

Abstract
Purpose of review
There remains a dire need for therapies that impact the clinical course of patients with idiopathic pulmonary fibrosis (IPF). Indeed, there is a surge of interest in IPF therapeutics, with many candidate agents in various stages of development. Optimal design and implementation of the appropriate prospective clinical trials are essential to demonstrate clinical efficacy of promising drugs for the treatment of IPF. A key element in the success of such clinical trials is the choice of the best endpoint(s) to match the design of the study.
Recent findings
Although the results of many IPF clinical trials have been disappointing, these trials have provided valuable insights into the epidemiology and natural history of the disease and have sparked debate into the best clinical trial designs and endpoints.
Summary
This review will discuss the various clinical trial endpoints that have been used or proposed with a focus on their potential utility, as well as possible pitfalls that investigators should consider in the design of such studies.
Video abstract
http://links.lww.com/COPM/A13
Keywords: clinical trials as topic, pulmonary fibrosis, research design, respiratory function tests
INTRODUCTION
Many randomized, controlled clinical trials in idiopathic pulmonary fibrosis (IPF) have been implemented and completed over the last decade and a half [1]. Unfortunately, most of these trials have ended with the frustration of failure despite early hope for success that was based on biologic plausibility, promising phase 1 and 2 data, and subgroup analyses from phase 3 studies. Since study interpretation is based upon demonstrating a significant difference in the primary endpoint, choosing the correct endpoint is integral to demonstrating success of any therapeutic agent. What constitutes the best primary endpoint has generated much controversy. Indeed, articulate and convincing arguments have been espoused for and against some commonly used and proposed metrics [2–4]. This review will discuss the most commonly used and proposed endpoints with a focus on their utility and potential pitfalls (Table 1).
Table 1.
Clinical trial endpoints (summary of advantages and potential pitfalls)
| Test | Test parameter | Advantages | Potential issues and disadvantages |
| FVC [4,5,▪, 7–9] | FVC categorical change | Greater sensitivity or specificity | Dependent upon predefined change threshold |
| FVC absolute change | Traditional measurement of choice | Greater decrease in FVC required to meet 10% threshold | |
| FVC relative change | ‘Autocorrects’ for baseline FVC | ||
| FVC slope of change | Includes all FVC measurements obtained during course of study (decreases effect of intrinsic variability) | ||
| DLco [13] | DLco raw value (Kco) | Measure of gas exchange | May be difficult to perform |
| Better prognostic indicator than FVC | Somewhat more variable than FVC | ||
| Can be affected by significant cardiac dysfunction | |||
| DLco adjusted for alveolar volume (DL/VA) | May track a disease domain not captured by lung volume measurements | The presence of coexistent emphysema may confound this measurement. Value of serial measurements affected by the variability of individual components | |
| DLco adjusted for blood hemoglobin concentration | Adjusts for factors such as anemia or polycythemia | ||
| 6MWT [14–22] | 6MWT distance | Baseline and change in 6MWT distance correlates with outcome | Change in 6MWT distance may be influenced by other factors |
| 6MWT oxygen saturation profile | May be more clinically relevant than 6MWT distance | Subject to more variability | |
| 6MWT composite | Distance-saturation product incorporates two factors | Not validated for IPF | |
| 6MWT pulse rate recovery | Potentially better prognostic indicator than distance or saturation | ||
| Correlates with coexistent pulmonary hypertension | |||
| Hospitalization [23,24▪▪, 25] | All-cause hospitalization (nonelective) | Validation is lacking | Nonrespiratory events will confound the signal |
| Practice habits can be highly variable | |||
| Hospitalization for a respiratory event | Available data suggest a significant impact on subsequent outcomes | Early events may not reflect effects/benefit of the study drug | |
| Mortality [27,28▪▪, 29] | All-cause | Considered to be a robust endpoint | Longer study duration and more patients required |
| May be better suited for studies of patients with more advanced disease | Can be affected by significant co-morbid conditions | ||
| High cost | |||
| Respiratory | More specific to the effects of the disease | Difficult to adjudicate | |
| HRCT [31,32] | HRCT scoring | Improved imaging and computer-based scoring have increased accuracy | Accuracy, precision, and reproducibility have yet to be determined and validated |
| Questionnaire [33–35] | Patient-reported outcomes | Can capture quality of life, dyspnea, cough, or global burden of disease measures | IPF-specific instruments have not been adequately validated |
| Blood tests [36–39] | Blood biomarkers | Study length may be able to be shortened If disease progression is accurately reflected | Reproducibility and precision as predictors of disease progression have not been validated |
| May be useful to stratify patients | |||
| Composites [30] | (see Table 2) | May enable shorter event-driven studies Global reflection of disease progression | May be compromised if measures are tightly linked to each other (e.g. co-linearity of pulmonary function measures) |
| Components: May be imbalance in importance and occurrence of individual measures. |
6MWT, 6-min walk test; FVC, forced vital capacity; HRCT, high-resolution computed tomography; IPF, idiopathic pulmonary fibrosis; VA, alveolar volume.
Box 1.

no caption available
FORCED VITAL CAPACITY
The forced vital capacity (FVC) has been the most commonly employed and accepted endpoint in clinical trials of IPF to date [5▪]. It has many advantages that include being relatively easy to measure and reproduce. It is also commonly regarded as reflecting the burden of the fibrotic disease process. Although both the baseline FVC as well as FVC change have been shown to be associated with subsequent survival [6–8], it remains controversial as to whether the FVC is, can, or should be regarded as a surrogate for survival [2,3].
The change in the FVC over time is the outcome measure of interest, but how best to evaluate the change remains unresolved. Typically, it has been the mean change in FVC for the patient cohorts that have been reported. However, mandating a categorical or threshold change in FVC has the advantage of reducing any ‘noise’ due to the inherent variability of the test. This has commonly been regarded as approximately 10% and any change that breaches this boundary has previously been regarded as the minimal difference that defines a true change. However, there are data to suggest that even a change as small as 5% is associated with increased mortality [7,8]. Whether an absolute change or a relative change in the FVC provides the best measure of FVC change over time remains unresolved [9]. Whereas the absolute change has been used traditionally, the relative change does ‘autocorrect’ for the baseline FVC. A more recently proposed method for evaluating the change in the FVC is to measure the slope of change [10]. This has the advantage of incorporating all FVC measurements obtained for the duration of the study, rather than evaluating change between two predefined time points. Furthermore, this method may mute the influence of the intrinsic variability of the test. This concept also raises the issue of how often the FVC should be measured during the course of a study. Traditionally, this has been every 3 months, but there are no data to support this as the optimal period. A subsequent consequence is that a number of patients will progress and die within this timeframe without prior documented significant change in their FVC [11]. These ‘missed’ events might also be one of the reasons that FVC has not been universally accepted as an adequate surrogate for mortality.
The results of the recently released ASCEND study of pirfenidone provide support for the use of the FVC as a valid IPF study endpoint and as a surrogate for mortality [12]. Specifically, the ASCEND study demonstrated a positive treatment effect on the rate of change in the FVC in association with a mortality benefit. The significant mortality benefit was confirmed by the prespecified combined analysis of the ASCEND dataset with the two prior CAPACITY (Clinical Studies Assessing Pirfenidone in Idiopathic Pulmonary Fibrosis: Research of Efficacy and Safety Outcomes) studies of pirfenidone [12,13]. The total number of patients required to demonstrate the mortality benefit from the three pooled studies was approximately 1250, which underscores the difficulty and cost of studies with mortality as the primary endpoint.
THE SINGLE BREATH DIFFUSING CAPACITY FOR CARBON MONOXIDE
Although there are data to suggest that the single breath diffusing capacity for carbon monoxide (DLco) performs better as a prognostic indicator than the FVC [6], it is more difficult to measure, requires a breath hold that can be difficult for more symptomatic patients and has greater intrinsic variability. The variability has commonly been recognized as being as high as 15%, which is the threshold that has typically been utilized to signify a significant change. When the DLco has been used in clinical trials, it has mostly been the absolute DLco measure without correction for the alveolar volume. Therefore, the DLco will tend to track with the FVC if it is used uncorrected for lung volumes, which raises the issue of colinearity between these two pulmonary function measurements. Therefore, the Kco value (transfer coefficient for carbon monoxide), which is actually the primary measurement and represents the DLco adjusted for the alveolar volume, might be the better parameter to serve as an endpoint [14].
THE SIX-MINUTE WALK TEST
The 6-min walk test (6MWT) is commonly employed to provide a measure of a patient's functional status. There is a growing body of literature attesting to its use in IPF with the baseline 6MWT distance having been shown to correlate with outcomes [15–18]. The minimally important difference has been examined in numerous studies and defined as anywhere from approximately 24–45 m [19,20]. Therefore, similar to the change in the FVC, it might be better to prespecify a categorical change. Apart from the distance, the oxygen saturation profile, Borg dyspnea score and pulse rate might also provide very useful ancillary information. While patients have been noted to continue to desaturate upon completion of the test, there is increasing recognition that it is the pulse rate recovery (PRR) that imparts important prognostic information [21,22]. The PRR is defined as the difference between the pulse rate at the end of the walk period and after 1 min of rest during the recovery phase. The smaller the pulse rate change, the worse the prognosis, with a cut point of 13 beats per minute proposed as best discriminating outcomes [21]. The PRR has been demonstrated to be a better prognostic indicator than any other 6MWT parameter, and it has also been shown to correlate closely with the presence of underlying pulmonary hypertension [22].
Standardization of the 6MWT has also been an issue of ongoing debate. The optimal method to manage the amount of supplemental oxygen utilized during the course of the walk is uncertain. However, the most important irrefutable concept is that every patient should be walked on the same amount of oxygen throughout the study. Whether there should be a ‘safety net’ of a low saturation as a stop signal or whether patients should be relied upon to halt if they get too symptomatic are also open to debate. Other issues include the role of a ‘training’ 6MWT, the number of baseline tests to perform, and whether it should be the average of the baseline tests or the best result that are used for analysis purposes [23].
HOSPITALIZATION
Hospitalization is undoubtedly a very important patient-related outcome. Not only does hospitalization have mortality implications, but it also has healthcare resource utilization ramifications. A consensus panel has proposed that all-cause nonelective hospitalization should be regarded as one of the only two suitable endpoints for IPF clinical trials, with the other endpoint being mortality itself [2].
Whether a hospitalization endpoint is best represented by all-cause, respiratory-related, acute exacerbation or suspected acute exacerbation events remains unanswered (Brown et al., in preparation) [24▪]. Details that meet criteria for an acute exacerbation versus other respiratory complications as a cause of hospitalization can be very difficult to tease out. Indeed, in the recently completed INPULSIS studies of nintedanib, only about one half of the 69 investigator-reported acute exacerbation events were ultimately deemed to be an acute or suspected acute exacerbation by an independent adjudication committee [10].
There are data to suggest that both all-cause and respiratory hospitalizations are associated with poor subsequent outcomes (Brown et al., in preparation). All-cause hospitalizations may be regarded as a composite between nonrespiratory and respiratory events. It is conceivable that the majority of the events might be nonrespiratory in nature, and a positive signal might be ‘lost’ in the context of this composite. Another issue is that practice habits may vary amongst providers, regions and countries where there are differing thresholds and criteria for hospital admissions.
The time lag from the onset of drug administration to the hospitalization event might also be an important consideration. The beneficial effects of an antifibrotic agent's effects might take months to years to become manifested, and, therefore, an early hospital admission event might not reflect drug activity and may have a greater likelihood of reflecting drug toxicity. Although this would obviously be important to report, this information is best captured as a serious adverse event, rather than as an endpoint.
In summary, whereas hospitalization events may well be a valid endpoint, there are many nuances that need to be considered depending on the distribution of study sites, the nature of the drug and the patient population being studied.
MORTALITY
Mortality is commonly regarded as the most robust endpoint for assessing efficacy in IPF clinical trials. Although mortality has been examined in many studies, a positive benefit has not been demonstrated in any single clinical trial to date. In the only studies that have shown an apparent mortality difference, this has paradoxically been driven by an untoward increased mortality rate in the active treatment arm [25,26]. Most studies of antifibrotic therapies have appropriately targeted patients with mild-to-moderate disease. However, it has recently been demonstrated that the mortality rate in the context of IPF studies is lower than expected with recently described 1- and 2-year mortality rates of 6.6 and 13.7%, respectively [27,28▪▪]. Therefore, mortality studies will, by necessity, be longer in their duration and not provide the opportunity for participants to receive open-label drug based on clinical worsening. Whereas patients might agree to participate when they have early disease and are less symptomatic, the ability to retain patients who deteriorate might become difficult. A significant drop-out rate would have deleterious consequences for the integrity and interpretation of any such mortality study. IPF may have a composite of causes for mortality. It has been estimated that approximately 44% of patients with IPF die from another cause, whereas in the context of clinical studies, approximately 17–23% of IPF study participants may die from another cause [27,29]. This lower estimate likely reflects the exclusion of participants with significant comorbid conditions from clinical trial participation. In summary, although mortality may be regarded as the gold standard endpoint for clinical trials in IPF, there are numerous pitfalls that need to be accounted for in the design of any such studies. Mortality studies might best be suited for patients with more advanced disease, patients who are having an acute exacerbation or other groups of patients who are deemed to be at high risk of disease progression.
COMPOSITE ENDPOINTS
Composite endpoints have been gaining favor in many different diseases and offer a number of advantages [30]. In diseases that manifest a multitude of deleterious consequences, they enable different important domains to be captured in one predetermined endpoint. This therefore represents a more global representation of the effects of the drug. Since a wider net is being cast in terms of possible outcomes, more events may be anticipated. The benefits of this are that fewer patients and less time might be needed to complete the study in the context of an event-driven study design.
There are a number of prerequisites for the ideal composite. There should be minimal colinearity between its components; in other words, they should not provide variations of the same information. Ideal composite endpoints should have equal weighting between components. This holds true for how meaningful the endpoint is as well as the numerical distribution of events. Indeed, an inequitable distribution of events can lead to misinterpretation of the study results. In the context of IPF, time to clinical worsening could be captured in a composite. Components to consider for building a composite include: categorical change in the FVC, categorical change in the 6MWT distance, respiratory hospitalization event, change in functional class, transplantation or death. Potential components of composite endpoints are shown in Fig. 1.
FIGURE 1.
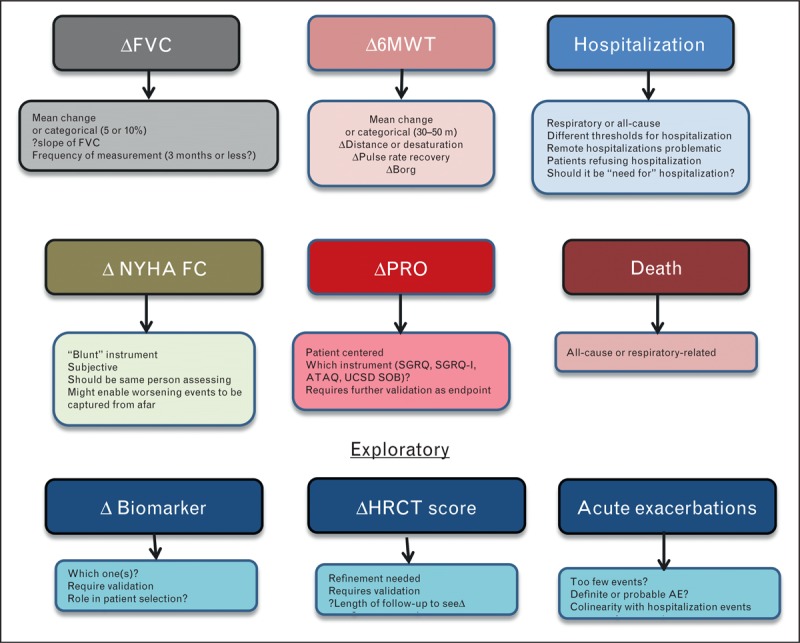
Potential components for a composite endpoint in IPF clinical trials with their attributes and drawbacks. HRCT, high-resolution computed tomography scan; NYHA-FC, New York Heart Association functional class; PVR, pulmonary vascular resistance; SpO2, percentage oxygen saturation recorded by pulse oximeter.
Lung transplantation has its own inherent pitfalls as an endpoint or component of a composite. First, not all patients with IPF will be transplant candidates, and, therefore, the younger patients and those without comorbidities will have another endpoint imposed, which will result in a clinical worsening bias against these patients. Similar to hospitalization, there are many institutional, regional and international differences not only in who is listed, but also donor organ prioritization as well as duration of time on the lung transplant wait list.
Composite endpoints can be made up of physiologic parameters alone. For example, change in the FVC and change in the 6MWT distance appear to reflect different disease domains. Whereas there is inevitably some colinearity between the two, they can also worsen independently. This might be due to the FVC reflecting progressive fibrosis, whereas pulmonary vascular involvement, specifically interceding pulmonary hypertension, might be best captured by a change in the 6MWT.
The FVC and DLco can also be used in combination as an endpoint. Although there is significant colinearity between the two, each can be used to validate smaller decrements in the other, thereby enabling lower thresholds to be set to define disease worsening. These individual criteria may be applied in a synchronous or serial fashion (Fig. 2). A summary of composites that have been used in prior studies is shown in Table 2.
FIGURE 2.
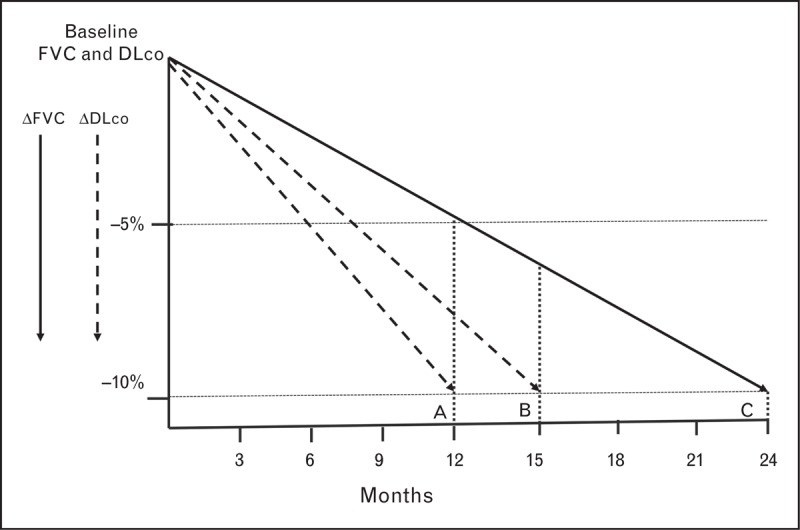
Theoretic construct of how a smaller change in the forced vital capacity validated by a change in the DLco may enable a shorter time interval to disease worsening. (A) A decrease in the FVC of 5% accompanied by a 10% decrement in the DLco results in the theoretic patient meeting the study endpoint at 12 months. (B) A patient who met the FVC criterion of a 5% decrease at 12 months has a 10% decrease in the DLco by 15 months and therefore meets the study endpoint. (C) Assuming a linear rate of decrement, in the absence of consideration of the DLco change, the patient meets the threshold of a 10% decrease in the FVC at 24 months. DLco, single breath diffusing capacity for carbon monoxide; FVC, forced vital capacity.
Table 2.
Constituents of composite endpoints from prior clinical trials in IPF
| Study name (yeara) | Drug | Nomenclature | Death | FVC | DLco | O2 | 6MWT | Respiratory decompensation | Lung transplant | AE |
| (2004) | Interferonδ | Progression-free survivalb | ↓>10% | P(A-a)O2 ↑>5 mmHg | ||||||
| (2010) | Imatinib | Disease progressionb | Yes | ↓>10% | ||||||
| BUILD 3 (2011) | Bosentan | IPF worsening or all-cause mortalityb | All-cause | ↓>10% + ↓>15% | Yes | |||||
| (2011) | Everolimus | Disease progressionb | ↓>10% (or TLC) | ↓>15% | ↓4% SpO2 rest | |||||
| Capacity studies | Pirfenidone | Progression-free survivalc | All-cause | ↓>10% | ↓>15% | |||||
| (2011) | Worsening IPFc | yes | Respiratory Hospitalization | Yes | Yes | |||||
| Panther (2012) | Azathioprine/prednisone/ | Death or hospitalizationc | Yes | All-cause hospitalization | ||||||
| NAC | Death or disease progressionc | Yes | ↓>10% | |||||||
| ACE (2012) | Coumadin | Compositeb | Yes | ↓>10% | All-cause hospitalization nonelective, nonbleeding | |||||
| Artemis-IPF (2013) | Ambrisentan | IPF disease progressionb | Yes | ↓≥10% + ↓≥5% | Respiratory Hospitalization (adjudicated) | |||||
| ↓≥5% + ↓≥15% | ||||||||||
| Music (2013) | Macitentan | IPF worsening or death c | All-cause | ↓>10% + ↓>15% | Yes | |||||
| Ascend (2014) | Pirfenidone | Progression free survivalc | Yes | ↓>10% | ↓50 meters | |||||
6MWT, 6-min walk test; AE, acute exacerbations; FVC, forced vital capacity; IPF, idiopathic pulmonary fibrosis.
aYear of publication or data release.
bPrimary endpoint.
cSecondary endpoint.
COMPUTED TOMOGRAPHY SCORING
With the advent of high-resolution computed tomography (CT) scanners and sophisticated software programs, there are endeavors to now include objective CT-based scoring systems as endpoints for clinical trials in IPF [31]. Previous attempts to characterize image-based disease burden have used more crude subjective scoring systems [32]. Although it makes intuitive sense to score change in the extent of disease via high-resolution imaging, the accuracy, reproducibility and precision of these imaging modalities remain to be determined.
PATIENT-REPORTED OUTCOMES
Patient-reported outcomes (PROs) are important constituents for any study in IPF [33]. There are a number of quality-of-life (QOL) measures that have been employed previously in the context of clinical trials, but most of these have not been IPF-specific instruments, such as the Saint George's Respiratory Questionnaire (SGRQ), the 36-Item Short Form Health Survey (SF-36) and the San Diego shortness of breath questionnaire (SDSQ). There have been attempts to develop IPF-specific instruments that capture the global burden of the disease, and it makes intuitive sense that such instruments should be further validated and employed in future studies [34,35]. In patients with very advanced disease in which stage symptom palliation is the primary objective, the use of such instruments as primary outcome measures might make sense.
BLOOD BIOMARKERS
A biomarker is defined as an objectively measured indicator of a normal or abnormal biological process that may also track progression of disease and/or the response to a therapeutic intervention. There are, however, no validated biomarkers that track progression of disease in IPF, and none have yet been shown to track response to therapy. A number of serum protein biomarkers have been shown to be increased [e.g. Krebs von den Lungen 6 glycoprotein (KL-6), surfactant protein A (SPA), chemokine (C-C motif) ligand 18 (CCL 18), matrix metalloproteinase-7 (MMP-7), intercellular adhesion molecule-1 (ICAM-1), interleukin (IL)-8, vascular cell adhesion molecule-1 (VCAM-1) and S100 calcium binding protein A12 (S100A12)] or decreased (albumin) in IPF and to be predictors of survival [36–39]. However, the reproducibility of these measures and the precision of serial change in predicting subsequent outcomes have yet to be established. Beyond proteins, other serum biomarkers may be useful, including the red cell distribution width, which is readily available on routine complete blood counts and has been shown to correlate with outcomes in IPF [40]. An increased level of brain natriuretic peptide (BNP), which may reflect the presence of underlying pulmonary hypertension or heart failure, has also been shown to be associated with worse outcomes [41–43]. BNP or pro-NT BNP levels may be useful to track as secondary endpoints in IPF studies that target the treatment of IPF-associated pulmonary hypertension. Many other cytokines and chemokines have been recently identified that are elevated as a consequence of the pathogenic cascade in IPF [44,45]. Indeed, proteomic and transcriptomic biomarkers have been identified that appear to be highly predictive of disease progression and mortality, and these may prove to be useful to stratify study participants enrolled in clinical trials [46,47▪]. However, whether serial change in any of these biomarkers ultimately proves to be a useful prognostic biomarker or study endpoint remains to be established.
PULMONARY VASCULAR MARKERS
There is increasing recognition of the role of pulmonary vascular changes in the clinical course of patients with IPF. The prevalence of pulmonary hypertension has been reported as ranging anywhere from 10 to 85% [48]. The presence of pulmonary hypertension, even when mild in nature, has a strong association with subsequent outcomes. Whether treating pulmonary hypertension is a worthwhile approach and which patient phenotype to study with this form of targeted therapy also remain to be determined. Additionally, the presence of pulmonary hypertension or right-ventricular dysfunction might be important to establish during patient selection for any study in order to either stratify patients or identify a group at higher risk for disease progression and mortality.
CONCLUSION
There are many necessary components to the implementation and completion of a successful IPF trial, not least of which is the chosen endpoint. However, consensus on the best IPF endpoint for clinical trials remains elusive. Whereas mortality has come to be commonly regarded as the ‘gold standard’ endpoint in this deadly disease, the implementation and successful completion of such studies are prone to many potential pitfalls. Because of the inherent difficulties and costs of mortality studies, other endpoints need to be considered not only for earlier phase trials, but also for pivotal phase 3 studies. Event-driven studies and composites of events that capture the full spectrum of untoward outcomes represent attractive, pragmatic approaches. Finally, the investigational agent and the patient phenotype being studied are important considerations in choosing the best endpoint for any given study.
Acknowledgements
None.
Conflicts of interest
S.D.N. has consulted for Bayer, Boerhinger-Ingelheim, Gilead Sciences, InterMune and Roche in the field of IPF. He has also received research funding for IPF studies from Boerhinger-Ingelheim, Gilead Sciences, InterMune and Roche.
K.C.M. has consulted for InterMune and Gilead Sciences in the field of IPF. He has also received funding for IPF clinical trials from InterMune, Boerhinger-Ingelheim, Actelion, Fibrogen, Bristol Myers Squibb, Wyeth, and Roche.
REFERENCES AND RECOMMENDED READING
Papers of particular interest, published within the annual period of review, have been highlighted as:
▪ of special interest
▪▪ of outstanding interest
Supplementary Material
REFERENCES
- 1.du Bois RM. Strategies for treating idiopathic pulmonary fibrosis. Nat Rev Drug Discov 2010; 9:129–140 [DOI] [PubMed] [Google Scholar]
- 2.Raghu G, Collard HR, Anstrom KJ, et al. Idiopathic pulmonary fibrosis: clinically meaningful primary endpoints in phase 3 clinical trials. Am J Respir Crit Care Med 2012; 185:1044–1048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Du Bois RM, Nathan SD, Richeldi L, et al. Idiopathic pulmonary fibrosis: lung function is a clinically meaningful endpoint for phase 3 trials. Am J Respir Crit Care Med 2012; 186:712–715 [DOI] [PubMed] [Google Scholar]
- 4.Wells AU. Forced vital capacity as a primary endpoint in idiopathic pulmonary fibrosis treatment trials: making a silk purse from a sow's ear. Thorax 2013; 68:309–310 [DOI] [PubMed] [Google Scholar]
- 5▪.Saketkoo LA, Mittoo S, Huscher D, et al. Connective tissue disease related interstitial lung diseases and idiopathic pulmonary fibrosis: provisional core sets of domains and instruments for use in clinical trials. Thorax 2014; 69:428–436 [DOI] [PMC free article] [PubMed] [Google Scholar]; The authors of this article employed the Delphi process to engage experts in the field as well as patients to define and develop a consensus on which outcomes measures were felt to be most important and valid in IPF (and connective tissue disease associated interstitial lung disease) clinical trials.
- 6.Nathan SD, Shlobin OA, Weir N, et al. Long-term course and prognosis of idiopathic pulmonary fibrosis in the modern era. Chest 2011; 140:221–229 [DOI] [PubMed] [Google Scholar]
- 7.Zappala CJ, Latsi PI, Nicholson AG, et al. Marginal decline in forced vital capacity is associated with a poor outcome in idiopathic pulmonary fibrosis. Eur Respir J 2010; 35:830–836 [DOI] [PubMed] [Google Scholar]
- 8.du Bois RM, Weycker D, Albera C, et al. Forced vital capacity in patients with idiopathic pulmonary fibrosis: test properties and minimal clinically important difference. Am J Respir Crit Care Med 2011; 184:1382–1389 [DOI] [PubMed] [Google Scholar]
- 9.Richeldi L, Ryerson CJ, Lee JS, et al. Relative versus absolute change in forced vital capacity in idiopathic pulmonary fibrosis. Thorax 2012; 67:407–411 [DOI] [PubMed] [Google Scholar]
- 10.Richeldi L, du Bois RM, Raghu G, et al. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med 2014; 370:2071–2082 [DOI] [PubMed] [Google Scholar]
- 11.Martinez FJ, Safrin S, Weycker D, et al. The clinical course of patients with idiopathic pulmonary fibrosis. Ann Intern Med 2005; 142:963–967 [DOI] [PubMed] [Google Scholar]
- 12.King TE, Bradford WZ, Castro-Bernardini S, et al. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N Engl J Med 2014; 370:2083–2092 [DOI] [PubMed] [Google Scholar]
- 13.Noble PW, Albera C, Bradford WZ, et al. Pirfenidone in patients with idiopathic pulmonary fibrosis (CAPACITY): two randomized trials. Lancet 2011; 377:1760–1769 [DOI] [PubMed] [Google Scholar]
- 14.Corte TJ, Wort SJ, MacDonald PS, et al. Pulmonary function vascular index predicts prognosis in idiopathic interstitial pneumonia. Respirology 2012; 17:674–680 [DOI] [PubMed] [Google Scholar]
- 15.Lama VN, Flaherty KR, Toews GB, et al. Prognostic value of desaturation during a 6-min walk test in idiopathic interstitial pneumonia. Am J Respir Crit Care Med 2003; 168:1084–1090 [DOI] [PubMed] [Google Scholar]
- 16.Lederer DJ, Arcasoy SM, Wilt JS, et al. Six-minute-walk distance predicts waiting list survival in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2006; 174:659–664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Caminati A, Bianchi A, Cassandro R, et al. Walking distance on 6-MWT is a prognostic factor in idiopathic pulmonary fibrosis. Res Med 2009; 103:117–123 [DOI] [PubMed] [Google Scholar]
- 18.Lettieri CJ, Browning RF, Shorr AF, et al. The distance saturation product: a novel measure for mortality prediction in idiopathic pulmonary fibrosis. Respir Med 2006; 100:1734–1741 [DOI] [PubMed] [Google Scholar]
- 19.Du Bois RM, Weycker D, Albera C, et al. Six-minute-walk test in idiopathic pulmonary fibrosis: test validation and minimal clinically important difference. Am J Respir Crit Care Med 2011; 183:1231–1237 [DOI] [PubMed] [Google Scholar]
- 20.Swigris JJ, Wamboldt FS, Behr J, et al. The 6 min walk in idiopathic pulmonary fibrosis: longitudinal changes and minimum important difference. Thorax 2010; 65:173–217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Swigris JJ, Swick J, Wamboldt FS, et al. Heart rate recovery after 6-minute walk test predicts survival in patients with idiopathic pulmonary fibrosis. Chest 2009; 136:841–848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Swigris JJ, Olson A, Shlobin OA, et al. Heart rate recovery after 6MWT predicts pulmonary hypertension in patients with IPF. Respirology 2011; 16:439–445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dolmage TE, Hill K, Evans RA, Goldstein RS. Has my patient responded? Interpreting clinical measurements such as the 6-min-walk test. Am J Respir Crit Care Med 2011; 184:642–646 [DOI] [PubMed] [Google Scholar]
- 24▪.Collard HR, Yow E, Richeldi L, et al. Suspected acute exacerbation of idiopathic pulmonary fibrosis as an outcome measure in clinical trials. Respir Res 2013; 14:73. [DOI] [PMC free article] [PubMed] [Google Scholar]; In this retrospective evaluation of the STEP-IPF study the authors describe that suspected acute exacerbations are indistinguishable from definite acute exacerbations of IPF and therefore inclusion of both are recommended as a clinical trial endpoint.
- 25.Noth I, Anstrom KJ, Calvert SB, et al. A placebo-controlled randomized trial of warfarin in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2012; 186:88–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.The Idiopathic Pulmonary Fibrosis Clinical Research Network Prednisone, azathioprine and N-acetylcysteine for pulmonary fibrosis. N Engl J Med 2012; 366:1968–1977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ley B, Collard HR, King TE. Clinical course and prediction of survival in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2011; 183:431–440 [DOI] [PubMed] [Google Scholar]
- 28▪▪.King TE, Jr, Albera C, Bradford WZ, et al. All-cause mortality rate in patients with idiopathic pulmonary fibrosis: implications for the design and execution of clinical trials. Am J Respir Crit Care Med 2014; 189:825–831 [DOI] [PubMed] [Google Scholar]; The authors collate the mortality rate of IPF patients with mild-to-moderate disease from three prior randomized controlled studies. They then performed power calculations to estimate the number of patients required for a clinical trial with all-cause mortality as the primary endpoint. They conclude that the size, duration and cost of such studies would likely be prohibitive.
- 29.Panos R. Clinical deterioration in patients with idiopathic pulmonary fibrosis: causes and assessment. Am J Med 1990; 88:396–404 [DOI] [PubMed] [Google Scholar]
- 30.Kaul S, Diamond GA. Trial and error: how to avoid commonly encountered limitations of published clinical trials. J Am Coll Cardiol 2010; 55:415–427 [DOI] [PubMed] [Google Scholar]
- 31.Kim HJ, Brown MS, Elashoff R, et al. Quantitative texture-based assessment of one-year changes in fibrotic reticular patterns on HRCT in scleroderma lung disease treated with oral cyclophosphamide. Eur Radiol 2011; 21:2455–2465 [DOI] [PubMed] [Google Scholar]
- 32.Kazerooni EA, Martinez FJ, Flint A, et al. Thin-section CT obtained at 10-mm increments versus limited three-level thin-section CT for idiopathic pulmonary fibrosis: correlation with pathologic scoring. AJR 1997; 169:977–983 [DOI] [PubMed] [Google Scholar]
- 33.Swigris JJ, Fairclough D. Patient-reported outcomes in idiopathic pulmonary fibrosis research. Chest 2012; 142:291–297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yorke J, Jones PW, Swigris JJ. Development and validity testing of an IPF-specific version of the St George's Respiratory Questionnaire. Thorax 2010; 65:921–926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Swigris JJ, Wilson SR, Green KE, et al. Development of the ATAQ-IPF: a tool to assess quality of life in IPF. Health Qual Life Outcomes 2010; 8:77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zisman DA, Kawut SM, Lederer DJ, et al. Serum albumen concentration and waiting list mortality in idiopathic interstitial pneumonia. Chest 2009; 135:929–935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yokoyama A, Kondo K, Nakajima M, et al. Prognostic value of circulating kl-6 in idiopathic pulmonary fibrosis. Respirology 2006; 11:164–168 [DOI] [PubMed] [Google Scholar]
- 38.Kinder BW, Brown KK, McCormack FX, et al. Serum surfactant protein-A is a strong predictor of early mortality in idiopathic pulmonary fibrosis. Chest 2009; 135:1557–1563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Richards TJ, Kaminski N, Baribaud F, et al. Peripheral blood proteins predict mortality in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2012; 185:67–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nathan SD, Reffett T, Brown AW, et al. The red cell distribution width as a prognostic indicator in IPF. Chest 2013; 143:1692–1698 [DOI] [PubMed] [Google Scholar]
- 41.Song JW, Song JK, Kim DS. Echocardiography and brain natriuretic peptide as prognostic indicators in idiopathic pulmonary fibrosis. Res Med 2009; 103:180–186 [DOI] [PubMed] [Google Scholar]
- 42.Leuchte HH, Baumgartner RA, Nounou ME, et al. Brain natruiretic peptide is a prognostic parameter in chronic lung disease. Am J Respir Crit Care Med 2006; 173:744–750 [DOI] [PubMed] [Google Scholar]
- 43.Corte TJ, Wort SJ, Gatzoulis MA, et al. Elevated brain natriuretic peptide predicts mortality in interstitial lung disease. Eur Respir J 2010; 36:819–825 [DOI] [PubMed] [Google Scholar]
- 44.Travis WD, Costabel U, Hansell DM, et al. ATS/ERS Committee on Idiopathic Interstitial Pneumonias. An official American Thoracic Society/European Respiratory Society statement: update of the international multidisciplinary classification of the idiopathic interstitial pneumonias. Am J Respir Crit Care Med 2013; 188:733–748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Martinez FJ. Meyer KC, Nathan SD. Idiopathic pulmonary fibrosis clinical trials: evolving concepts. Idiopathic pulmonary fibrosis: a comprehensive clinical guide. Respiratory medicine 9. New York:Springer; 2014. 403–426 [Google Scholar]
- 46.Richards TJ, Kaminski N, Baribaud F, et al. Peripheral blood proteins predict mortality in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2012; 185:67–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47▪.Herazo-Maya JD, Noth I, Duncan SR, et al. Peripheral blood mononuclear cell gene expression profiles predict poor outcome in idiopathic pulmonary fibrosis. Sci Transl Med 2013; 5:205ra136. [DOI] [PMC free article] [PubMed] [Google Scholar]; The authors evaluated gene expression of peripheral blood mononuclear cells and evaluated the performance characteristics of these in predicting transplant-free survival. They identified four genes that appeared to be predictive of outcomes.
- 48.Nathan SD, Noble PW, Tuder RM. Idiopathic pulmonary fibrosis and pulmonary hypertension: connecting the dots. Am J Respir Crit Care Med 2007; 175:875–880 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.