

Abstract
This study aimed to examine the contributions of brain-derived neurotrophic factor (BDNF) at the injury site toward neuroma formation and nerve regeneration after inferior alveolar nerve transection. Histological analysis confirmed neuroma formation at 2 weeks after complete transection of the inferior alveolar nerve. A local administration of an antibody to BDNF inhibited connective tissue proliferation at the injury site and promoted nerve fiber integrity. Fluorogold labeling showed a significantly higher number of labeled cells in the trigeminal ganglion in the anti-BDNF-treated group compared with the vehicle control group. In-situ hybridization histochemistry showed intense signals for tropomyosin receptor kinase B mRNA in the area of the injury site containing fibrous or granular tissue in the anti-BDNF-treated group. In contrast, these signals were close to the detection limit in the area of the perineurium in intact nerve trunks, indicating that the signals were expressed by fibroblasts within the connective tissue. These findings suggest that antagonization of endogenous BDNF induced by nerve injury reduces neuroma formation, without inhibiting damaged axon regeneration.
Keywords: brain-derived neurotrophic factor, inferior alveolar nerve, nerve injury, neuroma, tropomyosin receptor kinase B
Introduction
The inferior alveolar nerve (IAN) is a branch of the mandibular nerve, which is the third branch of the trigeminal nerve, and consists of sensory fibers that innervate the mandibular (lower jaw) teeth. The IAN is sometimes injured during dental treatments and/or oral surgeries, leading to an unpleasant abnormal sensation called dysesthesia that can persist long after the injury 1–4. During surgical procedures such as IAN or lingual nerve surgeries, neuroma formation is sometimes observed at the injury site of the nerve 5. Thus, it is conceivable that trauma can trigger neuroma formation 2,3,6–8, and this notion is supported by the finding that collected neuromas from human lingual nerves with pain show expression of Nav1.8 sodium channels 9. However, the factors that contribute toward neuroma formation following nerve injury remain unclear 4. Thus, the mechanism of such neuroma formation needs to be clarified for the development of a new prophylactic method for peripheral nerve damage.
In animal studies, it has been reported that peripheral nerve injury induces brain-derived neurotrophic factor (BDNF) expression in the dorsal root ganglion 10. In addition, endogenous BDNF promotes neurite extension in injured sensory neurons, meaning that BDNF has the potential to regenerate injured neurons 11,12. In contrast, exogenous BDNF has been shown to have variable effects on the regeneration of injured nerves, ranging from dose-dependent inhibition to facilitation 13–16. These findings suggest the possibility that nerve injury induces BDNF expression upon regeneration of injured sensory neurons, resulting in neuroma formation. Therefore, it is important to elucidate the effects of endogenous BDNF expression triggered by nerve injury on neuroma formation. In this study, we examined the possible involvement of BDNF in neuroma formation by investigating the effects of local application of an anti-BDNF antibody at the injury site.
Materials and methods
Male Sprague–Dawley rats weighing 160–280 g (Charles River Laboratories Japan Inc., Yokohama, Japan) were used. The animal experiments were approved by the Niigata University Institutional Animal Use and Care Committee (approval number #42). The animals were assigned randomly to three groups: naive (no nerve transection; n=6); anti-BDNF antibody treatment (n=18); and vehicle control (n=13). After anesthesia with 8% chloral hydrate (400 mg/kg, intraperitoneal), a small incision was made to the skin of the left cheek and the surface of the mandible was removed to expose the IAN. The IAN was transected completely with a microscissor at 7 mm proximal to the angle of the alveolar canal and then immediately brought back to the original position for the animals in the anti-BDNF-treated and vehicle control groups. Following the nerve transection, the animals in the anti-BDNF-treated group received a 1 μl injection of anti-BDNF antibody (1 mg/ml, Ab6201; Abcam, Cambridge, UK) into the transected IAN lesion using a Hamilton syringe (Hamilton Company, Reno, Nevada, USA). The vehicle control animals received a 1 μl injection of physiological saline. When the anti-BDNF antibody or physiological saline was injected, Spongel (Astellas Pharma Inc., Tokyo, Japan) was used to prevent any spreading of the liquid from the injection site and bone wax (Angiotech Pharmaceuticals Inc., Vancouver, Canada) was used to cover the lesion of the mandible. The animals in the naive group received an operation for IAN exposure without nerve resection and were euthanized at 2 weeks after the surgery.
For histopathological examination of the nerve regeneration processes, the animals were deeply anesthetized and perfused transcardially with 4% paraformaldehyde in 0.1 M phosphate buffer. The mandibles, including the IAN, from the anti-BDNF-treated (n=18), vehicle control (n=13), and naive (n=6) groups were decalcified with 5% formic acid and embedded in paraffin. Sections (7 μm thickness) were subjected to azocarmine and aniline blue (Azan) trichrome staining to distinguish fibrous tissue and collagen fibers from cellular elements. Immunohistochemistry for a neuronal marker, protein gene product 9.5 (PGP 9.5), was used to identify the neural elements. The sections were initially incubated with a rabbit polyclonal antiserum against human PGP 9.5 (RA-95101, 1 : 2000; Ultraclone Ltd, Wellow, Isle of Wight, UK), followed by biotinylated goat anti-rabbit IgG (JIR711-096-152, 1000; Vector Laboratories, Burlingame, California, USA) and peroxidase-conjugated avidin (ABC kit; Vector Laboratories) at room temperature. The immunoreactive sites were visualized by incubation in 0.05 M Tris-HCl buffer (pH 7.6) containing 0.04% 3,3′-diaminobenzidine and 0.03% hydrogen peroxide. The immunostained sections were counterstained with 0.03% methylene blue. Photomicrographs were taken using a color digital camera (AxioCam HRc; Carl Zeiss, Oberkochen, Germany) mounted on an Axio Imager M1 microscope (Carl Zeiss).
For fluorogold (FG) tracing, five rats were selected randomly from each group. Under anesthesia with sevoflurane (Mylan Pharmaceutical Co. Ltd, Osaka, Japan), each rat received a subcutaneous injection of 4% FG solution (10 μl of hydroxystilbamidine; Biotium, Hayward, California, USA) into the skin of the mental nerve region at a depth of 5 mm from the lower lip surface, as described previously 17. At 1 day after the FG injection, the animals were perfusion fixed as described above. The removed trigeminal ganglion was postfixed overnight in the same fixative, and then stored in 30% sucrose for 2 days. Cryostat sections of the trigeminal ganglion (20 μm) were directly mounted using Vectashield mounting medium containing propidium iodide (PI) (Vector Laboratories) and examined at the root of the third branch of the trigeminal ganglion. To determine the proportion of regenerated trigeminal ganglion neurons among the surviving neurons, the total numbers of both PI-labeled and FG-labeled neurons were counted under a fluorescence microscope (three sections for each animal). The ratios of FG-labeled neurons to all neurons in the trigeminal ganglion were compared among the groups and statistical significance was considered at the P value of less than 0.05 level (one-way analysis of variance).
In-situ hybridization (ISH) histochemistry was performed to investigate the presence of the tropomyosin receptor kinase B (trkB) receptor mRNA at the transection site. Paraffin-embedded sections were processed using a commercially available mRNA ISH kit (RNAscope 2.0 FFPE Assay Red; Advanced Cell Diagnostics, Hayward, California, USA) in accordance with the manufacturer’s instructions. A rat trkB probe (MM-NTRK2 NP_036; Advanced Cell Diagnostics) was used and the RNA transcripts were visualized using Fast red (Advanced Cell Diagnostics). Images were captured using a high-resolution digital camera (AxioCam HRc) mounted on a microscope and saved in a computer. In each specimen, four to five sections per animal were taken from across the width of the nerve injury part to quantify the counts of the trkB signal pixels (0.17×0.17 μm). The signals were analyzed using the free software Image-J (http://imagej.nih.gov/ij/) and the mean value was calculated as representative data for each specimen.
Statistical comparisons were carried out using Sigmaplot ver.11.0 (Systat Software Inc., San Jose, California, USA). The ratios of FG-labeled neurons to all neurons in the trigeminal ganglion were compared among the groups using one-way analysis of variance, whereas the counts of trkB receptor mRNA signals were compared between the anti-BDNF-treated group (n=3) and the vehicle control group (n=3) using Student’s t-test. A significant difference was considered at the P value less than 0.05 level.
Results
IAN transection induced the formation of an enlarged complex composed of scar tissue and neuroma at the injury site at 2 weeks postoperatively. Azan staining of the injured area showed that a large amount of connective tissue, rich in collagen fibers, had proliferated to invade the region between the proximal and distal stumps, indicating that the enlarged tissue was equivalent to a neuroma (Fig. 1). The injured animals showed discontinuous PGP 9.5-positive nerve fibers and these PGP 9.5-positive nerve fibers ran for short distances in various directions to form a neuroma at the distal site. Local administration of the anti-BDNF antibody markedly inhibited the proliferation of connective tissue at the injury site (Fig. 1). The intact IAN passed through the inferior alveolar canal in the naive group and immunohistochemistry for PGP 9.5 also indicated nerve fiber integrity in the anti-BDNF-treated group.
Fig. 1.
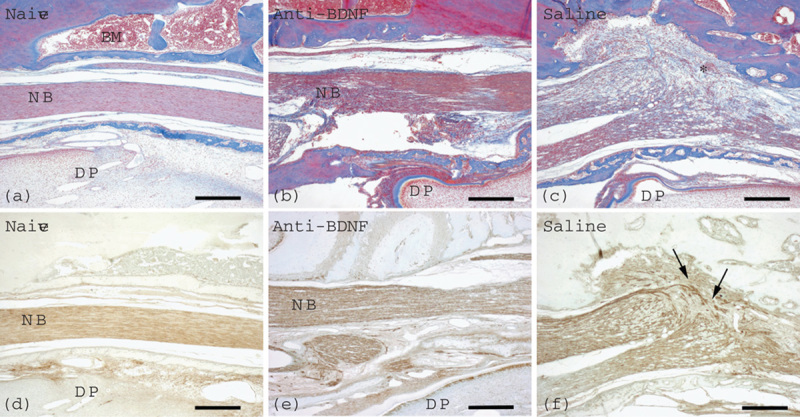
Effects of local application of an anti-BDNF antibody or physiological saline immediately after IAN transection on neuroma formation. Azan staining (a–c) and immunohistochemistry for PGP 9.5 (d–f) are presented. Samples were obtained at 2 weeks after injection of an anti-BDNF antibody (b, e) or physiological saline (c, f). In the naive group (a, d), the IAN bundle shows no damage and nerve fiber integrity as confirmed by Azan staining (a) and PGP 9.5 immunostaining (d). Neither neuroma formation nor proliferation of connective tissue is recognizable in the anti-BDNF-treated group (b, e), whereas neuroma formation with connective tissue proliferation (asterisk) is found in the vehicle control group (c, f). PGP 9.5 immunostaining shows disorganization of nerve fibers (arrows) in the vehicle control group (f), in contrast to the nerve fiber integrity in the anti-BDNF-treated group (e). Naive, anti-BDNF, and saline indicate the groups with no nerve transection, anti-BDNF antibody treatment, and nerve transection with vehicle control treatment, respectively. BDNF, brain-derived neurotrophic factor; BM, bone marrow; DP, dental pulp; IAN, inferior alveolar nerve; NB, nerve bundle; PGP 9.5, protein gene product 9.5. Scale bars=200 μm. The right and left sides in each picture indicate the proximal and the distal directions of the IAN, respectively.
PI staining identified neurons in the trigeminal ganglion of all groups (Fig. 2). Application of FG to the mental region enabled visualization and enumeration of the numbers of trigeminal ganglion neurons that had regenerated their axons. Many FG-labeled neurons were localized in the root of the third branch of the trigeminal nerve in the naive group. No labeled neurons were observed in the root of the second branch in any of the groups. At postoperative week 2, the number of FG-labeled neurons observed in the anti-BDNF-treated group was greater than that in the vehicle control group. The ratios of FG-labeled neurons to all neurons in the trigeminal ganglion are shown in Fig. 2. The ratios were 96.2±2.7% in the naive group (n=532 neurons), 74.9±4.9% in the vehicle control group (n=417 neurons), and 86.7±6.5% in the anti-BDNF-treated group (n=416 neurons). The percentage of FG-labeled neurons in the naive group was significantly higher than those in the anti-BDNF-treated group (Holm–Sidak method, P=0.01) and vehicle control (saline) group (Holm–Sidak method, P<0.001). A significant difference was also observed in the percentage of FG-labeled neurons between the vehicle control group and the anti-BDNF-treated group (Holm–Sidak method, P=0.003).
Fig. 2.
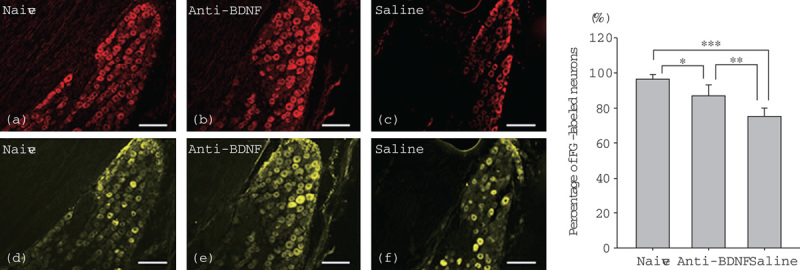
Changes in the expression of FG-positive cells after FG administration at the distal site after IAN transection. Images of PI staining in red (a–c) and FG staining in yellow (d–f) in the trigeminal ganglion of the naive (a, d), anti-BDNF-treated (b, e), and vehicle control (c, f) groups are presented. In the trigeminal ganglion of the naive group, almost all neurons are labeled with FG (d). Note the difference in the number of FG-labeled trigeminal neurons between the anti-BDNF-treated group (e) and the saline (vehicle control) group (f) (scale bars=100 μm). The right graph shows the percentages of FG-labeled neurons in the trigeminal ganglion of the three groups (*P=0.01, **P=0.003, and ***P<0.001, significant difference between the linked data by the Holm–Sidak method). BDNF, brain-derived neurotrophic factor; FG, fluorogold; IAN, inferior alveolar nerve; PI, propidium iodide.
ISH histochemistry showed a few isolated signals for trkB mRNA, which were close to the detection limit, in the nerve trunks of the naive group. The mean counts of trkB mRNA signals in the region corresponding to the injury site in the vehicle control group and the anti-BDNF-treated group were 57.0±5.6 and 340.7±184.9, respectively. A significant difference was observed between the two groups (Student’s t-test, P<0.05) (Fig. 3).
Fig. 3.
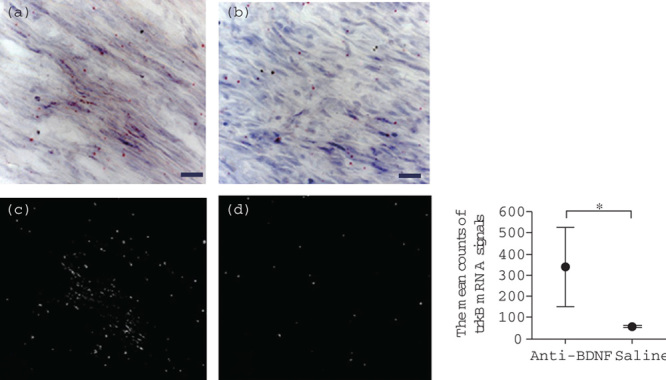
Effects of anti-BDNF administration on trkB mRNA expression in the trigeminal ganglion after IAN transection. Photographs showing the expression of trkB mRNA at postoperative week 2 in the anti-BDNF-treated (a) and saline (vehicle) control (b) groups evaluated by ISH histochemistry are shown. A few isolated signals for trkB mRNA are observed in the regenerating nerve fibers in the anti-BDNF-treated group. The red dots indicate trkB mRNA signals (scale bars=20 μm). (c and d) taken in the dark field indicate the signals of trkB mRNA in the anti-BDNF and the saline groups, respectively. Quantitative analysis obtained from the dark field images shows a comparison of the mean counts of the trkB mRNA signals between these groups (the right panel). These mean counts at the injury site in the saline (vehicle) control and the anti-BDNF-treated groups show 57.0±5.6 and 340.7±184.9, respectively. A significant difference exists between the two groups (Student’s t-test, *P<0.05). BDNF, brain-derived neurotrophic factor; IAN, inferior alveolar nerve; ISH, in-situ hybridization; trkB, tropomyosin receptor kinase B.
Discussion
In this study, we examined the possible contribution of BDNF toward neuroma formation caused by peripheral nerve injury. Local application of an anti-BDNF antibody clearly inhibited neuroma formation after nerve transection, whereas the vehicle control group showed rich production of fibrous tissue and neuroma formation. Therefore, it was concluded that neutralization of BDNF through local administration of an anti-BDNF antibody at the injury site exerted inhibitory effects on fibrous tissue generation as well as facilitative effects on nerve regeneration. We further found lower expression of BDNF receptor trkB mRNA at the injury site in the vehicle control group compared with the BDNF-neutralized animals. These findings indicate a contribution of endogenous BDNF toward neuroma formation following nerve injury.
Neurotrophic factors exert facilitative effects on neurite outgrowth during peripheral nerve regeneration, causing BDNF upregulation in neurons after axotomy and in denervated Schwann cells at the injury site 18,19. This localized gradient of BDNF leads to neurite sprouting at the injury site, resulting in radial proliferation of neurites from the injury site. TrkB-deficient mice show neuroma generation after peripheral nerve transection, suggesting an accumulative role of BDNF receptors in neuroma formation 15. These findings indicate that neurite proliferation can influence neuroma generation triggered by peripherally produced BDNF.
Several studies have investigated methods for neuroma prevention, including local and endogenous antagonization of neurotrophins and cytokines 20–22. However, the antagonization of neurotrophins showed opposite results after peripheral nerve injury 20,21. A low dose of exogenous BDNF was reported to have a facilitative effect on axonal regeneration, in contrast to the inhibitory effect observed after administration at high doses 13. The present findings using FG labeling suggested that local application of an anti-BDNF antibody promoted regeneration of the injured IAN. This indicates that antagonization of BDNF was equivalent to a residual low dose of endogenous BDNF. This also means that even after neutralizing the BDNF action, there remains some BDNF effect on the survival and/or elongation of the axon in the damaged nerve, further facilitating the effects on connective tissue proliferation at the injury site. These findings are consistent with a previous report 15, in which antagonization of trkB led to accumulation of BDNF and marked increases in the levels of inflammatory cells and connective tissue proliferation.
ISH histochemistry indicated that trkB mRNA was localized at the injury site. Although it was difficult to identify the cell types expressing the mRNA for trkB, the cells were situated in an area of the perineurium in the vicinity of intact nerve fibers. These observations indicated that the signals corresponded to the area of fibroblasts encountered within the connective tissue, suggesting that the fibroblasts synthesize, secrete, and bind BDNF in an autocrine/paracrine manner. Therefore, the anti-BDNF antibody antagonized BDNF produced by the fibroblasts in the perineurium by trkB, thereby inhibiting the proliferation of connective tissue. This effect may be explained by the involvement of fibroblast growth factor-2 and fibroblast growth factor receptor, both of which are constitutively expressed in peripheral nerves and upregulated in the proximal and distal nerve stumps after peripheral nerve lesion formation 23. These observations suggest that their main sources are fibroblasts, Schwann cells, and macrophages that can invade the injury site. In addition to these peripheral actions, another possibility for the source of locally existing BDNF is the trigeminal ganglion, on the basis of a previous report suggesting that peripheral inflammation upregulates BDNF in the dorsal root ganglia 24. In contrast, it was reported that a decrease in the expression of trkB receptors occurs in the trigeminal ganglion after primary afferent axotomy 25. Given that exhaustion of trkB receptors by binding endogenous BDNF results in a decrease in trkB receptor expression at the lesion site, this further supports our ISH results, showing decreased trkB receptor expression in the vehicle control group compared with the anti-BDNF-treated group.
Conclusion
Antagonization by local application of an anti-BDNF antibody inhibited the proliferation of connective tissue containing collagen fibers at the injury site, resulting in the prevention of neuroma formation, but did not suppress the regeneration of injured axons. Therefore, we propose the hypothesis that endogenously expressed BDNF, which is triggered immediately by nerve injury at the injured site, facilitates neuroma formation, and may subsequently lead to intractable chronic pain after nerve injury.
Acknowledgements
The authors are grateful to Dr F. Harada (Division of Oral Anatomy, Niigata University Graduate School of Medical and Dental Sciences) for her assistance with analysis of the ISH data.
This study was supported by a Scientific Grant from the Japan Society for the Promotion of Science (23390401).
Conflicts of interest
There are no conflicts of interest.
References
- 1.Arribas-García I, Alcalá-Galiano A, Gutiérrez R, Montalvo-Moreno JJ. Traumatic neuroma of the inferior alveolar nerve: a case report. Med Oral Patol Oral Cir Bucal 2008;13:E-186–E-188 [PubMed] [Google Scholar]
- 2.Katre C, Triantafyllou A, Shaw RJ, Brown JS. Inferior alveolar nerve damage caused by bone wax in third molar surgery. Int J Oral Maxillofac Surg 2010; 39:511–513 [DOI] [PubMed] [Google Scholar]
- 3.Tay ABG, Zuniga JR. Clinical characteristics of trigeminal nerve injury referrals to a university centre. Int J Oral Maxillofac Surg 2007;36:922–927 [DOI] [PubMed] [Google Scholar]
- 4.Zimmermann M. Pathobiology of neuropathic pain. Eur J Pharmacol 2001;429:23–37 [DOI] [PubMed] [Google Scholar]
- 5.Robinson PP, Loescher AR, Yates JM, Smith KG. Current management of damage to the inferior alveolar and lingual nerves as a result of removal of third molars. Br J O Maxillofac Surg 2004;42:285–292 [DOI] [PubMed] [Google Scholar]
- 6.Juodzbalys G, Wang HL, Sabalys G. Injury of the inferior alveolar nerve during implant placement: a literature review. J Oral Maxillofac Res 2011;2:e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Renton T, Yilmaz Z, Gaballah K. Evaluation of trigeminal nerve injuries in relation to third molar surgery in a prospective patient cohort. Recommendations for prevention. Int J Oral Maxillofac Surg 2012;41:1509–1518 [DOI] [PubMed] [Google Scholar]
- 8.Seo K, Tanaka Y, Terumitsu M, Someya G. Characterization of different paresthesias following orthognathic surgery of the mandible. J Oral Maxillofac Surg 2005;63:298–303 [DOI] [PubMed] [Google Scholar]
- 9.Bird EV, Christmas CR, Loescher AR, Smith KG, Robinson PP, Black JA, et al. Correlation of Nav1.8 and Nav1.9 sodium channel expression with neuropathic pain in human subjects with lingual nerve neuromas. Mol Pain 2013;9:52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Karchewski LA, Gratto KA, Wetmore C, Verge VM. Dynamic patterns of BDNF expression in injured sensory neurons: differential modulation by NGF and NT-3. Eur J Neurosci 2002;16:1449–1462 [DOI] [PubMed] [Google Scholar]
- 11.Fukuoka T, Kondo E, Dai Y, Hashimoto N, Noguchi K. Brain-derived neurotrophic factor increases in the uninjured dorsal root ganglion neurons in selective spinal nerve ligation model. J Neurosci 2001;21:4891–4900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Geremia NM, Pettersson LM, Hasmatali JC, Hryciw T, Danielsen N, Schreyer DJ, Verge VM. Endogenous BDNF regulates induction of intrinsic neuronal growth programs in injured sensory neurons. Exp Neurol 2010; 223:128–142 [DOI] [PubMed] [Google Scholar]
- 13.Boyd JG, Gordon T. A dose-dependent facilitation and inhibition of peripheral nerve regeneration by brain-derived neurotrophic factor. Eur J Neurosci 2002;15:613–626 [DOI] [PubMed] [Google Scholar]
- 14.Kerr BJ, Bradbury EJ, Bennett DL, Trivedi PM, Dassan P, French J, et al. Brain-derived neurotrophic factor modulates nociceptive sensory inputs and NMDA-evoked responses in the rat spinal cord. J Neurosci 1999;19:5138–5148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kotulska K, Larysz-Brysz M, Marcol W, Grajkowska W, Jóźwiak S, Lewin-Kowalik J. The role of trkB receptor in the formation of post-traumatic neuroma. Folia Neuropathol 2006;44:221–227 [PubMed] [Google Scholar]
- 16.Marcol W, Kotulska K, Larysz-Brysz M, Kowalik JL. BDNF contributes to animal model neuropathic pain after peripheral nerve transection. Neurosurg Rev 2007;30:235–243 [DOI] [PubMed] [Google Scholar]
- 17.Saito K, Hitomi S, Suzuki I, Masuda Y, Kitagawa J, Tsuboi Y, et al. Modulation of trigeminal spinal subnucleus caudalis neuronal activity following regeneration of transected inferior alveolar nerve in rats. J Neurophysiol 2008;99:2251–2263 [DOI] [PubMed] [Google Scholar]
- 18.Gordon T. The role of neurotrophic factors in nerve regeneration. Neurosurg Focus 2009;26:E3. [DOI] [PubMed] [Google Scholar]
- 19.Thompson SW, Bennett DL, Bradbury EJ, McMahon SB. Brain-derived neurotrophic factor is an endogenous modulator of nociceptive responses in the spinal cord. Proc Natl Acad Sci USA 1999;96:7714–7718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Atkins S, Smith KG, Loescher AR, Boissonade FM, Ferguson MWJ, Robinson PP. The effect of antibodies to TGF-beta1 and TGF-beta2 at a site of sciatic nerve repair. J Peripher Nerv Syst 2006;11:286–293 [DOI] [PubMed] [Google Scholar]
- 21.Kryger GS, Kryger Z, Zhang F, Shelton DL, Lineaweaver WC, Buncke HJ. Nerve growth factor inhibition prevents traumatic neuroma formation in the rat. J Hand Surg Am 2001;26:635–644 [DOI] [PubMed] [Google Scholar]
- 22.Mantyh PW, Koltzenburg M, Mendell LM, Tive L, Shelton DL. Antagonism of nerve growth factor-TrkA signaling and the relief of pain. Anesthesiology 2011;115:189–204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Grothe C, Nikkhah G. The role of basic fibroblast growth factor in peripheral nerve regeneration. Anat Embryol (Berl) 2001;204:171–177 [DOI] [PubMed] [Google Scholar]
- 24.Lin YT, Ro LS, Wang HL, Chen JC. Up-regulation of dorsal root ganglia BDNF and trkB receptor in inflammatory pain: an in vivo and in vitro study. J Neuroinflamation 2011;8:126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bergman E, Fundin BT, Ulfhake B. Effects of aging and axotomy on the expression of neurotrophin receptors in primary sensory neurons. J Comp Neurol 1999;410:368–386 [DOI] [PubMed] [Google Scholar]