

Abstract
Abnormal choline phospholipid metabolism is an emerging hallmark of cancer, which is implicated in carcinogenesis and tumor progression. The malignant metabolic phenotype is characterized by high levels of phosphocholine (PC) and relatively low levels of glycerophosphocholine (GPC) in aggressive breast cancer cells. Phosphorus Magnetic Resonance Spectroscopy (31P MRS) is able to noninvasively detect these water-soluble metabolites of choline as well as ethanolamine phospholipid metabolism. Here we have investigated the effects of stably silencing glycerophosphoester diesterase domain containing 5 (GDPD5), which is an enzyme with glycerophosphocholine phosphodiesterase activity, in MDA-MB-231 breast cancer cells and orthotopic tumor xenografts. Tumors in which GDPD5 was stably silenced with GDPD5-specific shRNA contained increased levels of GPC and phosphoethanolamine (PE) compared to control tumors.
Keywords: 31P MRS, metabolism, in vivo, breast cancer, glycerophosphoesterdiesterase, GDPD5, silencing
Introduction
Altered choline phospholipid metabolism is an emerging hallmark of cancer, which is associated with oncogenesis and tumor progression (1). Oncogenic transformation of cells changes the expression and activity of enzymes that control anabolic and catabolic pathways in membrane phospholipid metabolism, thereby altering the levels of choline- and ethanolamine-containing precursors and breakdown products (1,2) as shown in Figure 1. These choline- and ethanolamine-containing metabolites can be monitored in vivo by 31P magnetic resonance spectroscopy (MRS), which detects phosphomonoesters (PME) predominantly in the anabolic pathway and phosphodiesters (PDE) in the catabolic pathway (Fig.1) (3-5). In breast and ovarian cancer cells, a switch from high glycerophosphocholine (GPC) and low phosphocholine (PC) in normal cells to high PC and low GPC in malignant cells has been observed, and the PC/GPC ratio increases with cancer aggressiveness (6,7).
Figure 1.
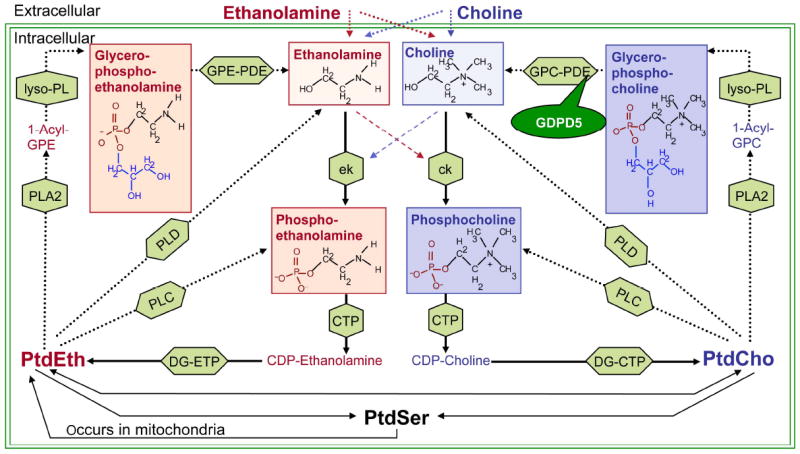
Network of ethanolamine and choline phospholipid metabolism. MRS-detectable metabolites are shown in red (ethanolamine) and blue (choline) boxes. Anabolic enzyme reactions are depicted by solid arrows and catabolic enzyme reactions by dotted arrows. Enzymes are shown in green boxes. GDPD5, which is examined in this study, is a GPC-PDE. Silencing of GPDP5 is hypothesized to increase glycerophosphocholine levels and decrease free choline levels.
Abbreviations of metabolites: PtdSer, phosphatidylserine; PtdEth, phosphatidylethanolamine; PtdCho, phosphatidylcholine; 1-acyl-GPE, 1-acyl-glycerophosphoethanolamine; 1-acyl-GPC, 1-acyl-glycerophosphocholine. Abbreviations of enzymes: ek, ethanolamine kinase; ck, choline kinase; CTP, phosphocholine cytidylyltransferase; DG-CTP, diacylglycerol choline phosphotransferase; DG-ETP, diacylglycerol ethanolamine phosphotransferase; PLC, phospholipase C; PLC, phospholipase D; PLA2, phospholipase A2; lyso-PL, lysophospholipase; GPC-GPE, glycerophosphocholine phosphodiesterase; GPE-GPE, glycerophosphoethanolamine phosphodiesterase; GDPD5, glycerophosphodiester phosphodiesterase domain containing 5.
Increased PC levels are associated with increased proliferation, and complex reciprocal interactions exist between oncogenic signaling and choline phospholipid metabolism (1). Since MRS noninvasively detects choline-containing compounds, an increase in these compounds can be used as a noninvasive biomarker of transformation and staging (6,8). A decrease in these compounds can indicate response to therapy, including novel therapies that target oncogenic signaling pathways (9,10).
Enhanced levels of PE and GPE have also been detected in various tumor tissues (4,8). However, the role of ethanolamine phospholipid metabolism in cancer cells has not yet been studied in great molecular detail, although phosphatidylethanolamine (PtdEth) constitutes 20%–40% of all phospholipids in mammalian cell membranes (11). Apart from its structural roles in cellular membranes, PtdEth also acts as the donor of the ethanolamine moiety that covalently links glycosylphosphatidylinositol membrane anchors to terminal carboxyl groups of proteins attached to the surface of cells (12). Choline kinase alpha and beta have the ability to also use ethanolamine as a substrate to produce PE (13), which could be a reason for the high PE levels in tumors.
The metabolites in the choline and ethanolamine pathways (red and blue shaded boxes in Fig.1) can be studied with 1H and 31P MRS. When using 1H MRS, these signals overlap even when studied ex vivo with 1H MRS at high field strengths (14). When using 31P MRS at high field strengths, the larger spectral separation of the 31P-containing choline and ethanolamine metabolites makes it possible to study them individually in vivo, however, at the cost of lower sensitivity (15,16).
Several enzymes in phospholipid metabolism (green arrows in Fig.1) could serve as potential targets for image-guided cancer therapy. The goal of phospholipid-metabolism-targeted therapy of breast cancer would be to revert the malignant metabolic phenotype of high PC and low GPC back to the normal metabolic phenotype of low PC and high GPC. Silencing and inhibition of choline kinase alpha has been shown to reduce PC levels in aggressive breast cancer cells and tumor xenografts and thereby reduce proliferation and tumor growth (10,17,18). A drug that inhibits choline kinase activity is currently being tested as cancer therapy in clinical trials (19).
Another possibility to lower the PC/GPC ratio of aggressive breast cancer cells would be to target enzymes with glycerophosphocholine phosphodiesterase (GPC-PDE) activity (20). Enzymes with GPC-PDE activity break down GPC into choline and glycerol-3-phosphate, thereby supplying the cancer cell with free choline, which is then quickly metabolized to PC due to the high activity of choline kinase alpha (20,21). We have recently shown that glycerophosphodiester phosphodiesterase domain containing 5 (GDPD5) is significantly overexpressed in highly malignant estrogen receptor negative breast cancer cells and breast tumors from patients (21). GDPD5 positively correlated with the total choline-containing metabolite level and PC/GPC ratio in human breast tumors, and showed a trend towards a negative correlation with the GPC level (21). We therefore hypothesize that silencing GDPD5 could lead to higher GPC levels and simultaneously to a reduction of free choline in the cell and therefore a reduced availability of substrate for choline kinase alpha to produce PC. This could in turn lead to a lower PC/GPC ratio, which is characteristic of the non-malignant metabolic phenotype of breast epithelial cells. To this end, we have studied the effects of stably silencing GDPD5 on the phospholipid metabolite levels in MDA-MB-231 breast cancer cells and MDA-MB-231 breast tumor xenografts in mice by using 31P MRS.
Materials and methods
Cells and cell culture conditions
Triple negative MDA-MB-231 breast cancer cells, which were derived from a metastatic lesion of a breast adenocarcinoma in the mammary gland, were purchased from the American Type Culture Collection (ATCC, Manassas, VA) and used within 6 months of obtaining from ATCC. This cell line was tested and authenticated by ATCC using two independent methods: the ATCC cytochrome C oxidase I PCR assay, and short tandem-repeat profiling using multiplex PCR. Cells were cultured in RPMI-1640 supplemented with 10% fetal bovine serum (FBS; Sigma), 100 units/mL penicillin, and 100 μg/mL streptomycin (Invitrogen, Calabasas, CA). RPMI-1640 contains 21.43 μmol/L of choline and no ethanolamine. Cells were maintained in a humidified atmosphere of 5% CO2 in air at 37C°.
Cloning and lentivirus preparation
A pLKO.1 vector containing lentiviral shRNA against GDPD5 with the hairpin sequence 5’CCGGGCTCTCCGTATGTTCAGACAACTCGAGTTGTCTGAACATACGGAGAGCTTTTT-3’ was digested with SacII and Nde1. The isolated and purified shRNA insert against GDPD5 was cloned between Nde1 and EcoRV into a human U6 promoter-driven pRRL vector containing enhanced green fluorescent protein (EGFP) as a reporter gene driven by a phosphoglycerate kinase (pGK) promoter as previously described (10). An empty vector control lacking any shRNA, but expressing pGK-driven EGFP, was used as control (vector control) (22). Infectious viral supernatants (DMEM (Mediatech, Manassas, VA) with 1% FBS) were derived by transient cotransfection of 293T cells (6 × 106 in 100-mm cell culture dishes) using Lipofectamine 2000 (Invitrogen, Calabasas, CA). A total of 19.5 μg of plasmid in the proportion of 12 μg of lentiviral vector carrying shRNA, 6 μg of packaging plasmid pCMVDR8.2 DVPR (VPR deleted; (23)), and 1.5 μg of pCMV-VSVG were used, and viral supernatant was collected at 48, 72, and 96 h after transfection. Pooled supernatants were concentrated using an Amicon Ultra-15 100K cutoff filter device (Millipore, Billerica, MA). The viral titer of the concentrated supernatant was determined by performing a p24 ELISA kit (Cell Biolabs, Inc. San Diego, CA) to detect the HIV-p24 core protein of the vector.
Generation of stably GDPD5-silenced breast cancer cell
For the transduction of MDA-MB-231 cells, 1×106 cells were plated in 100-mm cell culture dishes. Viral supernatants were centrifuged at 3,000 × g for 20 min at 4°C. Five mL of viral supernatant with 1 mg/mL polybrene (Sigma, St. Louis, MO) was added to the cells for 4 h. This procedure was repeated for three consecutive days. The transduction efficiency was assessed by EGFP expression. Photomicrographs were taken with a Nikon TS100 inverted microscope. GDPD5 mRNA expression in transduced MDA-MB-231-GDPD5-shRNA cells relative to vector control cells was assessed by qRT-PCR as described below. The expression of target RNA was calculated relative to the housekeeping gene hypoxanthine phosphoribosyltransferase 1 (HPRT1).
Generation of stably GDPD5-silenced breast cancer tumor xenografts
Approximately 2×106 MDA-MB-231-GDPD5-shRNA cells or empty vector control MDA-MB-231 (MDA-MB-231-vector control) cells in 50 μL HBSS (Mediatech) were inoculated in the upper right thoracic mammary fat pad of female athymic nude mice. Tumor volumes were measured weekly by caliper measurements. All surgical procedures and animal handling was performed in accordance with protocols approved by the Johns Hopkins University Institutional Animal Care and Use Committee, and conformed to the Guide for the Care and Use of Laboratory Animals published by the NIH.
RNA isolation, cDNA synthesis, and quantitative RT-PCR
MDA-MB-231-GDPD5-shRNA cells and empty vector control (MDA-MB-231-vector control) cells were cultured in 75-mm cell culture dishes in triplicate. When the cells were 70% confluent, they were washed quickly with DEPC-treated water twice, followed by RNA isolation using QIAshredder and RNeasy mini kit (Qiagen, Valencia, CA). cDNA was synthesized using qScript cDNA supermix (Quanta Biosciences, Gaithersburg, MD). 2μl of 1:10 diluted cDNA was used for real-time PCR performed in an iCycler IQTM real-time PCR machine (Bio-Rad). The GDPD5 specific primers used for PCR were as follows: sense strand 5’-CTACAACCCTGAGCAGAT-3’; anti-sense strand 5’-AACATACGGAGAGCACAT-3’. Normalization was performed with respect to the housekeeping genes hypoxanthine phosphoribosyltransferase 1 (HPRT1). The Primers for HPRT1 were as follows: sense strand 5’-CCTGGCGTCGTGATTAGTGATG-3’, antisense strand 5’-CAGAGGGCTACAATGTGATGGC-3’. The acquired data were analyzed in MS-Excel using the ΔΔct method. The relative fold change in gene expression of MDA-MB-231-GDPD5-shRNA cells was calculated based on the threshold cycle (ct) as R=2-Δ(Δct), where Δct=ct( GDPD5 - hPRT) and ΔΔct= Δct(GDPD5MDA-MB-231-GDPD5-shRNA)- Δct(GDPD5MDA-MB-231-vector control).
Dual-phase extraction of cells and tumors
For cell extracts, cells were grown to 70% confluence in standard cell culture medium, which contained 21.43 μmol/L of choline and no ethanolamine. Approximately 108 cells per extract were harvested by trypsinization with 0.25 % Trypsin-EDTA Solution (Sigma-Aldrich). Cells were counted in a dilution of trypan blue as a vital stain for quantification and subjected to dual-phase extraction as described below. For tumor extracts obtained directly after the in vivo measurements, mice were sacrificed and tumors were removed immediately. The entire tumor was quickly freeze-clamped, pulverized by grinding over liquid N2, and homogenized with a tissue homogenizer in 4 mL of ice-cold methanol. Both lipid and water-soluble cell and tumor extract fractions were obtained using a dual-phase extraction method based on methanol/chloroform/water (1:1:1; v/v/v) as previously described (18,24).
High-resolution 31P MRS (31P HR MRS) studies
The water-soluble and lipid fractions were dissolved in deuterated solvents containing 0.24×10-6 mol 3-(trimethylsilyl)propionic-2,2,3,3-d4 acid (TSP; Sigma-Aldrich) and 6.0×10-6 mol phenylphosphonic acid (PPA; Sigma-Aldrich) as internal concentration and chemical shift standards. 31P HR-MRS was performed on a Bruker Avance 500 (11.7 T) spectrometer (Bruker BioSpin Corp.) using a 10-mm broadband probe tuned to the 31P frequency. The MR spectra were acquired using the following acquisition parameters: 90° flip angle for tumor extracts, 60° flip angle for cell extracts, 10,162 Hz sweep width, 0.8 s time domain, 8K data points, repetition time of 3 seconds for cell extracts and 15 seconds for tumor extracts, 2000 averages for cell extracts, and 300 averages for tumor extracts. All 31P HR MR spectra were processed using the MestReC 4.9.9.6 software (MestReLab Research). Lorentzian lines were fitted to the signals of PPA, PE, PC, GPE, and GPC. The areas under the curve were corrected for differences in T1 relaxation time and possible saturation effects owing to the relatively large flip angle. Metabolite levels were normalized to cell number or tumor weight. T1 relaxation times of the metabolites were measured with a progressive saturation series in phantom solutions of PC, PE, GPC in D2O.
Non-invasive in vivo 31P MRS studies
In vivo 31P MRS was performed on a 9.4T Bruker Biospec spectrometer. A double tuned (1H and 31P frequency) solenoid coil with an inner diameter of 12 mm was used (MRcoils BV, Drunen, The Netherlands). Mice were anesthetized by breathing a mixture of air and isoflurane (2%) through a nose cone. The tumor was hanging into the coil while the animal lay on a cradle with an opening for the coil. Body temperature was maintained during the experiment by using a blanket with circulating warm water. Breathing rate was monitored throughout all MR measurements with a movement sensor, which was attached at the mouse’s abdomen. A 3D RARE image was acquired with the following parameters: echo time (TE) of 7.2ms, repetition time (TR) of 500ms, RARE factor of 4, flip angle of 900, field of view (FOV) of 1cm×1cm×1cm, 64 phase encode steps (64×64×64), and number of averages (NA) of 4. Total acquisition time was 13 minutes. Shimming of 1st and 2nd order B0 gradient fields was performed manually by iteratively assessing the water line width obtained from the entire tumor. Non-localized 31P MR spectra were acquired with an adiabatic excitation (BIR4 45°, 200μs, 120ppm band width), repetition time of 1 second, and 1800 averages. A saturation slab (adiabatic full passage pulse driven at half the amplitude to achieve excitation with fully dispersed phase (25)) covered the mouse body to eliminate signals from muscles in the body. The combination of the drop off of 31P radiofrequency (RF) field strength perpendicular to the solenoid coil in which the tumor was placed, and the saturation slab positioned on the mouse body ensured that we acquired signal from tumor tissue only.
Analysis of in vivo 31P MRS data
Lorentzian lines were fitted to the 31P MRS data using JMRUI 4.0 software (26) and the AMARES algorithm (27). The resonance of phosphocreatine (PCr) was set to 0 ppm. In the fitting, the line widths of phosphomonoesters and phosphodiesters were constrained to the line width of PCr, and the frequency difference between PC and phosphoethanolamine (PE) and between GPC and glycerophosphoethanolamine (GPE) was fixed to 100 Hz. Metabolite levels were quantified as ratios with respect to β-nucleotide triphosphate (NTP). Metabolite levels were corrected for differences in T1 relaxation. Metabolite T1 values were measured in vivo by progressive saturation series in normal MDA-MB-231 tumors (n=4).
Statistical analysis of all data
A two-sided t-test assuming unequal variances between the MDA-MB-231-GDPD5-shRNA group (N=5) and the vector control group (N=5) was performed to test for statistically significant differences.
Results
The knockdown of GDPD5 was verified by qRT-PCR analysis of the mRNA expression of GDPD5. MDA-MB-231-GDPD5-shRNA cells had significantly decreased gene expression levels of GDPD5 mRNA compared to the MDA-MB-231-vector control cells (Fig. 2a). In 31P HR MR spectra of cell extracts (n=3), no signals for GPE and GPC could be detected in MDA-MB-231-vector control cells, whereas MDA-MB-231-GDPD5-shRNA cells clearly displayed GPC and GPE signals at a concentration of 0.057 fmol per cell and 0.048 fmol per cell respectively, resulting in an increase of both upon GDPD5-silencing (p=0.18 for GPC, p=0.24 for GPE). We also observed a borderline significant decrease in PC levels (p=0.06) in MDA-MB-231-GDPD5-shRNA cells compared to vector controls.
Figure 2.
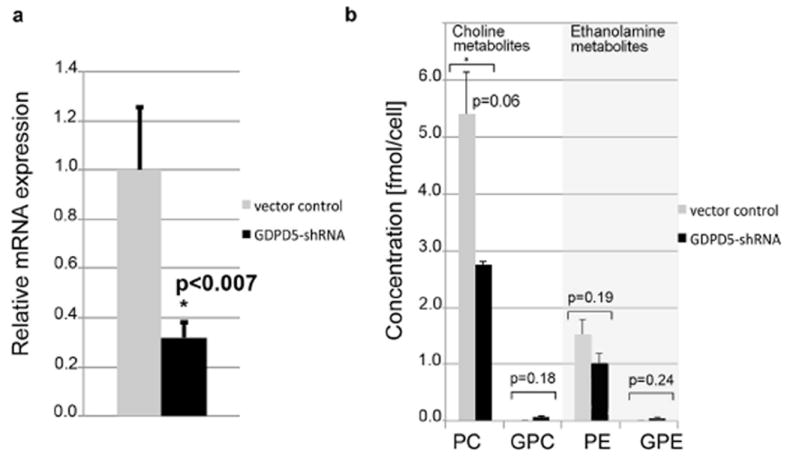
(a) qRT-PCR demonstrating significant GDPD5 knockdown in MDA-MB-231-GDPD5-shRNA cells compared to vector controls. MDA-MB-231 cells containing empty vector, which lacked any shRNA expression served as vector controls. (b) High resolution 31P MRS of MDA-MB-231-GDPD5-shRNA and MDA-MB-231 vector control cells showed a trend towards decreased PC and PE levels in MDA-MB-231-GDPD5-shRNA cells compared to vector controls. GPE was not detectable in MDA-MB-231 vector control cells. Average and standard error of 3 cell extracts are shown for each group.
In vivo 31P MR measurements detected PME and PDE signals, as well as inorganic phosphate (Pi), phosphocreatine (PCr), and α-, β-, and γ-NTP in both the MDA-MB-231-GDPD5-shRNA and MDA-MB-231-vector control tumors (Fig. 3a). Since the magnetic susceptibilities of water and fat are different from each other, the frequently found water-lipid transitions in heterogeneous tumor tissue lead to broad line widths in in vivo MR spectra. The average and standard deviation of the line width of PCr was 94 ± 8 Hz and 98 ± 16 Hz respectively in MDA-MB-231-GDPD5-shRNA and MDA-MB-231-vector control tumors. However, it was still possible to distinguish the individual signals of PE, PC, GPE, and GPC in the in vivo 31P MR spectra. We detected a trend towards higher ratios of GPC/β-NTP and PE/β-NTP in the MDA-MB-231-GDPD5-shRNA tumors, which did not reach statistical significance (Fig. 3b).
Figure 3.
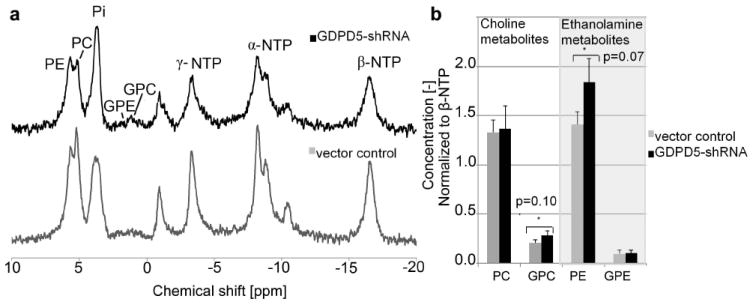
In vivo data of MDA-MB-231-GDPD5-shRNA and vector control tumor xenografts. (a) In vivo 31P MR spectra at 9.4 Tesla. (b) Quantified metabolite levels of PE, PC, GPE and GPC. Average and standard error of 6 tumors per group is shown. Metabolite levels were normalized to β-NTP.
The PME and PDE signals were clearly separated in the 31P HR MR spectra of tumor extracts (Fig. 4a). When normalized to tumor weight and the internal reference PPA, significantly increased levels of PE were detected in MDA-MB-231-GDPD5-shRNA tumors as compared to vector controls (Fig. 4b). This confirmed the in vivo findings of elevated PE and GPC levels in MDA-MB-231-GDPD5-shRNA tumors compared to vector control tumors.
Figure 4.
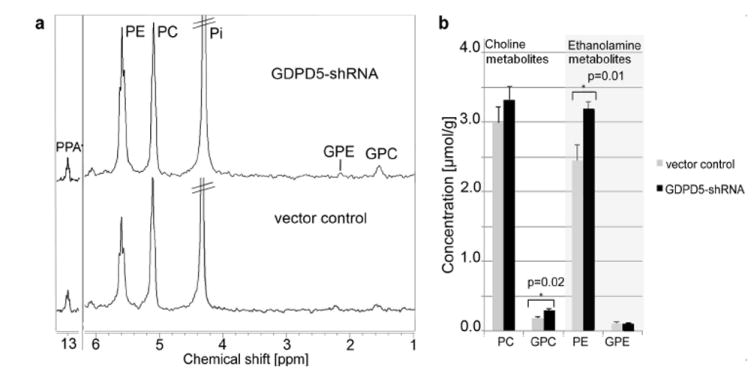
(a) Ex vivo 31P HR MRS of water-soluble extract fractions obtained from MDA-MB-231-GDPD5-shRNA and vector control tumor xenografts. (b) Corresponding metabolite quantification from MDA-MB-231-GDPD5-shRNA and vector control tumor (average and standard error, n=6 each).
Discussion
Stable silencing of GDPD5 in MDA-MB-231 breast cancer cells resulted in higher levels of GPC and PE in extracts of MDA-MB-231-GDPD5-shRNA tumors compared to MDA-MB-231-vector control tumors. The trends towards increased PE and GPC levels in MDA-MB-231-GDPD5-shRNA tumors xenografts relative to vector controls measured in vivo were consistent with the significant increases observed in tumor extracts using ex vivo 31P HR MRS. Ex vivo 31P HR MRS provided a good method to validate in vivo 31P MRS results. Such changes in phospholipid metabolite levels observed by in vivo 31P MRS might be large enough to be clinically relevant.
Silencing of GDPD5, which is an enzyme with GPC-PDE activity (28,29), was expected to increase GPC levels. This was indeed the case as confirmed by a higher GPC/β-NTP ratio in MDA-MB-231-GDPD5-shRNA tumors and a higher GPC level in MDA-MB-231-GDPD5-shRNA tumor extracts (see Fig. 3b and 4b). We also observed an increased GPC level in the MDA-MB-231-GDPD5-shRNA cells, which did not reach statistical significance (Fig. 2). The increase in GPC was not as drastic as expected when considering the good efficiency of the GDPD5 knockdown, which resulted in about 68% reduction in GDPD5 mRNA levels in stably transduced MDA-MB-231-GDPD5-shRNA cells. However, there is a possibility that 32% of GDPD5 is still sufficient to hydrolyze the majority of the produced GPC. It is likely that GDPD5 is not the only GPC-PDE enzyme responsible for the breakdown of GPC. For example, it was recently shown that GDPD6, i.e. EDI3, is also responsible for breaking down GPC in breast cancer cells (30). This was demonstrated by transient siRNA silencing of GDPD6 in MDA-MB-231 breast cancer cells, which significantly increased the GPC/PC ratio by a factor of 3.3 (30). Our GDPD5-knockdown data are from stably silenced cells, in which shRNA against GDPD5 is continuously produced in cells that are able to survive. This approach results in adaptations in stably silenced cells that cannot be observed during transient siRNA knockdown, which is why compensatory mechanisms in phospholipid metabolism might play an important role in our stably GDPD5-silenced cells, which nevertheless displayed an increase of GPC levels from undetectable to 0.057 fmol/cell. GDPD isozymes such as GDPD1, 2, 3, 4, or 6 may partially compensate for the reduction in GDPD5 and, to some extent, counteract the GPC increase caused by GDPD5 silencing. It is also possible that breast cancer cells have other compensatory mechanisms that are counteracting the knockdown of GDPD5. The significant increase in the level of PE/β-NTP in the MDA-MB-231-GDPD5-shRNA tumors compared to vector control tumors could be due to ethanolamine phosphorylation by choline and/or ethanolamine kinases as compensatory mechanism for the reduced PC levels in these cells. However, the absolute increase in GPC by 0.057 fmol/cell following stable GPDP5 silencing is relatively small compared to the total cellular PC content of 2.75 fmol/cell. Unaltered PC levels in GDPD5-silenced versus vector control tumors in vivo, which in turn resulted in unaltered PC/GPC ratios in vivo, do not necessarily indicate that GDPD5-silencing does not influence in vivo tumor aggressiveness. An increased PC/GPC ratio was shown to be associated with elevated ovarian and breast cancer cell aggressiveness (6,7). However, more recent studies have shown that patient-derived animal models of basal-like breast cancer and tumor tissue from patients with triple-negative breast cancer contained high GPC levels and, as a consequence, relatively low PC/GPC levels (31,32). This emphasizes the fact that GPC levels in breast cancers are not yet well understood and require further investigation. In addition, the unaltered PC/GPC ratios in our GDPD5-silenced tumor xenografts may also be due to compensatory mechanisms in these stably silenced cell lines that where selected for survival.
In cultured cells, PC decreased as a consequence of GDPD5 silencing, which was not the case when growing the same cells as orthotopic tumor xenografts. This finding indicates that the tumor microenvironment, e.g. stromal cells (33), tumor pH (34), or hypoxia (35), may have increased tumor PC and PE levels by modulating enzymes in choline phospholipid metabolism such as choline kinase alpha (35). Choline kinase alpha and beta both have the ability to also use ethanolamine as a substrate to produce PE (13). It is well known that phospholipid metabolite levels in cells are also controlled by culture conditions (36-39). Our cell culture experiments were performed with MDA-MB-231 cells growing in logarithmic growth phase with 21.43 μM choline and no ethanolamine. However, since mammalian cells are unable to synthesize ethanolamine de novo, ethanolamine must be provided from the diet or from degradation of PtdEth made by the phosphatidylserine decarboxylase pathway (40), which could explain the elevated PE levels in tumor xenografts compared to the same cells in cell culture.
In vivo 31P MRS at 9.4T enabled the assessment of the individual phosphorylated metabolites in choline and ethanolamine phospholipid metabolism. Shimming of the tumor tissue was challenging because frequent water-lipid transitions in the tumor caused changes in magnetic susceptibility on a microscopic scale. These susceptibility changes on a microscopic scale led to an inhomogeneous B0 field that could not be corrected by shimming, where smooth first, second, and third order magnetic field gradients were applied to compensate for the variations in B0. Therefore, the line width obtained in in vivo MRS of the tumors in our study likely reflected the heterogeneity of the tumor tissue. The line width was broader in tumors with more necrotic tissue.
Tumor heterogeneity is commonly found in different types of tumors and could result from genetic as well as microenvironmental differences within the tumor tissue (41). Therefore, a next step would be to use 31P chemical shift imaging (CSI) to investigate the spatial distribution of phospholipid metabolites inside the tumor tissue. So far, mostly 1H CSI has been used in tumor models to study total choline levels (tCho = PC+GPC+Choline) (42). In general, higher tCho levels have been associated with increased breast cancer aggressiveness (6,43). Treatment response to chemotherapy (44) or targeted anticancer therapies that disrupt oncogenic signaling pathways (45-49) can be detected by means of a decrease in tCho level within a short time period after treatment, e.g. after 24 hours of treatment in some cases (50). However, some novel molecular anticancer treatments that inhibit Hsp90 (51) or histone deacetylase (HDAC) (52) were shown to increase tCho, emphasizing the fact that the molecular pathways that lead to a detectable change in tCho need to be investigated for every drug for which tCho will be monitored as a biomarker of treatment response. With the use of 31P MRS, a change in tCho can be narrowed down to changes in PC and/or GPC, which is molecularly specific. In regard to the question if GDPD5 could be a potential anticancer target in breast cancer cells, it would be premature to draw any conclusions based on the presented data on stable GDPD5 silencing. Further molecular studies are ongoing in our laboratory to answer this question.
In conclusion, silencing of GDPD5 increased the PE and GPC levels in MDA-MB-231-GDPD5-shRNA tumors, indicating that GDPD5 is an enzyme with GPC-PDE activity whose expression is important in choline and ethanolamine phospholipid metabolism of breast cancer, which can be studied by 31P MRS in vivo. Phosphorus HR MRS of tumor extracts can be used to validate in vivo 31P MRS measurements and may provide prior knowledge for in vivo data analysis. Phosphorus MRS is a powerful tool for studying cancer metabolism and can potentially be used for monitoring novel therapies that target enzymes in choline/ethanolamine phospholipid metabolism
Acknowledgments
We thank Drs. Artemov and Chacko for help with the experimental set-up of the MR scanners.
Grant support
We gratefully acknowledge financial support from the National Institutes of Health (NIH) grant R01 CA134695 and the Niels Stensen Foundation.
Alphabetical list of nonstandard abbreviations
- B1
applied magnetic field
- BIR
B1 independent rotation
- cDNA
complementary deoxyribonucleic acid
- ck
choline kinas CTP: phosphocholine cytidylyltransferase
- DG-CTP
diacylglycerol choline phosphotransferase
- DG-ETP
diacylglycerol ethanolamine phosphotransferase
- EDTA
ededic acid (ethyl-enediaminetetraacetic acid)
- EGFP
enhanced green fluorescent protein
- ek
ethanolamine kinase
- FBS
fetal bovine serum
- FOV
field of view
- GPC
glycerophosphocholine
- GPC-PDE
glycerophocholine phosphodiesterase
- GDPD
glycerophosphodiester phosphodiesterase domain containing
- GPE
glycerophosphoethanolamine
- HPRT 1
hypoxanthine phosphoribosyltransferase
- HR-MRS
high resolution magnetic resonance spectroscopy
- HSP
heat shock protein
- lyso-PL
lysophospholipase
- MR
Magnetic Resonance
- MRI
Magnetic Resonance Imaging
- MRS
Magnetic Resonance Spectroscopy
- NMR
Nuclear Magnetic Resonance
- NA
number of averages
- NTP
nucleotide triphosphate
- PC
phosphocholine
- PCr
phosphocreatine
- PCR
polymerase chain reaction
- PDE
phosphodiesters
- PE
phosphoethanolamine
- pGK
phosphoglycerate kinase
- Pi
inorganic phosphate
- PME
phosphomonoesters
- PLC
phospholipase C
- PLC
phospholipase D
- PLA2
phospholipase A2
- ppm
parts per million
- PtdEth
phosphatidyl ethanolamine
- qRT-PCR
quantitative Reverse Transcription PCR
- RARE
rapid acquisition with relaxation enhancement
- RF
radiofrequency
- RNA
Ribonucleic acid
- ROI
region of interest
- RPMI
Roswell Park Memorial Institute
- shRNA
stably silenced Ribonucleic acid
- SNR
signal to noise ratio
- T
tesla
- tCho
total choline
- TE
echo time
- TR
repetition time
Footnotes
Conflicts of interest: We have no conflicts of interest regarding the work described in this paper.
References
- 1.Glunde K, Bhujwalla ZM, Ronen SM. Choline metabolism in malignant transformation. Nature reviews Cancer. 2011;11(12):835–848. doi: 10.1038/nrc3162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Podo F, Carpinelli G, Di Vito M, Giannini M, Proietti E, Fiers W, Gresser I, Belardelli F. Nuclear magnetic resonance analysis of tumor necrosis factor-induced alterations of phospholipid metabolites and pH in Friend leukemia cell tumors and fibrosarcomas in mice. Cancer research. 1987;47(24 Pt 1):6481–6489. [PubMed] [Google Scholar]
- 3.Negendank W. Studies of human tumors by MRS: a review. NMR Biomed. 1992;5(5):303–324. doi: 10.1002/nbm.1940050518. [DOI] [PubMed] [Google Scholar]
- 4.Podo F. Tumour phospholipid metabolism. NMR Biomed. 1999;12(7):413–439. doi: 10.1002/(sici)1099-1492(199911)12:7<413::aid-nbm587>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- 5.Arias-Mendoza F, Payne GS, Zakian KL, Schwarz AJ, Stubbs M, Stoyanova R, Ballon D, Howe FA, Koutcher JA, Leach MO, Griffiths JR, Heerschap A, Glickson JD, Nelson SJ, Evelhoch JL, Charles HC, Brown TR. In vivo 31P MR spectral patterns and reproducibility in cancer patients studied in a multi-institutional trial. NMR Biomed. 2006;19(4):504–512. doi: 10.1002/nbm.1057. [DOI] [PubMed] [Google Scholar]
- 6.Aboagye EO, Bhujwalla ZM. Malignant transformation alters membrane choline phospholipid metabolism of human mammary epithelial cells. Cancer research. 1999;59(1):80–84. [PubMed] [Google Scholar]
- 7.Iorio E, Mezzanzanica D, Alberti P, Spadaro F, Ramoni C, D’Ascenzo S, Millimaggi D, Pavan A, Dolo V, Canevari S, Podo F. Alterations of choline phospholipid metabolism in ovarian tumor progression. Cancer research. 2005;65(20):9369–9376. doi: 10.1158/0008-5472.CAN-05-1146. [DOI] [PubMed] [Google Scholar]
- 8.Podo F, Canevari S, Canese R, Pisanu ME, Ricci A, Iorio E. MR evaluation of response to targeted treatment in cancer cells. NMR Biomed. 2011 Jul;24(6):648–672. doi: 10.1002/nbm.1658. [DOI] [PubMed] [Google Scholar]
- 9.Bathen TF, Jensen LR, Sitter B, Fjosne HE, Halgunset J, Axelson DE, Gribbestad IS, Lundgren S. MR-determined metabolic phenotype of breast cancer in prediction of lymphatic spread, grade, and hormone status. Breast Cancer Res Treat. 2007;104(2):181–189. doi: 10.1007/s10549-006-9400-z. [DOI] [PubMed] [Google Scholar]
- 10.Krishnamachary B, Glunde K, Wildes F, Mori N, Takagi T, Raman V, Bhujwalla ZM. Noninvasive detection of lentiviral-mediated choline kinase targeting in a human breast cancer xenograft. Cancer research. 2009;69(8):3464–3471. doi: 10.1158/0008-5472.CAN-08-4120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vance JE, Vance DE. Phospholipid biosynthesis in mammalian cells. Biochem Cell Biol. 2004;82(1):113–128. doi: 10.1139/o03-073. [DOI] [PubMed] [Google Scholar]
- 12.Menon AK, Stevens VL. Phosphatidylethanolamine is the donor of the ethanolamine residue linking a glycosylphosphatidylinositol anchor to protein. J Biol Chem. 1992;267(22):15277–15280. [PubMed] [Google Scholar]
- 13.Gallego-Ortega D, Ramirez de Molina A, Ramos MA, Valdes-Mora F, Barderas MG, Sarmentero-Estrada J, Lacal JC. Differential role of human choline kinase alpha and beta enzymes in lipid metabolism: implications in cancer onset and treatment. PLoS One. 2009;4(11):e7819. doi: 10.1371/journal.pone.0007819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gribbestad IS, Sitter B, Lundgren S, Krane J, Axelson D. Metabolite composition in breast tumors examined by proton nuclear magnetic resonance spectroscopy. Anticancer Res. 1999;19(3A):1737–1746. [PubMed] [Google Scholar]
- 15.Klomp DW, van de Bank BL, Raaijmakers A, Korteweg MA, Possanzini C, Boer VO, van de Berg CA, van de Bosch MA, Luijten PR. (31) P MRSI and (1) H MRS at 7 T: initial results in human breast cancer. NMR Biomed. 2011;24 doi: 10.1002/nbm.1696. [DOI] [PubMed] [Google Scholar]
- 16.Wijnen JP, van der Kemp WJ, Luttje MP, Korteweg MA, Luijten PR, Klomp DW. Quantitative 31P magnetic resonance spectroscopy of the human breast at 7 T. Magnetic resonance in medicine : official journal of the Society of Magnetic Resonance in Medicine / Society of Magnetic Resonance in Medicine. 2012;68(2):339–348. doi: 10.1002/mrm.23249. [DOI] [PubMed] [Google Scholar]
- 17.Lacal JC. Choline kinase: a novel target for antitumor drugs. IDrugs. 2001;4(4):419–426. [PubMed] [Google Scholar]
- 18.Glunde K, Raman V, Mori N, Bhujwalla ZM. RNA interference-mediated choline kinase suppression in breast cancer cells induces differentiation and reduces proliferation. Cancer research. 2005;65(23):11034–11043. doi: 10.1158/0008-5472.CAN-05-1807. [DOI] [PubMed] [Google Scholar]
- 19.TCDPharma. 2012 http://currentcancer.com/tcd-announces-first-patient-treated-in-its-tcd-717-phase-i-trial.html.
- 20.Dopkens M, Greenwood T, Vesuna F, Raman V, Leibfritz D, Glunde K. GDPD5 inhibition alters the choline phospholipid metabolite profile of breast cancer cells toward a less malignant metabolic profile. Biomedical Spectroscopy and Imaging. 2012;1:3–15. [Google Scholar]
- 21.Cao MD, Dopkens M, Krishnamachary B, Vesuna F, Gadiya MM, Lonning PE, Bhujwalla ZM, Gribbestad IS, Glunde K. Glycerophosphodiester phosphodiesterase domain containing 5 (GDPD5) expression correlates with malignant choline phospholipid metabolite profiles in human breast cancer. NMR Biomed. 2012 doi: 10.1002/nbm.2766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zheng JY, Chen D, Chan J, Yu D, Ko E, Pang S. Regression of prostate cancer xenografts by a lentiviral vector specifically expressing diphtheria toxin A. Cancer gene therapy. 2003;10(10):764–770. doi: 10.1038/sj.cgt.7700629. [DOI] [PubMed] [Google Scholar]
- 23.An DS, Morizono K, Li QX, Mao SH, Lu S, Chen IS. An inducible human immunodeficiency virus type 1 (HIV-1) vector which effectively suppresses HIV-1 replication. J Virol. 1999;73(9):7671–7677. doi: 10.1128/jvi.73.9.7671-7677.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tyagi RK, Azrad A, Degani H, Salomon Y. Simultaneous extraction of cellular lipids and water-soluble metabolites: evaluation by NMR spectroscopy. Magnetic resonance in medicine : official journal of the Society of Magnetic Resonance in Medicine / Society of Magnetic Resonance in Medicine. 1996;35(2):194–200. doi: 10.1002/mrm.1910350210. [DOI] [PubMed] [Google Scholar]
- 25.Choi IY, Tkac I, Gruetter R. Single-shot, three-dimensional “non-echo” localization method for in vivo NMR spectroscopy. Magn Reson Med. 2000;44(3):387–394. doi: 10.1002/1522-2594(200009)44:3<387::aid-mrm8>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- 26.Naressi A, Couturier C, Devos JM, Janssen M, Mangeat C, de Beer R, Graveron-Demilly D. Java-based graphical user interface for the MRUI quantitation package. Magn Reson Mater Phys. 2001;12(2-3):141–152. doi: 10.1007/BF02668096. [DOI] [PubMed] [Google Scholar]
- 27.Vanhamme L, van den Boogaart A, van Huffel S. Improved method for accurate and efficient quantification of MRS data with use of prior knowledge. J Magn Reson. 1997;129(1):35–43. doi: 10.1006/jmre.1997.1244. [DOI] [PubMed] [Google Scholar]
- 28.Gallazzini M, Ferraris JD, Burg MB. GDPD5 is a glycerophosphocholine phosphodiesterase that osmotically regulates the osmoprotective organic osmolyte GPC. Proc Natl Acad Sci U S A. 2008;105(31):11026–11031. doi: 10.1073/pnas.0805496105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gallazzini M, Heussler GE, Kunin M, Izumi Y, Burg MB, Ferraris JD. High NaCl-induced activation of CDK5 increases phosphorylation of the osmoprotective transcription factor TonEBP/OREBP at threonine 135, which contributes to its rapid nuclear localization. Mol Biol Cell. 2011;22(5):703–714. doi: 10.1091/mbc.E10-08-0681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stewart JD, Marchan R, Lesjak MS, Lambert J, Hergenroeder R, Ellis JK, Lau CH, Keun HC, Schmitz G, Schiller J, Eibisch M, Hedberg C, Waldmann H, Lausch E, Tanner B, Sehouli J, Sagemueller J, Staude H, Steiner E, Hengstler JG. Choline-releasing glycerophosphodiesterase EDI3 drives tumor cell migration and metastasis. Proc Natl Acad Sci U S A. 2012;109(21):8155–8160. doi: 10.1073/pnas.1117654109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Moestue SA, Borgan E, Huuse EM, Lindholm EM, Sitter B, Borresen-Dale AL, Engebraaten O, Maelandsmo GM, Gribbestad IS. Distinct choline metabolic profiles are associated with differences in gene expression for basal-like and luminal-like breast cancer xenograft models. BMC cancer. 2010;10:433. doi: 10.1186/1471-2407-10-433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Moestue SA, Giskeodegard GF, Cao MD, Bathen TF, Gribbestad IS. Glycerophosphocholine (GPC) is a poorly understood biomarker in breast cancer. Proc Natl Acad Sci U S A. 2012;109(38):E2506. doi: 10.1073/pnas.1208226109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mao Y, Keller ET, Garfield DH, Shen K, Wang J. Stromal cells in tumor microenvironment and breast cancer. Cancer metastasis reviews. 2013;32(1-2):303–315. doi: 10.1007/s10555-012-9415-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gillies RJ, Raghunand N, Garcia-Martin ML, Gatenby RA. pH imaging. A review of pH measurement methods and applications in cancers. IEEE Eng Med Biol Mag. 2004;23(5):57–64. doi: 10.1109/memb.2004.1360409. [DOI] [PubMed] [Google Scholar]
- 35.Glunde K, Shah T, Winnard PT, Jr, Raman V, Takagi T, Vesuna F, Artemov D, Bhujwalla ZM. Hypoxia regulates choline kinase expression through hypoxia-inducible factor-1 alpha signaling in a human prostate cancer model. Cancer research. 2008;68(1):172–180. doi: 10.1158/0008-5472.CAN-07-2678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shedd SF, Lutz NW, Hull WE. The influence of medium formulation on phosphomonoester and UDP-hexose levels in cultured human colon tumor cells as observed by 31P NMR spectroscopy. NMR Biomed. 1993;6(4):254–263. doi: 10.1002/nbm.1940060405. [DOI] [PubMed] [Google Scholar]
- 37.Franks SE, Kuesel AC, Lutz NW, Hull WE. 31P MRS of human tumor cells: effects of culture media and conditions on phospholipid metabolite concentrations. Anticancer Res. 1996;16(3B):1365–1374. [PubMed] [Google Scholar]
- 38.Delikatny EJ, Lander CM, Jeitner TM, Hancock R, Mountford CE. Modulation of MR-visible mobile lipid levels by cell culture conditions and correlations with chemotactic response. International journal of cancer Journal international du cancer. 1996;65(2):238–245. doi: 10.1002/(SICI)1097-0215(19960117)65:2<238::AID-IJC18>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 39.Daly PF, Lyon RC, Faustino PJ, Cohen JS. Phospholipid metabolism in cancer cells monitored by 31P NMR spectroscopy. J Biol Chem. 1987;262(31):14875–14878. [PubMed] [Google Scholar]
- 40.Vance JE, Tasseva G. Formation and function of phosphatidylserine and phosphatidylethanolamine in mammalian cells. Biochim Biophys Acta. 2013;1831(3):543–554. doi: 10.1016/j.bbalip.2012.08.016. [DOI] [PubMed] [Google Scholar]
- 41.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 42.Jiang L, Greenwood TR, Artemov D, Raman V, Winnard PT, Jr, Heeren RM, Bhujwalla ZM, Glunde K. Localized hypoxia results in spatially heterogeneous metabolic signatures in breast tumor models. Neoplasia. 2012;14(8):732–741. doi: 10.1593/neo.12858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bolan PJ, Meisamy S, Baker EH, Lin J, Emory T, Nelson M, Everson LI, Yee D, Garwood M. In vivo quantification of choline compounds in the breast with 1H MR spectroscopy. Magn Reson Med. 2003;50(6):1134–1143. doi: 10.1002/mrm.10654. [DOI] [PubMed] [Google Scholar]
- 44.Sharma U, Baek HM, Su MY, Jagannathan NR. In vivo 1H MRS in the assessment of the therapeutic response of breast cancer patients. NMR Biomed. 2011;24(6):700–711. doi: 10.1002/nbm.1654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ronen SM, Jackson LE, Beloueche M, Leach MO. Magnetic resonance detects changes in phosphocholine associated with Ras activation and inhibition in NIH 3T3 cells. British journal of cancer. 2001;84(5):691–696. doi: 10.1054/bjoc.2000.1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Beloueche-Babari M, Jackson LE, Al-Saffar NM, Eccles SA, Raynaud FI, Workman P, Leach MO, Ronen SM. Identification of magnetic resonance detectable metabolic changes associated with inhibition of phosphoinositide 3-kinase signaling in human breast cancer cells. Molecular cancer therapeutics. 2006;5(1):187–196. doi: 10.1158/1535-7163.MCT-03-0220. [DOI] [PubMed] [Google Scholar]
- 47.Beloueche-Babari M, Jackson LE, Al-Saffar NM, Workman P, Leach MO, Ronen SM. Magnetic resonance spectroscopy monitoring of mitogen-activated protein kinase signaling inhibition. Cancer research. 2005;65(8):3356–3363. doi: 10.1158/10.1158/0008-5472.CAN-03-2981. [DOI] [PubMed] [Google Scholar]
- 48.Al-Saffar NM, Troy H, Ramirez de Molina A, Jackson LE, Madhu B, Griffiths JR, Leach MO, Workman P, Lacal JC, Judson IR, Chung YL. Noninvasive magnetic resonance spectroscopic pharmacodynamic markers of the choline kinase inhibitor MN58b in human carcinoma models. Cancer research. 2006;66(1):427–434. doi: 10.1158/0008-5472.CAN-05-1338. [DOI] [PubMed] [Google Scholar]
- 49.Su JS, Woods SM, Ronen SM. Metabolic consequences of treatment with AKT inhibitor perifosine in breast cancer cells. NMR Biomed. 2012;25(2):379–388. doi: 10.1002/nbm.1764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Meisamy S, Bolan PJ, Baker EH, Bliss RL, Gulbahce E, Everson LI, Nelson MT, Emory TH, Tuttle TM, Yee D, Garwood M. Neoadjuvant chemotherapy of locally advanced breast cancer: predicting response with in vivo (1)H MR spectroscopy--a pilot study at 4 T. Radiology. 2004;233(2):424–431. doi: 10.1148/radiol.2332031285. [DOI] [PubMed] [Google Scholar]
- 51.Brandes AH, Ward CS, Ronen SM. 17-allyamino-17-demethoxygeldanamycin treatment results in a magnetic resonance spectroscopy-detectable elevation in choline-containing metabolites associated with increased expression of choline transporter SLC44A1 and phospholipase A2. Breast cancer research : BCR. 2010;12(5):R84. doi: 10.1186/bcr2729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ward CS, Eriksson P, Izquierdo-Garcia JL, Brandes AH, Ronen SM. HDAC Inhibition Induces Increased Choline Uptake and Elevated Phosphocholine Levels in MCF7 Breast Cancer Cells. PLoS One. 2013;8(4):e62610. doi: 10.1371/journal.pone.0062610. [DOI] [PMC free article] [PubMed] [Google Scholar]