

Background: Factor C is autocatalytically activated on lipopolysaccharides.
Results: The N-terminal Arg of factor C is essential for the autocatalytic activation.
Conclusion: The N-terminal Arg of factor C is required to be in sufficiently close vicinity for the autocatalytic activation.
Significance: The recombinant factor C with restricted and homogeneous N-glycans may be useful for further structure-function studies.
Keywords: Glycoprotein, Lipopolysaccharide (LPS), Protein-protein Interaction, Recombinant Protein Expression, Serine Protease, Chemical Cross-linker, Coagulation Cascade, Proteolytic Cleavage
Abstract
Factor C, a serine protease zymogen involved in innate immune responses in horseshoe crabs, is known to be autocatalytically activated on the surface of bacterial lipopolysaccharides, but the molecular mechanism of this activation remains unknown. In this study, we show that wild-type factor C expressed in HEK293S cells exhibits a lipopolysaccharide-induced activity equivalent to that of native factor C. Analysis of the N-terminal addition, deletion, or substitution mutants shows that the N-terminal Arg residue and the distance between the N terminus and the tripartite of lipopolysaccharide-binding site are essential factors for autocatalytic activation, and that the positive charge of the N terminus may interact with an acidic amino acid(s) of the molecule to convert the zymogen into an active form. Chemical cross-linking experiments indicate that the N terminus is required to form a complex of the factor C molecules in a sufficiently close vicinity to be chemically cross-linked on the surface of lipopolysaccharides. We propose a molecular mechanism of the autocatalytic activation of the protease zymogen on lipopolysaccharides functioning as a platform to induce specific protein-protein interaction between the factor C molecules.
Introduction
Factor C is a serine protease zymogen involved in lipopolysaccharide-induced innate immune responses in horseshoe crabs, such as hemolymph coagulation, activation of the complement system, and hemocyte exocytosis (1–3). Factor C consists of six types of domains, and contains six potential N-linked glycosylation sites (Fig. 1) (4). The N-glycosylation plays a pivotal role in its secretion in Drosophila S2 cells (5). Factor C triggers hemolymph coagulation in response to the stimulation of bacterial lipopolysaccharides: the resulting activated factor C activates factor B, which in turn converts the proclotting enzyme into the clotting enzyme to promote the conversion of coagulogen to coagulin gel (6). The lipopolysaccharide-induced autocatalytic cleavage occurs within the -Phe737-Ile738- bond in the C-terminal catalytic domain of factor C, and this peptide bond is also cleaved by chymotrypsin in vitro to trigger the coagulation cascade under sterile conditions (7). A lipopolysaccharide-binding site is located in the N-terminal Cys-rich domain, and it has been established that a tripeptide sequence (-Arg36-Trp37-Arg38-) in the N-terminal Cys-rich domain is important for lipopolysaccharide recognition (3, 8).
FIGURE 1.
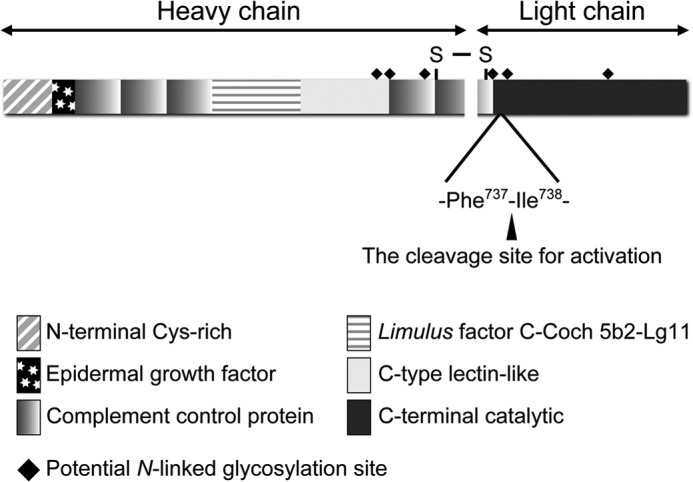
A schematic domain structure of factor C.
Proteases involved in proteolytic cascades also reside as latent precursors in the mammalian blood coagulation (9): the intrinsic pathway begins with the activation of factor XII by the interaction of high molecular weight kininogen and prekallikrein on negatively charged surfaces, and the extrinsic one begins with the activation of factor VII by interaction with tissue factor on lipid surfaces at injured sites. The precise activation mechanism of factor XII or factor VII remains unknown, whereas tissue factor is known to trap one of the conformational states of factor VII, converting its zymogen-like state into an active state (10). The classical molecular mechanism of the zymogen activation of serine proteases in trypsinogen activation has been established (11). Trypsinogen is activated by proteolytic cleavage of the -Arg15-Ile16- bond in chymotrypsinogen numbering, following an insertion of the newly appearing N-terminal Ile16 into the activation pocket known as the Ile16 cleft to form a salt bridge between the α-amino group of Ile16 and the side chain of Asp194, which triggers conformational changes of the substrate binding site and the oxyanion hole required for catalysis.
Based on the importance of the salt bridge formation at the Ile16 cleft in zymogen activation, a molecular sexuality model has been proposed for non-proteolytic activation of plasminogen by streptokinase secreted from Streptococcus pyogenes (12). However, the mechanism of the molecular sexuality model by means of a protein-protein interaction remains unknown, except for non-proteolytic activation of prothrombin by staphylocoagulase (13–15). Staphylocoagulase, a non-enzymatic protein secreted by Staphylococcus aureus, binds to prothrombin to form a one to one complex, and inserts its N-terminal portion (Ile1-Val2-) into the Ile16 cleft of bound prothrombin to induce a thrombin-like conformation, providing the first structural evidence for the molecular sexuality mechanism of non-proteolytic zymogen activation.
On the other hand, zymogens of intracellular cysteine proteases including caspases-1, -8, and -9 are activated in the signaling platforms of the protein complexes known as inflammasomes, the death including signaling complex, and apoptosomes, respectively (16). The death including signaling complex recruits pro-caspase-8 to a platform composed of Fas antigen and Fas-associated death domain protein, which leads to the autocatalytic activation of pro-caspase-8 (16, 17). Likewise, binding of monomers of pro-caspase-9 to an oligomeric platform of apoptosome comprising cytochrome c and Apaf-1 by the N-terminal domain of pro-caspase-9 results in dimerization between the C-terminal catalytic domains to allow the dimer to induce an active state without proteolytic cleavage (18). One of the two potential active sites in the dimer of pro-caspase-9 closely resembles the active form, whereas in the second potential active site, expulsion of the active loop disrupts the catalytic machinery (19). To clarify the molecular mechanism of the autocatalytic activation of factor C on lipopolysaccharides, we prepared several recombinants expressed in a HEK293 mutant cell line lacking N-acetylglucosaminyltransferase I (HEK293S GnTI−),5 which produces recombinants with a restricted and homogeneous N-glycan of Man5GlcNAc2 (20). Here, we show that the N-terminal Arg and the distance between the N terminus and the lipopolysaccharide-binding site are essential factors for the autocatalytic activation.
EXPERIMENTAL PROCEDURES
Materials
HEK293S GnTI− cells were obtained from ATCC. Lipopolysaccharides from Salmonella minnesota R595 (Re) were purchased from List Biological Laboratories Inc. (Campbell, CA). Biotinylation of S. minnesota R595 (Re) lipopolysaccharides was performed using EZ-Link Sulfo-NHS-LC-Biotin (ThermoFisher Scientific). N-tert-Butoxycarbonyl-Val-Pro-Arg-p-nitroanilide (Boc-Val-Pro-Arg-pNA) was provided from Seikagaku Corporation, Tokyo, Japan. Boc-Val-Pro-Arg-4-methylcoumaryl-7-amide (MCA) was purchased from Peptide Institute Inc. (Osaka, Japan). A homobifunctional cross-linker dimethyl adipimidate (spacer arm length 8.6 Å) was purchased from Tokyo Chemical Industry Co. Ltd. (Tokyo, Japan). The molecular weights of lipopolysaccharides and Triton X-100 were assumed to be 17,000 and 647, respectively.
Cloning and Mutagenesis
To construct an expression vector of wild-type factor C, plasmid pPSB8/factor C, which contains a DNA encoding the full-length of factor C, was used as a template to amplify DNA of factor C by PCR. The amplified DNA fragment was subcloned into the vector pCA7 (21). As a secretion signal sequence, a sequence derived from pHLsec (22) or an original signal sequence of factor C was used. Mutations in wild-type factor C were introduced using site-directed mutagenesis by inverse PCR.
Expression of Recombinant Proteins
Recombinant proteins were expressed in HEK293S GnTI− cells, and secreted into the culture medium. HEK293S GnTI− cells were maintained in DMEM supplemented with 1% glutamine, 1% penicillin/streptomycin, and 10% fetal bovine serum at 37 °C under 5% CO2. DMEM supplemented with wild-type or mutant factor C plasmid (4 μg), polyethylenimine (10 μg), 1% glutamine, 1% penicillin/streptomycin, and 2% fetal bovine serum was used to transfect HEK293S GnTI− cells at 80–90% confluence in a six-well plate. Culture media were collected 5 days after transfection, and centrifuged at 6,000 × g for 30 min. The resulting supernatant was filtrated, and the concentration of each recombinant protein was determined by ELISA, using a monoclonal antibody 2C12 against factor C (23). Culture media containing recombinant proteins were dialyzed with 10 mm MES-NaOH, pH 6.0, and applied to a CM-Sepharose CL-6B column. After washing with the same buffer, the recombinant proteins were eluted by stepwise elution using 20–200 mm NaCl in the same buffer. Eluted fractions were subjected to SDS-PAGE, followed by electroblotting to a PVDF membrane. The membrane was stained with Coomassie Brilliant Blue R-250, and the N-terminal sequences of the stained bands were analyzed by Edman degradation (Genostaff, Tokyo, Japan) (Table 1).
TABLE 1.
The N-terminal sequence analyses of wild-type factor C, ETG-factor C, and R1K-factor C
| Cycle No. | Wild-type |
+ETG-factor C |
R1K-factor C |
|||
|---|---|---|---|---|---|---|
| Amino acid | Yield | Amino acid | Yield | Amino acid | Yield | |
| pmol | pmol | pmol | ||||
| 1 | Arg | 1.4 | Glu | 0.7 | Lys | 1.1 |
| 2 | Gly | 0.4 | Thr | 0.6 | Gly | 1.4 |
| 3 | Val | 0.9 | Gly | 0.6 | Val | 0.9 |
| 4 | Arg | 0.5 | Asp | 0.7 | ||
| 5 | Gly | 0.7 | Leu | 1.0 | ||
ELISA
The microtiter plates were coated with the monoclonal antibody 2C12 by incubating overnight at 4 °C in 50 mm sodium carbonate, pH 9.6. After washing with 20 mm Tris-HCl, pH 7.5, containing 150 mm NaCl (TBS1), the plates were blocked with 2.5% casein in TBS1, and serial dilutions of the samples were added, incubated at 37 °C for 1 h, and washed with TBS1. Polyclonal antibodies against factor C were added and the plates were incubated at 37 °C for 1 h. After washing with TBS1, HRP-conjugated goat anti-rabbit IgG antibody (Bio-Rad) was added and the plates were again incubated at 37 °C for 1 h. The enzyme activity of HRP was detected with o-phenylenediamine at 492 nm with a microplate reader model 3550 (Bio-Rad) or Multiskan GO (Thermo Fisher Scientific).
Binding Assay to Lipopolysaccharide-immobilized Sepharose
Sepharose coupled to streptavidin was incubated for 2 h at 4 °C in the presence or absence of 10 μg of biotinylated lipopolysaccharides in 1.0 ml of TBS1, and Sepharose, obtained by centrifugation at 6,000 × g for 5 min was washed with TBS1. The resulting lipopolysaccharide-immobilized Sepharose or control Sepharose was incubated with culture media containing 4.3 nm recombinant proteins for 1 h at 4 °C, and washed three times with TBS1. Sepharose reacted with recombinant proteins was obtained by centrifugation and subjected to Western blotting.
Amidase Activity of Recombinant Proteins Activated by Lipopolysaccharides
Recombinant proteins were incubated in the presence of various concentrations of lipopolysaccharides in 100 mm Tris-HCl, pH 8.0, at 37 °C for 20 min. Boc-Val-Pro-Arg-pNA was then added to the solutions at the final concentration of 0.4 mm and incubated at 37 °C for 5 min, and acetic acid was added to terminate the reaction. The rate of hydrolysis was calculated from the absorbance at 405 nm. The unit of the amidase activity was defined as micromole of the digested substrate per min. The specific activity was expressed as units per nmol of factor C.
Purification of Recombinants by Dextran Sulfate CL-6B Sepharose Column Chromatography
Culture media containing recombinants were applied to a dextran sulfate-Sepharose CL-6B column (1.0 × 1.5 cm) prepared previously (24). The column was washed with 20 mm Tris-HCl, pH 8.0, containing 150 mm NaCl (TBS2), and the recombinant proteins were eluted with the same buffer containing 500 mm NaCl.
Amidase Activity of Recombinant Proteins Activated by Chymotrypsin
Recombinant proteins (17.2 nm) purified by dextran sulfate CL-6B Sepharose column chromatography were incubated with 80 nm α-chymotrypsin in TBS2 at 37 °C for 30 min, and 0.1 mm tosyl-l-phenylalanine chloromethyl ketone was added to stop the reaction. An aliquot of the recombinants treated with chymotrypsin was incubated with 0.4 mm Boc-Val-Pro-Arg-pNA in TBS2 containing 1.5 μm bovine serum albumin for 5 min at 37 °C, and acetic acid was added to terminate the reaction.
Kinetic Analysis
Stock solutions of Boc-Val-Pro-Arg-MCA were prepared in dimethylformamide. Activated recombinant proteins (10 μl) were added to a cuvette containing 480 μl of TBS2 containing 1.5 μm bovine serum albumin thermostatted at 37 °C. The initial rate of hydrolysis was measured fluorometrically with excitation at 380 nm and emission at 440 nm after adding the substrate solution. Kinetic parameters were determined by direct linear plotting (25).
Lipopolysaccharide-binding Assay with a Surface Plasmon Sensor
Biotinylated lipopolysaccharides (2.3 μm in 10 mm Hepes-NaOH, pH 7.4, containing 150 mm NaCl) were immobilized on a sensor chip SA of a BIAcore X system (GE Healthcare), according to the manufacturer's specifications. Recombinant proteins were injected at a flow rate of 30 μl/min in the running buffer, 10 mm Hepes-NaOH, pH 7.4, containing 150 mm NaCl. The change in the mass concentration on the sensor chip was monitored as a resonance signal by using the program supplied by the manufacturer. Sensorgrams of the interactions obtained by using the various concentrations of recombinant proteins (5–20 nm) were analyzed by the BIAEVALUATION program, version 3.0.
Cross-linking of Recombinant Proteins by Dimethyladipimidate
Recombinant proteins (8.6 nm) purified by dextran sulfate-Sepharose CL-6B chromatography were incubated with 4 mm dimethyl adipimidate in 100 mm triethanolamine-HCl, pH 8.0, at 37 °C for 1 h after mixture with various concentrations of lipopolysaccharides and subjected to Western blotting using the monoclonal antibody 2C12 for factor C heavy chain.
Western Blotting
Samples were subjected to SDS-PAGE in 6, 8, or 10% slab gel, and transferred to a PVDF membrane. After blocking with 5% skim milk, the membrane was incubated with the monoclonal antibody 2C12 and then with the secondary antibody (HRP-conjugated goat anti-mouse IgG; Bio-Rad), followed by development with Chemi-Lumi One (Nacalai Tesque, Kyoto, Japan). Precision plus protein standards (Bio-Rad) were used to determine the apparent molecular masses.
RESULTS
Addition of Amino Acids at the N terminus of Factor C Prevents Autocatalytic Activation
A recombinant factor C was expressed in HEK293S GnTI− cells; the recombinant had an extra tripeptide of Glu-Thr-Gly derived from the expression vector at the native N terminus (+ETG-factor C), as confirmed by protein sequencing (Table 1). Unexpectedly, +ETG-factor C did not show amidase activity against a chromogenic synthetic substrate, Boc-Val-Pro-Arg-pNA, in the presence of lipopolysaccharides (Fig. 2A), suggesting that the extra N-terminal peptide inhibits autocatalytic activation of factor C on lipopolysaccharides. Therefore, the secretion signal sequence of the expression vector was replaced by an original secretion signal sequence of factor C, and the N-terminal sequence of the resulting recombinant factor C (wild-type factor C) was confirmed to be consistent with that of native factor C by protein sequencing (Table 1). Wild-type factor C exhibited amidase activity against the pNA substrate in the presence of lipopolysaccharides, and the dose-dependent curve of lipopolysaccharides for the autocatalytic activation of wild-type factor C was indistinguishable from that for the autocatalytic activation of native factor C isolated from hemocytes (Fig. 2A) (26). The specific amidase activity of wild-type factor C activated under optimum conditions in Fig. 2A was sufficient compared with that previously reported for native factor C (1). Kinetic parameters of wild-type factor C activated by lipopolysaccharides or chymotrypsin were determined against a fluorescence peptide substrate, Boc-Val-Pro-Arg-MCA. The factor C molecules activated by lipopolysaccharides and chymotrypsin were, respectively, designated α-factor C and β-factor C. Although Km values of the two active forms are indistinguishable, the kcat value of β-factor C was about 40% lower than that of α-factor C (Table 2).
FIGURE 2.
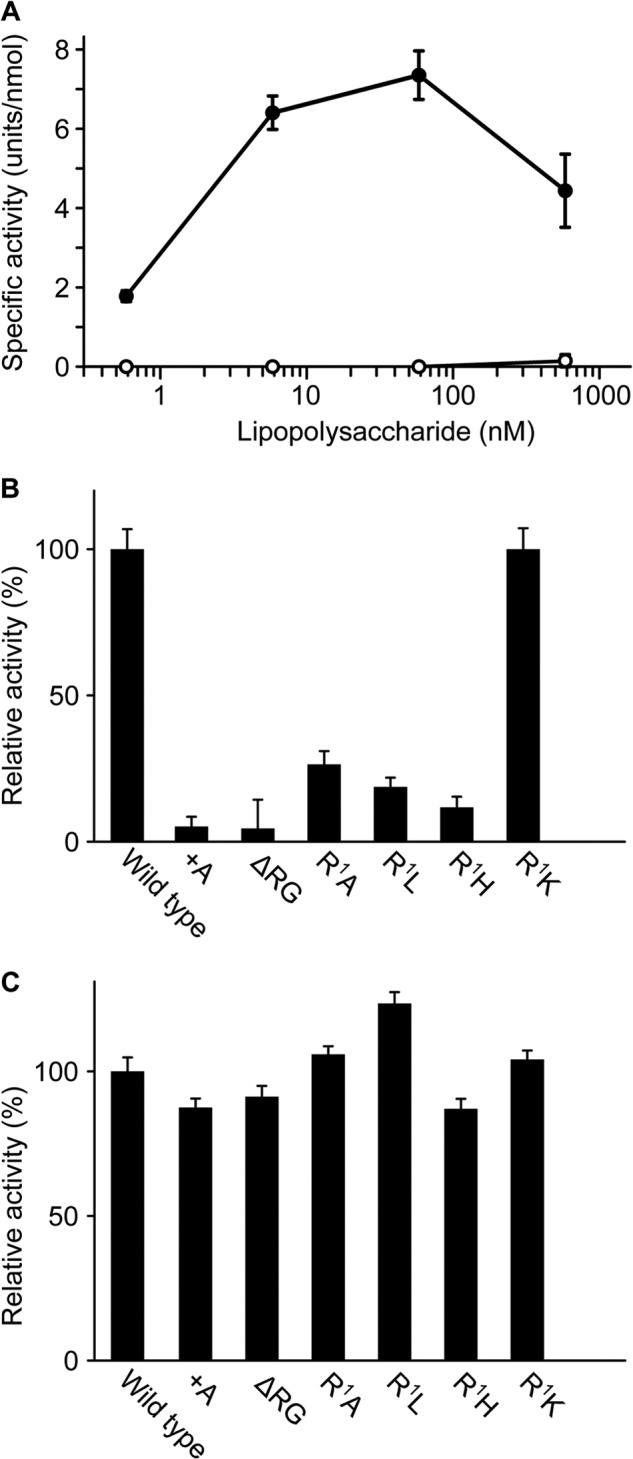
The native N terminus is required for autocatalytic activation of factor C on lipopolysaccharides. A, the wild-type and +ETG-factor C (1.7 nm) were incubated with various concentrations of lipopolysaccharides and assayed with Boc-Val-Pro-Arg-pNA. The closed circles and open circles indicate wild-type and +ETG-factor C, respectively. The unit of the amidase activity was defined as micromole of the digested substrate per min. The specific activity was expressed as units per nmol of factor C. B, each recombinant factor C (1.7 nm) was incubated with 12 nm lipopolysaccharides and assayed with Boc-Val-Pro-Arg-pNA. C, each recombinant factor C was incubated with α-chymotrypsin and assayed with Boc-Val-Pro-Arg-pNA. The vertical axes in B and C show relative activities in percentage terms against the activity of wild-type factor C. The values are the mean ± S.D. of three independent experiments.
TABLE 2.
Kinetic parameters of wild-type factor C activated by lipopolysaccharides (α-factor C) or chymotrypsin (β-factor C) for Boc-Val-Pro-Arg-MCA
| Km | kcat | kcat/Km | |
|---|---|---|---|
| μm | s−1 | m−1 s−1 | |
| α-Factor C | 84.2 ± 6.6 | 7.80 ± 1.09 | 92,200 ± 6,400 |
| β-Factor C | 71.2 ± 9.4 | 4.53 ± 0.64 | 63,600 ± 2,700 |
The N-terminal Arg Is Essential for Autocatalytic Activation
To examine the effects of addition or deletion of amino acids at the N terminus on the lipopolysaccharide-induced autocatalytic activation, an addition mutant of Ala (+A-factor C) or a deletion mutant of Arg-Gly (ΔRG-factor C) was expressed (Table 3). In addition, to investigate the significance of the N-terminal Arg, it was replaced by Ala (R1A-factor C), Leu (R1L-factor C), His (R1H-factor C), or Lys (R1K-factor C) (Table 3). When these mutants were incubated with lipopolysaccharides, only R1K-factor C, but not the other mutants, exhibited significant amidase activity (Fig. 2B). However, all mutants could be proteolytically activated by chymotrypsin, and their specific activities were comparable with that of wild-type factor C (Fig. 2C). These results indicate that the N-terminal Arg of wild-type factor C is essential for lipopolysaccharide-induced autocatalytic activation. When the N-terminal Arg was replaced with Lys, but not when it was replaced with another basic amino acid, His, it showed amidase activity equivalent to that of lipopolysaccharide-induced autocatalytic activation of wild-type factor C, suggesting that the specific interaction of the N-terminal Arg with a target amino acid(s) on factor C is required to trigger autocatalytic activation. In addition, to elucidate the importance of the distance between the N-terminal Arg and the tripartite lipopolysaccharide-binding site (-Arg36-Trp37-Arg38-) for the autocatalytic activation, the addition of mutants Arg (+R-factor C) and Arg-Thr-Gly (+RTG-factor C) or deletion mutants of Gly (ΔGly2-factor C), Gly-Val (ΔGly2-Val3-factor C), and Gly-Val-Asp (ΔGly2-Val3-Asp4-factor C) were expressed (Table 4). When these mutants were incubated with lipopolysaccharides, their amidase activities were not detectable, except that +R-factor C exhibited 30% relative activity compared with wild-type factor C (Table 4). These results indicate that the distance between the N-terminal Arg and the lipopolysaccharide-binding site is also an essential factor for autocatalytic activation of factor C.
TABLE 3.
The N-terminal sequences of recombinant of factor C
| −3 | −2 | −1 | 1 | 2 | 3 | 4 | |
|---|---|---|---|---|---|---|---|
| Wild type | R | G | V | D | |||
| +ETG-factor C | E | T | G | R | G | V | D |
| +A-factor C | A | R | G | V | D | ||
| ΔRG-factor C | V | D | |||||
| R1A-factor C | A | G | V | D | |||
| R1L-factor C | L | G | V | D | |||
| R1H-factor C | H | G | V | D | |||
| R1K-factor C | K | G | V | D |
TABLE 4.
Importance of the distance between the N-terminal Arg residue and the tripartite of the lipopolysaccharide-binding site
Relative activities were expressed in percentage terms against the activity of wild-type factor C.
| −3 | −2 | −1 | 1 | 2 | 3 | 4 | 5 | Relative activity | |
|---|---|---|---|---|---|---|---|---|---|
| % | |||||||||
| Wild type | R | G | V | D | L | 100 | |||
| ΔGly2-factor C | R | V | D | L | NDa | ||||
| ΔGly2-Val3-factor C | R | D | L | ND | |||||
| ΔGly2-Val3-Asp4-factor C | R | L | ND | ||||||
| +R-factor C | R | R | G | V | D | L | 30 ± 1.9 | ||
| +RTG-factor C | R | T | G | R | G | V | D | L | ND |
a ND, not detectable.
A Lipopolysaccharide-binding Capacity of Factor C Is Necessary but Not Sufficient for Lipopolysaccharide-induced Autocatalytic Activation
We previously reported that the N-terminal Cys-rich domain of factor C contains the tripartite lipopolysaccharide-binding site, and that a double point mutation in this domain (R36E/R38E-factor C) has no significant lipopolysaccharide-binding activity (8, 27). The dose-dependent curve of lipopolysaccharides for the autocatalytic activation of R36E/R38E-factor C was examined, and as expected, R36E/R38E-factor C was dysfunctional for lipopolysaccharide-induced autocatalytic activation (Fig. 3A). To examine whether the N-terminal dysfunctional mutants, including R1H-, R1A-, and R1L-factor C, have a lipopolysaccharide binding capacity, these mutants were incubated with lipopolysaccharide-immobilized Sepharose beads. Unexpectedly, all these N-terminal dysfunctional mutants as well as the functional mutant of R1K-factor C exhibited lipopolysaccharide-binding activity comparable with that of wild-type factor C (Fig. 3B). To confirm the lipopolysaccharide-binding activity of these mutants quantitatively, the lipopolysaccharide-binding parameters were determined by surface plasmon resonance analysis. These three mutants with dysfunction for lipopolysaccharide-induced autocatalytic activation and the functional mutant R1K-factor C retained an association constant (Ka = 0.4 × 109 m−1 ∼ 1.1 × 109 m−1) comparable with wild-type factor C (Ka = 1.3 × 109 m−1) (Table 5). These results indicate that the lipopolysaccharide binding capacity of factor C is necessary but not sufficient for lipopolysaccharide-induced autocatalytic activation.
FIGURE 3.
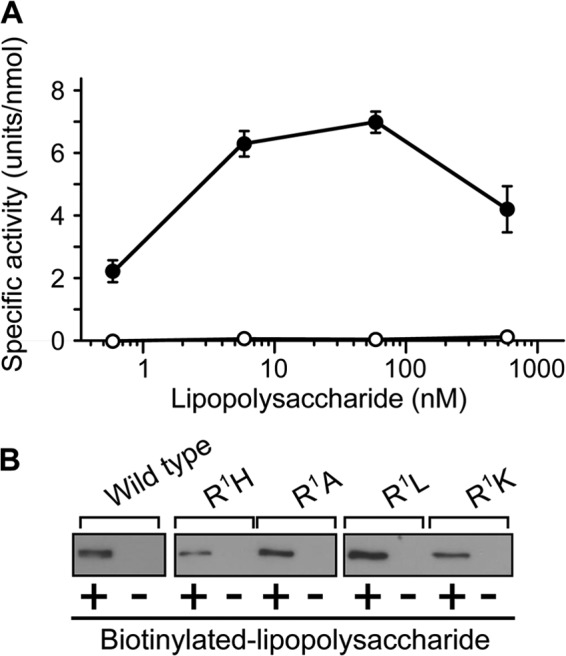
Lipopolysaccharide-binding activities of the N-terminal substituted mutants of factor C. A, wild-type and R36E/R38E-factor C (1.7 nm) were incubated with various concentrations of lipopolysaccharides and assayed with Boc-Val-Pro-Arg-pNA. The unit of the amidase activity was defined as micromole of the digested substrate per min. The specific activity was expressed as units per nmol of factor C. Closed and open circles indicate wild-type and R36E/R38E-factor C, respectively. Data are the mean ± S.D. of three independent experiments. B, Sepharose beads coupled to streptavidin was incubated with recombinants of factor C in the presence (+) or absence (−) of biotinylated lipopolysaccharides. Precipitates were subjected to Western blotting using the monoclonal antibody 2C12. Data are representative of three independent experiments.
TABLE 5.
Lipopolysaccharide-binding parameters and amidase activities of the N-terminal substitution mutants of factor C
| ka | kd | Ka | Activation by lipopolysaccharides | |
|---|---|---|---|---|
| m−1s−1 | s−1 | m−1 | ||
| Wild-type | 4.29 × 106 ± 2.50 × 106 | 3.25 × 10−3 ± 0.42 × 10−3 | 1.32 × 109 | Yes |
| R1H-factor C | 8.26 × 106 ± 2.76 × 106 | 1.41 × 10−2 ± 0.92 × 10−2 | 0.59 × 109 | No |
| R1A-factor C | 6.41 × 106 ± 3.87 × 106 | 1.74 × 10−2 ± 0.87 × 10−2 | 0.37 × 109 | No |
| R1L-factor C | 5.80 × 106 ± 4.15 × 106 | 5.79 × 10−3 ± 0.30 × 10−3 | 1.00 × 109 | No |
| R1K-factor C | 6.85 × 106 ± 3.73 × 106 | 6.30 × 10−3 ± 1.28 × 10−3 | 1.09 × 109 | Yes |
Factor C Is Autocatalytically Activated with Close Vicinity to be Chemically Cross-linked on Lipopolysaccharides
There was an optimum concentration of lipopolysaccharides for the autocatalytic activation of factor C, namely an optimum molar ratio of factor C to lipopolysaccharides, suggesting that an opportunity to form a complex between at least two molecules of factor C with an adequate molecular orientation on lipopolysaccharides is required to trigger autocatalytic activation (Fig. 2A) (26). To capture the lipopolysaccharide-supported competent complex, wild-type factor C was incubated with the homobifunctional cross-linker dimethyl adipimidate in the presence of lipopolysaccharides, and the resulting cross-linked factor C molecules were detected by Western blotting under reducing conditions, using the monoclonal antibody 2C12 that recognizes the heavy chain of factor C. The cross-linked factor C molecules with molecular masses of 160, 280, and ∼500 kDa were detected on SDS-PAGE at 5.9 and 59 nm lipopolysaccharides, and cross-linking of factor C molecules was prevented at 590 nm lipopolysaccharides (Fig. 4A). The dose dependence of lipopolysaccharides for cross-linking of factor C molecules was consistent with that for autocatalytic activation, suggesting that factor C molecules in sufficiently close vicinity leading to autocatalytic activation were specifically cross-linked by dimethyl adipimidate (Fig. 4, B and C). Based on the finding that the heavy chain had an apparent molecular mass of 80 kDa on SDS-PAGE, the dimer and its multiple species of wild-type factor C seem to be cross-linked (Fig. 4A). To examine the role of the N-terminal amino acid for cross-linking of factor C molecules, the N-terminal substitution mutants (R1H-, R1A-, and R1L-factor C) and the functional N-terminal mutant (R1K-factor C) were incubated with dimethyl adipimidate in the presence of lipopolysaccharides. Western blotting showed that the amounts of cross-linked factor C molecules of the dysfunctional N-terminal mutants (R1H-, R1A-, and R1L-factor C) were significantly lower than those of wild-type factor C and the functional N-terminal mutant of R1K-factor C (Fig. 5).
FIGURE 4.
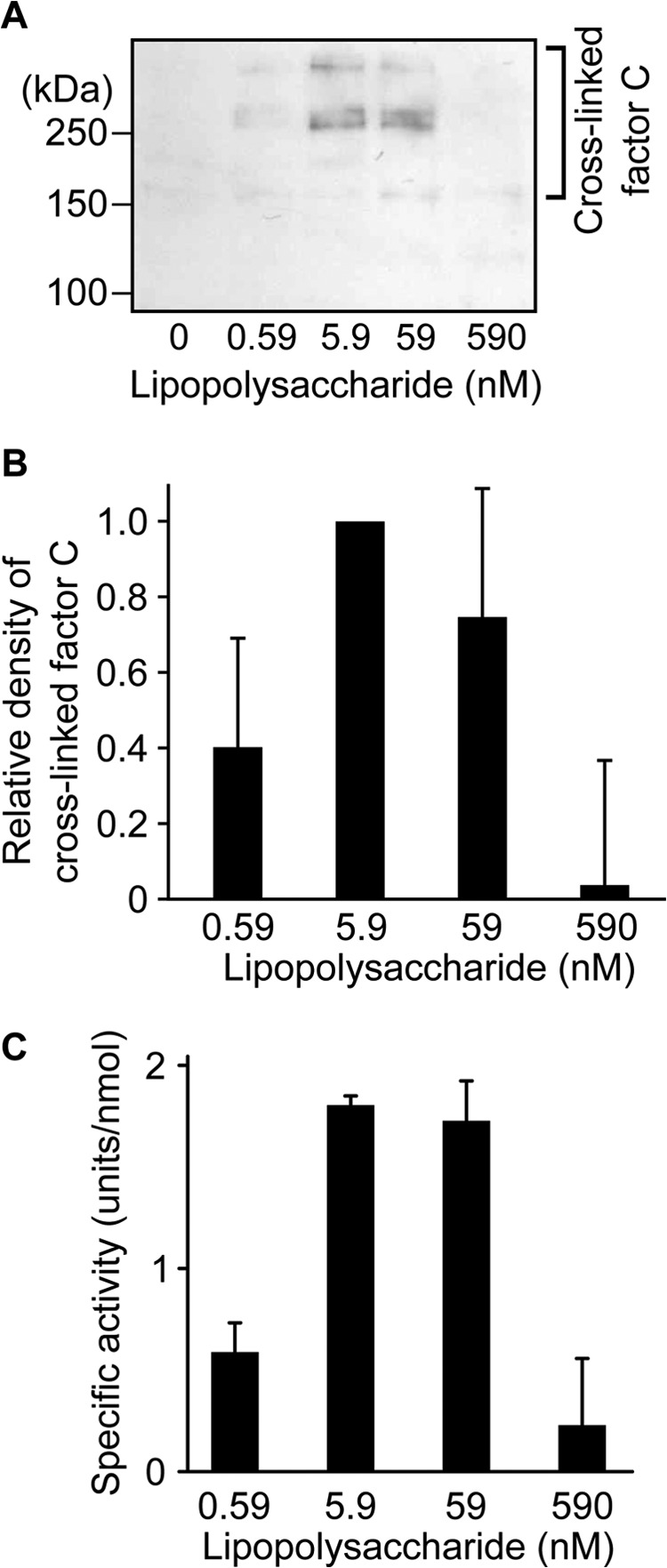
Chemical cross-linking of factor C with dimethyl adipimidate on lipopolysaccharides. A, wild-type factor C (8.6 nm) was cross-linked with dimethyl adipimidate at several concentrations of lipopolysaccharides and subjected to Western blotting. Data are representative of three independent experiments. B, the density of cross-linked wild-type factor C molecules on the PVDF membrane was analyzed with Image J software. The vertical axis shows relative densities against that of wild-type factor C cross-linked at 5.9 nm lipopolysaccharides. Data are the mean ± S.D. of three independent experiments. C, wild-type factor C (8.6 nm) was incubated with several concentrations of lipopolysaccharides and assayed with Boc-Val-Pro-Arg-pNA. The unit of the amidase activity was defined as micromole of the digested substrate per min. The specific activity was expressed as units per nmol of factor C. Data are the mean ± S.D. of three independent experiments.
FIGURE 5.
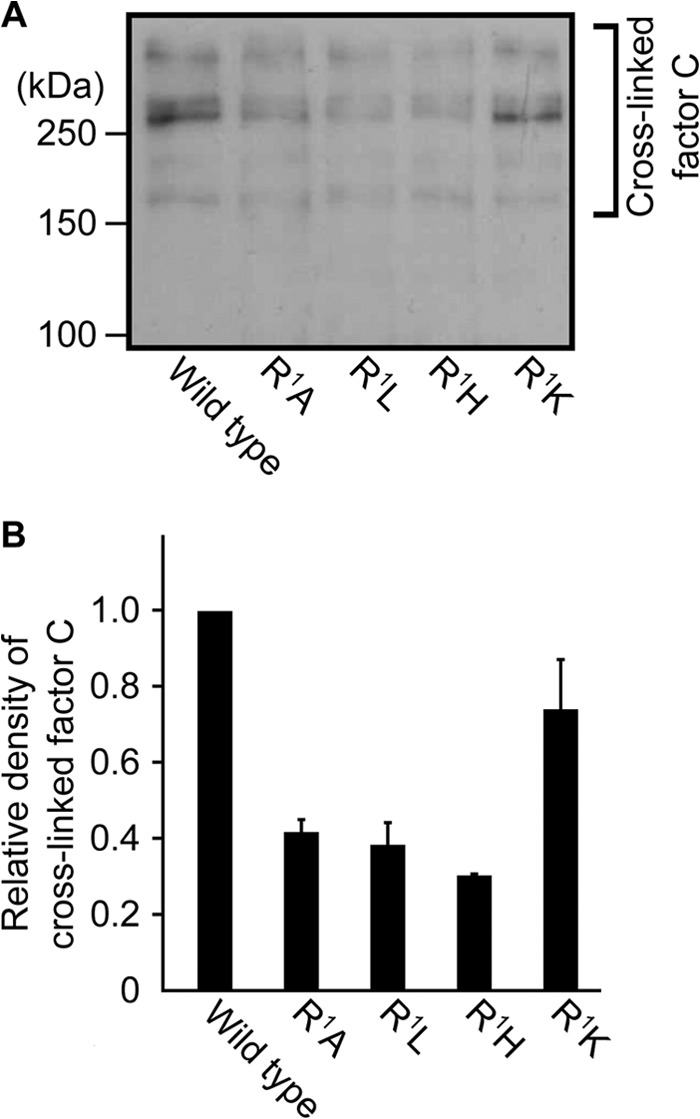
Chemical cross-linking of the N-terminal-substituted mutants with dimethyl adipimidate. A, each recombinant of factor C (8.6 nm) was treated by dimethyl adipimidate in the presence of lipopolysaccharides (59 nm) and subjected to Western blotting. Data are representative of two independent experiments. B, density of cross-linked factor C molecules on the PVDF membrane was analyzed with Image J software. The vertical axis shows relative densities against that of wild-type factor C. Data are the mean ± S.D. of two independent experiments.
An Inhibitory Effect of Triton X-100 on Lipopolysaccharide-induced Autocatalytic Activation
Autocatalytic activation of native factor C on lipopolysaccharides is inhibited in the presence of a synthetic surfactant Triton X-100 (28). To determine whether autocatalytic activation of recombinant factor C on lipopolysaccharides is inhibited by Triton X-100, wild-type factor C was incubated with various concentrations of Triton X-100 in the presence of lipopolysaccharides (59 nm). Autocatalytic activation of wild-type factor C on lipopolysaccharides was inhibited by adding Triton X-100, especially above the critical micelle concentration of Triton X-100 (0.24 mm), suggesting that Triton X-100 is mixed into lipopolysaccharides to form an ineffective platform for the autocatalytic activation of factor C (Fig. 6).
FIGURE 6.
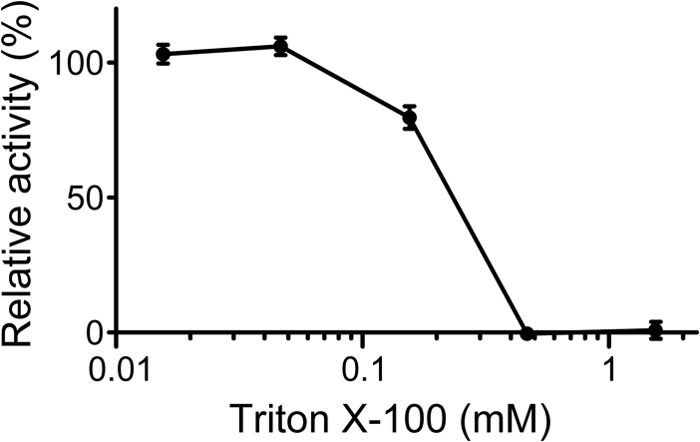
Inhibition of autocatalytic activation of factor C by Triton X-100. Wild-type factor C (8.6 nm) was incubated with lipopolysaccharides (59 nm) under various concentrations of Triton X-100 and assayed with Boc-Val-Pro-Arg-pNA. The vertical axis shows relative activities in percentage terms against the activity of wild-type factor C in the absence of Triton X-100. Data are the mean ± S.D. of three independent experiments.
Effect of Substitution of Acidic Amino Acids in the C-terminal Catalytic Domain
We found an acidic region clustered with three repetitive Asp residues, -Asp78-Asp79-Asp80-, in chymotrypsinogen numbering in the C-terminal catalytic domain of factor C (Fig. 7), and the corresponding region of trypsinogen or thrombin was exposed to the surface of the molecule (29). To examine whether the region interacts with the N-terminal Arg1, each Asp was replaced by Asn (D814N-factor C, D815N-factor C, and D816N-factor C). When these three mutants were incubated with lipopolysaccharides, they all exhibited a level of amidase activity equivalent to that of the wild-type factor C (Fig. 8).
FIGURE 7.

The partial sequences of factor C, chymotrypsinogen A, trypsin, elastase, and thrombin corresponding to that around the three repetitive Asp residues of factor C. The identical residues are boxed. The numbers at the top indicate chymotrypsinogen numbering. The three repetitive Asp residues of factor C are highlighted in gray.
FIGURE 8.
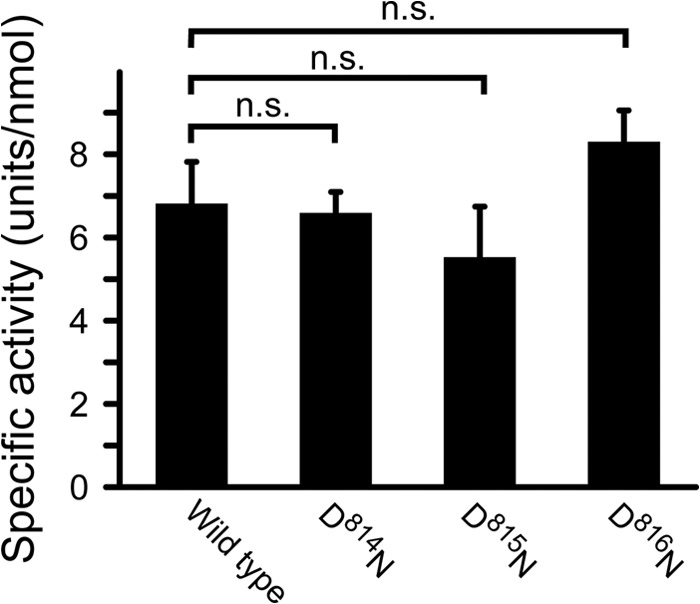
Effects of point mutations at the three-tandem Asp region in the C-terminal catalytic domain on lipopolysaccharide-induced autocatalytic activation. D814N-factor C, D815N-factor C, and D816N-factor C (1.7 nm) were incubated with lipopolysaccharides (59 nm) and assayed with Boc-Val-Pro-Arg-pNA. The unit of the amidase activity was defined as micromole of the digested substrate per min. The specific activity was expressed as units per nmol of factor C. Data are the mean ± S.D. of three independent replicates. n.s., not significant.
DISCUSSION
Wang et al. (5) reported that factor C expressed in Spodoptera frugiperda Sf9 cells, but not expressed in Drosophila S2 cells, exhibits lipopolysaccharide-induced autocatalytic activation; N-glycans of factor C expressed with Sf9 cells contains sialic acid with an α(2–6) linkage to galactose more abundantly than that with an α(2–3) linkage, whereas N-glycans of factor C expressed in S2 cells is rich in sialic acid with an α(2–3) linkage, suggesting that the complexity of N-glycans influences the lipopolysaccharide-induced autocatalytic activation. We reported here that a recombinant factor C with homogeneous N-glycan of Man5GlcNAc2 expressed in HEK293S GnTI− cells exhibited a level of lipopolysaccharide-induced amidase activity equivalent to that of native factor C isolated from hemocytes (Fig. 2A). These results suggest that complex N-glycans have little or no effect on the lipopolysaccharide-induced autocatalytic activation of factor C.
Occasionally, recombinant proteins are expressed with extra amino acid sequences at the N-terminal or C-terminal end, such as epitope tags or reluctant sequences derived from an initiating Met and multicloning sites. In this study, +ETG-factor C with an extra tripeptide and factor C mutants with amino acid additions or deletions at the N terminus were incompetent for the lipopolysaccharide-induced autocatalytic activation, but competent for the chymotrypsin-catalyzed activation (Fig. 2). Moreover, N-terminal substitution mutants of R1A-, R1L-, and R1H-factor C but not R1K-factor C were also incompetent for the lipopolysaccharide-induced autocatalytic activation (Fig. 2), indicating that the N-terminal Arg or Lys were essential for autocatalytic activation and its positive charge may interact with an acidic amino acid(s) of the factor C molecule to convert the zymogen into an active form on lipopolysaccharides. The dysfunctional mutants, i.e. R1A-, R1L-, and R1H- factor C, showed lipopolysaccharide-binding activity with an affinity constant equivalent to that of wild-type factor C (Table 5), indicating that the lipopolysaccharide-binding activity of factor C is required but not sufficient for lipopolysaccharide-induced autocatalytic activation. Wild-type factor C was chemically cross-linked by dimethyl adipimidate under an optimum concentration of lipopolysaccharides to form a dimer and its multiple factor C molecules, and the amount of cross-linked factor C molecules for the dysfunctional mutants of the N-terminal substitutions was significantly lower than that for wild-type factor C or R1K-factor C (Figs. 4 and 5).
The surface of lipopolysaccharides appears to function as a platform to induce specific protein-protein interaction between the factor C molecules. Lipopolysaccharides mixed with Triton X-100 did not induce the autocatalytic activation of wild-type factor C as well as native factor C (Fig. 6), suggesting that lipopolysaccharides mixed with Triton X-100 failed to maintain the platform to induce autocatalytic activation of factor C. The effective autocatalytic activation of factor C requires an optimum ratio of factor C to lipopolysaccharides, and thus increasing the amount of lipopolysaccharides reduces the opportunity for the protein-protein interaction of factor C on the surface of lipopolysaccharides. Interestingly, the kcat value of the recombinant activated by chymotrypsin (β-factor C) was 40% lower than that activated by lipopolysaccharides (α-factor C) (Table 2), suggesting that proteolytic cleavage of zymogen factor C by chymotrypsin may occur not only within the -Phe737-Ile738- bond but also within other site(s), causing the reduction of the kinetic parameter of β-factor C.
Here we show that the N-terminal Arg and the distance between the N terminus and the lipopolysaccharide-binding site are essential factors for autocatalytic activation on the surface of lipopolysaccharides. The tripartite lipopolysaccharide-binding site is essential to maintain binding activity with lipopolysaccharide with the association constant (Ka) ∼109 m−1 (Table 5). On the other hand, the N-terminal Arg with a constant length from the tripartite lipopolysaccharide-binding site does not contribute to lipopolysaccharide binding affinity but appears to be essential to induce specific protein-protein interaction between the factor C molecules, leading to autocatalytic activation on lipopolysaccharides. However, the possibility of the molecular sexuality mechanism established by staphylocoagulase is unlikely, because the basic side chains of Arg and Lys, but not the hydrophobic ones, are essential for the lipopolysaccharide-induced autocatalytic activation of factor C (Fig. 2B). As another possibility, a three-tandem Asp region in the C-terminal catalytic domain may interact with the N-terminal Arg on lipopolysaccharides (Fig. 7), because the sequence and structure of α-thrombin (29) suggest that this region might locate on the surface of the C-terminal catalytic domain. However, autocatalytic activation of substitution mutants D79N-factor C, D78N-factor C, and D80N-factor C was sufficient (Fig. 8). A triple-point mutation in the region might suggest that this site would interact with the N terminus, but the expression of this mutant was too low to determine its lipopolysaccharide-induced activation. Thus, we need another approach to elucidate the region interacting with the N-terminal Arg. The recombinant factor C with restricted and homogeneous N-glycans produced in this study may be useful for further structure-function studies. Interestingly, tissue-type plasminogen activator, a physiological initiator in fibrinolysis, possesses an intrinsic activity residing in a single-chain form (30). Renatus et al. (31) reported that the ϵ-amino group of Lys156 in chymotrypsinogen numbering located in the inside of the C-terminal catalytic domain of the tissue-type plasminogen activator forms a salt bridge with the side group of Asp194, promoting an active conformation in the zymogen form. However, the Lys156 residue in the tissue-type plasminogen activator is replaced by the Gln156 residue in factor C just as in the case of chymotrypsinogen (Fig. 9). Therefore, factor C may have no intrinsic proteolytic activity residing in the zymogen form. We propose here that the N-terminal Arg of factor C is essential to induce specific protein-protein interaction between the factor C molecules in close vicinity with a sufficiently proper orientation on lipopolysaccharides, leading to autocatalytic activation.
FIGURE 9.

The partial sequences of chymotrypsinogen A and factor C corresponding to that around the Lys156 in tissue-type plasminogen activator. Identical residues are boxed. The Lys156 residue in the tissue-type plasminogen activator and the corresponding residues in factor C and chymotrypsinogen A are highlighted in gray.
Acknowledgments
We are grateful to Dr. Y. Yanagi (Kyushu University, Fukuoka) for providing plasmid vector pHLsec and pCA7. We thank Dr. S. Higashi (Yokohama City University, Yokohama) for helpful discussion on autocatalytic activation of the mammalian blood coagulation system.
This work was supported by the joint research fund from Seikagaku Corporation.
- GnTI
- N-acetylglucoaminyltransferase I
- Boc
- butoxycarbonyl
- pNA
- p-nitoroanilide
- MCA
- 4-methylcoumaryl-7-amide.
REFERENCES
- 1. Nakamura T., Morita T., Iwanaga S. (1986) Lipopolysaccharide-sensitive serine-protease zymogen (factor C) found in Limulus hemocytes. Eur. J. Biochem. 154, 511–521 [DOI] [PubMed] [Google Scholar]
- 2. Ariki S., Takahara S., Shibata T., Fukuoka T., Ozaki A., Endo Y., Fujita T., Koshiba T., Kawabata S. (2008) Factor C acts as a lipopolysaccharide-responsive C3 convertase in horseshoe crab complement activation. J. Immunol. 181, 7994–8001 [DOI] [PubMed] [Google Scholar]
- 3. Ariki S., Koori K., Osaki T., Motoyama K., Inamori K., Kawabata S. (2004) A serine protease zymogen functions as a pattern-recognition receptor for lipopolysaccharides. Proc. Natl. Acad. Sci. U.S.A. 101, 953–958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Muta T., Miyata T., Misumi Y., Tokunaga F., Nakamura T., Toh Y., Ikehara Y., Iwanaga S. (1991) Limulus factor C: an endotoxin-sensitive serine protease zymogen with a mosaic structure of complement-like, epidermal growth factor-like, and lectin-like domains. J. Biol. Chem. 266, 6554–6561 [PubMed] [Google Scholar]
- 5. Wang J., Tan N. S., Ho B., Ding J. L. (2002) Modular arrangement and secretion of a multidomain serine protease: evidence for involvement of proline-rich region and N-glycans in the secretion pathway. J. Biol. Chem. 277, 36363–36372 [DOI] [PubMed] [Google Scholar]
- 6. Kawabata S. (2010) in Invertebrate Immunity (Söderhäll K., ed) pp. 122–136, Springer, New York [Google Scholar]
- 7. Tokunaga F., Nakajima H., Iwanaga S. (1991) Further studies on lipopolysaccharide-sensitive serine protease zymogen (factor C): its isolation from Limulus polyphemus hemocytes and identification as an intracellular zymogen activated by α-chymotrypsin, not by trypsin. J. Biochem. 109, 150–157 [DOI] [PubMed] [Google Scholar]
- 8. Koshiba T., Hashii T., Kawabata S. (2007) A structural perspective on the interaction between lipopolysaccharide and factor C, a receptor involved in recognition of Gram-negative bacteria. J. Biol. Chem. 282, 3962–3967 [DOI] [PubMed] [Google Scholar]
- 9. Davie E. W., Fujikawa K., Kurachi K., Kisiel W. (1979) in Advances in Enzymology and Related Areas of Molecular Biology (Meister A., ed) Vol. 48, pp. 277–318, John Wiley & Sons, Inc., New York: [DOI] [PubMed] [Google Scholar]
- 10. Higashi S., Matsumoto N., Iwanaga S. (1997) Conformation of factor VIIa stabilized by a labile disulfide bond (Cys310–Cys329) in the protease domain is essential for interaction with tissue factor. J. Biol. Chem. 272, 25724–25730 [DOI] [PubMed] [Google Scholar]
- 11. Bode W., Huber R. (1976) Induction of the bovine trypsinogen: trypsin transition by peptides sequentially similar to the N-terminus of trypsin. FEBS Lett. 68, 231–236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Reddy K. N., Markus G. (1972) Mechanism of activation of human plasminogen by streptokinase presence of active center in streptokinase-plasminogen complex. J. Biol. Chem. 247, 1683–1691 [PubMed] [Google Scholar]
- 13. Kawabata S., Morita T., Miyata T., Iwanaga S., Igarashi H. (1986) Isolation and characterization of staphylocoagulase chymotryptic fragment. Localization of the procoagulant- and prothrombin-binding domain of this protein. J. Biol. Chem. 261, 1427–1433 [PubMed] [Google Scholar]
- 14. Friedrich R., Panizzi P., Fuentes-Prior P., Richter K., Verhamme I., Anderson P. J., Kawabata S., Huber R., Bode W., Bock P. E. (2003) Staphylocoagulase is a prototype for the mechanism of cofactor-induced zymogen activation. Nature 425, 535–539 [DOI] [PubMed] [Google Scholar]
- 15. Panizzi P., Friedrich R., Fuentes-Prior P., Bode W., Bock P. E. (2004) The staphylocoagulase family of zymogen activator and adhesion proteins. Cell Mol. Life Sci. 61, 2793–2798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Park H. H. (2012) Structural features of caspase-activating complexes. Int. J. Mol. Sci. 13, 4807–4818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Muzio M., Stockwell B. R., Stennicke H. R., Salvesen G. S., Dixit V. M. (1998) An induced proximity model for caspase-8 activation. J. Biol. Chem. 273, 2926–2930 [DOI] [PubMed] [Google Scholar]
- 18. Stennicke H. R., Deveraux Q. L., Humke E. W., Reed J. C., Dixit V. M., Salvesen G. S. (1999) Caspase-9 can be activated without proteolytic processing. J. Biol. Chem. 274, 8359–8362 [DOI] [PubMed] [Google Scholar]
- 19. Renatus M., Stennicke H. R., Scott F. L., Liddington R. C., Salvesen G. S. (2001) Dimer formation drives the activation of the cell death protease caspase 9. Proc. Natl. Acad. Sci. 98, 14250–14255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Reeves P. J., Callewaert N., Contreras R., Khorana H. G. (2002) Structure and function in rhodopsin: high-level expression of rhodopsin with restricted and homogeneous N-glycosylation by a tetracycline-inducible N-acetylglucosaminyltransferase I-negative HEK293S stable mammalian cell line. Proc. Natl. Acad. Sci. U.S.A. 99, 13419–13424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hashiguchi T., Kajikawa M., Maita N., Takeda M., Kuroki K., Sasaki K., Kohda D., Yanagi Y., Maenaka K. (2007) Crystal structure of measles virus hemagglutinin provides insight into effective vaccines. Proc. Natl. Acad. Sci. U.S.A. 104, 19535–19540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Aricescu A. R., Lu W., Jones E. Y. (2006) A time- and cost-efficient system for high-level protein production in mammalian cells. Acta Crystallogr. D Biol. Crystallogr. 62, 1243–1250 [DOI] [PubMed] [Google Scholar]
- 23. Miura Y., Tokunaga F., Miyata T., Moriyasu M., Yoshikawa K., Iwanaga S. (1992) Preparation and properties of monoclonal antibodies against lipopolysaccharide-sensitive serine protease zymogen, factor C, from horseshoe crab (Tachypleus tridentatus) hemocytes. J. Biochem. 112, 476–481 [DOI] [PubMed] [Google Scholar]
- 24. Nakamura T., Morita T., Iwanaga S. (1985) Intracellular proclotting enzyme in Limulus (Tachypleus tridentatus) hemocytes: its purification and propertied. J. Biochem. 97, 1561–1574 [DOI] [PubMed] [Google Scholar]
- 25. Cornish-Bowden A., Eisenthal R. (1978) Estimation of Michaelis constant and maximum velocity from the direct linear plot. Biochim. Biophys. Acta 523, 268–272 [DOI] [PubMed] [Google Scholar]
- 26. Nakamura T., Tokunaga F., Morita T., Iwanaga S., Kusumoto S., Shiba T., Kobayashi T., Inoue K. (1988) Intracellular serine-protease zymogen, factor C, from horseshoe crab hemocytes. Eur. J. Biochem. 176, 89–94 [DOI] [PubMed] [Google Scholar]
- 27. Kawabata S., Koshiba T., Shibata T. (2009) The lipopolysaccharide-activated innate immune response network of the horseshoe crab. Invertebrate Surviv. J. 6, 59–77 [Google Scholar]
- 28. Nakamura T., Tokunaga F., Morita T., Iwanaga S. (1988) Interaction between lipopolysaccharide and intracellular serine protease zymogen, factor C, from horseshoe crab (Tachypleus tridentatus) hemocytes. J. Biochem. 103, 370–374 [DOI] [PubMed] [Google Scholar]
- 29. Bode W., Mayr I., Baumann U., Huber R., Stone S. R., Hofsteenge J. (1989) The refined 1.9-Å crystal structure of human alpha-thrombin: interaction with d-Phe-Pro-Arg chloromethyl ketone and significance of the Tyr-Pro-Pro-Trp insertion segment. EMBO J. 8, 3467–3475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Rånby M., Bergsdorf N., Nilsson T. (1982) Enzymatic properties of the one- and two-chain form of tissue plasminogen activator. Thromb. Res. 27, 175–183 [DOI] [PubMed] [Google Scholar]
- 31. Renatus M., Engh R. A., Stubbs M. T., Huber R., Fischer S., Kohnert U., Bode W. (1997) Lysine 156 promotes the anomalous proenzyme activity of tPA: x-ray crystal structure of single-chain human tPA. EMBO J. 16, 4797–4805 [DOI] [PMC free article] [PubMed] [Google Scholar]