

Background: Loss of phospholamban (PLB) C-terminal residues causes cardiomyopathy in humans.
Results: Deletion or mutation of C-terminal residues significantly altered PLB structure and the structure of the PLB-SERCA regulatory complex.
Conclusion: PLB C-terminal residues determine the localization of the N terminus of PLB on SERCA.
Significance: PLB C terminus is an important structural determinant that affects localization of PLB N terminus at a remote location on SERCA.
Keywords: Calcium ATPase, Confocal Microscopy, Fluorescence Resonance Energy Transfer (FRET), Fret, Heart Failure, Membrane Biophysics, Membrane Protein, Sarcoplasmic Reticulum, Sarcoplasmic Reticulum (SR)
Abstract
To determine the structural and regulatory role of the C-terminal residues of phospholamban (PLB) in the membranes of living cells, we fused fluorescent protein tags to PLB and sarco/endoplasmic reticulum calcium ATPase (SERCA). Alanine substitution of PLB C-terminal residues significantly altered fluorescence resonance energy transfer (FRET) from PLB to PLB and SERCA to PLB, suggesting a change in quaternary conformation of PLB pentamer and SERCA-PLB regulatory complex. Val to Ala substitution at position 49 (V49A) had particularly large effects on PLB pentamer structure and PLB-SERCA regulatory complex conformation, increasing and decreasing probe separation distance, respectively. We also quantified a decrease in oligomerization affinity, an increase in binding affinity of V49A-PLB for SERCA, and a gain of inhibitory function as quantified by calcium-dependent ATPase activity. Notably, deletion of only a few C-terminal residues resulted in significant loss of PLB membrane anchoring and mislocalization to the cytoplasm and nucleus. C-terminal truncations also resulted in progressive loss of PLB-PLB FRET due to a decrease in the apparent affinity of PLB oligomerization. We quantified a similar decrease in the binding affinity of truncated PLB for SERCA and loss of inhibitory potency. However, despite decreased SERCA-PLB binding, intermolecular FRET for Val49-stop (V49X) truncation mutant was paradoxically increased as a result of an 11.3-Å decrease in the distance between donor and acceptor fluorophores. We conclude that PLB C-terminal residues are critical for localization, oligomerization, and regulatory function. In particular, the PLB C terminus is an important determinant of the quaternary structure of the SERCA regulatory complex.
Introduction
Heart failure is a leading cause of mortality, affecting an estimated 26 million people worldwide and up to 6 million people in the United States (1). Dilated cardiomyopathy and hypertrophic cardiomyopathy are the primary causes of heart failure (2, 3) and are often associated with mutations in genes encoding cardiac calcium (Ca2+)-handling proteins including phospholamban (PLB)5 (3–12). PLB is a homopentameric, integral sarcoplasmic reticulum membrane protein that upon deoligomerization into active monomers reversibly inhibits sarco/endoplasmic reticulum calcium ATPase (SERCA) (13–15), thereby directly regulating cardiac Ca2+ kinetics and contractility (16–21). PLB has a “helix-loop-helix” tertiary structure consisting of the N-terminal cytosolic domain IA (residues 1–16), flexible linker (residues 17–22), domain IB (residues 23–30), and C-terminal transmembrane (TM) domain II (residues 31–52) (22, 23). The C-terminal TM domain is highly conserved among species (13) and may be important for PLB oligomerization (24, 25) and SERCA regulation (26). A naturally occurring missense mutation in PLB caused by substitution of a stop codon for Leu39 (L39X) results in loss of PLB protein in humans, resulting in dysregulation of sarcoplasmic reticulum Ca2+ cycling, dilated cardiomyopathy, hypertrophic cardiomyopathy, heart failure, and premature death (5–7). We have shown previously that truncating the C terminus of PLB midway through its TM domain by L39X mutation greatly reduced PLB oligomerization and SERCA binding (27). In the present study, we investigated the role of C-terminal residues of PLB in membrane anchoring, localization, PLB oligomerization, and SERCA regulation. The results provide insight into the structural and functional consequences of mutating or truncating the TM domain and reveal an unexpected role for PLB C-terminal residues in determining the quaternary conformation of the PLB-SERCA regulatory complex.
EXPERIMENTAL PROCEDURES
Molecular Biology and Cell Culture
A series of truncation mutants of PLB were constructed by introducing stop codons at residues 52, 51, 50, 49, 48, 39, 38, or 33 (Fig. 1). Individual or multiple alanine (Ala) substitution mutants were generated by replacing the residues 52, 51, 50, and/or 49 by Ala. mCerulean (Cer) or enhanced yellow fluorescent protein (YFP) was fused to the N terminus of canine PLB or canine SERCA2a as described previously (15, 27, 28). Deletion or Ala substitution mutants of PLB were generated using the QuikChange IIXL site-directed mutagenesis kit (Stratagene, La Jolla, CA) and custom oligonucleotide primers (Eurofins MWG Operon). The nucleotide sequences of all the constructs were verified by DNA sequencing (ACGT, Inc.). AAV-293 cells were cultured in complete DMEM growth medium with 10% fetal bovine serum, 1% l-glutamine and incubated at 37 °C under 5% CO2. Transient transfection of cultured AAV-293 cells was performed using the MBS mammalian transfection kit (Stratagene, La Jolla, CA). Cells were co-transfected with plasmids encoding Cer-PLB and YFP-PLB or Cer-SERCA and YFP-PLB with a molar ratio of 1:5 or 1:20, respectively (15, 27, 28). Following transfection, the cells were subjected to mild trypsinization, plated on poly-d-lysine-coated glass bottom dishes, and allowed to adhere for 2 h before imaging as described previously (29).
FIGURE 1.

Schematic representation of C-terminal mutants of PLB. A, the amino acid sequence of PLB-WT showing the N-terminal cytosolic domain IA (residues 1–16), flexible linker (residues 17–22), domain IB (residues 23–30), and the C-terminal TM domain (residues 31–52). Cer or YFP was fused to the N terminus. B, the structure of PLB monomer highlighting the mutation sites. The thickness of the lipid bilayer is ∼40 Å.
Quantification of PLB/SERCA Function
SERCA and PLB were co-reconstituted as described previously (30). SERCA1a was purified from rabbit skeletal muscle sarcoplasmic reticulum using affinity chromatography (31, 32), and recombinant PLB was purified from Escherichia coli using a two-step method (30). The purified proteoliposomes yielded a final molar ratio of 1:4.5:120 SERCA:PLB:lipids. The SERCA and PLB concentrations were determined by quantitative SDS-PAGE, and lipid concentration was determined by phosphate assay (33, 34). Ca2+-dependent ATPase activities of SERCA co-reconstituted with PLB were determined using a coupled enzyme assay as described previously (35). All co-reconstituted PLB Ala and truncation mutants were compared with a negative control (SERCA alone) and a positive control (SERCA with wild-type PLB). A minimum of three independent reconstitutions and activity assays were performed for each mutant, and the ATPase activity was measured over a range of Ca2+ concentrations (0.1–10 μm). The Ca2+ concentration at half-maximal activity (KCa) and the maximal activity (Vmax) were calculated based on non-linear least square fitting of the activity data to the Hill equation using Sigma Plot (SPSS Inc., Chicago, IL).
Fluorescence Resonance Energy Transfer (FRET) Quantification
PLB oligomerization and interaction with SERCA were quantified by acceptor sensitization FRET (E-FRET) (36) as described previously (28, 29). MetaMorph software was used to acquire a montage of 48 images using a motorized stage (Prior, Rockland, MA). Focus was automatically maintained by an optical feedback system (Perfect Focus System, Nikon), and images were acquired using a 40× objective having a numerical aperture of 0.75. The cells were automatically selected based on criteria including diameter between 40 and 100 μm, minimum fluorescent area of 50 μm2, and average intensity of 100 counts above background. The average intensities of each channel, Cer, YFP, and FRET (Cer excitation/YFP emission), were then transferred to a spreadsheet for quantification of FRET efficiency. After subtracting the background, FRET efficiency was calculated according to the following formula: E = [IDA − a(IAA) − d(IDD)]/[IDA − a(IAA) + (G − d) (IDD)] where IDD is the intensity of fluorescence emission from the donor channel (472/30 nm) with excitation of 427/10 nm, IAA is the intensity of fluorescence emission from the acceptor channel (542/27 nm) with excitation of 504/12 nm, and IDA is the intensity of fluorescence emission detected in the FRET channel (542/27 nm) with excitation of 427/10 nm. The constants a and d are cross-talk coefficients determined from acceptor-only or donor-only control samples, respectively: a = IDA/IAA, and d = IDA/IDD. We obtained values of 0.083 and 0.69 for a and d, respectively. G represents the ratio of the sensitized emission to the corresponding amount of donor recovery, which was determined to be 4.3. Automated quantification of fluorescence intensity was done using the Multi Wavelength Cell Scoring application module in MetaMorph (29).
“In-cell” Binding Assay
An in-cell binding assay was performed to estimate the parameters related to structure and binding affinity as described previously (9, 27–29, 37). Briefly, the FRET efficiency of individual cells coexpressing Cer-PLB/YFP-PLB or Cer-SERCA/YFP-PLB was plotted against relative protein concentration quantified from the observed YFP fluorescence intensities. The concentration dependence of FRET was fit to a hyperbolic curve of the form y = (FRETmax)x/(KD + x) with all parameters independently fit where y is the observed FRET efficiency and x is the protein concentration in the cell in arbitrary units. FRETmax is the intrinsic FRET of the protein complex and a measure of average distances between the binding partners, providing structural information. KD is the protein concentration that yields half-FRETmax and represents the dissociation constant of the protein complex, providing an estimate of the apparent binding affinity. KD1 is the apparent dissociation constant of the PLB-PLB oligomer, and KD2 is the apparent dissociation constant of the PLB-SERCA regulatory complex. Each binding curve was developed by using an average of ∼2000 cells. Probe separation distance, the distance between donor and acceptor fluorophores (R), for the SERCA-PLB regulatory complex was calculated using the Förster equation (38): R = (R0)[(1/E) −1]1/6 where R0 is the Förster radius and E is the measured FRETmax. Intrapentameric probe separation distance was calculated from FRETmax using the MatLab application assuming an oligomer subunit number of 5 (pentamer) as described previously (27). The average acceptor molar fraction was calculated to be 0.92 ± 0.01 for WT-PLB, 0.89 ± 0.02 for the alanine substitution mutants, and 0.88 ± 0.01 for the truncation mutants. For estimation of probe separation distances for both the pentamer and regulatory complex, the Förster radius of 49.8 Å was used for the Cer-YFP pair (39), and 4% nonspecific FRET was subtracted from the measured FRETmax values. Previously we estimated nonspecific FRET to be 4% as determined from competition with unlabeled PLB or with a fluorescently tagged PLB that is unable to participate in FRET (9, 27).
Fluorescence Microscopy
To quantify the membrane localization of PLB, cells were cotransfected with Cer-SERCA and YFP-PLB truncation mutants at a 1:1 ratio and subjected to confocal imaging using an inverted Leica TCS SP5 confocal microscope with a 63× water immersion objective. Cer and YFP were sequentially excited at 458 and 514 nm, respectively. The membrane partitioning of YFP-PLB truncation mutants was evaluated by comparison with Cer-SERCA. The apparent membrane partition coefficient for PLB and all the truncation mutants of PLB was quantified as the ratio of YFP fluorescence intensity in the endoplasmic reticulum (ER) region (perinuclear) to nuclear fluorescence intensity. Solubilization of PLB was also quantified using wide field fluorescence microscopy to measure the loss of fluorescence from cells permeabilized with 100 μg/ml saponin. Cell-impermeant nuclear stain propidium iodide was used to verify permeabilization. Cells expressing Cer-SERCA and YFP-PLB truncation mutants were preincubated with 2 μg/ml propidium iodide and imaged for Cer, YFP, and propidium iodide fluorescence during the course of saponin permeabilization.
Computational Modeling
Molecular dynamics simulation studies of PLB embedded in a lipid bilayer were performed for full-length PLB wild-type (WT) pentamer and heart failure mutant L39X. The NMR structure of the wild-type PLB pentamer was obtained from the Research Collaboratory for Structural Bioinformatics Protein Data Bank (40) (Protein Data Bank code 2KYV (23)). Molecular dynamics simulations were performed with GROMACS (42, 43) using the CHARMM all-atom parameter set 27 (44, 45). The pentamer was energy-minimized in vacuum to eliminate unfavorable interactions and placed into a palmitoyloleoylphosphatidylcholine lipid bilayer. Lipids overlapping with the pentamer were removed, and periodic boundary conditions were applied. The particle mesh Ewald method was used to calculate the electrostatic interactions (46, 47). van der Waals interactions were reduced to zero by switch truncation applied from 10 to 12 Å. The molecular dynamics simulation was carried out with an integration time step of 2 fs. To reach the target temperature (300 K) and pressure (1 bar), the Berendsen method was used with relaxation times of 0.1 ps (48). After 1-ns equilibration, the production run was performed in the NPT ensemble using the Nose-Hoover thermostat (49, 50) and the Parrinello-Rahman barostat (51, 52) with relaxation times of 1.0 ps. The coordinates of the trajectory were saved every 1 ps. The production run was carried out for 100 ns.
Statistical Analysis
All experiments were independently repeated three or four times for each sample. Errors are reported as standard error, and statistical significance was evaluated using Student's t test where p < 0.05 was considered significant. The comparison of KCa and Vmax was carried out using one-way analysis of variance (between subjects) followed by the Holm-Sidak test for pairwise comparisons.
RESULTS
Functional Role of C-terminal Residues in Regulation of SERCA Function
To investigate the role of C-terminal residues (Fig. 1) in determining the inhibitory potency of PLB, we reconstituted SERCA with WT or with C-terminal Ala substitutions or truncations of PLB. Previous Ala substitution of individual residues in this region (Val49-Met-Leu-Leu52) revealed little change in inhibitory potency for the L52A or M50A mutants and gain of function for the L51A and V49A mutants (53). This gain-of-function effect of the individual Ala substitutions is visible in Fig. 2 as an increase in KCa for L51A and V49A compared with WT-PLB. Multiple Ala mutations in this C-terminal region also maintained PLB inhibitory function with a gain-of-function effect for Ala substitution of all 4 residues 49–52. Interestingly, there appeared to be a nexus for gain of function at Val49 for both individual and multiple alanine substitutions. In contrast, deletion of these same residues, which shortens the PLB TM helix, resulted in a loss of function. Deletion of only 1 C-terminal residue, Leu52, yielded 50% of the inhibitory potency of wild-type PLB, and further deletions caused complete loss of PLB function. Deletion of the last 2–3 residues decreased PLB inhibitory potency such that the observed KCa values were not significantly different from SERCA alone (Fig. 2 and Table 1). The data indicate that the C-terminal residues of PLB are important determinants for SERCA inhibition and that even small deletions have a particularly deleterious effect.
FIGURE 2.

C-terminal alanine substitutions resulted in gain of PLB regulatory function and truncation of C-terminal residues resulted in loss of PLB regulatory function. Calcium-dependent ATPase activity assays of SERCA co-reconstituted with WT and mutants of PLB were performed. The horizontal line at the bottom represents KCa of SERCA reconstituted alone, and the horizontal line at the top represents KCa of SERCA co-reconstituted with WT-PLB. Error bars represent S.E.
TABLE 1.
Summary of kinetic parameters of calcium-dependent ATPase activity of SERCA co-reconstituted with WT and mutant constructs of PLB: effect of C-terminal alanine substitutions and truncation mutations on KCa and Vmax
| Kinetic parameters of calcium-dependent ATPase activity of SERCA |
||
|---|---|---|
| KCa | Vmax | |
| μm | μmol mg−1 min−1 | |
| Controls | ||
| SERCA | 0.46 ± 0.02 | 4.1 ± 0.1 |
| WT-PLB | 0.88 ± 0.03 | 6.1 ± 0.1 |
| Individual alanine substitutions | ||
| L52A | 0.69 ± 0.04 | 5.0 ± 0.1 |
| L51A | 1.12 ± 0.08 | 4.3 ± 0.1 |
| M50A | 0.69 ± 0.02 | 4.6 ± 0.1 |
| V49A | 0.99 ± 0.05 | 5.1 ± 0.1 |
| Multiple alanine substitutions | ||
| L51A/L52A | 0.72 ± 0.04 | 3.4 ± 0.1 |
| M50A/L51A/L52A | 0.85 ± 0.09 | 2.3 + 0.1 |
| V49A/M50A/L51A/L52A | 1.14 ± 0.11 | 1.7 ± 0.2 |
| Sequential truncation mutations | ||
| L52X | 0.72 ± 0.03 | 4.9 ± 0.1 |
| L51X | 0.45 ± 0.06 | 3.0 ± 0.1 |
| M50X | 0.52 ± 0.02 | 3.6 ± 0.1 |
Ala Substitution Mutations of PLB C-terminal Residues Alter PLB Pentamer Structure and Oligomerization Affinity
To investigate the role of the C-terminal residues in determining the structure and affinity of the PLB pentamer, we quantified intraoligomeric FRET for mixed pentamers of Cer- and YFP-PLB. Replacement of C-terminal residues with a single Ala substitution generally decreased average FRET efficiency compared with WT (Fig. 3A and Table 2). To determine the relative contributions of pentamer structure change or a change in the degree of PLB oligomerization to the observed changes in average FRET, we performed an in-cell binding assay in which FRET is quantified from a heterogeneous population of transfected cells expressing a wide range of concentrations of PLB. Fig. 3B shows that cells expressing high concentrations of PLB exhibited higher FRET than cells with a low expression level. FRET increased with [protein] to a maximum level (FRETmax), and this relationship was well described by a hyperbolic fit. FRETmax was modestly increased for L52A, the last residue of the helical TM domain of PLB (Fig. 3C, red). L51A also showed a small increase in FRETmax (Fig. 3C, blue), but this value must be viewed with caution as the FRET versus [protein] relationship never achieved maximal FRET for this mutant (Fig. 3B). A failure to saturate is often observed for highly monomeric mutants of PLB as a result of increased nonspecific FRET between non-interacting monomers. Thus, the fitted value of FRETmax is compromised by this nonspecific FRET contribution. Consistent with this non-saturating FRET relationship, we observed a right shift of the L51A FRET versus [protein] binding curve (Fig. 3B), suggesting a decreased oligomerization affinity. KD1 was significantly increased for all Ala mutants, indicating a decreased oligomerization affinity. We observed a >1.5-fold increase in KD1 for L52A and a >4-fold increase in KD1 for L51A compared with WT (Fig. 3D) as quantified from a hyperbolic fit of the data in Fig. 3B. Next, moving up the TM helix, we investigated the effect of an M50A substitution. This mutant showed no change in pentamer structure (Fig. 3C) but a 1.5-fold increase in KD1 (Fig. 3D) as seen from the right-shifted binding curve of M50A (Fig. 3B, pink) relative to WT (Fig. 3B, black) without a change in maximal FRET. The data suggest that the mutant destabilizes oligomerization without altering the structure of the pentamer. Finally V49A showed a very large decrease in FRETmax (Fig. 3C) consistent with an increase in the average separation of N-terminal fluorescent tags of 3 Å and a 40% increase in the affinity of oligomerization for this mutant (Fig. 3D). The results are summarized in Table 2. The data demonstrate that the position of the fluorescent protein, fused to the N terminus (on the cytoplasmic side of the bilayer), is altered by substitution of Leu or Val with Ala at remote sites in the C terminus (on the luminal side of the bilayer).
FIGURE 3.

C-terminal alanine substitutions alter the oligomerization affinity and structure of PLB pentamer. Shown are the effects of C-terminal alanine substitution mutants of PLB on average intrapentameric FRET efficiency (A), in-cell intrapentameric FRET efficiency (B), FRETmax for the pentamer (C), and KD1 (D). *, p < 0.05. Error bars represent S.E. AU, arbitrary units.
TABLE 2.
Summary of quantitative FRET data: effect of C-terminal alanine substitutions and truncation mutations on PLB intrapentameric FRET efficiency and SERCA-PLB FRET efficiency
AU, arbitrary units; ND, not determined.
| PLB-PLB FRET |
SERCA-PLB FRET |
|||||||
|---|---|---|---|---|---|---|---|---|
| Average FRET | FRETmax | KD1 | R | Average FRET | FRETmax | KD2 | R | |
| % | % | AU | Å | % | % | AU | Å | |
| PLB alanine mutants | ||||||||
| WT | 35.4 ± 0.2 | 42.2 ± 0.4 | 1.8 ± 0.1 | 61.7 ± 0.4 | 12.5 ± 0.2 | 23.8 ± 0.6 | 9.7 ± 0.6 | 62.9 ± 0.5 |
| L52A | 37.3 ± 0.4 | 47.8 ± 0.6a | 2.8 ± 0.2a | 59.6 ± 0.6a | 12.3 ± 0.2 | 24.8 ± 0.8 | 10.6 ± 0.8 | 62.2 ± 0.3 |
| L51A | 28.8 ± 0.4a | 46.2 ± 1.0a | 7.7 ± 0.5a | 60.3 ± 0.3a | 15.7 ± 0.3a | 38.7 ± 1.4a | 17.9 ± 1.3a | 55.4 ± 0.9a |
| M50A | 29.3 ± 0.3a | 39.6 ± 0.6 | 2.7 ± 0.2a | 62.1 ± 0.6 | 12.8 ± 0.3a | 26.8 ± 1.1 | 11.0 ± 1.0 | 61.1 ± 1.2 |
| V49A | 23.6 ± 0.3a | 32.0 ± 0.5a | 2.5 ± 0.2a | 64.6 ± 0.2a | 17.3 ± 0.2a | 29.3 ± 0.5a | 2.6 ± 0.1a | 59.6 ± 0.4a |
| PLB truncation mutants | ||||||||
| WT | 42.4 ± 0.3 | 50.8 ± 0.5 | 0.3 ± 0.02 | 58.4 ± 0.4 | 13.7 ± 0.2 | 25.4 ± 0.7 | 9.3 ± 0.6 | 61.9 ± 0.4 |
| L52X | 40.5 ± 0.5 | 53.3 ± 0.6 | 0.6 ± 0.03a | 57.4 ± 0.1 | 13.8 ± 0.3 | 30.8 ± 0.9a | 16.4 ± 1.0a | 58.9 ± 0.4a |
| L51X | 39.0 ± 0.4 | 54.2 ± 0.6 | 0.8 ± 0.04a | 56.4 ± 0.1a | 17.1 ± 0.4a | 41.3 ± 1.5a | 18.8 ± 1.4a | 54.3 ± 0.9a |
| M50X | 25.1 ± 0.4a | 50.9 ± 1.0 | 2.5 ± 0.1a | 57.7 ± 0.2 | 19.6 ± 0.5a | 43.4 ± 1.2a | 13.8 ± 0.8a | 53.5 ± 0.3a |
| V49X | 23.7 ± 0.4a | 56.6 ± 2.2 | 3.6 ± 0.3a | 55.9 ± 0.5a | 16.3 ± 0.5 | 51.7 ± 2.5a | 22.5 ± 2.0a | 50.6 ± 0.7a |
| I48X | 9.0 ± 0.3a | 19.9 ± 1.3a | ND | ND | 5.8 ± 0.3a | 25.1 ± 3.4 | ND | ND |
| L39X | 5.0 ± 0.2a | 9.2 ± 0.8a | ND | ND | 3.5 ± 0.1a | 5.63 ± 0.3a | ND | ND |
| I38X | 8.0 ± 0.1a | 11.0 ± 0.4a | ND | ND | 3.4 ± 0.1a | 5.73 ± 0.3a | ND | ND |
| I33X | 9.0 ± 0.1a | 13.0 ± 0.4a | ND | ND | 6.6 ± 0.2a | 7.71 ± 0.3a | ND | ND |
a p < 0.05.
Mutation of PLB C-terminal Residues Alters Regulatory Complex Quaternary Structure and PLB-SERCA Binding Affinity
To determine how N-terminal residues affect the structure and affinity of the PLB-SERCA regulatory complex, we measured FRET from the N-terminal Cer tag on SERCA2a to YFP-PLB. Fig. 4A shows that average FRET was increased by L51A and V49A mutants. In-cell binding assays revealed the relative changes in binding affinity and structure for these mutants (Fig. 4B). As observed in the oligomerization binding assay (Fig. 3B), L51A showed poor saturation (Fig. 4B) consistent with nonspecific FRET from an increased population of monomers (increased KD1; Fig. 3D). Thus, the FRETmax value for this mutant is not a clear representation of regulatory complex structure. The other mutant with a significant change in SERCA binding was V49A, which showed a 23% increase in FRETmax, suggesting a very compact regulatory complex conformation, and a 73% decrease in KD2 (Fig. 4D), indicating an increase in the apparent affinity for SERCA. The observed increase in SERCA binding is in harmony with previous reports that suggest increased inhibitory potency for this mutant (54, 55).
FIGURE 4.
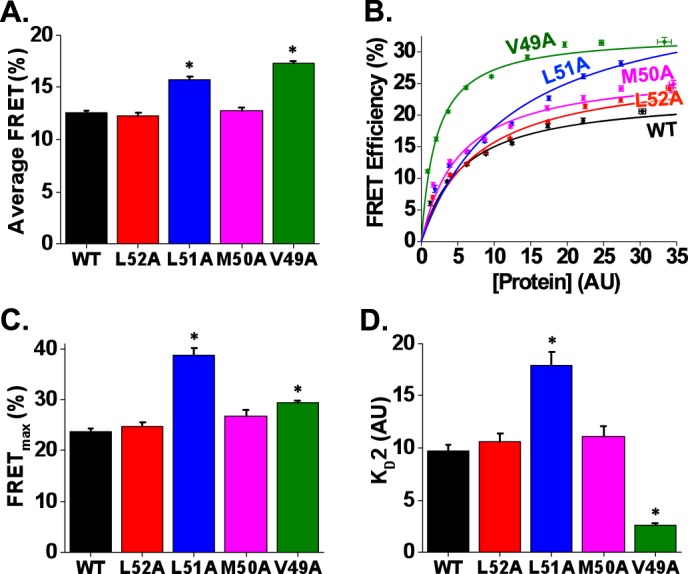
C-terminal alanine substitutions alter binding affinity and structure of SERCA-PLB regulatory complex. Shown are the effects of C-terminal alanine substitution mutants of PLB on average SERCA-PLB FRET efficiency (A), in-cell SERCA-PLB FRET efficiency (B), FRETmax for the SERCA-PLB complex (C), and KD2 (D). *, p < 0.05. Error bars represent S.E. AU, arbitrary units.
C-terminal Residues Are Critical for PLB Membrane Localization
We previously observed solubilization of PLB by the human heart failure missense mutation L39X (27), and we anticipated that smaller C-terminal deletions of the PLB TM domain could likewise disrupt anchoring of the protein in the membrane. To verify this, the relative partitioning of PLB in the aqueous cytoplasm and the ER bilayer was assessed with confocal microscopy. Although single Ala substitutions did not significantly alter the localization of PLB expressed in AAV-293 cells (not shown), we observed a significant degree of mislocalization of PLB with C-terminal truncations as shown in Fig. 5. An overlay of images revealed co-localization of Cer-SERCA (Fig. 5A, cyan) and YFP-WT-PLB truncation mutants (Fig. 5A, yellow). Successive truncations caused mislocalization of PLB to the cytoplasm and nucleus (Fig. 5A) as quantified from the apparent membrane partition coefficient (ratio of ER (perinuclear) fluorescence to nuclear fluorescence)) (Fig. 5B). Interestingly, deleting only 1 C-terminal residue of PLB of the 22 TM domain residues resulted in a significant loss of ER membrane localization, which was nearly abolished by deletion of more than 4 residues. Cer-SERCA localization was not changed (Fig. 5A). To determine whether the observed mislocalization was due to solubilization of PLB, we selectively permeabilized the plasma membrane of cells using saponin (27). Cer-SERCA ER localization was not changed during the course of saponin permeabilization, but truncated YFP-PLBL39X rapidly diffused out of the cell into the surrounding medium (Fig. 5C and supplemental Video 1). As expected, the degree of PLB solubilization depended upon the severity of the PLB truncation. Longer PLB constructs left residual fluorescence in cells after permeabilization (Fig. 5D). The apparent membrane localization of PLB mutants was quantified by dividing the ER fluorescence (membrane-bound PLB) by the total fluorescence (nuclear + ER) or by dividing the residual PLB fluorescence after saponin permeabilization by total PLB fluorescence before permeabilization. Partition equilibrium constants estimated by the localization and permeabilization methods were referred to as KL and KP, respectively. Truncations of PLB C terminus decreased both KL and KP (Fig. 5E). The alternative methods were in good agreement with a linear relationship between KL and KP (Fig. 5F), although KL exhibited a non-zero offset due to the contribution of non-membrane-bound PLB to the ER fluorescence signal. The data are summarized in Table 3.
FIGURE 5.

Progressive truncation of the C-terminal residues of PLB resulted in increased mislocalization to the cytoplasm and nucleus. A, confocal microscopic images of AAV-293 cells expressing Cer-SERCA (top) cotransfected with YFP-PLB truncation mutants (middle). Bottom, overlay of Cer-SERCA and YFP-PLB truncation mutants. Cer-SERCA served as a marker for ER membrane localization. Scale bar, 5 μm. B, successive truncations caused a progressive decrease in the apparent membrane partition coefficient (ratio of ER fluorescence/nuclear fluorescence). Error bars represent cell-to-cell variability; an average of 40–50 cells was used for each mutant. C, plasma membrane permeabilization resulted in complete loss of YFP-PLBL39X, no change in Cer-SERCA fluorescence, and increased propidium (Prop.) iodide staining. D, diffusion of YFP-PLB truncation mutants from cells after saponin (Sap) permeabilization. E, successive truncations lead to a decrease in partition equilibrium constants estimated either by localization (KL) or permeabilization (KP). Error bars represent S.E. App., apparent; AU, arbitrary units. F, there was good agreement between KL and KP.
TABLE 3.
Summary of quantitative localization data: effect of deleting the C-terminal residues of PLB on partition equilibrium constants estimated either by localization (KL) or permeabilization (KP)
| PLB truncation mutants | PLB C-terminal residues deleted | Partition equilibrium constant |
|
|---|---|---|---|
| Localization (KL) | Permeabilization (KP) | ||
| WT | 0 | 1 ± 0.002 | 1 ± 0.03 |
| L52X | 1 | 0.98 ± 0.004 | 0.96 ± 0.04 |
| L51X | 2 | 0.98 ± 0.002 | 0.97 ± 0.03 |
| M50X | 3 | 0.92 ± 0.01 | 0.82 ± 0.02 |
| V49X | 4 | 0.87 ± 0.01 | 0.85 ± 0.03 |
| I48X | 5 | 0.71 ± 0.01 | 0.47 ± 0.06 |
| L39X | 14 | 0.44 ± 0.01 | 0.05 ± 0.02 |
| I38X | 15 | 0.43 ± 0.01 | 0.03 ± 0.01 |
| I33X | 20 | 0.39 ± 0.01 | 0.04 ± 0.01 |
Molecular Dynamics of PLB Pentamer Structure
To investigate the role of C-terminal residues in PLB structure and membrane anchoring, we performed all-atom molecular dynamics simulations and quantified changes in root mean square deviation, van der Waals and electrostatic interactions, and hydrogen bonding. The root mean square deviation of Cα atoms of the heart failure truncation mutant L39X (Fig. 6A, colored traces) was markedly increased compared with WT, which consistently showed a stable structure with little deviation from the NMR solution (Fig. 6A, black traces). We observed no statistically significant difference in the coulombic interactions but found that L39X lacks van der Waals interactions needed for stabilizing the PLB oligomer (Fig. 6B). Because most of the residues in TM helices are hydrophobic, hydrogen bonding between PLB monomers is not a significant contributor to pentamer stability. Consistent with expectation, the TM domain of the WT protein was retained in the bilayer, but the L39X mutant TM domain translocated out of the bilayer within a few nanoseconds and was fully solubilized by the end of the simulation (Fig. 6C and supplemental Video 2).
FIGURE 6.

Molecular dynamics simulation showed instability of L39X-PLB membrane insertion and oligomer structure compared with WT. A, 100-ns time trajectories show changes in root mean square deviation (RMSD) of Cα atoms. L39X, colored traces; WT, black traces. B, van der Waals and coulombic interactions. *, p < 0.05. Error bars represent S.E. C, translocation of the L39X mutant out of the bilayer.
C-terminal Residues Are Also Important for PLB Oligomerization
Deletion of any of the PLB C-terminal residues greatly decreased PLB-PLB binding in live cells as quantified by FRET between Cer and YFP fused to PLB N termini. Fig. 7A shows that PLB average intrapentameric FRET efficiency was progressively decreased upon truncating the C-terminal residues of PLB. Truncation of 4 C-terminal residues of the 22-amino acid TM domain of PLB (V49X) resulted in a 44% decrease in the average FRET efficiency (Fig. 7A). FRET was largely abolished by truncation of more than 4 residues. Thus, the FRET data underscore the importance of Val49 in determining PLB structure/function. In particular, we used the in-cell binding assay to quantify the relative contributions of altered binding and pentamer conformational change to the observed FRET change. Truncating the C-terminal residues of PLB resulted in a progressive decrease in PLB intrapentameric FRET efficiency as shown in Fig. 7B. FRETmax was unchanged for L52X, L51X, M50X, or V49X compared with WT, indicating that PLB quaternary structure was not significantly affected upon truncating up to 4 C-terminal residues (Fig. 7C and Table 2). This is in contrast with Ala substitution mutants, several of which profoundly affected PLB oligomer structure (Fig. 3C and Table 2). Truncating more than 4 residues of the 22-amino acid-long TM domain significantly decreased FRETmax. We attribute this to loss of membrane anchoring rather than a change in pentamer structure. C-terminal truncations also progressively increased the pentamer dissociation constant KD1, indicating a decrease in the affinity of PLB oligomerization (Fig. 7D). Truncating only 1 C-terminal residue resulted in a 2-fold increase in KD1 compared with WT, and binding affinity worsened with each additional residue that was deleted. We quantified a 12-fold increase in KD1 for V49X compared with WT, indicating a decrease in oligomerization affinity. The affinity for the shortest truncation mutants was too low to measure. The data are summarized in Table 2. PLB-PLB binding was reduced by loss of C-terminal residues, and this loss of binding paralleled the loss of bilayer anchoring. As predicted, colocalization of PLB protomers in the membrane was prerequisite to oligomerization.
FIGURE 7.

C-terminal truncations of PLB result in progressive loss of PLB oligomerization. A, progressive truncation of the C terminus of PLB resulted in a decrease in average FRET efficiency. B, in-cell intrapentameric FRET efficiency measurements for WT and truncation mutant constructs of PLB. C, truncating more than 4 residues resulted in a decrease in FRETmax. D, C-terminal truncations resulted in a progressive increase in KD1. *, p < 0.05. Error bars represent S.E. AU, arbitrary units; Con, control.
PLB C-terminal Residues Are Critical for Regulatory Complex Structure and Function
To determine how the C-terminal truncation of PLB affects its interaction with SERCA, we quantified FRET from Cer-SERCA to YFP-tagged truncation mutants of PLB. Surprisingly, truncation of up to 4 C-terminal residues (of 22 residues in the TM domain) significantly increased SERCA-PLB FRET efficiency (Fig. 8, A and B). This was not due to increased binding of PLB to SERCA because KD2 predictably increased with deletion of C-terminal residues (decreased binding affinity) (Fig. 8D). Truncating only 1 residue resulted in a nearly 2-fold increase in KD2, truncating 4 residues (V49X) caused an ∼2.5-fold increase in KD2, and removing more than 4 residues resulted in a binding affinity that was too low to measure (Fig. 8D). It appears that, despite the decrease in binding of truncated PLB for SERCA, the average FRET is still increased because the remaining regulatory complexes have a higher intrinsic FRET efficiency (FRETmax). Deleting only 1 C-terminal residue of PLB resulted in a 1.2-fold increase in FRETmax, and truncating 4 C-terminal residues (V49X) resulted in a greater than 2-fold increase in FRETmax (Fig. 8C) consistent with an increase in the average separation of N-terminal fluorescent tags of 11.3 Å. The data suggest that that C-terminal truncations alter the structure of PLB-SERCA regulatory complex (Table 2). The structural effect of C-terminal deletions mimics the regulatory complex conformational change due to V49A (Fig. 4C), which also decreased the separation of fluorescent protein tags positioned at least 40 Å away on the other side of the bilayer.
FIGURE 8.

C-terminal truncations of PLB alter binding affinity and structure of SERCA-PLB regulatory complex. A, truncation of up to 4 C-terminal residues of PLB resulted in increased average FRET efficiency. B, in-cell SERCA-PLB FRET efficiency measurements for WT and truncation mutant constructs of PLB. Truncation of up to 4 C-terminal residues of PLB resulted in increased FRETmax (C) and increased KD2 (D). *, p < 0.05. Error bars represent S.E. AU, arbitrary units.
DISCUSSION
Alanine substitution has been a common mutagenesis strategy for understanding how residues of PLB influence the functional regulation of SERCA (26, 53, 56). This method effectively removes the side chain of individual residues, thereby providing information on their role in SERCA inhibition by PLB. Two of the most common human mutations in PLB linked to heart failure involve a single residue deletion (Arg14 deletion) (10–12) and a multiple residue truncation (Leu39-stop) (5–7). Because a large portion of the C terminus of PLB is missing in this latter truncation variant, we wished to understand the comparative effects of mutations and truncations in this region. Because of the dramatic effects we observed on PLB function, only the last 4 residues were considered (Val49-Met-Leu-Leu52). In measurements of SERCA ATPase activity in the absence and presence of PLB, individual or multiple Ala substitutions revealed a nexus for gain of function at Val49 of PLB (Fig. 2A). The gain of function was an unexpected outcome, and it contrasted sharply with the deleterious effects of truncating these last few residues. Removal of just 1 residue severely reduced PLB function, and removal of 2 or more residues completely eliminated PLB function. Given the extreme sensitivity of PLB to truncation, we wished to understand the impact on PLB oligomerization and the regulatory interaction with SERCA.
The present observations are summarized in the schematic model in Fig. 9 with reversible equilibria indicated by black arrows and effects of mutations highlighted with red arrows. Indeed, deletion and substitution mutations had distinct effects on PLB oligomerization. Although deletions decreased PLB-PLB binding (Fig. 9B, i) without altering the pentamer structure, alanine substitutions increased or decreased PLB pentamer FRETmax consistent with a structural change (Fig. 9A, i) that alters the distance between FRET pairs. The structural details of this putative change in pentamer quaternary conformation are not clear. One possibility is that C-terminal substitutions alter PLB topology or perturb the structural equilibrium of PLB between “tense” and “relaxed” states (57, 58). It is noteworthy that the relatively conservative substitution of Val49 to Ala exerts an effect over such a long distance, altering the position of fluorescent probes that are more than 40 Å away (Fig. 1).
FIGURE 9.

Proposed model for the effect of PLB C-terminal substitution and deletion mutations on membrane localization, oligomerization, and interaction with SERCA. The WT PLB protomers and SERCA are shown in gray, and the alanine substitution (V49A) or the deletion mutant (V49X) PLB protomers are highlighted in red. The reversible equilibria are indicated by black arrows, and effects of mutations are highlighted with red arrows. A, we propose that C-terminal alanine substitution mutations of PLB result in altered pentamer structure and decreased oligomerization affinity (i), increased SERCA-PLB binding affinity (ii), and altered quaternary conformation of the SERCA-PLB regulatory complex (iii). B, in addition, C-terminal deletions of PLB result in decreased oligomerization affinity (i), decreased SERCA-PLB binding affinity (ii), altered quaternary conformation of the SERCA-PLB regulatory complex (iii), and decreased membrane anchoring (iv).
Although determining the true quantitative affinity of monomeric PLB for the pump is complicated by oligomerization (27) and differential protein localization, it is clear that substitution and deletion mutations exert opposite effects on the apparent affinity of PLB for SERCA. V49A in particular shows a significant increase in SERCA binding (Fig. 9A, ii), whereas deletions of residues progressively decreases regulatory complex formation (Fig. 9B, ii). Both deletions and substitutions of C-terminal residues caused large changes in the regulatory complex structure (Fig. 9, A and B, iii). Deleting only 1 C-terminal residue of PLB resulted in a 1.2-fold increase in FRETmax, which corresponds to a distance change from 61.9 to 58.9 Å (Table 2). Truncating 4 C-terminal residues (V49X) increased FRETmax by more than 2-fold, which corresponds to an 11.3 Å decrease in the distance between donor and acceptor fluorophores (Table 2). Similarly, some Ala substitutions increased regulatory complex intrinsic FRET with V49A showing a 23% increase in FRETmax, corresponding to a decrease in the donor-acceptor distance of 3.3 Å. The magnitude of these changes in FRET distance may be appreciated by considering that functionally significant phosphomimetic mutations altered probe separation distance by only 4 Å (37). One possible explanation for the observed structure change is that substitutions and deletions may cause misregistration of PLB in the inhibitory cleft (Fig. 9, A and B, iii). Translocation of the TM domain could permit cytoplasmic domain repositioning, accounting for the observed increase in FRET from a donor fluorophore on the SERCA N terminus. Val49 appears to be particularly important in setting the registration of the TM helix in the bilayer, affecting the disposition of the adjacent TM domain and the more distant PLB cytoplasmic domain positioned on the other side of the bilayer more than 40 Å away.
C-terminal residues are also important for membrane anchoring of PLB. Although substitutions appeared to be benign for localization, we observed that C-terminal deletions decreased PLB membrane anchoring (Fig. 9B, iv), releasing a fraction of the PLB into the cytoplasm where it can no longer participate in regulatory interactions. Previously, the C-terminal region of PLB region has been shown to be important for subcellular trafficking as increasing the length of the TM domain of PLB by adding 4 extra leucine residues to the C terminus resulted in mistargeting to the plasma membrane (59). Similarly, the C-terminal RSYQY sequence of the related SERCA regulator sarcolipin was shown to mediate its retention in the ER (60). The present results demonstrate an additional role for the PLB C terminus in protein localization. Despite the presence of many other hydrophobic residues in the PLB TM domain, luminal residues nearest the PLB C terminus are critical for membrane anchoring and structure determination of both the PLB pentamer and the PLB-SERCA regulatory complex. It is noteworthy that small deletions are so poorly tolerated. Loss of only 1 C-terminal residue from the 22-amino acid TM domain of PLB resulted in significant disruption of localization and consequently PLB regulatory interactions. Although SERCA coexpression has been shown to improve localization of sarcolipin truncation mutants (60), PLB localization was not improved by coexpression of SERCA in the present work or in previous studies (59, 61).
The most striking difference between the substitution and deletion mutants is the disparate effects on PLB inhibitory function. The loss-of-function character of the deletion mutants may be due in part to decreased membrane anchoring (Fig. 9B, iv) and decreased SERCA binding (Fig. 9B, ii). Alternatively, the putative misregistration of the PLB TM domain in the SERCA regulatory cleft may result in a non-inhibitory interaction. We and others have previously provided evidence that PLB and SERCA can interact in a non-inhibitory complex in the presence of Ca2+ (29, 62, 63) or after PLB phosphorylation (64). This physiological relief of inhibition also alters the affinity and structure of the PLB-SERCA complex (29, 62–64). In contrast to deletion mutants or physiological regulation, the structure change induced by L51A or V49A mutation is strongly gain of function (Fig. 2 and Table 1). This observation is compatible with other groups' previous observations. Although V49A was initially reported to be a loss-of-function mutation (41, 65), subsequent studies have demonstrated that V49A is a gain-of-function mutation (54). A possible mechanism for this was revealed by x-ray crystallography, which suggests that Val49 encounters steric hindrance from SERCA residue Val89 (55). The present data support the hypothesis that replacing PLB Val49 with a smaller residue can alleviate this hindrance, increasing the affinity and potency of the inhibitory interaction (19, 54, 55). It is noteworthy that deleting Val49 did not produce the same result; rather the affinity of the interaction was decreased, and even small deletions of the C-terminal residues resulted in loss of inhibitory function.
In summary, we conclude that C-terminal residues of PLB are important structural determinants as mutations of the C-terminal amino acids had significant effects on protein-protein interactions and PLB quaternary structures. It is noteworthy that both deletions and Ala substitutions of PLB C-terminal residues increased SERCA-PLB FRET but had opposite effects on SERCA inhibition by PLB. Future detailed structural studies of the PLB-SERCA regulatory complex may reveal how substitutions and deletions at the C-terminal end of PLB can strongly alter the disposition of the cytoplasmic domain on the other side of the membrane.
Supplementary Material
Acknowledgments
We are grateful for technical assistance from Chris Stefonowicz, Ryan Himes, and Marsha Pribadi and helpful discussions with Gianluigi Veglia. Molecular dynamics simulations were performed with the Loyola Research Computing Core, supported by National Institutes of Health Grant 1G20RR030939, and the Extreme Science and Engineering Discovery Environment, supported by National Science Foundation Grant OCI-1053575.
This work was supported, in whole or in part, by National Institutes of Health Grant R01 HL092321. This work was also supported by a gift from the McCormick Foundation to Loyola University Chicago.

This article contains supplemental Movies 1 and 2.
- PLB
- phospholamban
- ER
- endoplasmic reticulum
- SERCA
- sarco/endoplasmic reticulum calcium ATPase
- X
- stop codon
- L39X
- substitution of a stop codon for Leu39
- TM
- transmembrane
- Cer
- mCerulean
- KL
- partition equilibrium constant estimated by localization
- KP
- partition equilibrium constant estimated by permeabilization
- FRETmax
- maximal FRET efficiency
- KD1
- apparent dissociation constant of the PLB-PLB oligomer
- KD2
- apparent dissociation constant of the PLB-SERCA regulatory complex
- R
- distance between donor and acceptor fluorophores
- KCa
- Ca2+ concentration at half-maximal activity.
REFERENCES
- 1. Lopez-Sendon J. (2011) The heart failure epidemic. Medicographia 33, 363–369 [Google Scholar]
- 2. Towbin J. A., Bowles N. E. (2002) Molecular diagnosis of myocardial disease. Expert Rev. Mol. Diagn. 2, 587–602 [DOI] [PubMed] [Google Scholar]
- 3. Kimura A. (2008) Molecular etiology and pathogenesis of hereditary cardiomyopathy. Circ. J. 72, Suppl. A, A38–A48 [DOI] [PubMed] [Google Scholar]
- 4. Medeiros A., Biagi D. G., Sobreira T. J., de Oliveira P. S., Negrão C. E., Mansur A. J., Krieger J. E., Brum P. C., Pereira A. C. (2011) Mutations in the human phospholamban gene in patients with heart failure. Am. Heart J. 162, 1088–1095.e1 [DOI] [PubMed] [Google Scholar]
- 5. Haghighi K., Kolokathis F., Pater L., Lynch R. A., Asahi M., Gramolini A. O., Fan G. C., Tsiapras D., Hahn H. S., Adamopoulos S., Liggett S. B., Dorn G. W., 2nd, MacLennan D. H., Kremastinos D. T., Kranias E. G. (2003) Human phospholamban null results in lethal dilated cardiomyopathy revealing a critical difference between mouse and human. J. Clin. Investig. 111, 869–876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chiu C., Tebo M., Ingles J., Yeates L., Arthur J. W., Lind J. M., Semsarian C. (2007) Genetic screening of calcium regulation genes in familial hypertrophic cardiomyopathy. J. Mol. Cell. Cardiol. 43, 337–343 [DOI] [PubMed] [Google Scholar]
- 7. Landstrom A. P., Adekola B. A., Bos J. M., Ommen S. R., Ackerman M. J. (2011) PLN-encoded phospholamban mutation in a large cohort of hypertrophic cardiomyopathy cases: summary of the literature and implications for genetic testing. Am. Heart J. 161, 165–171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Schmitt J. P., Kamisago M., Asahi M., Li G. H., Ahmad F., Mende U., Kranias E. G., MacLennan D. H., Seidman J. G., Seidman C. E. (2003) Dilated cardiomyopathy and heart failure caused by a mutation in phospholamban. Science 299, 1410–1413 [DOI] [PubMed] [Google Scholar]
- 9. Ha K. N., Masterson L. R., Hou Z., Verardi R., Walsh N., Veglia G., Robia S. L. (2011) Lethal Arg9Cys phospholamban mutation hinders Ca2+-ATPase regulation and phosphorylation by protein kinase A. Proc. Natl. Acad. Sci. U.S.A. 108, 2735–2740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Haghighi K., Kolokathis F., Gramolini A. O., Waggoner J. R., Pater L., Lynch R. A., Fan G. C., Tsiapras D., Parekh R. R., Dorn G. W., 2nd, MacLennan D. H., Kremastinos D. T., Kranias E. G. (2006) A mutation in the human phospholamban gene, deleting arginine 14, results in lethal, hereditary cardiomyopathy. Proc. Natl. Acad. Sci. U.S.A. 103, 1388–1393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. DeWitt M. M., MacLeod H. M., Soliven B., McNally E. M. (2006) Phospholamban R14 deletion results in late-onset, mild, hereditary dilated cardiomyopathy. J. Am. Coll. Cardiol. 48, 1396–1398 [DOI] [PubMed] [Google Scholar]
- 12. Posch M. G., Perrot A., Geier C., Boldt L. H., Schmidt G., Lehmkuhl H. B., Hetzer R., Dietz R., Gutberlet M., Haverkamp W., Ozcelik C. (2009) Genetic deletion of arginine 14 in phospholamban causes dilated cardiomyopathy with attenuated electrocardiographic R amplitudes. Heart Rhythm 6, 480–486 [DOI] [PubMed] [Google Scholar]
- 13. Simmerman H. K., Jones L. R. (1998) Phospholamban: protein structure, mechanism of action, and role in cardiac function. Physiol. Rev. 78, 921–947 [DOI] [PubMed] [Google Scholar]
- 14. Karim C. B., Stamm J. D., Karim J., Jones L. R., Thomas D. D. (1998) Cysteine reactivity and oligomeric structures of phospholamban and its mutants. Biochemistry 37, 12074–12081 [DOI] [PubMed] [Google Scholar]
- 15. Robia S. L., Campbell K. S., Kelly E. M., Hou Z., Winters D. L., Thomas D. D. (2007) Forster transfer recovery reveals that phospholamban exchanges slowly from pentamers but rapidly from the SERCA regulatory complex. Circ. Res. 101, 1123–1129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Park W. J., Oh J. G. (2013) SERCA2a: a prime target for modulation of cardiac contractility during heart failure. BMB Rep. 46, 237–243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kranias E. G., Hajjar R. J. (2012) Modulation of cardiac contractility by the phospholamban/SERCA2a regulatome. Circ. Res. 110, 1646–1660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. MacLennan D. H., Kranias E. G. (2003) Phospholamban: a crucial regulator of cardiac contractility. Nat. Rev. Mol. Cell Biol. 4, 566–577 [DOI] [PubMed] [Google Scholar]
- 19. Haghighi K., Schmidt A. G., Hoit B. D., Brittsan A. G., Yatani A., Lester J. W., Zhai J., Kimura Y., Dorn G. W., 2nd, MacLennan D. H., Kranias E. G. (2001) Superinhibition of sarcoplasmic reticulum function by phospholamban induces cardiac contractile failure. J. Biol. Chem. 276, 24145–24152 [DOI] [PubMed] [Google Scholar]
- 20. Bluhm W. F., Kranias E. G., Dillmann W. H., Meyer M. (2000) Phospholamban: a major determinant of the cardiac force-frequency relationship. Am. J. Physiol. Heart Circ. Physiol. 278, H249–H255 [DOI] [PubMed] [Google Scholar]
- 21. Zvaritch E., Backx P. H., Jirik F., Kimura Y., de Leon S., Schmidt A. G., Hoit B. D., Lester J. W., Kranias E. G., MacLennan D. H. (2000) The transgenic expression of highly inhibitory monomeric forms of phospholamban in mouse heart impairs cardiac contractility. J. Biol. Chem. 275, 14985–14991 [DOI] [PubMed] [Google Scholar]
- 22. Zamoon J., Mascioni A., Thomas D. D., Veglia G. (2003) NMR solution structure and topological orientation of monomeric phospholamban in dodecylphosphocholine micelles. Biophys. J. 85, 2589–2598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Verardi R., Shi L., Traaseth N. J., Walsh N., Veglia G. (2011) Structural topology of phospholamban pentamer in lipid bilayers by a hybrid solution and solid-state NMR method. Proc. Natl. Acad. Sci. U.S.A. 108, 9101–9106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Simmerman H. K., Kobayashi Y. M., Autry J. M., Jones L. R. (1996) A leucine zipper stabilizes the pentameric membrane domain of phospholamban and forms a coiled-coil pore structure. J. Biol. Chem. 271, 5941–5946 [DOI] [PubMed] [Google Scholar]
- 25. Fujii J., Maruyama K., Tada M., MacLennan D. H. (1989) Expression and site-specific mutagenesis of phospholamban. Studies of residues involved in phosphorylation and pentamer formation. J. Biol. Chem. 264, 12950–12955 [PubMed] [Google Scholar]
- 26. Kimura Y., Kurzydlowski K., Tada M., MacLennan D. H. (1996) Phospholamban regulates the Ca2+-ATPase through intramembrane interactions. J. Biol. Chem. 271, 21726–21731 [DOI] [PubMed] [Google Scholar]
- 27. Kelly E. M., Hou Z., Bossuyt J., Bers D. M., Robia S. L. (2008) Phospholamban oligomerization, quaternary structure, and sarco(endo)plasmic reticulum calcium ATPase binding measured by fluorescence resonance energy transfer in living cells. J. Biol. Chem. 283, 12202–12211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hou Z., Robia S. L. (2010) Relative affinity of calcium pump isoforms for phospholamban quantified by fluorescence resonance energy transfer. J. Mol. Biol. 402, 210–216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bidwell P., Blackwell D. J., Hou Z., Zima A. V., Robia S. L. (2011) Phospholamban binds with differential affinity to calcium pump conformers. J. Biol. Chem. 286, 35044–35050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Trieber C. A., Douglas J. L., Afara M., Young H. S. (2005) The effects of mutation on the regulatory properties of phospholamban in co-reconstituted membranes. Biochemistry 44, 3289–3297 [DOI] [PubMed] [Google Scholar]
- 31. Eletr S., Inesi G. (1972) Phospholipid orientation in sarcoplasmic membranes: spin-label ESR and proton MNR studies. Biochim. Biophys. Acta 282, 174–179 [DOI] [PubMed] [Google Scholar]
- 32. Stokes D. L., Green N. M. (1990) Three-dimensional crystals of CaATPase from sarcoplasmic reticulum. Symmetry and molecular packing. Biophys. J. 57, 1–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Young H. S., Jones L. R., Stokes D. L. (2001) Locating phospholamban in co-crystals with Ca2+-ATPase by cryoelectron microscopy. Biophys. J. 81, 884–894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Young H. S., Rigaud J. L., Lacapère J. J., Reddy L. G., Stokes D. L. (1997) How to make tubular crystals by reconstitution of detergent-solubilized Ca2+-ATPase. Biophys. J. 72, 2545–2558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Warren G. B., Toon P. A., Birdsall N. J., Lee A. G., Metcalfe J. C. (1974) Reconstitution of a calcium pump using defined membrane components. Proc. Natl. Acad. Sci. U.S.A. 71, 622–626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zal T., Gascoigne N. R. (2004) Photobleaching-corrected FRET efficiency imaging of live cells. Biophys. J. 86, 3923–3939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hou Z., Kelly E. M., Robia S. L. (2008) Phosphomimetic mutations increase phospholamban oligomerization and alter the structure of its regulatory complex. J. Biol. Chem. 283, 28996–29003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Förster T. (1948) Intermolecular energy migration and fluorescence. Ann. Phys. 2, 55–75 [Google Scholar]
- 39. Gadella T. W. J. (2009) FRET and FLIM Techniques, 1st Ed., p. 193, Elsevier, Amsterdam [Google Scholar]
- 40. Bernstein F. C., Koetzle T. F., Williams G. J., Meyer E. F., Jr., Brice M. D., Rodgers J. R., Kennard O., Shimanouchi T., Tasumi M. (1977) The Protein Data Bank: a computer-based archival file for macromolecular structures. J. Mol. Biol. 112, 535–542 [DOI] [PubMed] [Google Scholar]
- 41. Minamisawa S., Hoshijima M., Chu G., Ward C. A., Frank K., Gu Y., Martone M. E., Wang Y., Ross J., Jr., Kranias E. G., Giles W. R., Chien K. R. (1999) Chronic phospholamban-sarcoplasmic reticulum calcium ATPase interaction is the critical calcium cycling defect in dilated cardiomyopathy. Cell 99, 313–322 [DOI] [PubMed] [Google Scholar]
- 42. Hess B., Kutzner C., van der Spoel D., Lindahl E. (2008) GROMACS 4: algorithms for highly efficient, load-balanced, and scalable molecular simulation. J. Chem. Theory Comput. 4, 435–447 [DOI] [PubMed] [Google Scholar]
- 43. Pronk S., Páll S., Schulz R., Larsson P., Bjelkmar P., Apostolov R., Shirts M. R., Smith J. C., Kasson P. M., van der Spoel D., Hess B., Lindahl E. (2013) GROMACS 4.5: a high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics 29, 845–854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. MacKerell A. D., Jr., Feig M., Brooks C. L., 3rd (2004) Improved treatment of the protein backbone in empirical force fields. J. Am. Chem. Soc. 126, 698–699 [DOI] [PubMed] [Google Scholar]
- 45. Mackerell A. D., Jr., Feig M., Brooks C. L., 3rd (2004) Extending the treatment of backbone energetics in protein force fields: limitations of gas-phase quantum mechanics in reproducing protein conformational distributions in molecular dynamics simulations. J. Comput. Chem. 25, 1400–1415 [DOI] [PubMed] [Google Scholar]
- 46. Darden T., York D., Pedersen L. (1993) Particle mesh Ewald—an n.log(n) method for Ewald sums in large systems. J. Chem. Phys. 98, 10089–10092 [Google Scholar]
- 47. Essmann U., Perera L., Berkowitz M. L., Darden T., Lee H., Pedersen L. G. (1995) A smooth particle mesh Ewald method. J. Chem. Phys. 103, 8577–8593 [Google Scholar]
- 48. Berendsen H. J., Postma J. P., Gunsteren W. F., DiNola A., Haak J. R. (1984) Molecular dynamics with coupling to an external bath. J. Chem. Phys. 81, 3684–3690 [Google Scholar]
- 49. Hoover W. G. (1985) Canonical dynamics—equilibrium phase-space distributions. Phys. Rev. A 31, 1695–1697 [DOI] [PubMed] [Google Scholar]
- 50. Nose S. (1984) A molecular dynamics method for simulations in the canonical ensemble. Mol. Phys. 52, 255–268 [Google Scholar]
- 51. Nose S., Klein M. L. (1983) Constant pressure molecular dynamics for molecular systems. Mol. Phys. 50, 1055–1076 [Google Scholar]
- 52. Parrinello M., Rahman A. (1981) Polymorphic transitions in single crystals—a new molecular dynamics method. J. Appl. Phys. 52, 7182–7190 [Google Scholar]
- 53. Trieber C. A., Afara M., Young H. S. (2009) Effects of phospholamban transmembrane mutants on the calcium affinity, maximal activity, and cooperativity of the sarcoplasmic reticulum calcium pump. Biochemistry 48, 9287–9296 [DOI] [PubMed] [Google Scholar]
- 54. Chen Z., Akin B. L., Stokes D. L., Jones L. R. (2006) Cross-linking of C-terminal residues of phospholamban to the Ca2+ pump of cardiac sarcoplasmic reticulum to probe spatial and functional interactions within the transmembrane domain. J. Biol. Chem. 281, 14163–14172 [DOI] [PubMed] [Google Scholar]
- 55. Akin B. L., Hurley T. D., Chen Z., Jones L. R. (2013) The structural basis for phospholamban inhibition of the calcium pump in sarcoplasmic reticulum. J. Biol. Chem. 288, 30181–30191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Ceholski D. K., Trieber C. A., Young H. S. (2012) Hydrophobic imbalance in the cytoplasmic domain of phospholamban is a determinant for lethal dilated cardiomyopathy. J. Biol. Chem. 287, 16521–16529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Karim C. B., Zhang Z., Howard E. C., Torgersen K. D., Thomas D. D. (2006) Phosphorylation-dependent conformational switch in spin-labeled phospholamban bound to SERCA. J. Mol. Biol. 358, 1032–1040 [DOI] [PubMed] [Google Scholar]
- 58. Traaseth N. J., Thomas D. D., Veglia G. (2006) Effects of Ser16 phosphorylation on the allosteric transitions of phospholamban/Ca2+-ATPase complex. J. Mol. Biol. 358, 1041–1050 [DOI] [PubMed] [Google Scholar]
- 59. Butler J., Lee A. G., Wilson D. I., Spalluto C., Hanley N. A., East J. M. (2007) Phospholamban and sarcolipin are maintained in the endoplasmic reticulum by retrieval from the ER-Golgi intermediate compartment. Cardiovasc. Res. 74, 114–123 [DOI] [PubMed] [Google Scholar]
- 60. Gramolini A. O., Kislinger T., Asahi M., Li W., Emili A., MacLennan D. H. (2004) Sarcolipin retention in the endoplasmic reticulum depends on its C-terminal RSYQY sequence and its interaction with sarco(endo)plasmic Ca2+-ATPases. Proc. Natl. Acad. Sci. U.S.A. 101, 16807–16812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Stenoien D. L., Knyushko T. V., Londono M. P., Opresko L. K., Mayer M. U., Brady S. T., Squier T. C., Bigelow D. J. (2007) Cellular trafficking of phospholamban and formation of functional sarcoplasmic reticulum during myocyte differentiation. Am. J. Physiol. Cell Physiol. 292, C2084–C2094 [DOI] [PubMed] [Google Scholar]
- 62. Mueller B., Karim C. B., Negrashov I. V., Kutchai H., Thomas D. D. (2004) Direct detection of phospholamban and sarcoplasmic reticulum Ca-ATPase interaction in membranes using fluorescence resonance energy transfer. Biochemistry 43, 8754–8765 [DOI] [PubMed] [Google Scholar]
- 63. Li J., Bigelow D. J., Squier T. C. (2004) Conformational changes within the cytosolic portion of phospholamban upon release of Ca-ATPase inhibition. Biochemistry 43, 3870–3879 [DOI] [PubMed] [Google Scholar]
- 64. Pallikkuth S., Blackwell D. J., Hu Z., Hou Z., Zieman D. T., Svensson B., Thomas D. D., Robia S. L. (2013) Phosphorylated phospholamban stabilizes a compact conformation of the cardiac calcium-ATPase. Biophys. J. 105, 1812–1821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Kimura Y., Kurzydlowski K., Tada M., MacLennan D. H. (1997) Phospholamban inhibitory function is activated by depolymerization. J. Biol. Chem. 272, 15061–15064 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.