

Background: Myocardial ischemia and cardioprotection induce signal networks mediated by post-translational modification.
Results: Phosphorylation sites altered by ischemia and/or cardioprotection were influenced by inhibition of lysine acetylation consistent with cross-talk.
Conclusion: Lysine acetylation induces proximal dephosphorylation in a kinase motif by salt bridge inhibition.
Significance: Cross-talk is likely to be an important aspect of signaling during ischemia and cardioprotection.
Keywords: Heart, Mass Spectrometry (MS), Myocardial Infarction, Phosphorylation, Post-translational Modification (PTM), Proteomics, Signaling, Cardioprotection, Lysine Acetylation
Abstract
Myocardial ischemia and cardioprotection by ischemic pre-conditioning induce signal networks aimed at survival or cell death if the ischemic period is prolonged. These pathways are mediated by protein post-translational modifications that are hypothesized to cross-talk with and regulate each other. Phosphopeptides and lysine-acetylated peptides were quantified in isolated rat hearts subjected to ischemia or ischemic pre-conditioning, with and without splitomicin inhibition of lysine deacetylation. We show lysine acetylation (acetyl-Lys)-dependent activation of AMP-activated protein kinase, AKT, and PKA kinases during ischemia. Phosphorylation and acetyl-Lys sites mapped onto tertiary structures were proximal in >50% of proteins investigated, yet they were mutually exclusive in 50 ischemic pre-conditioning- and/or ischemia-associated peptides containing the KXXS basophilic protein kinase consensus motif. Modifications in this motif were modeled in the C terminus of muscle-type creatine kinase. Acetyl-Lys increased proximal dephosphorylation by 10-fold. Structural analysis of modified muscle-type creatine kinase peptide variants by two-dimensional NMR revealed stabilization via a lysine-phosphate salt bridge, which was disrupted by acetyl-Lys resulting in backbone flexibility and increased phosphatase accessibility.
Introduction
Prolonged blockage of coronary blood flow (ischemia), even with subsequent restoration (reperfusion), may result in cardiomyocyte apoptosis and necrosis. Ischemia/reperfusion (I/R)4 thus represents a significant clinical burden. IPC, a series of short I/R cycles prior to extended ischemia, elicits a degree of cardioprotection against I/R injury. The oxygen supply/demand imbalance upon ischemia reduces oxidative metabolism and results in depletion of intracellular ATP accompanied by a time-dependent rise in AMP. Anaerobiosis also generates pyruvate conversion to lactate to maintain oxidized NAD+ levels, which leads to intracellular acidosis and promotes Na+ and Ca2+ overload (1, 2). Activation of signal pathways in ischemia and IPC is established (3, 4) and includes mitogen-activated protein kinases (MAPKs) (5), protein kinase C (PKC) (6–8), phosphatidylinositol 3′-hydroxykinase/AKT (PI3K/AKT) (9), and cyclic AMP-dependent protein kinase A (PKA) (10, 11).
The ischemia-associated increase in AMP:ATP also activates a network initiated in part by AMP-activated protein kinase (AMPK). AMPK regulates fatty acid metabolism, where AMPK-mediated phosphorylation inactivates acetyl-CoA carboxylase (ACC) resulting in a decrease in cytosolic malonyl-CoA (12) and an increase in fatty acid uptake and oxidation in the mitochondria (13–15). AMPK activation also modulates energy consumption (16), cell survival, and regulation of the fuel-sensing sirtuin (SIRT) family of protein deacetylases (17). AMPK and SIRT1 regulate each other, which suggests an AMPK/SIRT1 sensing cycle with phosphorylation/acetylation cross-talk on common substrates (18). The SIRTs are a family of seven NAD+-dependent enzymes, whose activity is influenced by cellular energy and redox status (19). SIRT1 overexpression confers myocardial protection during oxidative stress (20) and prevents apoptosis via p53 inactivation (21), whereas splitomicin-mediated inhibition of SIRT1 attenuates IPC (22), which collectively provides strong evidence that lysine acetylation (acetyl-Lys)/deacetylation contributes to cardioprotection and cell survival during ischemia.
Post-translational modification (PTM) cross-talk is best exemplified in the “chromatin code” (23–26). Cross-talk can include the following: (i) competitive modification of the same site; (ii) modification influencing additional PTM at a proximal site; or (iii) a PTM activating/deactivating the modifying enzyme for a second modification. Studies of histone PTM cross-talk identified a modification that subsequently resulted in the alteration of a secondary cis (intramolecular) modification, i.e. phosphorylation of Ser-10 on histone H3 increases acetylation of Lys-14 (27, 28). This process was also observed at a trans (inter-molecular) level, i.e. ubiquitylation of histone H2B stimulates histone H3 methylation (29). Cross-talk can also influence PTM kinetics (30–33). Alternatively, PTMs may activate/deactivate modifying enzymes, including (de)acetylation of kinases (34–37), for example SIRT1 promotion of AKT activity (38), (de)phosphorylation of deacetylases/acetyltransferases (39–42), or a PTM-modifying enzyme regulating global levels of another PTM (43, 44). Cross-talk between phosphorylation and acetyl-Lys can include the recruitment of kinases/phosphatases by acetyl-Lys (45, 46), the recruitment of histone deacetylases/acetyltransferases by phosphorylation (47–49), or where either phosphorylation or acetyl-Lys is required for the other to occur at a proximal site (50). Examples of site-specific competition include the following: between acetylation and phosphorylation of serine in the activation loop of MAP2K6 (51); O-GlcNAcylation and phosphorylation of Thr-58 in c-Myc (52); Ser-1177 in endothelial nitric-oxide synthase (53); and Ser-733 in IKKβ (54).
Peptide-centric enrichment combined with quantitative tandem mass spectrometry (MS/MS) has become the gold standard for generating global phosphopeptide profiles under a variety of stimuli and disease states (55–57), although antibody-based enrichment of acetylated peptides followed by MS/MS has also proven successful (58–63). We utilized sequential enrichment to globally quantify phosphorylated and lysine-acetylated peptides in isolated rat hearts subjected to ischemia and IPC, in the presence or absence of splitomicin inhibition of lysine deacetylation, using isobaric labeling and MS/MS. These data enabled an evaluation of sequence motifs displaying evidence of PTM cross-talk, which showed that phosphorylation and acetyl-Lys are mutually exclusive in 50 ischemia/IPC-regulated peptides containing a basophilic protein kinase motif (KXXS). We determined the rates of cross-talk between PTM in the C-terminal region of muscle-type creatine kinase (CKM) and demonstrated the formation of a phosphate-lysine salt bridge. Acetyl-Lys disrupts the phosphate-lysine interaction and results in increased de-phosphorylation at this site.
EXPERIMENTAL PROCEDURES
Rat Heart Isolation
Male Lewis rats (∼200 g) were euthanized with an intraperitoneal injection of sodium pentobarbital (0.5 mg/g; University of Sydney Animal Ethics Committee approval K20/6-2009/3/5078). The aorta was cut ∼1 cm above the aortic valve; the heart removed was and placed in ice-cold saline solution (0.9% (w/v) NaCl, pH 7.4). Hearts were subjected to Langendorff perfusion using oxygenated Krebs-Henseleit buffer (118 mm NaCl, 25 mm NaHCO3, 1.2 mm KH2PO4, 4.8 mm KCl, 1.2 mm MgSO4, 11 mm glucose, 2.0 mm CaCl2, pH 7.4) at 37 °C, as described (64). Heart rates and left ventricular developed pressure (LVDP) were recorded (LabChart, ADInstuments, Bella Vista, Australia) and measured as a rate pressure product (RPP; beats/min × LVDP in mm Hg). For ischemia and IPC, hearts were equilibrated for a baseline period of 15 min with normoxic perfusion and then assigned to 12 groups as follows: (i) 0 min; (ii) 2 min; (iii) 10 min; and (iv) 20 min of no-flow ischemia (n = 3 for each time point); (v) 0 min; (vi) 2 min; (vii) 10 min; and (viii) 20 min of no-flow ischemia following 15 min of equilibration with 10 μm splitomicin (n = 3 for each time point); (ix) acute IPC with three 2-min cycles of I/R (n = 3); (x) acute IPC with three 2-min I/R following 15 min of equilibration with 10 μm splitomicin (n = 3); (xi) 20 min of normoxic perfusion (n = 3); and (xii) 20 min of normoxic perfusion with 10 μm splitomicin. Hearts were snap-frozen and stored at −80 °C. All hearts achieved a, RPP of 25,000 (±5000), a flow rate of 9 ml/min, and a perfusion pressure of 80 mm Hg at the end of the baseline period. Myocardial function was determined by comparing the RPP for additional hearts subjected to 20 min of ischemia, followed by 30 min of reperfusion, to allow determination of functional recovery. The extent of necrosis was determined by staining hearts with triphenyltetrazolium chloride (64). Following normoxic perfusion, no significant difference in the functional performance of the hearts was observed (RPP = 92.2% of baseline; see supplemental Fig. S1). Hearts subjected to 20 min of ischemia and 30 min of reperfusion showed loss of hemodynamic performance (RPP = ∼30% of baseline; p value <0.05) and evidence of necrosis (17.8 ± 3.6% negative triphenyltetrazolium chloride staining of left ventricular mass compared with 0.4 ± 0.1% for baseline hearts) (supplemental Fig. S1). Splitomicin had no significant effect on normoxic heart function, as reported (22).
Cell Culture
Rat L6 myoblasts were 2-plex SILAC-labeled in α-minimal essential medium containing 5.5 mm glucose (Invitrogen) and 10% fetal bovine serum (Hyclone, Logan UT). Following five passages, cells at 80% confluence were transfected with Lipofectamine-2000 and 15 μg of plasmid DNA. The plasmid consisted of human CKM cloned into a pCMV5 plasmid containing an N-terminal FLAG tag and was generously provided by Prof. Yong-Bin Yan and Prof. Hai-Meng Zhou (School of Life Sciences, Tsinghua University, China). Site-directed mutagenesis was performed with the QuikChange site-directed mutagenesis kit (Invitrogen) to generate a K369Q acetyl-Lys mimic and an S372E phospho-mimic using the primers 5′GAGAAGAAGTTGGAGCAAGGCCAGTCCATCG and 5′GAAGTTGGAGAAAGGCCAGGAAATCGACGACATGATCCCCG, respectively. Cells were transfected for 24 h and harvested after an additional 24 h.
Myocardial Peptide Preparation
Myocardial tissue was ground under liquid nitrogen, and 100 mg of powdered tissue from each group was pooled (i.e. n = 3, total 300 mg). Samples were homogenized in 3-fold the weight (i.e. 1 mg/3 μl) of lysis buffer (20 mm Tris, 1 mm dithiothreitol (DTT), 0.5% SDS, protease inhibitor mixture (Roche Applied Science), phosphatase inhibitor mixture III (Calbiochem), 10 μm trichostatin A, 10 mm nicotinamide, 50 mm butyric acid, pH 7.5) at 4 °C using an Omni homogenizer (Omni International, Kennesaw GA) followed by tip-probe sonication. The homogenate was centrifuged at 12,000 × g for 15 min at 4 °C, and the supernatant was precipitated with chloroform/methanol. Proteins were resuspended in 6 m urea, 2 m thiourea, 50 mm triethylammonium biocarbonate, pH 8.0, and reduced with 10 mm DTT for 1 h at 25 °C followed by alkylation with 25 mm iodoacetamide for 30 min at 25 °C in the dark. The reaction was diluted 1:10 with 50 mm triethylammonium biocarbonate and digested with trypsin (Promega, Madison WI; 50:1 substrate/enzyme) for 12 h at 30 °C, acidified to 2% (v/v) formic acid (FA), centrifuged at 12,000 × g for 15 min, and the supernatant removed. Peptides were desalted with hydrophilic-lipophilic balance solid phase extraction columns (Waters Corp., Milford MA), eluted with 80% acetonitrile (ACN), 0.1% trifluoroacetic acid (TFA), and dried by vacuum centrifugation. The entire procedure was performed in duplicate.
Stable Isotope Labeling with Tandem Mass Tags (TMT) and Peptide Enrichment
200 μg of peptide from each group was labeled with TMT (Thermo Scientific, San Jose CA) according to the manufacturer's instructions. The first replicate was labeled as follows: TMT6-126: 0 min ischemia; TMT6-127: 0 min ischemia with 10 μm splitomicin; TMT6-128: 20 min ischemia; TMT6-129: 20 min ischemia with 10 μm splitomicin; TMT6-130: IPC; TMT6-131: IPC with 10 μm splitomicin. The second replicate was labeled in reverse. Peptides were resuspended in 50 mm MOPS, 10 mm NaH2PO4, 50 mm NaCl (immunoprecipitation buffer; pH 7.2), supplemented with 50 μl of anti-acetyl-Lys antibody-conjugated agarose (ImmuneChem Pharmaceuticals, Canada), and rotated at 4 °C for 16 h. The suspension was centrifuged at 1000 × g for 1 min and the supernatant collected. The resin was washed with 4× 1 ml immunoprecipitation buffer followed by 2× 1 ml H2O. Lysine-acetylated peptides were eluted with 2× 200 μl 0.2% (v/v) TFA and purified using a C18/POROS Oligo R2/R3 reversed phase (RP) micro-column, dried by vacuum centrifugation, and stored at −20 °C. The supernatants from the IP flow-through and each wash were combined and diluted 10-fold in 1 m glycolic acid in 80% ACN, 5% TFA (TiO2 loading buffer), as described (65). This solution was incubated with 3 mg of TiO2 beads for 15 min at room temperature with gentle agitation. The suspension was then centrifuged at 1000 × g for 1 min. Supernatant was removed, and beads were washed with TiO2 loading buffer followed by 80% ACN, 2% TFA and finally 20% ACN, 0.2% TFA. Phosphopeptides were eluted with 1% ammonium hydroxide, pH 11.3, and purified using a C18/POROS Oligo R3 RP micro-column, dried by vacuum centrifugation, and stored at −20 °C. Enriched peptides were fractionated into 10 fractions for phosphopeptides and six fractions for acetyl-Lys peptides, respectively, using TSKgel Amide-80 HILIC capillary HPLC (66).
Reversed Phase Nano-Liquid Chromatography-Electrospray Ionization Tandem Mass Spectrometry
Fractionated TMT-labeled peptides were resuspended in 0.1% FA and separated by reversed phase chromatography on an in-house 25-cm × 75-μm Reprosil-Pur C18-AQ column (3 μm; Dr. Maisch, Germany) using an EASY nLC nano-HPLC (Proxeon, Odense, Denmark). The HPLC gradient was 0–40% solvent B (solvent A, 0.1% FA; solvent B, 95% ACN, 0.1% FA) over 120 min at a flow of 250 nl/min. MS was performed using an LTQ-Orbitrap Velos (Thermo Scientific). An MS scan (400–2000 m/z; MS AGC 1 × 106) was recorded in the Orbitrap set at a resolution of 30,000 at 400 m/z followed by data-dependent higher energy collision dissociation (HCD) MS/MS analysis of the seven most intense precursor ions. Parameters for HCD were as follows: activation time 0.1 ms; normalized energy 45 (48 for phosphopeptides); dynamic exclusion enabled with repeat count 1; resolution 7500; exclusion duration 30 s; maximum injection time 500 ms; and MSn AGC 1 × 105. Collected data are shown in supplemental Table S1.
SILAC-labeled rat L6 myoblast peptides were resuspended in 0.5% acetic acid and separated by reversed-phase chromatography on an in-house 40-cm × 75-μm Reprosil-Pur C18-AQ column (1.9 μm; Dr. Maisch) using an EASY nLC-1000 nano-UHPLC (Proxeon). The HPLC gradient was 0–40% solvent B (solvent A, 0.5% acetic acid; solvent B, 90% ACN, 0.5% acetic acid) over 90 min at a flow of 250 nl/min. MS was performed using a Q-Exactive (Thermo Scientific). An MS scan (300–1750 m/z; MS AGC 3 × 106) was recorded in the Orbitrap set at a resolution of 70,000 at 200 m/z followed by data-dependent HCD MS/MS of the 12 most intense precursor ions. Parameters for HCD were as follows: normalized energy 25; dynamic exclusion enabled; resolution 35,000; exclusion duration 60 s, maximum injection time 120 ms, and MSn AGC 1 × 106.
Metabolite Quantification by Ion Pairing LC-SRM
Metabolites were extracted from 50 mg of tissue using a modified boiling water extraction (67). Tissue was added to 150 μl of 1 mm EDTA, 1 mm HEPES, pH 7.2, 4 °C and vortexed for 20 s. The extract was boiled for 90 s, snap-frozen in liquid nitrogen, incubated at 25 °C for 5 min, and then snap-frozen for 1 min. A second incubation at 25 °C for 5 min was followed by incubation at −20 °C for 1 h. The extract was incubated at 4 °C for 10 min and centrifuged at 20,000 × g for 15 min at 4 °C. Aliquots of 10 μl of supernatant were analyzed in triplicate. Metabolites were separated by reversed-phase chromatography on an in-house 320-μm × 15-cm C18-AQ (3 μm; Dr. Maisch) column. The HPLC gradient was 0–20% solvent B over 5 min, 20% for 15 min, 20–35% over 2.5 min, 35% for 2.5 min, 35–60% over 2.5 min, 60–95% over 2.5 min, 95% for 7.5 min (solvent A, 10 mm tributylamine adjusted to pH 4.9 with 15 mm acetic acid; solvent B, methanol) at 6 μl/min. MS was performed using a 5500 QTRAP with Turbo Ion spray source in negative ion MRM mode. Parameters were as follows: GS1 20 p.s.i., curtain gas 20, ion spray voltage = −4500 V, unit resolution. Acquisition was performed by unscheduled MRM with a dwell time for each transition set to 100 ms. Synthetic metabolites (Sigma) were prepared in 10 mm stock solutions. Each metabolite was directly infused into the mass spectrometer to select and optimize transitions and to determine retention times. A 10 μm standard mixture was used for quality control and standard curves. LLOQ and ULOQ were assessed using a standard curve at 10 concentrations from 100 pm to 10 μm defined as R2 >0.95 and coefficient of variation <20%. Standard additions experiments were performed to validate metabolite identification based on retention times, but no standard addition curves were performed, and therefore relative quantification is presented (supplemental Tables S2 and S3). All data analysis was performed in MultiQuant Version 2.0 (AB Sciex).
Assessment of Splitomicin Stability by LC-SRM
To ensure stability, splitomicin solutions were analyzed on a 1 × 50-mm Luna Phenyl-Hexyl column (5 μm; Phenomenex, Torrance, CA). The HPLC gradient was 0–100% solvent B over 5 min and 100% for 5 min (solvent A, 0.1% acetic acid; solvent B, acetonitrile) at 100 μl/min. MS was performed using a 5500 QTRAP in positive ion MRM mode. Parameters were as follows: GS1 20 p.s.i., GS2 25 p.s.i., curtain gas 20, ion spray voltage = 5000 V, unit resolution. Acquisition was performed by unscheduled MRM with a dwell time for each transition set to 100 ms. Splitomicin (Sigma) was prepared in a primary stock solution of 10 mm in methanol. A 10 μm solution was prepared in 50% methanol, 0.1% acetic acid and used to select and optimize transitions. A 10 μm stock solution was prepared in Buffer A, and LLOQ and ULOQ were assessed using a standard curve at five concentrations from 1 nm to 10 μm defined as R2 > 0.95 (supplemental Fig. S2A). Next, 10 μm splitomicin in phosphate-buffered saline, pH 7.5, was incubated at 37 °C. At the specified times (0, 10, 20, 30, and 40 min), 10 μl was removed and diluted 10-fold with Buffer A, and 10 μl was immediately analyzed in triplicate (supplemental Fig. S2B). Data analysis was performed in MultiQuant Version 2.0 and manually verified for peak quality (supplemental Fig. S2C). Following 20 min of incubation (the specified baseline period for splitomicin in heart perfusions prior to ischemia), the splitomicin concentration was reduced from 10 to 5 μm, which is >10-fold the minimum inhibitory concentration (68). At 40 min (the total time of the longest Langendorff experiment, including ischemia), the concentration still remained above the minimum inhibitory concentration.
Western Blotting
20 μg of proteins were separated on 7-cm SDS-polyacrylamide gels followed by wet transfer to PVDF membranes. All blots were blocked with 5% bovine serum albumin in Tris-buffered saline, 0.1% Tween 20 overnight at 4 °C. Primary antibodies were obtained from Cell Signaling Technology (Danvers, MA) and were as follows: anti-phospho-AMPKα (Thr-172), anti-AMPKα, anti-phosphoacetyl-CoA carboxylase (Ser-79); anti-phospho-Akt (Ser-473); anti-Akt, anti-phospho-eIF4B (Ser-422); anti-eIF4B; anti-phospho-PKA substrate (RRX(pS/pT). Secondary antibody was anti-rabbit IgG-HRP. Equal protein loading was confirmed with Coomassie staining.
Analysis of Untargeted Mass Spectrometry Data
Raw data were processed using Proteome Discoverer Version 1.3β (Thermo Scientific) into .mgf files. TMT-labeled myocardial peptides were searched against the UniProt Rattus norvegicus database (March, 2012: 41,696 entries) using MASCOT (Version 1.12). SILAC-labeled L6 myoblast peptides were searched against the UniProt R. norvegicus database containing an additional CKM entry containing a K369Q mutation, using SEQUEST. Searches were performed with the following fixed parameters: precursor mass tolerance of 10 ppm, product ion tolerance of 0.02 Da, Cys-carboxyamidomethylation (TMT peptides), and one missed cleavage (three for acetyl-Lys enrichments). For TMT peptides, searches were conducted with variable modifications as follows: oxidation of Met; TMT labeling of N termini and Lys; phosphorylation of Ser, Thr, and Tyr, and acetylation of Lys. For SILAC peptides, variable modifications were as follows: oxidation of Met; SILAC labeling of Arg and Lys; and phosphorylation of Ser, Thr, and Tyr. Quantification was performed using Proteome Discoverer with HCD MS/MS reporter ion integration within a 50 ppm window for TMT labeling and mass precision set to 2 ppm for SILAC labeling-extracted ion chromatograms. All searches were filtered to a <1% false discovery rate using Percolator (69). Phospho-RS probabilities were used to calculate phosphorylation site localizations and a confident site reported as >75% localization probability (70). Phosphopeptide and acetyl-Lys peptide ratios were normalized by the median ratios of nonmodified peptides in each replicate. The global effect of ischemia, IPC, and splitomicin on phosphorylation and acetyl-Lys was assessed by calculating the median of the duplicate averaged ratios. The intensity of the observed reporter ions is known to influence quantification accuracy (71). To account for this, the standard deviation of the log2 ratios were calculated by ranking the peptide intensities, and a sliding window script in R-project using the Ringo Package enabled the calculation of a peptide intensity-dependent standard deviation (72). The p values were calculated using a two-sided t test and adjusted for multiple testing using a false discovery rate approach (73) with the Stats Package in R-project. Peptides were considered to be significantly regulated if their ratios were >2 S.D. (i.e. p value <0.05 following false discovery rate correction) and with a cutoff of ±1.5-fold change within each replicate. A less stringent analysis was also utilized to investigate regulated sites with only a cutoff ±1.5-fold change. Fuzzy c-means cluster analysis was performed using GProX (74). Kinase-substrate predictions were made using Scansite (75), and pathway analysis was performed with Ingenuity Pathway Analysis.
Peptide Synthesis
Peptide synthesis was performed by Fmoc chemistry (76). Briefly, peptides were prepared on a 0.25 mmol scale by manual stepwise solid-phase synthesis using benzotriazol-1-yl-1,1,3,3-tetramethyluronium/N,N-diisopropylethylamine on Rink-amide MBHA resin (substitution 0.78 mmol/g) (Novachem, Collingwood, Australia). Amino acid (5 eq) and N,N-diisopropylethylamine (10 eq) were employed in each coupling step for 10 min except for the synthesis of phosphorylated peptides, where the coupling of Fmoc-Ser(PO(OBzl)-OH)-OH was performed in 2.5 eq in the presence of 2-(7-aza-benzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate. Fmoc deprotections were achieved with 3× 3 min in an excess of 20% piperidine in dimethylformamide, and capping was achieved with 8% acetic anhydride in dimethylformamide for 10 min. Synthesis of lysine-acetylated peptides was achieved with Nα-9-fluorenylmethoxycarbonyl-Nϵ-4-metyltrityl-lysine (Fmoc-Lys(Mtt)-OH), which was deprotected after the final assembly with 5× 3 min in an excess of 2.5% TFA, 2.5% triisopropylsilane in chloroform followed by 3× 3 min in an excess of 10% N,N-diisopropylethylamine. Acetylation was performed with 2× 10 min in an excess of 8% acetic anhydride in dimethylformamide. The peptides were simultaneously deprotected and cleaved from the resin in 2.5% triisopropylsilane, 2.5% H2O in 95% TFA for 2 h. The solution was filtered and the filtrate dried under nitrogen. Peptides were precipitated with diethyl ether, resuspended in 50% ACN, lyophilized, resuspended in 0.1% TFA, and purified by HPLC on a 25-cm × 15.2-mm C18 column (10 μm; Phenomenex, Torrance, CA) using a GBC LC 1150 (Braeside, Australia). The HPLC gradient was 0–100% solvent B (solvent A, 0.1% TFA; solvent B, 90% ACN, 0.1% TFA) over 60 min at a flow of 3 ml/min with 1.5-ml fractions collected and UV detection at 210 nm. The nonacetylated peptides were purified to >99% (according to UV spectrophotometry), although acetylated peptides contained a minor (∼9%) amount of the nonacetylated form.
In Vitro Kinase/Phosphatase/Acetyltransferase/Deacetylase Assays by nLC-SRM
Hearts were equilibrated for 15 min with normoxic perfusion and then subjected to 20 min of ischemia using Langendorff perfusion as described above. Tissue (500–600 mg) was homogenized in 3-fold the weight (i.e. 1 mg/3 μl) of lysis buffer containing 20 mm Tris, 7.5 mm MgCl2, protease inhibitor mixture, pH 7.4, at 4 °C. For kinase assays, lysis buffer was supplemented with 5 mm ATP, phosphatase inhibitor mixture III, and 10 μm trichostatin A, 10 mm nicotinamide, and 50 mm butyric acid. For acetyltransferase assays, lysis buffer was supplemented with 1 mm acetyl-CoA containing phosphatase and deacetylase inhibitor mixtures. For phosphatase/deacetylase assays, lysis buffer was supplemented with 1 mm NAD. Lysates were adjusted to pH 7.4, kept at 4 °C, and assays performed within 30 min. Assays were performed as described for kinase activity assay for kinome profiling (KAYAK) with minor modifications (77, 78). Briefly, reactions were performed with 100 μg of lysate and 250 pmol of substrate in a final reaction volume of 20 μl. Reactions were performed at 28 °C and quenched with 20 μl of 20% trichloroacetic acid (TCA) to precipitate the lysate following 0, 10, 20, 30, and 60 min. The reactions were centrifuged at 16,000 × g for 15 min at 4 °C; supernatants were removed and diluted 1:1 with 0.2% TFA and desalted with C18/R2/R3 micro-columns. Peptides were resuspended in 100 μl of 0.1% FA and 5 μl analyzed by nLC-SRM. Peptides were trapped on a 0.5-cm × 300-μm Zorbax C18 column (5 μm; Agilent Technologies, Palo Alto, CA) at 5 μl/min for 5 min using a nano-LC 2Dplus system (Eksigent, Dublin, CA). Peptides were separated by reversed phase chromatography on an in-house 30-cm × 75-μm Reprosil-Pur C18-AQ (3 μm; Dr. Maisch) PicoFrit column (New Objective, Woburn, MA). The HPLC gradient was 0–40% solvent B (solvent A, 0.1% FA; solvent B, 90% ACN, 0.1% FA) over 10 min at a flow of 250 nl/min. MS was performed using a 5500 QTRAP operated in MRM mode. Parameters were as follows: GS1 20 p.s.i., curtain gas 20 p.s.i., interface temperature 150 °C, ion spray voltage 2200 V, unit resolution. All four peptides were monitored (unscheduled) for each reaction, and areas under the curve were obtained using Skyline (79) and expressed as % substrate conversion (areaproduct/(areasubstrate + areaproduct)). Transitions were Savitzky Golay transformed and integration boundaries manually verified. All reactions were analyzed in triplicate, and those with coefficient of variation >20% removed. Controls were performed without addition of lysate, and incubation of the substrates was at 28 °C for 60 min. No significant differences in substrate peak areas were observed. Five transitions/peptide were utilized (supplemental Table S4) (80). Validation of equimolar concentrations and transition selectivity were determined by creating a quality control mix of the four peptides (KXXS, KXXpS, AcKXXS, and AcKXXpS) and adding 50 fmol into a background myocardial lysate followed by TCA precipitation, desalting, and analysis by nLC-SRM (supplemental Fig. S3).
Immunoprecipitation of CKM WT, K369Q, and S372E and in Vitro Kinase/Phosphatase and Deacetylase Assays
L6 myoblast cells were lysed in Nonidet P-40 lysis buffer (25 mm Tris, 137 mm NaCl, 10% glycerol, 1% Nonidet P-40, protease inhibitor mixture, phosphatase inhibitor mixture, pH 7.4) and added to protein G beads pre-incubated with anti-FLAG M2 antibody (Sigma). Following incubation at 4 °C for 2 h, beads were washed three times with Nonidet P-40 lysis buffer, three times with PBS, and one time with either kinase buffer (20 mm Tris, 7.5 mm MgCl2, 5 mm ATP, 2 μm CaCl2, 1.2 μm calmodulin, pH 7.4) or deacetylase buffer (20 mm Tris, 4 mm MgCl2, 50 μm NAD, pH 7.4). For phosphatase assays, CKM was first phosphorylated by resuspending the beads in 20 μl of kinase buffer containing 80 units of CaMKII (New England Biolabs) and reacted for 60 min at 30 °C. The beads were washed three times with PBS and once with phosphatase buffer (20 mm Tris, 10 mm MnCl2, pH 7.4), and heavy labeled samples were treated with 5 units of protein phosphatase 1 (PP1; New England Biolabs) for 30 or 60 min at 30 °C. For deacetylase assays, the heavy labeled samples were resuspended in 20 μl of deacetylase buffer containing active FLAG-tagged SIRT1 (generously provided by Dr. Amanda Brandon, Diabetes and Obesity Program, Garvan Institute of Medical Research) and reacted for 30 or 60 min at 30 °C. Elution was performed with 8 μg/30 μl of FLAG peptide in PBS, and light and heavy samples were pooled. Proteins were separated by SDS-PAGE and stained with SYPRO Ruby (Invitrogen) (supplemental Fig. S4) followed by in-gel tryptic digestion. The experiment was performed in two biological replicates.
Peptide Analysis by NMR Spectroscopy and CD
Peptides were dissolved in 20 mm Tris-d11 (1 mm, 600 μl H2O/D2O 9:1) and adjusted to pH 7.2. One-dimensional 1H NMR spectra were acquired on a Bruker Avance III 600 spectrometer (Bruker Corp., Billerica, MA) with water suppression (decoupling in the presence of Scalar interactions) at 276–298 K. Two-dimensional 1H NMR spectra were acquired on a Bruker Avance III 800 spectrometer with water suppression (decoupling in the presence of Scalar interactions) at 276 K with a mixing time of 100 ms. 1H chemical shifts were referenced to 4,4-dimethyl-4-silapentane-1-sulfonic acid (δ 0.00 ppm) in water. Spectra were processed using Topspin (Bruker) and manually analyzed with Sparky Version 3.114. For CD analysis, peptides were dissolved in 20 mm Tris (100 μm, 300 μl) and adjusted to pH 7.2. Analysis was performed in a 1-mm quartz cell using a Jasco spectropolarimeter (Easton, MD) (λ 185–260 nm) at 50 nm/min with a bandwidth of 1.0 nm. Each spectrum represented an average of 5 scans.
Data Availability
MS data have been deposited into the PeptideAtlas repository (Identifier(s): PASS00283 for global quantification of phosphorylation and acetyl-Lys following ischemia and IPC; PASS00280 for quantification of phosphorylation and acetyl-Lys cross-talk on CKM synthetic peptides; and PASS00286 for quantification of dephosphorylation rates of Ser-372 on human CKM with and without acetyl-Lys.
RESULTS
Myocardial Phosphorylation and Acetyl-Lys Networks
To gain insights into signaling networks initiated by ischemia and IPC, we quantified protein phosphorylation and acetyl-Lys using isobaric labeling and MS/MS. Rat hearts (n = 3 per group) were isolated using an ex vivo Langendorff model and subjected to the following: (i) 0 min ischemia (0I); (ii) 20 min ischemia (20I); and (iii) an IPC protocol (3× 2 min I/R); all in the presence or absence of 10 μm splitomicin (Fig. 1A) (22). Lysates were trypsin-digested and peptides labeled with TMT. Phospho- and lysine-acetylated peptides were enriched by TiO2 chromatography and immunoprecipitation, respectively, and fractionated by HILIC. These fractions were analyzed by nano-LC-MS/MS, and the entire procedure was repeated with reverse labeling. Overall, 3101 protein groups were modified (supplemental Table S1). Phosphorylation and acetyl-Lys modified 2546 and 728 protein groups, respectively, and 331 were modified by both PTMs. Quantification at the protein level was obtained for 694 protein groups (>1 peptide), with 298 and 361 of these containing either phosphorylation or acetyl-Lys sites, respectively (supplemental Table S1). Protein abundance displayed very low variation (global log2 standard deviations measured at 0.31 and 0.34 for 20I and IPC, respectively), whereas larger variations were observed for phosphopeptides (log2 standard deviations of 0.58 and 0.64 for 20I and IPC) and lysine-acetylated peptides (log2 standard deviations of 0.48 and 0.59 for 20I and IPC), suggesting the myocardial response to acute ischemia and IPC is dominated by changes in PTMs rather than protein abundance (Fig. 1B). Perfusion of rat hearts with 10 μm splitomicin, without subsequent ischemia or IPC, increased global acetyl-Lys by ∼10% (median ratio for 0I + splitomicin:0I = 1.1) and phosphorylation by ∼2.5%. 20I increased global phosphorylation (median ratio for 20I:0I = 1.04) and acetyl-Lys (median ratio = 1.05), and both were further increased by splitomicin (median ratios compared with 0I of 1.08 for phosphorylation and acetyl-Lys, respectively). IPC decreased global acetyl-Lys by ∼10% (median ratio for IPC:0I = 0.91), and phosphorylation by ∼20% (median ratio for IPC:0I = 0.83). Splitomicin inhibited the reduction in acetyl-Lys (median ratio for IPC + splitomicin:0I = 0.98) but not phosphorylation (median ratio for IPC + splitomicin:0I = 0.81). For all conditions, total protein abundance showed median ratios that indicated a <1% change. These data suggest that ischemia and IPC influence global acetyl-Lys and phosphorylation and also show that modulation of acetyl-Lys with splitomicin can influence phosphorylation indicative of cross-talk.
FIGURE 1.

Phosphorylation and lysine acetylation analysis from ex vivo myocardial tissue. A, Langendorff perfused hearts were subjected to 0 or 20 min ischemia or IPC with or without the presence of 10 μm splitomicin during 20 min of baseline (nonischemic) perfusion. Quantification was performed by TMT isobaric labeling and sequential enrichment of lysine-acetylated peptides and phosphopeptides. Peptides were fractionated with HILIC and analyzed by reversed phase nLC-MS/MS. B, histograms showing the distribution of phosphopeptides (purple, phospho), lysine-acetylated peptides (blue, AcetylK), and total protein (orange) quantification expressed as log2 ratios relative to 0-min ischemia (0I).
We quantified 7437 unique phosphopeptides (5605 phosphosites with >75% localization probability; supplemental Table S1), 346 (271 phosphosites; supplemental Table S1) showing >1.5-fold up-regulation and 194 (154 phosphosites) showing >1.5-fold down-regulation following 20I. Of the regulated phosphosites, 69 were significant with p value <0.05 in two independent experiments and 272 with p value <0.05 in at least one experiment (two-sided t test). 402 unique phosphopeptides were regulated (299 phosphosites; supplemental Table S1) by >±1.5-fold following IPC (54 phosphosites with p value <0.05 in two independent experiments and 197 with p value <0.05 in at least one experiment). Surprisingly, 267 of these phosphopeptides were not regulated in 20I and were highlighted as IPC-specific. 2898 unique lysine-acetylated peptides were quantified, with 229 of these up- or down-regulated (>±1.5-fold) following either 20I (80 peptides) or IPC (159 peptides), which was consistent with the median ratios for these conditions (Fig. 1B and supplemental Table S1).
The large scale data were interrogated using Ingenuity Pathway Analysis (IPA) to identify pathways enriched in proteins containing peptides modified by phosphorylation, acetyl-Lys, or both (supplemental Fig. S5). Site-specific analysis also allowed us to highlight PTM sites differentially regulated between ischemia and IPC, and those influenced by splitomicin to determine potential targets of cross-talk. Several pathways were over-represented with regulated phospho- and acetyl-Lys sites in both ischemia and IPC (supplemental Fig. S5), including those associated with metabolism (fatty acid metabolism, TCA cycle, glycolysis; Fig. 2A), as well as those involved in contraction and mitochondrial function (supplemental Fig. S6). Fatty acid metabolism was enriched in both regulated phosphorylation and acetyl-Lys sites (Fig. 2A) during ischemia and IPC. Several acetyl-Lys sites showed differential modification when compared between 20I and IPC, for example Lys-214 of 3-ketoacyl-CoA thiolase (ACAA2) was significantly deacetylated (>1.5-fold down-regulated; p value <0.05) but only during IPC. Similar data were observed for mitochondrial trifunctional enzyme subunits α and β (HADHA and HADHB), which also display 3-ketoacyl-CoA thiolase activity. Splitomicin attenuated deacetylation of ACAA2 Lys-214 (p value <0.05), and additional sites on HADHA. Deacetylation events were observed more frequently in IPC than ischemia; however, this effect was not global, and we observed increased acetyl-Lys on a number of sites, including additional sites on ACAA2 and HADHB, suggesting multiple acetylases/deacetylases are active during IPC. Our results also show extensive modification of proteins associated with mitochondrial dysfunction (supplemental Fig. S6), including several involved in redox homeostasis and oxidative phosphorylation. IPC-specific deacetylation was observed on more than 10 subunits of complex I; however, none of these sites was attenuated by splitomicin. Complex V was extensively modified with both phosphorylation and acetyl-Lys. For example, increased acetylation of Lys-73 on mitochondrial ATP synthase subunit O (ATP5O) was observed with splitomicin treatment following both 20I and IPC. Additional pathways included the mitochondrial permeability transition pore (mPTP) complex and the contractile apparatus (supplemental Fig. S6). Tropomyosin (TPM1), in particular, contained numerous acetyl-Lys and phosphorylation sites that were either significantly deacetylated or dephosphorylated during IPC, with this effect significantly reversed by splitomicin, which suggests co-regulation between these PTM.
FIGURE 2.

Site-specific relative quantification of modified peptides and total protein (A) and metabolites (B) from pathways highlighted in ischemia and IPC, in the presence or absence of splitomicin. A, relative quantification (heat maps) of phosphopeptides (side bar, purple; phospho), lysine-acetylated peptides (side bar, blue; AcetylK), and total protein (side bar, orange) with quantification expressed as log2 ratios relative to 0-min ischemia (0I) for selected proteins clustered into the pathways “fatty acid metabolism,” “TCA cycle,” and “glycolysis.” B, selected metabolites quantified by LC-SRM; **, p < 0.001; *, p < 0.05; F-1,6-B, fructose 1,6-bisphosphate.
Because splitomicin attenuates IPC (22) and our data identified pathways influenced by splitomicin in ischemia and IPC, we wished to validate the functional basis for these changes. The production and utilization of high energy phosphates are metabolic processes intimately associated with ischemia and IPC. These pathways (e.g. TCA cycle and glycolysis) were over-represented as containing PTM sites responding to ischemia and/or IPC (Fig. 2A; supplemental Fig. S5). We attempted to validate our PTM data and pathway analysis using metabolomics assays quantified by LC selected reaction monitoring (LC-SRM) (supplemental Table S2). We monitored succinyl-CoA (tricarboxylic acid cycle), fructose 1,6-bisphosphate (glycolysis), and creatine phosphate (mitochondrial high energy phosphate essential for muscle contraction), as well as co-factors (NAD, NADH, and NADP) and lactate (Fig. 2B). Fructose 1,6-bisphosphate levels remained unaltered compared with 0I in both 20I and IPC, irrespective of splitomicin treatment. Succinyl-CoA, however, increased >5-fold following 20I, and this was attenuated by splitomicin. Several acetyl-Lys sites were identified on succinyl-CoA ligase (SUCLA2; Fig. 2A), yet only Lys-88 was deacetylated during ischemia and ameliorated to control levels by splitomicin in a manner consistent with the metabolite data. No alterations in succinyl-CoA levels were observed in IPC. Creatine phosphate was reduced by >40-fold after 20I, irrespective of splitomicin treatment; however, splitomicin in IPC resulted in creatine phosphate levels significantly higher than those observed in IPC alone. We identified acetyl-Lys sites in both muscle-type creatine kinase (CKM) and creatine kinase mitochondrion type 2 (CKMT2) that were regulated by ischemia and IPC, and a subset of these responded to splitomicin (supplemental Fig. S6). Ischemia induced accumulation of NADH, yet NAD+ and NADP were unaltered relative to 0I. As expected, lactate increased by >40-fold after 20I; however, splitomicin attenuated this significantly. Our data suggest this is unrelated to lactate oxidation, because splitomicin did not influence NADH levels.
Acetyl-Lys Influences Kinase Activation during Myocardial Ischemia and IPC
Cross-talk between phosphorylation and acetyl-Lys is likely mediated by kinases/phosphatases and/or acetylases/deacetylases. To test this, we employed splitomicin inhibition of lysine deacetylation and examined alterations in signal pathways and their kinases. First, phosphopeptides up-regulated by >1.5-fold in 20I were clustered into two groups as follows: (i) those unaltered and (ii) those responding to splitomicin (Fig. 3, A and B). These clusters were subjected to IPA analysis (supplemental Fig. S5, B and C). Enriched pathways containing proteins showing phosphorylation increases in 20I that were ameliorated by splitomicin (Fig. 3B; supplemental Fig. S5C) included AMPK signaling, insulin receptor signaling (taken as AKT), and PKA signaling, although those that did not respond to splitomicin were predominantly associated with metabolism (glycolysis/gluconeogenesis), aldosterone, and actin signaling, ubiquitination, and extracellular remodeling (supplemental Fig. S5B). Site-specific analysis was performed on phosphopeptides regulated during 20I that responded to splitomicin, and a subset of these mapped onto a network centered on AMPK, PKA, and AKT (Fig. 3C). Many substrates of these kinases were regulated by 20I, and the phosphorylation changes were ameliorated by splitomicin. This shows a role for acetyl-Lys in regulating AMPK and AKT during myocardial ischemia and is the first report to suggest a role for acetyl-Lys in PKA regulation.
FIGURE 3.

Kinase activation during myocardial ischemia is regulated by splitomicin-mediated inhibition of SIRT1-associated lysine deacetylation. A, fuzzy c-means clustering of phosphopeptides differentially regulated between 0 min of ischemia (0I) (or 0 min of ischemia with splitomicin (0ISPLIT)) and 20-min ischemia (20I) but not attenuated following 20 min of ischemia in the presence of splitomicin. B, fuzzy c-means clustering of phosphopeptides differentially regulated between 0 min of ischemia (0I) (or 0 min of ischemia with splitomicin (0ISPLIT)) and 20 min of ischemia (20I), and attenuated by 20 min of ischemia with splitomicin (20ISPLIT). C, site-specific analysis of phosphopeptides (>75% localization probability) clustered in B regulated by 20I and attenuated with splitomicin. Previously verified targets of AMPK (green), AKT (purple), and PKA (blue) with ischemia-induced increases in phosphorylation attenuated by splitomicin (supplemental Table S1). Immunoblotting shows splitomicin attenuates ischemia-associated phosphorylation of Thr-172 AMPKα, Ser-473 AKT, Ser-422 eIF4B, and Ser-79 ACC (D) and PKA substrate, relative to 0 min of ischemia and 20 min of normoxic time control perfusion (TC) (E). Blots are representative of n = 3. F and G, relative quantification of acetyl-CoA (F) and cAMP (G) by LC-SRM in myocardial tissue subjected to 20 min of ischemia (20I) or 20 min of ischemia with splitomicin (20ISPLIT) expressed relative to 0 min of ischemia (0I) (n = 3). *, p value < 0.05 (two-side t test); ns, not significant. Error bars show standard deviation.
Western blotting confirmed the effects of splitomicin on phosphorylation of AMPK, AKT, PKA, and their substrates (Fig. 3, D and E; supplemental Fig. S7). For these experiments, we generated hearts over a time course of ischemia (0, 2, 10, and 20 min ischemia; n = 3) compared with 20 min free perfusion (or “time control”; n = 3). Splitomicin attenuated ischemia-induced phosphorylation of Thr-172 on αAMPK and Ser-473 on AKT, and this effect was more pronounced at 20I (Fig. 3D). The downstream targets of AMPK (Ser-79 on ACC) and AKT (Ser-422 on eukaryotic initiation factor 4B) were regulated similarly to their respective kinases (Fig. 3D). Ser-79 phosphorylation inhibits ACC activity (carboxylation of acetyl-CoA to malonyl-CoA); however, splitomicin in 20I did not result in a concomitant decrease in acetyl-CoA, as determined by LC-SRM (Fig. 3F; supplemental Table S3), despite the reduction in ACC phosphorylation. This suggests acetyl-Lys/deacetylation may influence other enzymes with acetyl-CoA as a substrate or product (e.g. 3-ketoacyl-CoA thiolase). Citrate and other small molecules also allosterically modulate ACC activity (81). Ischemic phosphorylation of PKA substrates (RRX(pS/pT)) was also attenuated by splitomicin (Fig. 3E). This correlated with splitomicin attenuation of ischemia-associated increases in cAMP and provided evidence that PKA activation may be regulated by acetyl-Lys/deacetylation via modulation of cAMP levels (Fig. 3G; supplemental Table S3).
We next investigated kinase activation in IPC. Phosphopeptides up-regulated by >1.5-fold were functionally clustered using IPA (supplemental Fig. S5D). Enriched pathways included PKA, α-adrenergic, and calcium signaling. A common feature of these is MAPK signaling. We identified increased phosphorylation of Tyr-185 in the activation loop of MAPK1 and Tyr-205 on MAPK3, both of which are associated with IPC (82). We did not, however, observe altered phosphorylation of a variety of PKC isoforms or substrates, including PKCA/PKCB (Thr-497/Thr-550) and PKCD (Thr-505) nor the activation autophosphorylation site of PKCD (Ser-643), the activation C-terminal hydrophobic site of PKCD (Ser-662), or of PKCE (Ser-729). Phosphopeptides up-regulated by >1.5-fold in IPC were next clustered into two groups as follows: (i) those unaltered, and (ii) those ameliorated by splitomicin, and each was analyzed using IPA (supplemental Fig. S5, E and F). In contrast to the results from 20I, PKA signaling was the most enriched pathway unaffected by splitomicin treatment. Enriched pathways in the cluster containing phosphopeptides ameliorated by splitomicin were predominantly associated with cell junction signaling, as well as AMPK signaling (consistent with the results observed in 20I ± splitomicin).
Acetyl-Lys Promotes Dephosphorylation in a Basophilic Kinase Motif
Our large scale data show strong evidence for cross-talk as follows: phosphorylation and acetyl-Lys co-occurred on many proteins, both PTM could be influenced by splitomicin, and inhibition of lysine deacetylation influenced the activation of three kinases (AMPK, AKT, and PKA) and their downstream targets. We next examined the sequence context and structural basis of cross-talk by determining the proximity of PTM in tertiary structures (Protein Data Bank) of 45 proteins containing at least one confident ischemia or IPC-regulated phosphorylation/acetyl-Lys site. Of these, 25 structures were available in the mammalian taxonomy containing at least 80% sequence similarity to the Swiss-Prot R. norvegicus sequence. Three proteins contained an amino acid substitution at one site of PTM observed here. Fourteen proteins contained at least one observed phosphorylated residue within ∼10 Å of an identified acetylated lysine (supplemental Fig. S8). For example, the solution structure of TPM1 highlights head-to-tail interactions between individual molecules where the C-terminal phosphorylation on Ser-283 is in close proximity to an N-terminal acetylation on Lys-12. Proximal modifications were both adjacent (<5 amino acids) and distant (>5 amino acids) in the primary sequence.
We next attempted to identify sequence motifs in which phosphorylation and acetyl-Lys were over-represented. MS/MS sequences from PTM analysis of ischemia and IPC were combined, and those showing evidence of modification by both were considered further. We identified 50 peptides containing the basophilic protein kinase motif KXXS, modified with either acetyl-Lys or phosphoserine, but not both simultaneously (supplemental Table S5). For example, CKM was modified by acetyl-Lys at Lys-369 or phosphorylation at Ser-372, and both sites were regulated by >1.5-fold following 20I. These changes were partially attenuated by splitomicin (supplemental Table S1).
To investigate this cross-talk and the rates of modification induced by proximal PTM in the KXXS motif, we synthesized the C-terminal peptide from CKM (364EKKLEKGQSIDD375) as four variants: (i) nonmodified (KXXS); (ii) phosphorylated at Ser-9 (equivalent to Ser-372; KXXpS); (iii) acetylated at Lys-6 (Lys-369; AcKXXS); and (iv) acetylated at Lys-6 and phosphorylated at Ser-9 (Lys-369 and Ser-372; AcKXXpS). We performed in vitro kinase, phosphatase, acetyltransferase, and deacetylase reactions on all variants and monitored the substrates and products by nLC-SRM. Each variant was added into a separate lysate generated following 20I. Quantification was performed by comparing the area under the curve of the product/substrate and expressed as percent substrate conversion/min. Initially, kinase reactions were performed separately on the two nonphosphorylated substrates (± acetyl-Lys) in the presence of 5 mm ATP, phosphatase, and deacetylase inhibitors. Maximum phosphorylation of the KXXS substrate was observed after 20 min (13% substrate conversion of KXXS to KXXpS); however, no conversion of AcKXXS to AcKXXpS was observed (Fig. 4A and supplemental Table S6). Dephosphorylation was next measured for the two phosphorylated substrates (± acetyl-Lys) in the presence and absence of phosphatase and deacetylase inhibitors. The KXXpS substrate displayed 4% dephosphorylation to KXXS irrespective of the presence of inhibitors, whereas the AcKXXpS substrate showed 70% dephosphorylation to AcKXXS within 60 min (Fig. 4B and supplemental Table S6B) in the absence of phosphatase inhibition, and 33% dephosphorylation to AcKXXS in the presence of phosphatase inhibitors (Fig. 4C and supplemental Table S6C). No changes in acetylation/deacetylation were observed for any of the test peptides, even in the absence of deacetylase inhibitors and in the presence of 1 mm NAD, or with added 1 mm acetyl-CoA (supplemental Tables S6, D and E). These results suggest that acetyl-Lys at the “minus 3 amino acids” position (P-3) to the phosphorylation site promotes dephosphorylation. To further investigate this, the C-terminal CKM phosphopeptide was synthesized with the following: (i) Gln at Lys-6 (Lys to Gln; QXXpS) to mimic acetyl-Lys; and (ii) Arg at Lys-6 (Lys to Arg; RXXpS) to mimic the positively charged lysine. The RXXpS substrate displayed 7% dephosphorylation to RXXS, whereas the QXXpS acetyl-Lys mimic showed 43% dephosphorylation to QXXS, providing further evidence for the promotion of dephosphorylation by the presence of acetyl-Lys at P-3 (Fig. 4D and supplemental Table S6F).
FIGURE 4.
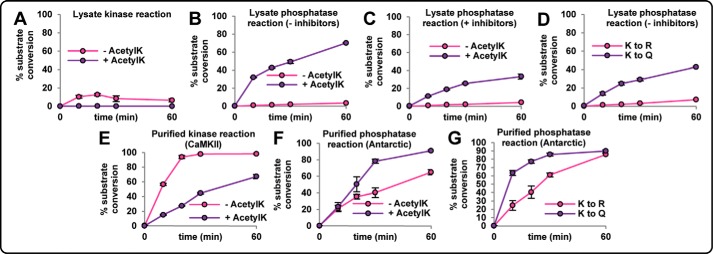
In vitro kinase and phosphatase assays demonstrate the kinetic effects of proximal phosphorylation and acetyl-Lys (AcetylK) on the CKM peptide 364EKKLEKGQSIDD375 as follows: (i) nonmodified (KXXS); (ii) phosphorylated at Ser-9 (Ser-372; KXXpS); (iii) acetylated at Lys-6 (Lys-369; AcKXXS); (iv) acetylated at Lys-6 and phosphorylated at Ser-9 (Lys-369 and Ser-372; AcKXXpS); (v) acetyl-Lys mimic Gln-6 and phosphorylated at Ser-9 (K369Q and Ser-372; QXXpS); and (vi) acetyl-Lys-negative Arg-6 and phosphorylated at Ser-9 (K369R and Ser-372; RXXpS). A, kinase assay using KXXS and AcKXXS added separately into lysates in the presence of phosphatase and deacetylase inhibitors. B, phosphatase assay using KXXpS and AcKXXpS added separately into lysates in the absence of phosphatase and deacetylase inhibitors. C, phosphatase assay using KXXpS and AcKXXpS added separately into lysates in the presence of phosphatase and deacetylase inhibitors. D, phosphatase assay using QXXpS and RXXpS added separately into lysates in the absence of phosphatase deacetylase inhibitors. E, kinase assay using KXXS and AcKXXS with purified recombinant CaMKII. F, phosphatase assay using KXXpS and AcKXXpS with purified recombinant antarctic phosphatase. G, phosphatase assay using QXXpS and RXXpS with purified recombinant antarctic phosphatase. Peak areas of all variants were simultaneously monitored for each reaction using nLC-SRM to obtain % substrate conversion (areaproduct/(areasubstrate + areaproduct)) at 0, 10, 20, 30, and 60 min (n = 3).
The hypothesis that dephosphorylation is promoted by proximal acetyl-Lys requires the protein to be initially modified, even transiently, at both sites. To investigate this, kinase and phosphatase assays were performed using purified recombinant enzymes. Scansite suggested that phosphorylation of CKM Ser-372 is most likely catalyzed by CaMKII. CaMKII-mediated reactions were performed using the KXXS and AcKXXS CKM peptides. Maximum phosphorylation of the KXXS substrate was observed after 60 min (98% substrate conversion to KXXpS), whereas conversion of the AcKXXS substrate proceeded at a slower rate (67% substrate conversion to AcKXXpS by 60 min) (Fig. 4E and supplemental Table S6G). These results show that simultaneous phosphorylation and acetyl-Lys can occur in vitro. Next, antarctic phosphatase reactions were performed on the two phosphorylated substrates (± acetyl-Lys). The KXXpS substrate displayed 65% dephosphorylation to KXXS, whereas AcKXXpS displayed a faster dephosphorylation rate (91% substrate conversion to AcKXXS within 60 min) (Fig. 4F and supplemental Table S6H). Reactions were also performed tk;4on the mimic phosphopeptides, with the RXXpS substrate displaying slower dephosphorylation compared with the QXXpS acetyl-Lys mimic (Fig. 4G and supplemental Table S6I).
We next investigated acetyl-Lys-induced dephosphorylation of full-length CKM. FLAG-tagged CKM wild-type (WT) and a K369Q acetyl-Lys mimic were overexpressed in L6 myoblasts grown in light and heavy stable isotope labeling by amino acids in cell culture (SILAC) (Fig. 5A). Following anti-FLAG immunoprecipitation, enriched WT and K369Q mimic from both light and heavy cells were subjected to in vitro CaMKII kinase reactions. Excess kinase was removed, and the heavy labeled WT and K369Q CKM were subjected to either 30- or 60-min phosphatase reactions using recombinant PP1. Heavy labeled, phosphatase-treated CKM was mixed with light labeled non-phosphatase-treated CKM and digested with trypsin, and the rate of dephosphorylation was measured by nLC-MS/MS. Dephosphorylation rates were then determined by comparing the phosphopeptide heavy/light ratios. WT CKM treated with PP1 for 30 and 60 min displayed very little dephosphorylation at Ser-372 (<1.1-fold by 60 min or phosphorylation decrease of only 3 ± 2%; Fig. 5B; supplemental Table S7). The K369Q acetyl-Lys mimic, however, displayed significant dephosphorylation of Ser-372 by 30 min (phosphorylation decrease of 68% ± 2%) and >2-fold dephosphorylation following 60 min (Fig. 5B). These assays show that acetyl-Lys in the KXXS kinase motif reduces the rate of phosphorylation and promotes rapid dephosphorylation.
FIGURE 5.

In vitro phosphatase and deacetylase assays using SILAC-labeled, immunoprecipitated full-length CKM wild-type (WT), acetyl-Lys mimic (K369Q), and phospho-mimic (S372E). A and C, overview of the approach. FLAG-tagged WT, K369Q (A) and S372E (C) CKM were overexpressed in light (L) and heavy (H) SILAC-labeled rat L6 myoblasts. Following immunoprecipitation, CKM from both light and heavy labeled cells was subjected to an in vitro kinase assay with CaMKII (A). Heavy labeled CKM was subsequently subjected to phosphatase assays with PP1 for 30 or 60 min or an in vitro deacetylase assay with immunoprecipitated FLAG-tagged SIRT1 for 30 or 60 min (C). Light and heavy labeled CKM were eluted, mixed, and digested, and the rates of dephosphorylation or deacetylation were measured between light and heavy labeled peptides using nLC-MS/MS. B, quantification of the relative abundance of the SILAC phosphopeptides GQSpIDDMIPAQK (WT) and KLEQGQSpIDDMIPAQK (K369Q) between light (untreated) and heavy (treated with PP1 for 30 or 60 min) showing the rate of dephosphorylation expressed as log2(H/L). D, quantification of the relative abundance of the SILAC acetylated peptide KLEK(Ac)GQSIDDMIPAQK (WT) and KLEK(Ac)GQEIDDMIPAQK (S372E) between light (untreated) and heavy (treated with SIRT1 for 30 or 60 min) showing the rate of deacetylation expressed as log2(H/L).
To investigate whether phosphorylation influenced deacetylation, FLAG-tagged CKM WT and an S372E phospho-mimic were overexpressed in L6 myoblasts grown in light and heavy SILAC (Fig. 5C). Following anti-FLAG immunoprecipitation, heavy labeled WT and S372E CKM were subjected to 30 and 60 min of incubation with immunoprecipitated FLAG-tagged SIRT1 deacetylase. Heavy labeled, deacetylase-treated CKM was mixed with light labeled nontreated CKM and digested with trypsin, and deacetylation was measured by nLC-MS/MS. S372E CKM treated with SIRT1 displayed only minor deacetylation at Lys-369 (1.1-fold by 60 min, acetyl-Lys decrease of ∼10% ± 1%; Fig. 5D and supplemental Table S8). WT CKM, however, displayed significant SIRT1-mediated deacetylation of Lys-369 (1.6-fold by 60 min, acetyl-Lys decrease of 38 ± 3%; Fig. 5D). These data suggest that phosphorylation inhibits SIRT1-mediated deacetylation within the KXXS motif. The mutually exclusive nature of these two PTMs in the large scale ex vivo dataset combined with the in vitro assays performed here suggests that acetyl-Lys promotes dephosphorylation, which in turn promotes deacetylation. The deacetylated form of the motif can then be phosphorylated, which is consistent with both our synthetic peptide and recombinant CKM protein kinase/phosphatase assays.
Acetyl-Lys Disrupts a Phosphate-Lysine Salt Bridge and Induces Peptide Backbone Flexibility
We next investigated the mechanism of acetyl-Lys-induced dephosphorylation. To examine whether peptide backbone conformational changes were induced by acetyl-Lys and/or phosphorylation, we analyzed the four CKM peptide variants by 1H NMR in the presence of 10% D2O in H2O at pH 7.2 (supplemental Fig. S9). One-dimensional 1H NMR of the NH region of the four variants highlighted only minor chemical shift differences between KXXS and AcKXXS with one additional peak arising from the acetyl amide proton (supplemental Fig. S9, A and B). Phosphorylation induced larger chemical shifts, and comparison of KXXpS and AcKXXpS highlighted differences that indicated significant changes in peptide backbone environments (supplemental Fig. S9, C and D). We investigated these by two-dimensional 1H NMR total correlation spectroscopy (TOCSY) and nuclear Overhauser effect spectroscopy (NOESY) (supplemental Fig. S10). An overlay of HA-HN TOCSY region of KXXpS and AcKXXpS highlighted chemical shift differences in residuesLys-6 to Asp-12, although Lys-3 to Glu-5 was unaltered (Fig. 6A). Overlay of the HB-HN TOCSY and NOESY region of AcKXXpS highlighted additional peaks in the TOCSY spectrum from residues I10-Asp-12 that are absent in the NOESY region, indicating that the C-terminal region may be in two conformations (Fig. 6B). These peaks were absent for KXXpS, indicating a more rigid conformation. Multiple long range 1H-1H NOEs between the Lys-6 and Ser(P)-9 side chains in KXXpS, which were absent in AcKXXpS, indicated that these two residues are in close proximity (Fig. 6C) and provide evidence for a phosphate-lysine interaction, which is inhibited by acetyl-Lys. The rigidity of the KXXpS variant prompted us to investigate the presence of secondary structure. Interrogation of the NOESY spectrum revealed no HA-HN or HA-HB 1H-1H NOEs between i and i + 2 or 3 amino acids (indicative of α-helices). This was further confirmed by analysis of the four variants with CD spectroscopy, which revealed no obvious secondary structure (supplemental Table S9). Taken together, these results suggest that the KXXpS variant is in a fixed conformation due to the formation of a phosphate-lysine salt bridge (Fig. 6D). Acetyl-Lys inhibits the salt bridge and results in a more flexible conformation, which is more accessible to phosphatases.
FIGURE 6.

Analysis of CKM C-terminal peptide 364EKKLEKGQSIDD375: (i) phosphorylated at Ser-9 (Ser-372; KXXpS) or (ii) acetylated at Lys-6 and phosphorylated at Ser-9 (Lys-369 and Ser-372; AcKXXpS) by two-dimensional 1H NMR total correlation spectroscopy (TOCSY) and NOESY. Overlays of HA-HN TOCSY region of acetylated (AcKXXpS) and nonacetylated (KXXpS) phosphopeptide are shown in A; HB-HN TOCSY and NOESY regions of the acetylated phosphopeptide (AcKXXpS) are shown in B; side chain NOESY region of the acetylated (AcKXXpS) and nonacetylated (KXXpS) phosphopeptide is shown in C. D, proposed conformation of the acetylated (AcKXXpS) and nonacetylated (KXXpS) phosphopeptide.
DISCUSSION
Large scale acetyl-Lys and phosphorylation analysis in myocardial ischemia and IPC identified PTM sites not previously associated with injury or cardioprotection, and confirmed that a specific response to each occurs at the signal level. Ischemia induces extensive metabolic changes and acetyl-Lys is emerging as a regulator of metabolism (58, 63), cell protection, and signal propagation (35, 38). Splitomicin treatment confirmed acetyl-Lys is involved in myocardial ischemia and IPC and is capable of cross-talk with phosphorylation-mediated signaling. We confirmed that acetyl-Lys influences AMPK and AKT activation, and we also revealed a role in PKA regulation, most likely via cAMP levels, although it remains unclear whether this occurs through alteration of adenylate cyclase activity. Ischemia-induced phosphorylation of PKA site Ser-16 on phospholamban was attenuated with splitomicin. Phospho-Ser-16 increases Ca2+ uptake into the sarcoplasmic reticulum by disrupting the inhibitory phospholamban·sarcoplasmic reticulum-Ca2+-ATPase complex (83, 84). In contrast, dephosphorylation of phospholamban Ser-16/Ser-17 was observed in IPC and was unaffected by splitomicin, along with other PKA targets, suggesting that the loss of cardioprotection associated with splitomicin is independent of PKA. Numerous other targets of PKA were identified in the ischemia cluster responding to splitomicin (e.g. Ser-156 on Bcl2 antagonist of cell death); however, not all PKA targets responded similarly; for example, cardiac myosin-binding protein C (MYBPC3) and cardiac troponin I (TNNI3) did not display increased phosphorylation in 20I nor reduced phosphorylation post-splitomicin treatment. PKA phosphorylation of Ser-434 on SIRT1 increases deacetylase activity independently of NAD+ (39), suggesting SIRT1 and PKA may regulate each other in a similar fashion to SIRT1 and AMPK (17), although unlike SIRT1/AMPK, the SIRT/PKA relationship appears to be specific to ischemia. Although our study used splitomicin to inhibit lysine deacetylation (22, 85), we cannot unequivocally state that acetyl-Lys sites altered by splitomicin are direct targets of SIRT1, nor whether other SIRTs (or non-SIRT acetylases/deacetylases) may be affected. For example, members of metabolic pathways containing ischemia or IPC-induced changes in acetyl-Lys may also be targets of SIRT3 (86). A detailed investigation of the IC50 values of splitomicin on other deacetylases has not been performed. Therefore, further validation of the regulated targets is required to unequivocally pinpoint the role of SIRT1.
Phosphoproteomics showed no direct evidence for PKC activation during IPC. Despite this, we did observe increased phosphorylation of MAPK variants, consistent with the model of IPC proposed in Ref. 82, which shows MAPK activation in IPC is PKC-dependent. Alternatively, our model did not examine I/R post-IPC. The PKCϵ isoform implicated in signal complex formation and IPC-mediated signaling (87) may be only transiently activated during one of the cycles of I/R that constitute IPC, or indeed it may be downstream of alternative signal pathways implicated in IPC or I/R; for example, PKC activation itself may be downstream of the ERK- and PI3K/AKT-associated reperfusion injury salvage kinase pathway (88) and thus may occur upon I/R after the protective IPC period is concluded.
Among the functional clusters containing members with ischemia- and/or IPC-associated peptides that were influenced by splitomicin were the contractile apparatus and mPTP. Regulation of contractility via phosphorylation has been demonstrated on many proteins, including cardiac MYBPC3, TPM1, troponins I and T, and myosin light chains 1 and 2. Our study suggests acetyl-Lys may play a similar role in regulating the contractile apparatus. For example, 28 acetyl-Lys sites were identified on TPM1, and the proximity of acetyl-Lys and phosphorylation in head-to-tail structures of TPM1 suggested PTM cross-talk is possible. Permeabilization and/or rupture of the mitochondrial membrane are linked to apoptotic and necrotic cell death, which are associated with the mPTP in ischemic injury. The molecular composition of the mPTP is still not fully unraveled; however, proposed members include hexokinase 2, ADP/ATP translocase (SLC25A), VDAC, CKMT2, mitochondrial apoptosis-induced channel, and peptidylprolyl isomerase (89–91). Genetic studies, however, suggest some of these may be dispensable for mitochondrion-associated cell death (92, 93). Recently, a novel composition of the mPTP has been proposed (94) and includes ATP synthase, which suggests a dual function for complex V as follows: (i) to produce energy and (ii) to control cell death. We identified extensive phosphorylation (>30 sites) and acetyl-Lys (33 sites) of mPTP members, many of which were regulated by ischemia and/or IPC. Splitomicin ameliorated changes to acetylated peptides observed in ischemia and IPC from CKMT2 and SLC25A4, providing further evidence of a widespread role for lysine deacetylation in mediating the functional effects of ischemia and cardioprotection.
Another cluster of modified peptides belonged to proteins involved in fatty acid metabolism. Unlike other tissues, the myocardium produces ∼60–70% of its energy from fatty acid oxidation. Ischemia results in lowered fatty acid oxidation and an accumulation of fatty acid intermediates, which have detrimental effects, including increased membrane instability, arrhythmias, and increased infarct size. Reducing fatty acid metabolism and promoting glucose oxidation has long been known to provide cardioprotection (95). Our data show that at least three enzymes (ACAA2, HADHA, and HADHB) with 3-ketoacyl-CoA thiolase activity contained post-translational modifications that were induced by ischemia and/or IPC, and ACAA2 acetyl-Lys was ameliorated in IPC by splitomicin. Inhibition of 3-ketoacyl-CoA thiolase activity with trimetazidine reduces β-oxidation of fatty acids, promotes glucose oxidation, and is a clinically effective anti-angina strategy (96). Differential regulation of acetyl-Lys between ischemia and IPC on enzymes displaying 3-ketoacyl-CoA thiolase activity provides insight into the potential mechanism of cardioprotection.
Ischemia and IPC induced different changes to pathways involved in metabolism and the production or utilization of high energy phosphates. Conversion to anaerobic glycolysis and production of lactate are well characterized in ischemia, and we were surprised that splitomicin ameliorated this to a significant extent. This suggests lysine deacetylation may be beneficial in ischemia, which is contradictory to the loss of cardioprotection (22) induced by splitomicin in IPC. Ischemia resulted in a similar rise in succinyl-CoA levels that were also attenuated by splitomicin. This correlated with attenuated deacetylation of at least one site in SUCLA2 suggesting a link between acetyl-Lys and SUCLA2 activity. The large reduction in creatine phosphate seen in both ischemia and IPC was only attenuated by splitomicin during cardioprotection. These results were consistent with several PTM on both CKM and CKMT2, which again suggests that individual modification sites may be critical for activity and be influential in contributing to the ischemic or cardioprotective phenotype. Changes in the function of metabolic enzymes, such as CKMT2 and SUCLA2, which utilize the high energy ATP pool that is rapidly depleted during ischemia, may thus also be involved in maintenance of ATP levels during IPC, with their functions at least partially regulated by PTM.
Methylation in kinase motifs (97–101) and acetylation (102) can occur mutually exclusively to phosphorylation. The structural basis for cross-talk is thought to be primarily via steric hindrance and/or disruption of salt bridges or hydrogen bonds between substrate and kinase (103). Contrary to this, acetylation of Lys-105 on CDC6 results in increased phosphorylation of adjacent Ser-106 by cyclin-dependent kinases (45), and co-modification of Lys-9 and Ser-10 on histone H3 also occurs (104). Therefore, these PTMs can co-occur, and cross-talk may mostly contribute to the kinetics of modification. A systematic investigation of neighboring modifications in the protein kinase KXXS motif was undertaken to determine the structural and kinetic constraints for cross-talk, because this motif was over-represented in PTM regulated by ischemia and IPC. Modeling of the motif using modified variants of the C-terminal CKM peptide (acetylation of Lys-369 and phosphorylation of Ser-372) showed that acetyl-Lys induced a >10-fold dephosphorylation suggesting the recruitment of one or more phosphatases. This effect was also observed on full-length CKM containing a K369Q acetyl-Lys mimic. Although the recruitment of a phosphatase by acetyl-Lys has been suggested (46), the molecular mechanism mediating this interaction has not previously been investigated. Our data confirmed that the KXXpS substrate contains a salt bridge interaction between lysine and phosphate and a peptide backbone conformational change compared with the doubly modified AcKXXpS substrate. We propose that the Lys-Ser(P) interaction inhibits phosphatase access, whereas acetylation neutralizes the charge, inhibits salt bridge formation, and induces a flexible peptide backbone facilitating phosphatase access.
In vitro SIRT1 deacetylase assays of CKM WT and an S372E phosphomimic further suggest that once dephosphorylated, the peptide is then more amenable to SIRT1-mediated deacetylation and hence may once again become available for phosphorylation, depending on the rate of protein turnover and the availability of the kinase(s) and acetylase(s)/deacetylase(s) that drive the modifications. Additionally, because the acetylase/deacetylase assays with CKM synthetic C-terminal peptides in complex lysates did not show a similar effect, other acetylases/deacetylases (beyond SIRT1) may be involved in maintaining the PTM balance in cardiac tissue during I/R and/or IPC.
Fifty peptides regulated in ischemia and/or IPC contained the KXXS motif. We did not, however, observe evidence for “reciprocal” regulation (i.e. increase in the acetylated form of a peptide corresponding to a decrease in the phosphorylated form or vice versa), nor did we see examples of any doubly modified peptides in these data. For the structural and enzymatic evidence we observed to be physiologically relevant, the doubly modified form must be able to occur in vivo. We proved that phosphorylation of both the CKM C-terminal peptide and full-length protein can occur when lysine-acetylated, albeit at a much slower rate than the native forms. These data, combined with the rapid dephosphorylation of lysine-acetylated synthetic peptide variants and full-length K369Q CKM, suggest any doubly modified forms within the KXXS motif would be highly transient, and therefore it is not surprising that these were not observed as they are likely to be present at extremely low abundance in the complex tissue lysates used for large scale analysis. The lack of apparent reciprocal regulation is also likely to be due to the sub-stoichiometric levels of these (and indeed most) PTMs in the context of the overall protein abundance. This means PTM cross-talk in this context is likely involved in signal “dampening” or “tuning” (e.g. to protect against uncontrolled signal pathway activation) rather than acting as a strict “on”/“off” switch for every copy of the protein, or by generating an extreme reversal of the PTM state. Cross-talk in this motif likely provides a mechanism to modulate the level of signal activation and thus ensure the maintenance of cellular balance. To our knowledge, this is the first study to perform a detailed investigation of intra-molecular cross-talk between phosphorylation and acetyl-Lys within a kinase consensus motif. We believe that the observed mechanism represents a previously unexplored level of regulation that is potentially widespread and is likely to involve other signaling proteins and PTMs.
Supplementary Material
Acknowledgments
We thank Lene Jakobsen, Melanie Schulz, Ann Kwan, Kiersten Liddy, Alistair Edwards, Ben Crossett, Nestor Solis, and Rima Chaudhuri for useful discussions and instrument support.
This work was supported in part by National Health and Medical Research Council of Australia Project Grant 571002 (to S. J. C. and B. D. H.), the Lundbeck Foundation Junior Group Leader Fellowship and LTQ-Orbitrap Velos (to M. R. L.), and the Danish Natural Science Research Council Grant 09-06-5989 (to M. R. L.).

This article contains supplemental Figs. S1–S10 and Tables S1–S9.
- I/R
- ischemia/reperfusion
- IPC
- ischemic pre-conditioning
- PTM
- post-translational modification
- CKM
- muscle-type creatine kinase
- AMPK
- AMP-activated protein kinase
- ACC
- acetyl-CoA carboxylase
- Fmoc
- N-(9-fluorenyl)methoxycarbonyl
- TMT
- tandem mass tag
- ACN
- acetonitrile
- FA
- formic acid
- mPTP
- mitochondrial permeability transition pore
- LC-SRM
- liquid chromatography-selected reaction monitoring
- SILAC
- stable isotope labeling by amino acids in cell culture
- HCD
- higher energy collision dissociation
- IPA
- Ingenuity Pathway Analysis
- 0I
- 0-min ischemia
- 20I
- 20-min ischemia
- CaMKII
- calcium/calmodulin-dependent protein kinase II
- TCA
- tricarboxylic acid
- SIRT
- sirtuin
- pp1
- protein phosphatase 1
- MRM
- multiple reaction monitoring.
REFERENCES
- 1. Karmazyn M., Moffat M. P. (1993) Na+/H+ exchange and regulation of intracellular Ca2+. Cardiovasc. Res. 27, 2079–2080 [DOI] [PubMed] [Google Scholar]
- 2. Liu B., Clanachan A. S., Schulz R., Lopaschuk G. D. (1996) Cardiac efficiency is improved after ischemia by altering both the source and fate of protons. Circ. Res. 79, 940–948 [DOI] [PubMed] [Google Scholar]
- 3. Armstrong S. C. (2004) Protein kinase activation and myocardial ischemia/reperfusion injury. Cardiovasc. Res. 61, 427–436 [DOI] [PubMed] [Google Scholar]
- 4. Hausenloy D. J., Yellon D. M. (2006) Survival kinases in ischemic preconditioning and postconditioning. Cardiovasc. Res. 70, 240–253 [DOI] [PubMed] [Google Scholar]
- 5. Rose B. A., Force T., Wang Y. (2010) Mitogen-activated protein kinase signaling in the heart: angels versus demons in a heart-breaking tale. Physiol. Rev. 90, 1507–1546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Barnett M. E., Madgwick D. K., Takemoto D. J. (2007) Protein kinase C as a stress sensor. Cell. Signal. 19, 1820–1829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Budas G. R., Churchill E. N., Mochly-Rosen D. (2007) Cardioprotective mechanisms of PKC isozyme-selective activators and inhibitors in the treatment of ischemia-reperfusion injury. Pharmacol. Res. 55, 523–536 [DOI] [PubMed] [Google Scholar]
- 8. Palaniyandi S. S., Sun L., Ferreira J. C., Mochly-Rosen D. (2009) Protein kinase C in heart failure: a therapeutic target? Cardiovasc. Res. 82, 229–239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Mullonkal C. J., Toledo-Pereyra L. H. (2007) Akt in ischemia and reperfusion. J. Invest. Surg. 20, 195–203 [DOI] [PubMed] [Google Scholar]
- 10. Diviani D., Dodge-Kafka K. L., Li J., Kapiloff M. S. (2011) A-kinase anchoring proteins: scaffolding proteins in the heart. Am. J. Physiol. Heart Circ. Physiol. 301, H1742–H1753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sanada S., Asanuma H., Tsukamoto O., Minamino T., Node K., Takashima S., Fukushima T., Ogai A., Shinozaki Y., Fujita M., Hirata A., Okuda H., Shimokawa H., Tomoike H., Hori M., Kitakaze M. (2004) Protein kinase A as another mediator of ischemic preconditioning independent of protein kinase C. Circulation 110, 51–57 [DOI] [PubMed] [Google Scholar]
- 12. Dyck J. R., Kudo N., Barr A. J., Davies S. P., Hardie D. G., Lopaschuk G. D. (1999) Phosphorylation control of cardiac acetyl-CoA carboxylase by cAMP-dependent protein kinase and 5′-AMP activated protein kinase. Eur. J. Biochem. 262, 184–190 [DOI] [PubMed] [Google Scholar]
- 13. Kudo N., Barr A. J., Barr R. L., Desai S., Lopaschuk G. D. (1995) High rates of fatty acid oxidation during reperfusion of ischemic hearts are associated with a decrease in malonyl-CoA levels due to an increase in 5′-AMP-activated protein kinase inhibition of acetyl-CoA carboxylase. J. Biol. Chem. 270, 17513–17520 [DOI] [PubMed] [Google Scholar]
- 14. Lopaschuk G. D., Spafford M. A., Davies N. J., Wall S. R. (1990) Glucose and palmitate oxidation in isolated working rat hearts reperfused after a period of transient global ischemia. Circ. Res. 66, 546–553 [DOI] [PubMed] [Google Scholar]
- 15. Oliver M. F., Opie L. H. (1994) Effects of glucose and fatty acids on myocardial ischaemia and arrhythmias. Lancet 343, 155–158 [DOI] [PubMed] [Google Scholar]
- 16. Kahn B. B., Alquier T., Carling D., Hardie D. G. (2005) AMP-activated protein kinase: ancient energy gauge provides clues to modern understanding of metabolism. Cell Metab. 1, 15–25 [DOI] [PubMed] [Google Scholar]
- 17. Cantó C., Gerhart-Hines Z., Feige J. N., Lagouge M., Noriega L., Milne J. C., Elliott P. J., Puigserver P., Auwerx J. (2009) AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature 458, 1056–1060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ruderman N. B., Xu X. J., Nelson L., Cacicedo J. M., Saha A. K., Lan F., Ido Y. (2010) AMPK and SIRT1: a long-standing partnership? Am. J. Physiol. Endocrinol. Metab. 298, E751–E760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lynn E. G., McLeod C. J., Gordon J. P., Bao J., Sack M. N. (2008) SIRT2 is a negative regulator of anoxia-reoxygenation tolerance via regulation of 14-3-3ζ and BAD in H9c2 cells. FEBS Lett. 582, 2857–2862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Alcendor R. R., Gao S., Zhai P., Zablocki D., Holle E., Yu X., Tian B., Wagner T., Vatner S. F., Sadoshima J. (2007) Sirt1 regulates aging and resistance to oxidative stress in the heart. Circ. Res. 100, 1512–1521 [DOI] [PubMed] [Google Scholar]
- 21. Alcendor R. R., Kirshenbaum L. A., Imai S., Vatner S. F., Sadoshima J. (2004) Silent information regulator 2α, a longevity factor and class III histone deacetylase, is an essential endogenous apoptosis inhibitor in cardiac myocytes. Circ. Res. 95, 971–980 [DOI] [PubMed] [Google Scholar]
- 22. Nadtochiy S. M., Redman E., Rahman I., Brookes P. S. (2011) Lysine deacetylation in ischaemic preconditioning: the role of SIRT1. Cardiovasc. Res. 89, 643–649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Berger S. L. (2007) The complex language of chromatin regulation during transcription. Nature 447, 407–412 [DOI] [PubMed] [Google Scholar]
- 24. Latham J. A., Dent S. Y. (2007) Cross-regulation of histone modifications. Nat. Struct. Mol. Biol. 14, 1017–1024 [DOI] [PubMed] [Google Scholar]
- 25. Lee J. S., Smith E., Shilatifard A. (2010) The language of histone cross-talk. Cell 142, 682–685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Strahl B. D., Allis C. D. (2000) The language of covalent histone modifications. Nature 403, 41–45 [DOI] [PubMed] [Google Scholar]
- 27. Cheung P., Tanner K. G., Cheung W. L., Sassone-Corsi P., Denu J. M., Allis C. D. (2000) Synergistic coupling of histone H3 phosphorylation and acetylation in response to epidermal growth factor stimulation. Mol. Cell 5, 905–915 [DOI] [PubMed] [Google Scholar]
- 28. Lo W. S., Trievel R. C., Rojas J. R., Duggan L., Hsu J. Y., Allis C. D., Marmorstein R., Berger S. L. (2000) Phosphorylation of serine 10 in histone H3 is functionally linked in vitro and in vivo to Gcn5-mediated acetylation at lysine 14. Mol. Cell 5, 917–926 [DOI] [PubMed] [Google Scholar]
- 29. Kim J., Guermah M., McGinty R. K., Lee J. S., Tang Z., Milne T. A., Shilatifard A., Muir T. W., Roeder R. G. (2009) RAD6-mediated transcription-coupled H2B ubiquitylation directly stimulates H3K4 methylation in human cells. Cell 137, 459–471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Guccione E., Bassi C., Casadio F., Martinato F., Cesaroni M., Schuchlautz H., Lüscher B., Amati B. (2007) Methylation of histone H3R2 by PRMT6 and H3K4 by an MLL complex are mutually exclusive. Nature 449, 933–937 [DOI] [PubMed] [Google Scholar]
- 31. Shi X., Hong T., Walter K. L., Ewalt M., Michishita E., Hung T., Carney D., Peña P., Lan F., Kaadige M. R., Lacoste N., Cayrou C., Davrazou F., Saha A., Cairns B. R., Ayer D. E., Kutateladze T. G., Shi Y., Côté J., Chua K. F., Gozani O. (2006) ING2 PHD domain links histone H3 lysine 4 methylation to active gene repression. Nature 442, 96–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wang Z., Zang C., Cui K., Schones D. E., Barski A., Peng W., Zhao K. (2009) Genome-wide mapping of HATs and HDACs reveals distinct functions in active and inactive genes. Cell 138, 1019–1031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zippo A., Serafini R., Rocchigiani M., Pennacchini S., Krepelova A., Oliviero S. (2009) Histone cross-talk between H3Ser-10ph and H4K16ac generates a histone code that mediates transcription elongation. Cell 138, 1122–1136 [DOI] [PubMed] [Google Scholar]
- 34. Fenton T. R., Gwalter J., Cramer R., Gout I. T. (2010) S6K1 is acetylated at lysine 516 in response to growth factor stimulation. Biochem. Biophys. Res. Commun. 398, 400–405 [DOI] [PubMed] [Google Scholar]
- 35. Lan F., Cacicedo J. M., Ruderman N., Ido Y. (2008) SIRT1 modulation of the acetylation status, cytosolic localization, and activity of LKB1. Possible role in AMP-activated protein kinase activation. J. Biol. Chem. 283, 27628–27635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Pillai V. B., Sundaresan N. R., Samant S. A., Wolfgeher D., Trivedi C. M., Gupta M. P. (2011) Acetylation of a conserved lysine residue in the ATP binding pocket of p38 augments its kinase activity during hypertrophy of cardiomyocytes. Mol. Cell. Biol. 31, 2349–2363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Sabò A., Lusic M., Cereseto A., Giacca M. (2008) Acetylation of conserved lysines in the catalytic core of cyclin-dependent kinase 9 inhibits kinase activity and regulates transcription. Mol. Cell. Biol. 28, 2201–2212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sundaresan N. R., Pillai V. B., Wolfgeher D., Samant S., Vasudevan P., Parekh V., Raghuraman H., Cunningham J. M., Gupta M., Gupta M. P. (2011) The deacetylase SIRT1 promotes membrane localization and activation of Akt and PDK1 during tumorigenesis and cardiac hypertrophy. Sci. Signal. 4, ra46. [DOI] [PubMed] [Google Scholar]
- 39. Gerhart-Hines Z., Dominy J. E., Jr., Blättler S. M., Jedrychowski M. P., Banks A. S., Lim J. H., Chim H., Gygi S. P., Puigserver P. (2011) The cAMP/PKA pathway rapidly activates SIRT1 to promote fatty acid oxidation independently of changes in NAD(+). Mol. Cell 44, 851–863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Liu Y., Denlinger C. E., Rundall B. K., Smith P. W., Jones D. R. (2006) Suberoylanilide hydroxamic acid induces Akt-mediated phosphorylation of p300, which promotes acetylation and transcriptional activation of RelA/p65. J. Biol. Chem. 281, 31359–31368 [DOI] [PubMed] [Google Scholar]
- 41. Nasrin N., Kaushik V. K., Fortier E., Wall D., Pearson K. J., de Cabo R., Bordone L. (2009) JNK1 phosphorylates SIRT1 and promotes its enzymatic activity. PLoS One 4, e8414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Pflum M. K., Tong J. K., Lane W. S., Schreiber S. L. (2001) Histone deacetylase 1 phosphorylation promotes enzymatic activity and complex formation. J. Biol. Chem. 276, 47733–47741 [DOI] [PubMed] [Google Scholar]
- 43. van Noort V., Seebacher J., Bader S., Mohammed S., Vonkova I., Betts M. J., Kühner S., Kumar R., Maier T., O'Flaherty M., Rybin V., Schmeisky A., Yus E., Stülke J., Serrano L., Russell R. B., Heck A. J., Bork P., Gavin A. C. (2012) Cross-talk between phosphorylation and lysine acetylation in a genome-reduced bacterium. Mol. Syst. Biol. 8, 571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Yao Q., Li H., Liu B. Q., Huang X. Y., Guo L. (2011) SUMOylation-regulated protein phosphorylation, evidence from quantitative phosphoproteomics analyses. J. Biol. Chem. 286, 27342–27349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Paolinelli R., Mendoza-Maldonado R., Cereseto A., Giacca M. (2009) Acetylation by GCN5 regulates CDC6 phosphorylation in the S phase of the cell cycle. Nat. Struct. Mol. Biol. 16, 412–420 [DOI] [PubMed] [Google Scholar]
- 46. Zhang T., Wang S., Lin Y., Xu W., Ye D., Xiong Y., Zhao S., Guan K. L. (2012) Acetylation negatively regulates glycogen phosphorylase by recruiting protein phosphatase 1. Cell Metab. 15, 75–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Govind C. K., Qiu H., Ginsburg D. S., Ruan C., Hofmeyer K., Hu C., Swaminathan V., Workman J. L., Li B., Hinnebusch A. G. (2010) Phosphorylated Pol II CTD recruits multiple HDACs, including Rpd3C(S), for methylation-dependent deacetylation of ORF nucleosomes. Mol. Cell 39, 234–246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ho P. C., Gupta P., Tsui Y. C., Ha S. G., Huq M., Wei L. N. (2008) Modulation of lysine acetylation-stimulated repressive activity by Erk2-mediated phosphorylation of RIP140 in adipocyte differentiation. Cell. Signal. 20, 1911–1919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Xiong S., Salazar G., San Martin A., Ahmad M., Patrushev N., Hilenski L., Nazarewicz R. R., Ma M., Ushio-Fukai M., Alexander R. W. (2010) PGC-1α serine 570 phosphorylation and GCN5-mediated acetylation by angiotensin II drive catalase down-regulation and vascular hypertrophy. J. Biol. Chem. 285, 2474–2487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Matsuzaki H., Daitoku H., Hatta M., Aoyama H., Yoshimochi K., Fukamizu A. (2005) Acetylation of Foxo1 alters its DNA-binding ability and sensitivity to phosphorylation. Proc. Natl. Acad. Sci. U.S.A. 102, 11278–11283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Mukherjee S., Keitany G., Li Y., Wang Y., Ball H. L., Goldsmith E. J., Orth K. (2006) Yersinia YopJ acetylates and inhibits kinase activation by blocking phosphorylation. Science 312, 1211–1214 [DOI] [PubMed] [Google Scholar]
- 52. Chou T. Y., Hart G. W., Dang C. V. (1995) c-Myc is glycosylated at threonine 58, a known phosphorylation site and a mutational hot spot in lymphomas. J. Biol. Chem. 270, 18961–18965 [DOI] [PubMed] [Google Scholar]
- 53. Musicki B., Kramer M. F., Becker R. E., Burnett A. L. (2005) Inactivation of phosphorylated endothelial nitric-oxide synthase (Ser-1177) by O-GlcNAc in diabetes-associated erectile dysfunction. Proc. Natl. Acad. Sci. U.S.A. 102, 11870–11875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Kawauchi K., Araki K., Tobiume K., Tanaka N. (2009) Loss of p53 enhances catalytic activity of IKKβ through O-linked β-N-acetyl glucosamine modification. Proc. Natl. Acad. Sci. U.S.A. 106, 3431–3436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Bodenmiller B., Wanka S., Kraft C., Urban J., Campbell D., Pedrioli P. G., Gerrits B., Picotti P., Lam H., Vitek O., Brusniak M. Y., Roschitzki B., Zhang C., Shokat K. M., Schlapbach R., Colman-Lerner A., Nolan G. P., Nesvizhskii A. I., Peter M., Loewith R., von Mering C., Aebersold R. (2010) Phosphoproteomic analysis reveals interconnected system-wide responses to perturbations of kinases and phosphatases in yeast. Sci. Signal. 3, rs4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Holt L. J., Tuch B. B., Villén J., Johnson A. D., Gygi S. P., Morgan D. O. (2009) Global analysis of Cdk1 substrate phosphorylation sites provides insights into evolution. Science 325, 1682–1686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Olsen J. V., Vermeulen M., Santamaria A., Kumar C., Miller M. L., Jensen L. J., Gnad F., Cox J., Jensen T. S., Nigg E. A., Brunak S., Mann M. (2010) Quantitative phosphoproteomics reveals widespread full phosphorylation site occupancy during mitosis. Sci. Signal. 3, ra3. [DOI] [PubMed] [Google Scholar]
- 58. Choudhary C., Kumar C., Gnad F., Nielsen M. L., Rehman M., Walther T. C., Olsen J. V., Mann M. (2009) Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science 325, 834–840 [DOI] [PubMed] [Google Scholar]
- 59. Kim S. C., Sprung R., Chen Y., Xu Y., Ball H., Pei J., Cheng T., Kho Y., Xiao H., Xiao L., Grishin N. V., White M., Yang X. J., Zhao Y. (2006) Substrate and functional diversity of lysine acetylation revealed by a proteomics survey. Mol. Cell 23, 607–618 [DOI] [PubMed] [Google Scholar]
- 60. Lundby A., Lage K., Weinert B. T., Bekker-Jensen D. B., Secher A., Skovgaard T., Kelstrup C. D., Dmytriyev A., Choudhary C., Lundby C., Olsen J. V. (2012) Proteomic analysis of lysine acetylation sites in rat tissues reveals organ specificity and subcellular patterns. Cell Rep. 2, 419–431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Wang Q., Zhang Y., Yang C., Xiong H., Lin Y., Yao J., Li H., Xie L., Zhao W., Yao Y., Ning Z. B., Zeng R., Xiong Y., Guan K. L., Zhao S., Zhao G. P. (2010) Acetylation of metabolic enzymes coordinates carbon source utilization and metabolic flux. Science 327, 1004–1007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Weinert B. T., Wagner S. A., Horn H., Henriksen P., Liu W. R., Olsen J. V., Jensen L. J., Choudhary C. (2011) Proteome-wide mapping of the Drosophila acetylome demonstrates a high degree of conservation of lysine acetylation. Sci. Signal. 4, ra48. [DOI] [PubMed] [Google Scholar]
- 63. Zhao S., Xu W., Jiang W., Yu W., Lin Y., Zhang T., Yao J., Zhou L., Zeng Y., Li H., Li Y., Shi J., An W., Hancock S. M., He F., Qin L., Chin J., Yang P., Chen X., Lei Q., Xiong Y., Guan K. L. (2010) Regulation of cellular metabolism by protein lysine acetylation. Science 327, 1000–1004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Parker B. L., Palmisano G., Edwards A. V., White M. Y., Engholm-Keller K., Lee A., Scott N. E., Kolarich D., Hambly B. D., Packer N. H., Larsen M. R., Cordwell S. J. (2011) Quantitative N-linked glycoproteomics of myocardial ischemia and reperfusion injury reveals early remodeling in the extracellular environment. Mol. Cell. Proteomics 10, M110.006833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Larsen M. R., Thingholm T. E., Jensen O. N., Roepstorff P., Jørgensen T. J. (2005) Highly selective enrichment of phosphorylated peptides from peptide mixtures using titanium dioxide microcolumns. Mol. Cell. Proteomics 4, 873–886 [DOI] [PubMed] [Google Scholar]
- 66. Engholm-Keller K., Birck P., Størling J., Pociot F., Mandrup-Poulsen T., Larsen M. R. (2012) TiSH–a robust and sensitive global phosphoproteomics strategy employing a combination of TiO(2), SIMAC, and HILIC. J. Proteomics 75, 5749–5761 [DOI] [PubMed] [Google Scholar]
- 67. Yanes O., Tautenhahn R., Patti G. J., Siuzdak G. (2011) Expanding coverage of the metabolome for global metabolite profiling. Anal. Chem. 83, 2152–2161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Hirao M., Posakony J., Nelson M., Hruby H., Jung M., Simon J. A., Bedalov A. (2003) Identification of selective inhibitors of NAD+-dependent deacetylases using phenotypic screens in yeast. J. Biol. Chem. 278, 52773–52782 [DOI] [PubMed] [Google Scholar]
- 69. Käll L., Canterbury J. D., Weston J., Noble W. S., MacCoss M. J. (2007) Semi-supervised learning for peptide identification from shotgun proteomics datasets. Nat. Methods 4, 923–925 [DOI] [PubMed] [Google Scholar]
- 70. Taus T., Köcher T., Pichler P., Paschke C., Schmidt A., Henrich C., Mechtler K. (2011) Universal and confident phosphorylation site localization using phosphoRS. J. Proteome Res. 10, 5354–5362 [DOI] [PubMed] [Google Scholar]
- 71. Karp N. A., Huber W., Sadowski P. G., Charles P. D., Hester S. V., Lilley K. S. (2010) Addressing accuracy and precision issues in iTRAQ quantitation. Mol. Cell. Proteomics 9, 1885–1897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Toedling J., Skylar O., Sklyar O., Krueger T., Fischer J. J., Sperling S., Huber W. (2007) Ringo–an R/Bioconductor package for analyzing ChIP-chip readouts. BMC Bioinformatics 8, 221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Benjamini Y., Hochberg Y. (1995) Controlling the false discovery rate–a practical and powerful approach to multiple testing. J. R. Stat. Soc. B Met. 57, 289–300 [Google Scholar]
- 74. Rigbolt K. T., Vanselow J. T., Blagoev B. (2011) GProX, a user-friendly platform for bioinformatics analysis and visualization of quantitative proteomics data. Mol. Cell. Proteomics 10, O110 007450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Obenauer J. C., Cantley L. C., Yaffe M. B. (2003) Scansite 2.0: Proteome-wide prediction of cell signaling interactions using short sequence motifs. Nucleic Acids Res. 31, 3635–3641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Shepherd N. E., Hoang H. N., Abbenante G., Fairlie D. P. (2005) Single turn peptide α helices with exceptional stability in water. J. Am. Chem. Soc. 127, 2974–2983 [DOI] [PubMed] [Google Scholar]
- 77. Kubota K., Anjum R., Yu Y., Kunz R. C., Andersen J. N., Kraus M., Keilhack H., Nagashima K., Krauss S., Paweletz C., Hendrickson R. C., Feldman A. S., Wu C. L., Rush J., Villén J., Gygi S. P. (2009) Sensitive multiplexed analysis of kinase activities and activity-based kinase identification. Nat. Biotechnol. 27, 933–940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Yu Y., Anjum R., Kubota K., Rush J., Villen J., Gygi S. P. (2009) A site-specific, multiplexed kinase activity assay using stable-isotope dilution and high-resolution mass spectrometry. Proc. Natl. Acad. Sci. U.S.A. 106, 11606–11611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. MacLean B., Tomazela D. M., Shulman N., Chambers M., Finney G. L., Frewen B., Kern R., Tabb D. L., Liebler D. C., MacCoss M. J. (2010) Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics 26, 966–968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Maclean B., Tomazela D. M., Abbatiello S. E., Zhang S., Whiteaker J. R., Paulovich A. G., Carr S. A., Maccoss M. J. (2010) Effect of collision energy optimization on the measurement of peptides by selected reaction monitoring (SRM) mass spectrometry. Anal. Chem. 82, 10116–10124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Martin D. B., Vagelos P. R. (1962) The mechanism of tricarboxylic acid cycle regulation of fatty acid synthesis. J. Biol. Chem. 237, 1787–1792 [PubMed] [Google Scholar]
- 82. Ping P., Zhang J., Cao X., Li R. C., Kong D., Tang X. L., Qiu Y., Manchikalapudi S., Auchampach J. A., Black R. G., Bolli R. (1999) PKC-dependent activation of p44/p42 MAPKs during myocardial ischemia-reperfusion in conscious rabbits. Am. J. Physiol. 276, H1468–H1481 [DOI] [PubMed] [Google Scholar]
- 83. Kirchberger M. A., Tada M., Repke D. I., Katz A. M. (1972) Cyclic adenosine 3′,5′-monophosphate-dependent protein kinase stimulation of calcium uptake by canine cardiac microsomes. J. Mol. Cell. Cardiol. 4, 673–680 [DOI] [PubMed] [Google Scholar]
- 84. Simmerman H. K., Collins J. H., Theibert J. L., Wegener A. D., Jones L. R. (1986) Sequence analysis of phospholamban. Identification of phosphorylation sites and two major structural domains. J. Biol. Chem. 261, 13333–13341 [PubMed] [Google Scholar]
- 85. Bedalov A., Gatbonton T., Irvine W. P., Gottschling D. E., Simon J. A. (2001) Identification of a small molecule inhibitor of Sir2p. Proc. Natl. Acad. Sci. U.S.A. 98, 15113–15118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Hebert A. S., Dittenhafer-Reed K. E., Yu W., Bailey D. J., Selen E. S., Boersma M. D., Carson J. J., Tonelli M., Balloon A. J., Higbee A. J., Westphall M. S., Pagliarini D. J., Prolla T. A., Assadi-Porter F., Roy S., Denu J. M., Coon J. J. (2013) Calorie restriction and SIRT3 trigger global reprogramming of the mitochondrial protein acetylome. Mol. Cell 49, 186–199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Baines C. P., Zhang J., Wang G. W., Zheng Y. T., Xiu J. X., Cardwell E. M., Bolli R., Ping P. (2002) Mitochondrial PKCϵ and MAPK form signaling modules in the murine heart: enhanced mitochondrial PKCϵ-MAPK interactions and differential MAPK activation in PKCϵ-induced cardioprotection. Circ. Res. 90, 390–397 [DOI] [PubMed] [Google Scholar]
- 88. Hausenloy D. J., Yellon D. M. (2004) New directions for protecting the heart against ischemia-reperfusion injury: targeting the reperfusion injury salvage kinase (RISK)- pathway. Cardiovasc. Res. 61, 448–460 [DOI] [PubMed] [Google Scholar]
- 89. Beutner G., Rück A., Riede B., Brdiczka D. (1998) Complexes between porin, hexokinase, mitochondrial creatine kinase and adenylate translocator display properties of the permeability transition pore. Implication for regulation of permeability transition by the kinases. Biochim. Biophys. Acta 1368, 7–18 [DOI] [PubMed] [Google Scholar]
- 90. Broekemeier K. M., Dempsey M. E., Pfeiffer D. R. (1989) Cyclosporin A is a potent inhibitor of the inner membrane permeability transition in liver mitochondria. J. Biol. Chem. 264, 7826–7830 [PubMed] [Google Scholar]
- 91. Pavlov E. V., Priault M., Pietkiewicz D., Cheng E. H., Antonsson B., Manon S., Korsmeyer S. J., Mannella C. A., Kinnally K. W. (2001) A novel, high conductance channel of mitochondria linked to apoptosis in mammalian cells and Bax expression in yeast. J. Cell Biol. 155, 725–731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Baines C. P., Kaiser R. A., Sheiko T., Craigen W. J., Molkentin J. D. (2007) Voltage-dependent anion channels are dispensable for mitochondrial-dependent cell death. Nat. Cell Biol. 9, 550–555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Kokoszka J. E., Waymire K. G., Levy S. E., Sligh J. E., Cai J., Jones D. P., MacGregor G. R., Wallace D. C. (2004) The ADP/ATP translocator is not essential for the mitochondrial permeability transition pore. Nature 427, 461–465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Giorgio V., von Stockum S., Antoniel M., Fabbro A., Fogolari F., Forte M., Glick G. D., Petronilli V., Zoratti M., Szabó I., Lippe G., Bernardi P. (2013) Dimers of mitochondrial ATP synthase form the permeability transition pore. Proc. Natl. Acad. Sci. U.S.A. 110, 5887–5892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Sodi-Pallares D., Testelli M. R., Fishleder B. L., Bisteni A., Medrano G. A., Friedland C., De Micheli A. (1962) Effects of an intravenous infusion of a potassium-glucose-insulin solution on the electrocardiographic signs of myocardial infarction. A preliminary clinical report. Am. J. Cardiol. 9, 166–181 [DOI] [PubMed] [Google Scholar]
- 96. Kantor P. F., Lucien A., Kozak R., Lopaschuk G. D. (2000) The antianginal drug trimetazidine shifts cardiac energy metabolism from fatty acid oxidation to glucose oxidation by inhibiting mitochondrial long-chain 3-ketoacyl coenzyme A thiolase. Circ. Res. 86, 580–588 [DOI] [PubMed] [Google Scholar]
- 97. Carr S. M., Munro S., Kessler B., Oppermann U., La Thangue N. B. (2011) Interplay between lysine methylation and Cdk phosphorylation in growth control by the retinoblastoma protein. EMBO J. 30, 317–327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Estève P. O., Chang Y., Samaranayake M., Upadhyay A. K., Horton J. R., Feehery G. R., Cheng X., Pradhan S. (2011) A methylation and phosphorylation switch between an adjacent lysine and serine determines human DNMT1 stability. Nat. Struct. Mol. Biol. 18, 42–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Guo Z., Zheng L., Xu H., Dai H., Zhou M., Pascua M. R., Chen Q. M., Shen B. (2010) Methylation of FEN1 suppresses nearby phosphorylation and facilitates PCNA binding. Nat. Chem. Biol. 6, 766–773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Sakamaki J., Daitoku H., Ueno K., Hagiwara A., Yamagata K., Fukamizu A. (2011) Arginine methylation of BCL-2 antagonist of cell death (BAD) counteracts its phosphorylation and inactivation by Akt. Proc. Natl. Acad. Sci. U.S.A. 108, 6085–6090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Yamagata K., Daitoku H., Takahashi Y., Namiki K., Hisatake K., Kako K., Mukai H., Kasuya Y., Fukamizu A. (2008) Arginine methylation of FOXO transcription factors inhibits their phosphorylation by Akt. Mol. Cell 32, 221–231 [DOI] [PubMed] [Google Scholar]
- 102. Cui Y., Zhang M., Pestell R., Curran E. M., Welshons W. V., Fuqua S. A. (2004) Phosphorylation of estrogen receptor α blocks its acetylation and regulates estrogen sensitivity. Cancer Res. 64, 9199–9208 [DOI] [PubMed] [Google Scholar]
- 103. Rust H. L., Thompson P. R. (2011) Kinase consensus sequences: a breeding ground for cross-talk. ACS Chem. Biol. 6, 881–892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Fischle W., Wang Y., Allis C. D. (2003) Binary switches and modification cassettes in histone biology and beyond. Nature 425, 475–479 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
MS data have been deposited into the PeptideAtlas repository (Identifier(s): PASS00283 for global quantification of phosphorylation and acetyl-Lys following ischemia and IPC; PASS00280 for quantification of phosphorylation and acetyl-Lys cross-talk on CKM synthetic peptides; and PASS00286 for quantification of dephosphorylation rates of Ser-372 on human CKM with and without acetyl-Lys.