N-Cap domain to control the rate of closure of the S6 activation gate via the C-linker. This interaction is likely to be transient, occurring only when the activation gate is open and stabilizing the open conformation of the channels such that the rate of S6 motion back to the closed conformation is markedly slowed.
Recent crystal structures of the cNBH domain from different KCNH channels show different orientations of the C-linker (39, 40), whereas some crystal structures do not even have the C-linker at all, perhaps because of difficulty in the expression, purification, and crystallization (18, 41). In the recent crystal structure of the PAS/cNBH domains of Kv10.1 channels (18), both the N-Cap and C-linker were not present, so direct structural evidence for the lack of this interaction in Kv10.1 channels is unavailable. However, even if we did possess crystal structures of the N-Cap/PAS domain plus the C-linker/cNBH domains of both Kv10.1 and Kv11.1 channels, these types of crystal structures rarely provide information about transient interactions.
Based on our data and homology model, we propose that Arg56 and Asp803 form a stable interaction that orients the N-Cap close to the C-linker/cNBH domains. The disordered region of the N-Cap domain, in particular the Arg4 and Arg5, is then in a position to form transient interactions with the C-linker to regulate slow channel deactivation. This proposal is supported by our observation that the perturbation caused by mutating both Arg4 and Arg5 to aspartic acid residues (R4D/R5D) has the same effect as the R56D or the triple mutant (R4D/R5D/R56D), i.e. in the absence of the Arg56–Asp803 interaction; then mutations to Arg4 and Arg5 have no additional effect on deactivation gating. Given the direct link between the C-linker and the S6 gate, it is plausible to suggest that the N-cap interactions with the C-linker are allosterically transmitted to the S6 gate. We suggest that the N-Cap binds to the C-linker in the open state, and only when this interaction is broken can the channel return to the closed conformation. The dynamic structure of the disordered region of the N-Cap domain is therefore likely to be essential to ensure that the interaction with the C-linker is transient rather than permanent and yet strong enough to slow deactivation of Kv11.1 channels.
Supplementary Material
This work was supported by Grant-in-Aid NHFA G11S 5829S from the Heart Foundation of Australia and National Health and Medical Research Council of Australia Project Grant 635520) and Fellowship Grants 459401 and 1019693 (to J. I. V.).

This article contains supplemental Tables S1 and S2.
- PAS
- Per-Arnt-Sim
- cNBH
- cyclic nucleotide binding homology.
REFERENCES
- 1. Warmke J. W., Ganetzky B. (1994) A family of potassium channel genes related to eag in Drosophila and mammals. Proc. Natl. Acad. Sci. U.S.A. 91, 3438–3442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Chiesa N., Rosati B., Arcangeli A., Olivotto M., Wanke E. (1997) A novel role for HERG K+ channels: spike-frequency adaptation. J. Physiol. 501, 313–318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Greenwood I. A., Yeung S. Y., Tribe R. M., Ohya S. (2009) Loss of functional K+ channels encoded by ether-a-go-go-related genes in mouse myometrium prior to labour onset. J. Physiol. 587, 2313–2326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ohya S., Horowitz B., Greenwood I. A. (2002) Functional and molecular identification of ERG channels in murine portal vein myocytes. Am. J. Physiol. Cell Physiol. 283, C866–C877 [DOI] [PubMed] [Google Scholar]
- 5. Schäfer R., Wulfsen I., Behrens S., Weinsberg F., Bauer C. K., Schwarz J. R. (1999) The erg-like potassium current in rat lactotrophs. J. Physiol. 518, 401–416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Barros F., del Camino D., Pardo L. A., Palomero T., Giráldez T., de la Peña P. (1997) Demonstration of an inwardly rectifying K+ current component modulated by thyrotropin-releasing hormone and caffeine in GH3 rat anterior pituitary cells. Pflugers Arch. 435, 119–129 [DOI] [PubMed] [Google Scholar]
- 7. Bauer C. K., Engeland B., Wulfsen I., Ludwig J., Pongs O., Schwarz J. R. (1998) RERG is a molecular correlate of the inward-rectifying K current in clonal rat pituitary cells. Receptors Channels 6, 19–29 [PubMed] [Google Scholar]
- 8. Pardo L. A., Sühmer W. (2008) Eag1 as a cancer target. Expert Opin. Ther. Targets 12, 837–843 [DOI] [PubMed] [Google Scholar]
- 9. Sanguinetti M. C., Jiang C., Curran M. E., Keating M. T. (1995) A mechanistic link between an inherited and an acquired cardiac arrhythmia: HERG encodes the IKr potassium channel. Cell 81, 299–307 [DOI] [PubMed] [Google Scholar]
- 10. Trudeau M. C., Warmke J. W., Ganetzky B., Robertson G. A. (1995) HERG, a human inward rectifier in the voltage-gated potassium channel family. Science 269, 92–95 [DOI] [PubMed] [Google Scholar]
- 11. Curran M. E., Splawski I., Timothy K. W., Vincent G. M., Green E. D., Keating M. T. (1995) A molecular basis for cardiac arrhythmia: HERG mutations cause long QT syndrome. Cell 80, 795–803 [DOI] [PubMed] [Google Scholar]
- 12. Gustina A. S., Trudeau M. C. (2012) HERG potassium channel regulation by the N-terminal eag domain. Cell Signal. 24, 1592–1598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wang J., Trudeau M. C., Zappia A. M., Robertson G. A. (1998) Regulation of deactivation by an amino terminal domain in human ether-à-go-go-related gene potassium channels. J. Gen. Physiol. 112, 637–647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gustina A. S., Trudeau M. C. (2009) A recombinant N-terminal domain fully restores deactivation gating in N-truncated and long QT syndrome mutant hERG potassium channels. Proc. Natl. Acad. Sci. U.S.A. 106, 13082–13087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ng C. A., Hunter M. J., Perry M. D., Mobli M., Ke Y., Kuchel P. W., King G. F., Stock D., Vandenberg J. I. (2011) The N-terminal tail of hERG contains an amphipathic alpha-helix that regulates channel deactivation. PLoS One 6, e16191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Morais Cabral J. H., Lee A., Cohen S. L., Chait B. T., Li M., Mackinnon R. (1998) Crystal structure and functional analysis of the HERG potassium channel N terminus: a eukaryotic PAS domain. Cell 95, 649–655 [DOI] [PubMed] [Google Scholar]
- 17. Gustina A. S., Trudeau M. C. (2011) hERG potassium channel gating is mediated by N- and C-terminal region interactions. J. Gen. Physiol. 137, 315–325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Haitin Y., Carlson A. E., Zagotta W. N. (2013) The structural mechanism of KCNH-channel regulation by the eag domain. Nature 501, 444–448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Adaixo R., Harley C. A., Castro-Rodrigues A. F., Morais-Cabral J. H. (2013) Structural properties of PAS domains from the KCNH potassium channels. PLoS One 8, e59265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Piper D. R., Varghese A., Sanguinetti M. C., Tristani-Firouzi M. (2003) Gating currents associated with intramembrane charge displacement in HERG potassium channels. Proc. Natl. Acad. Sci. U.S.A. 100, 10534–10539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Terlau H., Heinemann S. H., Stühmer W., Pongs O., Ludwig J. (1997) Amino terminal-dependent gating of the potassium channel rat eag is compensated by a mutation in the S4 segment. J. Physiol. 502, 537–543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ng C. A., Perry M. D., Tan P. S., Hill A. P., Kuchel P. W., Vandenberg J. I. (2012) The S4-S5 linker acts as a signal integrator for HERG K+ channel activation and deactivation gating. PLoS One 7, e31640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Tan P. S., Perry M. D., Ng C. A., Vandenberg J. I., Hill A. P. (2012) Voltage-sensing domain mode shift is coupled to the activation gate by the N-terminal tail of hERG channels. J. Gen. Physiol. 140, 293–306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lanigan M. D., Kalman K., Lefievre Y., Pennington M. W., Chandy K. G., Norton R. S. (2002) Mutating a critical lysine in ShK toxin alters its binding configuration in the pore-vestibule region of the voltage-gated potassium channel, Kv1.3. Biochemistry 41, 11963–11971 [DOI] [PubMed] [Google Scholar]
- 25. Schreiber G., Fersht A. R. (1995) Energetics of protein-protein interactions: analysis of the barnase-barstar interface by single mutations and double mutant cycles. J. Mol. Biol. 248, 478–486 [DOI] [PubMed] [Google Scholar]
- 26. Guex N., Peitsch M. C. (1997) SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis 18, 2714–2723 [DOI] [PubMed] [Google Scholar]
- 27. Bordoli L., Kiefer F., Arnold K., Benkert P., Battey J., Schwede T. (2009) Protein structure homology modeling using SWISS-MODEL workspace. Nat. Protoc. 4, 1–13 [DOI] [PubMed] [Google Scholar]
- 28. Arnold K., Bordoli L., Kopp J., Schwede T. (2006) The SWISS-MODEL Workspace: A web-based environment for protein structure homology modelling. Bioinformatics 22, 195–201 [DOI] [PubMed] [Google Scholar]
- 29. Huang C. C., Couch G. S., Pettersen E. F., Ferrin T. E. (1996) Chimera: an extensible molecular modeling application constructed using standard components. Pac. Symp. Biocomput. 1, 724 [Google Scholar]
- 30. Ng C. A., Ke Y., Perry M. D., Tan P. S., Hill A. P., Vandenberg J. I. (2013) C-terminal β9-strand of the cyclic nucleotide-binding homology domain stabilizes activated states of Kv11.1 channels. PLoS One 8, e77032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Case D. A., Darden T. A., Cheatham I., T. E., Simmerling C. L., Wang J., Duke R. E., Luo R., Walker R. C., Zhang W., Merz K. M., Roberts B., Hayik S., Roitberg A., Seabra G., Swails J., Goetz A. W., Kolossváry I., Wong K. F., Paesani F., Vanicek J., Wolf R. M., Liu J., Wu X., Brozell S. R., Steinbrecher T., Gohlke H., Cai Q., Ye X., Wang J., Hsieh M.-J., Cui G., Roe D. R., Mathews D. H., Seetin M. G., Salomon-Ferrer R., Sagui C., Babin V., Luchko T., Gusarov S., Kovalenko A., Kollman P. A. (2012) AMBER 12, University of California, San Francisco [Google Scholar]
- 32. Chen J., Zou A., Splawski I., Keating M. T., Sanguinetti M. C. (1999) Long QT syndrome-associated mutations in the Per-Arnt-Sim (PAS) domain of HERG potassium channels accelerate channel deactivation. J. Biol. Chem. 274, 10113–10118 [DOI] [PubMed] [Google Scholar]
- 33. Wang J., Myers C. D., Robertson G. A. (2000) Dynamic control of deactivation gating by a soluble amino-terminal domain in HERG K+ channels. J. Gen. Physiol. 115, 749–758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Al-Owais M., Bracey K., Wray D. (2009) Role of intracellular domains in the function of the herg potassium channel. Eur. Biophys. J. 38, 569–576 [DOI] [PubMed] [Google Scholar]
- 35. Gianulis E. C., Liu Q., Trudeau M. C. (2013) Direct interaction of eag domains and cyclic nucleotide-binding homology domains regulate deactivation gating in hERG channels. J. Gen. Physiol. 142, 351–366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. de la Peña P., Alonso-Ron C., Machín A., Fernández-Trillo J., Carretero L., Domínguez P., Barros F. (2011) Demonstration of physical proximity between the N terminus and the S4-S5 linker of the human ether-a-go-go-related gene (hERG) potassium channel. J. Biol. Chem. 286, 19065–19075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Li Q., Gayen S., Chen A. S., Huang Q., Raida M., Kang C. (2010) NMR solution structure of the N-terminal domain of hERG and its interaction with the S4-S5 linker. Biochem. Biophys. Res. Commun. 403, 126–132 [DOI] [PubMed] [Google Scholar]
- 38. Muskett F. W., Thouta S., Thomson S. J., Bowen A., Stansfeld P. J., Mitcheson J. S. (2011) Mechanistic insight into human ether-a-go-go-related gene (hERG) K+ channel deactivation gating from the solution structure of the EAG domain. J. Biol. Chem. 286, 6184–6191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Brelidze T. I., Gianulis E. C., DiMaio F., Trudeau M. C., Zagotta W. N. (2013) Structure of the C-terminal region of an ERG channel and functional implications. Proc. Natl. Acad. Sci. U.S.A. 110, 11648–11653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Brelidze T. I., Carlson A. E., Sankaran B., Zagotta W. N. (2012) Structure of the carboxy-terminal region of a KCNH channel. Nature 481, 530–533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Marques-Carvalho M. J., Sahoo N., Muskett F. W., Vieira-Pires R. S., Gabant G., Cadene M., Schönherr R., Morais-Cabral J. H. (2012) Structural, biochemical, and functional characterization of the cyclic nucleotide binding homology domain from the mouse EAG1 potassium channel. J. Mol. Biol. 423, 34–46 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.